

Abstract
The transcription factor and tumor suppressor protein p53 is frequently inactivated in human cancers. In many cases, p53 gene mutations result in high levels of inactive, full-length p53 protein with one amino acid change in the core domain that recognizes p53 DNA-binding sites. The ability to endow function to mutated p53 proteins would dramatically improve cancer therapy, because it would reactivate a central apoptotic pathway. By using genetic strategies and p53 assays in yeast and mammalian cells, we identified a global suppressor motif involving codons 235, 239, and 240. These intragenic suppressor mutations, either alone or in combination, restored function to 16 of 30 of the most common p53 cancer mutants tested. The 235-239-240 suppressor motif establishes that manipulation of a small region of the core domain is sufficient to activate a large number of p53 cancer mutants. Understanding the structural basis of the rescue mechanism will allow the pursuit of small compounds able to achieve a similar stabilization of p53 cancer mutants.
The transcription factor p53 exerts its tumor suppressor function, in part, by activating target genes after upstream stress signals, such as DNA damage, have been relayed. Final outcomes of p53 activation are DNA repair, cell-cycle arrest, and/or apoptosis (1-3). The 393-aa-long protein has an amino-terminal transactivation domain, a core domain of ≈200 amino acids (amino acids 96-292) that recognizes p53 DNA-binding sites (DBSs), and a carboxyl-terminal tetramerization domain. Estimates suggest that up to half of all human cancers carry p53 gene mutations, of which many have been documented in large international databases (4, 5). The International Agency for Research on Cancer (IARC) TP53 Mutation Database contains 18,585 somatic p53 mutations (R8, www.iarc.fr/p53; ref. 5). A total of 13,262, or 71%, of these mutations result in full-length protein with one amino acid substitution within the p53 core domain.
The important functions of p53 and the high frequency of p53 mutations have resulted in significant interest in exploiting the p53 pathway for novel cancer therapies (6, 7). One particularly appealing approach is the use of small compounds for the pharmacological restoration of p53 function to those cancers that carry full-length p53 protein with one amino acid change in the DNA-binding core domain (8). This strategy, if realized, should have several significant advantages. The number of patients that may potentially benefit is quite large. Based on estimates by the IARC of new cancer cases worldwide for the year 2000 (www-dep.iarc.fr/globocan/globocan.html; ref. 9), estimates for the frequency of p53 mutations in specific cancers (10) and the IARC TP53 Mutation Database (5), 10 million people are diagnosed with cancer every year, and 2.6 million of them have cancers with a core domain missense mutation. In addition, p53 mutations are frequently found in solid tumors, such as lung, colon, head and neck, and pancreas cancers, that are quite resistant to conventional therapies (4, 5). Such compounds should, in theory, only have an effect on cancer cells, because the core domains of WT p53 in normal cells are already structurally intact. Furthermore, the approach promises systemic delivery and lack of a host immune response.
The small compound strategy, however, poses formidable challenges. Such compounds simply may not exist, or, if they do, may be effective against only a small subset of the nearly 1,000 reported p53 core domain mutants. However, from a clinical perspective, the problem may not be quite as complex, because only 8 amino acid changes account for 30% and 50 amino acid changes for 55% of all core domain mutants with a single amino acid change (5). Thus, compounds active on a subset of p53 cancer mutants could benefit a large number of patients.
Furthermore, as proof of principle, several compounds, amifostine, ellipticine, CP-31398, and PRIMA-1, have already been identified that endow function to a subset of p53 cancer mutants (11-14). The exact rescue mechanisms of and number of p53 mutants rescued by these compounds are currently not known. CP-31398 and PRIMA-1 were isolated in large-scale drug screens that may be the key to this therapeutic challenge. However, screens may also fail to identify compounds with the broadest activity, because, for example, such compounds were not represented in the screened libraries. We have taken a different approach of first establishing the existence of a global rescue mechanism for p53 cancer mutants through the studies of intragenic suppressor mutations and then exploiting these findings for the design of small compounds. Intragenic suppressor mutations are very instructive, because they pinpoint key regions of the p53 core domain that, after modification, provide increased stability to p53 protein.
In a pilot study (15), we demonstrated that genetic strategies using a p53 yeast assay were feasible and yielded intragenic suppressor mutations. This approach resulted in the rescue of p53 cancer mutant V143A by suppressor amino acid N268D, G245S by T123P, N239Y or S240N and R249S by the combination of T123A and H168R. Two key questions, however, remained unanswered. Are there additional suppressor regions within the p53 core domain? Are the identified suppressor mutations at codons 123, 168, 239/240, and 268 important for the rescue of other frequent p53 cancer mutants? Improved tools allowed us to answer these central questions, and we report here the identification of a global suppressor motif involving amino acids 235, 239, and 240 that rescues more than half of the most common p53 cancer mutants tested.
Materials and Methods
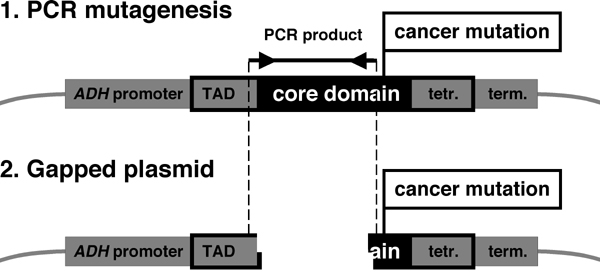
PCR Mutagenesis of the Upstream and Downstream Regions of p53 Cancer Mutations. PCR products covering the core domain regions upstream or downstream of a cancer mutation were obtained under mutagenic conditions (16, 17) and were transformed with appropriately gapped mutant p53 expression plasmids into the p53 yeast reporter strain RBy377. RBy377 contains two copies of the p53-dependent reporter gene 1cUAS53::URA and resulted from a mating of RBy41 (18, 19) and RBy171 (MAT α his3Δ200 leu2Δ1 1cUAS53::URA), selection for diploids on SC-His-Lys plates (lacking histidine and lysine) and segregation of pRB16. Yeast transformants that had repaired the p53 plasmid by homologous recombination were selected on plates lacking histidine (circular plasmid required) and uracil (expression of the URA3 reporter gene required). Single-colony-purified His+Ura+ yeast clones were tested for plasmid-dependency of the Ura+ phenotype. Retention of the cancer mutation on the p53 plasmid was verified by yeast colony PCR, followed by digestion of the PCR product to identify the unique restriction site provided by the cancer mutation codon. Positive plasmids were then rescued from yeast, were sequenced and were transformed again into RBy377 for phenotype confirmation (15). Between 1 × 106 to 1 × 107 yeast transformants per region were analyzed (see Supporting Materials and Methods, which is published as supporting information on the PNAS web site, for more details, including the construction and characterization of yeast and mammalian plasmids).
Oligonucleotide-Mediated Mutagenesis of p53 Codons 225-241. The saturation mutagenesis of codons 239 and 240 depended on our ability to create a short gap in p53 mutant plasmids by using restriction enzymes PflMI and NsiI. The second NsiI site in the backbone of pRS413 was therefore destroyed, and all expression cassettes for p53 mutants were transferred into this new backbone by using ApaI-SacI. Thirty-eight annealed primer pairs were generated, each of them representing a different amino acid change at codon 239 or 240. Equal amounts of each primer pair were cloned separately into the same gapped p53 plasmid, and the number of Escherichia coli clones was determined. Based on these results, the relative amounts of primer pairs were adjusted, and all 38 were combined to a library. This library was then cloned separately into the gapped expression plasmids of 30 p53 cancer mutants. The resultant plasmid libraries were transformed into RBy377, and yeast clones were processed as described above (15).
Mammalian Assays for p53 Cancer Mutants. H1299 and baby hamster kidney cell lines were grown and transfected as described (15). p53 cancer mutants with and without suppressor amino acids were evaluated in transient reporter gene assays by using the dualluciferase reporter assay system (Promega) in H1299 cells as described (15, 20, 21). Reporter gene plasmids with single p53 DBSs were based on pp53-TA-Luc (Clontech; ref 20). PGL3-Waf1-luc was the luciferase reporter plasmid for the p21 promoter (22, 23). Apoptosis assays were performed as described (15).
Results
Identification of Suppressor Amino Acids for the Eight Most Common p53 Cancer Mutants by Using PCR Mutagenesis. Our previous study (15) established that a functional assay for p53 in yeast was suitable for isolating intragenic suppressor mutations for p53 cancer mutants. In this assay, p53 binds to an artificial consensus p53 DBS and activates the reporter gene URA3, thus resulting in yeast growth on plates lacking uracil (15, 18, 19). To screen for suppressor mutations, the upstream and downstream regions of p53 cancer mutations are PCR-amplified under mutagenic conditions. The PCR products are then cotransformed into yeast with gapped expression plasmid for the p53 cancer mutant. Because of small overlapping regions at the ends, yeast can regenerate the complete expression plasmid by homologous recombination (Fig. 5, which is published as supporting information on the PNAS web site). Yeast cells are selected on plates lacking histidine (gapped plasmid was repaired) and uracil (functional p53 may be expressed). After several confirmatory steps, plasmids are rescued from yeast and are sequenced.
The pilot study (15) generated intriguing first results, but was limited in scope because of two significant experimental shortcomings. The natural p53 ORF provides very few restriction sites that appropriately gap the plasmids of p53 cancer mutants. More than 90% of the isolated yeast clones did not carry plasmids with the cancer and one or several suppressor mutations, because the cancer mutation codon had reverted to the WT codon. These falsepositives could only be eliminated at the very last step of the screens, sequencing of the plasmids, thus making screens very laborious.
We overcame the first obstacle by constructing an engineered p53 ORF with numerous silent restriction sites that allowed us to gap the p53 plasmid as needed. We addressed the problem of false-positive revertants to the WT codon by choosing, if possible, codons for the p53 cancer mutations that required two base pair changes to revert to the WT codon and whose retention could be checked for early on by yeast colony PCR and digestion of the PCR product to confirm the presence of a restriction site unique to a specific p53 cancer mutation.
A comprehensive search for intragenic suppressor mutations upstream and downstream of the eight most common p53 cancer mutations (5) (see Table 3, which is published as supporting information on the PNAS web site, for details on frequency of p53 cancer mutants) was unable to identify suppressor mutations for R175H, R248Q, R248W, and R282W, but isolated multiple suppressor mutation combinations for G245S, R249S, R273C, and R273H (Table 1 and Fig. 1A). Several interesting patterns emerged from this analysis. The known G245S suppressor amino acids T123P and N239Y (15) were reisolated in this screen, validating the approach. Another suppressor amino acid, F113L, was also identified. R249S is known to be rescued by a combination of T123A and H168R, but neither one alone, and H168R appears to directly compensate for the local structural perturbation caused by R249S (15, 24). In the current study, H168R was again found to be central to the rescue of R249S. Although T123A was not isolated in combination with H168R, two plasmids showed mutations in close proximity to this codon, T118M alone or V122I in combination with C124S. The significance of this region as a suppressor region is independently supported by the fact that mutation V122A rescues several p53 cancer mutants (25). Our experiments further established that H168R can also rescue R249S in combination with N239Y.
Table 1. Intragenic suppressor mutations obtained with PCR mutagenesis.
| Cancer mutation | No. | Suppressor mutations |
|---|---|---|
| G2455 | 1 | F113L |
| 2 | L114V + T123P* + V172I + A189V | |
| 3 | T123P* + A189V | |
| 4 | S227P + N239Y* | |
| 5 | N239Y* | |
| R249S | 6 | T118M + H168R* |
| 7 | V122I + C124S + H168R* | |
| 8 | K139R + H168R* + N239Y* | |
| 9 | H168R* + T231I | |
| R273C | 10 | T123A* + S240R |
| 11 | H178Y + S240R | |
| 12 | D281G + E285G + G325R + E343V | |
| 13 | E285K + E294G + E346G | |
| R273H | 14 | T81S + A83V + S240R |
| 15 | P87R + Q100L + Q104P + Q144L + S240R | |
| 16 | S183T + S240R | |
| 17 | E224G + S240R | |
| 18 | H233R + S240R | |
| 19 | S240R |
All clones shown are derived from independent PCR-mediated mutational events. In those instances where more than one codon change was isolated, one or more of the mutations may not be suppressor mutations, but rather, functionally silent mutations. For R249S and R273C, all isolated clones are shown. For G245S and R273H, only a subset of positive clones that were sequenced is shown. Mutations in bold led to our hypothesis of a suppressor motif around amino acids 239 and 240 of the p53 protein. For R175H, R248Q, R248W, and R282W, no suppressor mutations were isolated. Numbers in the second column (No.) correspond to the numbers in Fig. 1.
Mutations that were previously isolated, alone or in combination, with cancer mutations G245S or R249S (15).
Fig. 1.

The effect of suppressor amino acid changes on p53 cancer mutants in the p53 yeast assay. (A) PCR-mediated mutagenesis yielded suppressor mutation combinations that rescued transcriptional activity of four of the eight most common p53 cancer mutants, G245S, R249S, R273C, and R273H. SC-His plates select for the p53 expression plasmids and SC-Ura plates select for transcriptionally active p53. Pictures were taken after 2 days of incubation at 30°C. All yeast clones are represented by two patches. p53 cancer mutants are named (e.g., G245S), p53 mutants with cancer and suppressor combinations are numbered, and are crossreferenced with Table 1. (B) Design of the oligonucleotide-based mutagenesis. Expression plasmids for 30 of the most common p53 cancer mutants were gapped by using PflMI and NsiI. Pairs of annealed oligonucleotides representing all amino acid changes at codons 239 or 240 were cloned into the gap. The design also included background mutagenesis of the remaining codons between 225 and 241 that resulted in approximately one misincorporation per 100 nucleotides. (C) The global suppressor motif 235-239-240 rescues 16 of 30 of the most common p53 cancer mutants in the yeast p53 assay. All yeast transformants were processed and are shown as described for A. V272M was Ura+, but less so than V272M plus N239W. Identification of V272M plus N239W was likely possible, because the selection for rescued p53 cancer mutants (plating of transformants directly onto SC-His-Ura plates) was more stringent than subsequent confirmation of phenotypes for yeast patches by replica plating. V272M was transcriptionally inactive in mammalian reporter gene assays, whereas V272M plus N239W showed transcriptional activity (see Figs. 2 and 3). p53 mutants with cancer and suppressor combinations are numbered, and are crossreferenced with Table 2.
R273C and R273H were not previously analyzed. R273C was rescued by two combinations of downstream suppressor mutations. Both combinations contained mutations of codon 285, E285G, and E285K. Mutation of the adjacent codon 284, T284R, has been reported to rescue R248Q, R273C, and R273H under specific conditions (26). The most striking result was the fact that all other suppressor combinations for both of the 273 mutations contained S240R. Taken together with the results for G245S and R249S, half of all suppressor mutation combinations contained mutations in codons 239 or 240 (bold in Table 1).
Identification of a Suppressor Motif for 16 of 30 of the Most Common p53 Cancer Mutants. We hypothesized that the 239/240 area of the p53 core domain represents a suppressor motif that may also restore function to other p53 cancer mutants. We performed a saturation mutagenesis by constructing a library of annealed oligonucleotides that equally represented all possible 19 amino acid changes in codons 239 or 240. In addition, all other codons of the oligonucleotides (corresponding to codons 225-241) were synthesized under mutagenic conditions, resulting in approximately one misincorporation per 100 nucleotides. The annealed oligonucleotides were combined and were cloned into plasmids for 30 of the most common p53 cancer mutations (Fig. 1B; see Table 3 for details on frequency of p53 cancer mutants). The expression plasmid libraries were then transformed into the p53 yeast reporter strain and were analyzed as for the PCR mutagenesis. With this directed mutagenesis, more than half of the p53 cancer mutants tested was rescued (Table 2 and Fig. 1C). A total of 37 independent suppressor mutation combinations were isolated. Consistent with the design of the screen, all suppressor mutation combinations contained changes in codons 239 or 240. Ten suppressor combinations had concomitant changes of both codons 239 and 240, a significant number, considering that one of the codon changes was caused by the background mutagenesis. Twenty-four combinations had a change of codon 239 alone, whereas only three suppressor combinations contained a change of codon 240 alone. Eighteen of 34 codon 239 changes resulted in tyrosine. Nine of 13 codon 240 changes resulted in arginine. Even more striking was the overrepresentation of codon 235 changes caused by the background mutagenesis. Thirteen of 37 suppressor combinations contained a change of codon 235 to lysine, two additional ones showed a change to serine (Table 2). Considering the diversity of rescued p53 cancer mutants, these findings support the existence of a global suppressor motif within the p53 core domain.
Table 2. Intragenic suppressor mutations obtained with oligonucleotide-based mutagenesis.
| Cancer mutation
|
Codons
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | 227 S | 228 D | 229 C | 230 T | 232 T | 233 H | 234 Y | 235 N | 236 Y | 239 N | 240 S | |
| C141Y | 20 | A | K | M | ||||||||
| 21 | E | K | N | L | ||||||||
| 22 | V | R | F | K | L | |||||||
| 23 | K | T | ||||||||||
| V157F | 24 | F | L | |||||||||
| 25 | K | R | ||||||||||
| 26 | Y | |||||||||||
| R158L | 27 | Y | Y | |||||||||
| 28 | F | |||||||||||
| 29 | Y | |||||||||||
| 30 | Y | Q | ||||||||||
| Y163C | 31 | Y | K | Y | ||||||||
| 32 | K | Y | ||||||||||
| 33 | S | Y | N | |||||||||
| V173L | 34 | N | Y | |||||||||
| 35 | Y | |||||||||||
| V173M | 36 | K | Y | |||||||||
| Y205C | 37 | E | Y | |||||||||
| 38 | K | W | ||||||||||
| Y220C | 39 | K | N | |||||||||
| 40 | Y | |||||||||||
| G245C | 41 | Y | ||||||||||
| G245S | 42 | F | ||||||||||
| 43 | S | Y | ||||||||||
| R249M | 44 | T | K | Y | ||||||||
| 45 | Y | |||||||||||
| V272M | 46 | W | ||||||||||
| R273C | 47 | R | S | R | R | |||||||
| 48 | R | R | ||||||||||
| 49 | W | R | R | |||||||||
| 50 | L | R | ||||||||||
| 51 | F | R | ||||||||||
| R273H | 52 | V | H | R | ||||||||
| 53 | R | |||||||||||
| R273L | 54 | E | W | K | Y | R | ||||||
| 55 | Y | R | ||||||||||
| E286K | 56 | K | W | |||||||||
Numbers in the second column (No.) correspond to numbers of p53 mutants in Figs. 1, 2, 3, 4 and 6 and 7. All other columns represent codons of the p53 ORF for which mutations were identified after sequencing of Ura+ clones (the mutagenized codons were 225-241). Please see Supporting Materials and Methods for more details of the screens.
Effects of the 235-239-240 Suppressor Motif in Mammalian Assays. To confirm our findings for cancer and suppressor amino acids in mammalian assays, we first analyzed p53 mutants in p53-negative H1299 cells by using transient reporter gene assays for the p53 DBSs of the p53 target genes GML (27) or KILLER/DR5 (28). These DBSs represent important p53 target genes and are very similar to the consensus p53 DBS used in the yeast screens (19, 20). For 12 p53 cancer mutants, at least one suppressor combination restored transcriptional activity to 40-130% of WT p53 activity. Four mutants, R158L, V173M, Y205C, and Y220C, in combination with suppressor amino acids, showed only 20% of WT p53 activity (Fig. 2).
Fig. 2.
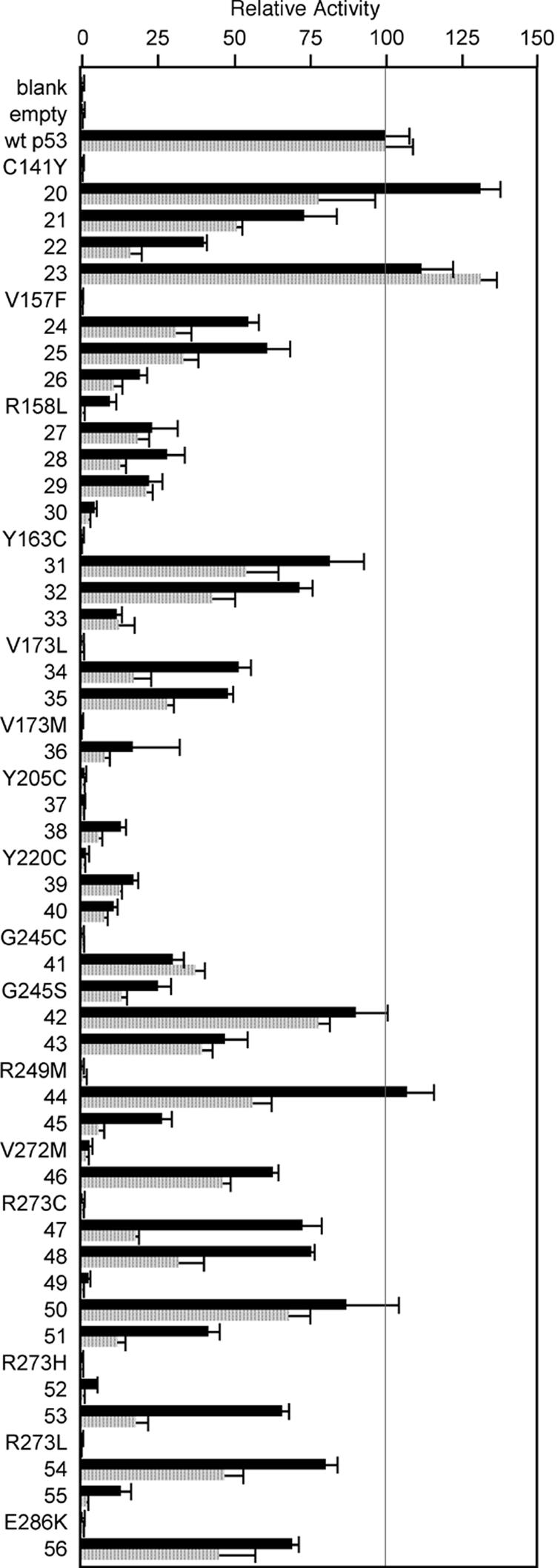
The global 235-239-240 suppressor motif rescues many common p53 cancer mutants in mammalian reporter gene assays for single p53 DBSs. The transcriptional activity of p53 cancer mutants with and without suppressor amino acids was evaluated in transient reporter gene assays in p53-negative H1299 cells. Luciferase activity, adjusted for transfection efficiency by using Renilla luciferase activity, was determined 24 h after transfection. The adjusted luciferase activity of cell lysates transfected with reporter plasmids and WT p53 expression plasmid was set as 100%. Shown are the mean and SD for three independent experiments. The reporter gene plasmids contained the DBSs of the p53 target genes GML (gray bars) or KILLER/DR5 (black bars) in the context of a heterologous yeast promoter. p53 cancer mutants are written out, and p53 mutants with cancer and suppressor amino acids are indicated by numbers that can be crossreferenced with Table 2 and Fig. 1C. The majority of p53 cancer mutants was rescued by at least one suppressor combination with transcriptional activities ranging from 40% to 130% of WT p53 activity. In the case of R158L, V173M, Y205C, and Y220C, the presence of suppressor amino acids resulted in only 20% of WT p53 activity. However, this result still represents a significant rescue effect, considering the lack of activity of the p53 cancer mutants alone.
We next investigated transcriptional activity of p53 cancer mutants with and without suppressor amino acids for the intact promoter of the p53 target gene p21 that is central to p53-mediated cell cycle arrest. Based on the results of Fig. 2, we chose one suppressor combination that had provided the best rescue effect for each of the p53 cancer mutants. Eleven of 16 p53 cancer mutants were rescued to activity levels as high as WT p53. The remaining five p53 cancer mutants showed at least 50% of WT p53 activity (Fig. 3). Thus, compared with single p53 DBSs (Fig. 2), the results with the p21 promoter showed a significantly stronger rescue effect. This result may be due to the fact that p53-specific coactivators are recruited to intact promoters of p53 target genes, but not to isolated p53 DBSs that are presented in the context of a heterologous yeast promoter.
Fig. 3.
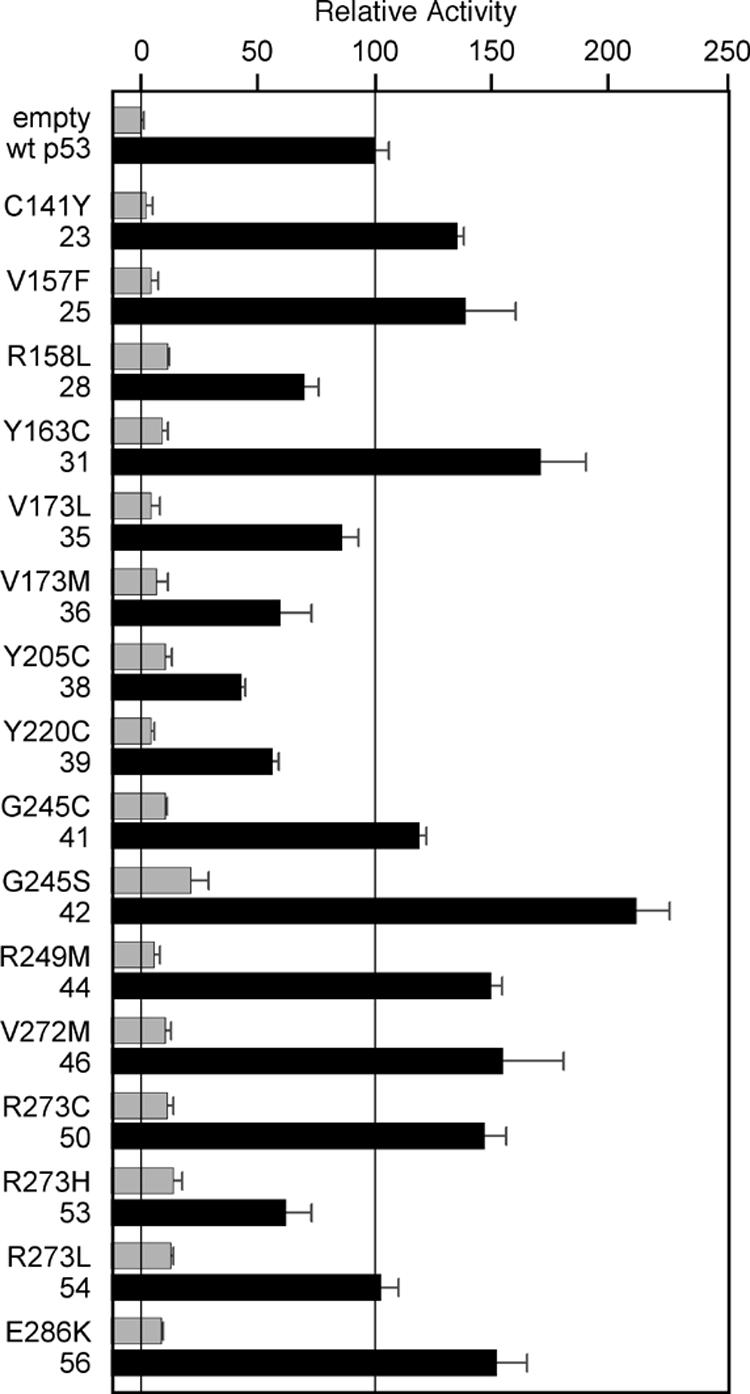
The global 235-239-240 suppressor motif rescues many common p53 cancer mutants in mammalian reporter gene assays for the p21 promoter. p53 cancer mutants with (black bars) and without (gray bars) suppressor amino acids were evaluated as described for Fig. 2, except that a reporter plasmid with the intact p21 promoter (2.3 kb) was used. The transcriptional activity of all p53 cancer mutants with suppressor amino acids was significantly better when compared with the results of Fig. 2, suggesting that p53 coactivators recruited to the p21 promoter can further enhance the effect of suppressor amino acids.
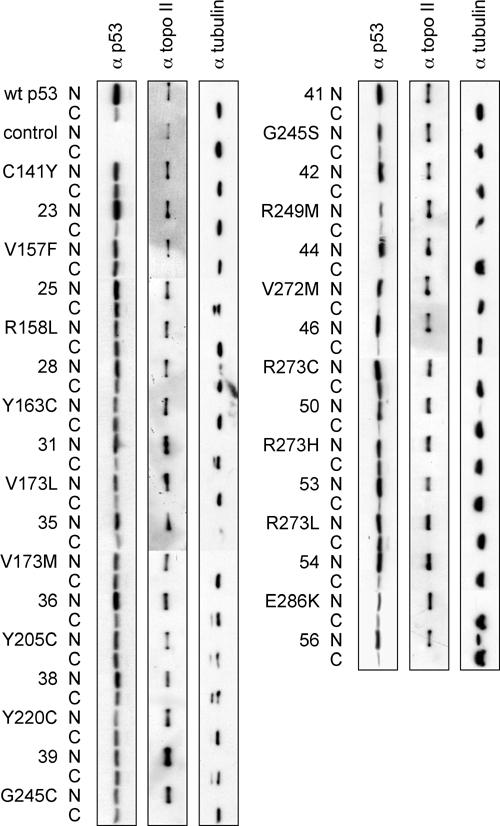
As expected, the protein levels for p53 cancer mutants of Fig. 3 were, in general, higher than for WT p53. The levels of p53 cancer mutants with suppressor amino acids were mostly similar to or slightly lower than the corresponding cancer mutants, indicating that the effect of suppressor amino acids is not explained by higher levels of p53 protein (Fig. 6, which is published as supporting information on the PNAS web site). Suppressor amino acids have been shown to restore function to p53 cancer mutants through at least two mechanisms, improving stability and/or restoring the conformation required for DNA binding (24). Our studies of cellular distribution of p53 mutants suggest that suppressor amino acids may not only contribute to improved binding to p53 DBSs (24), but also to correct localization of p53 mutants to the nucleus (Fig. 7, which is published as supporting information on the PNAS web site).
We determined the overall biological effects of suppressor mutations by assessing p53-mediated induction of apoptosis. We focused on the p53 cancer suppressor mutant combinations with less activity than WT p53 in the reporter gene assays for the p21 promoter (R158L, V173L, V173M, Y205C, Y220C, and R273H with and without suppressor mutations). We also included Y163C and G245S as examples of cancer mutants that were rescued to activity higher than WT p53. Baby hamster kidney cells were transfected with p53 and lacZ expression plasmids, transfected cells stained for β-galactosidase, and apoptotic cells scored by morphology (15). WT p53 induced apoptosis in 16.5% of transfected cells. All p53 cancer mutants showed activity similar to empty plasmid control, whereas cancer mutants with suppressor amino acids showed between 65% and 125% of WT p53 activity (Fig. 4). Our data indicate that mammalian reporter gene assays for p53 DBSs and p53-dependent promoters, with their focus on only one aspect of p53 activity, undervalue the rescue effect. In contrast, whole-cell assays, such as apoptosis assays, characterize the effect of suppressor mutations more accurately.
Fig. 4.

Induction of apoptosis by p53 cancer mutants with suppressor amino acids. Baby hamster kidney cells were transiently transfected with p53 and lacZ expression plasmids, transfected cells were fixed and were stained for β-galactosidase activity with X-gal 24 h later, and apoptotic cells were scored by cell morphology (small round blue cells) in a double-blind fashion. The bars show the percentage of apoptotic cells among the total number of counted blue cells. Two independent experiments performed in triplicate were combined. Between 2,461 and 3,972 cells were counted for p53 mutants (average, 3,090). For WT p53 and empty vector control, a combined 5,946 and 5,794 cells were counted, respectively. WT p53 induced apoptosis in 16.5% of cells above a background of 17%, which was likely due to transfection toxicity. Whereas p53 cancer mutants showed activity similar to empty vector control, p53 cancer mutants with suppressor amino acids showed between 65% and 125% of WT p53 activity.
Discussion
The ability to endow p53 cancer mutants with WT p53 activity holds the promise of significantly improving the therapeutic options for some of the most difficult to treat human cancers. In pursuit of this challenging goal, large-scale drug screens have resulted in the identification of two types of small molecules. Studies of their precise mechanisms of action and spectra of activity may make these compounds key to turning this therapeutic strategy into reality. We have taken a second, different approach of first studying mechanisms of p53 mutant stabilization through intragenic suppressor mutations. Although this strategy is one step further away from actual compounds, it has the potential to pinpoint one or more critical region(s) within the p53 core domain, whose manipulation results in reactivation of a large subset of p53 cancer mutants.
Studies for the eight most common p53 cancer mutants resulted in the identification of codons 239 and 240 as being central to the functional rescue of G245S, R249S, R273C, and R273H. We then pursued the hypothesis of a global suppressor motif around codons 239 and 240 through oligonucleotide-based mutagenesis. Our analysis evaluated 30 of the most common p53 cancer mutations and was not biased by any other factors, such as location of the p53 mutation within the p53 core domain.
We indeed confirmed codons 239 and 240 as an important global suppressor motif whose effect appears to be further enhanced by codon 235. With changes in these three amino acids, we rescued 16 of 30 p53 cancer mutants tested as judged by reporter gene and apoptosis assays. The rescued p53 mutations are located within the β-sandwich (codons 141, 157, 158, 163, 205, and 220), the L2 loop (codon 173), the L3 loop (codons 245 and 249), and the loop-sheet-helix motif (codons 272, 273, and 286; ref. 29), supporting the idea of a suppressor motif with a global rescue mechanism. The identification of a global suppressor mutation or motif is not unprecedented. For example, one suppressor mutation was able to rescue 16 of 30 missense mutations that affect different structural motifs of staphylococcal nuclease (30).
Structural studies of the WT p53 core domain and thermodynamic studies of several mutant p53 core domains indicate that missense mutations affecting the p53 core domain result in at least one of three general effects: loss of a DNA contact, disruption of a local structure, or denaturation of the entire core domain (8, 29, 31). Due to the significant number of p53 cancer mutants that we tested, several interesting patterns regarding the classes of suppressed p53 mutations can be discerned. Our set of 30 p53 mutants contained seven mutations that affect the β-sandwich. All but one of them (P151S) were reactivated, suggesting that the 235-239-240 suppressor motif will rescue a large fraction of these mutants. β-sandwich mutants account for 28% of p53 core domain missense mutations, resulting every year in ≈675,000 new cancer diagnoses worldwide or 300,000 in Europe and the U.S. alone (5, 9, 10).
The 235-239-240 suppressor motif also rescued p53 mutations affecting the L2 loop, the L3 loop, and the loop-sheet-helix motif. However, the current data set is too small to predict what mutations in these regions are likely to be rescued. Specific mutations in these areas may have divergent enough effects to allow for the rescue of some, but not others by the 235-239-240 suppressor motif.
Our data also suggests several types of p53 mutations that may not be rescued by the suppressor motif. Mutations affecting the zinc-binding site are one such group (R175H, C176F, H179R, and H179Y), which is consistent with the assessment that such mutants are globally denatured and require increased stability and DNA binding for rescue (8, 31). The effects of codon 248 mutations could also not be overcome. These mutations lead to loss of a crucial DNA contact and likely result in additional local structural changes (8, 31). Finally, mutations affecting the H2 α-helix of the loop-sheet-helix motif were also not rescued.
It is possible that p53 cancer mutants not rescued by the 235-239-240 suppressor motif alone will be rescued through the combined effect of suppressor regions. Our current and previous results indicate that additional suppressor regions exist. Particularly interesting are codons 113-124, because six of 13 suppressor combinations for G245S, R249S, and R273C contained changes in this region (see Table 1).
Pockets of resistance have been predicted and are probably inevitable (8, 31). However, they should not divert attention from the enormous clinical benefit the 235-239-240 suppressor motif may ultimately provide. Just the p53 cancer mutants that we successfully rescued amount to ≈18% of all core domain missense mutations. Thus, every year they alone account for ≈450,000 new cancer patients worldwide or 200,000 in Europe and the U.S. (5, 9, 10).
The 16 p53 cancer mutants were rescued by different combinations of suppressor mutations in codons 235, 239, and 240. This finding could theoretically reflect various rescue mechanisms. However, it appears much more likely that many of these combinations result in a very similar rescue mechanism, considering that 87% of codon 235 changes are to lysine, 61% of 239 changes are to either tyrosine or arginine, and 69% of 240 changes are to arginine. Amino acids 239 and 240 are close to DNA; mutations of these two codons may therefore add new DNA contacts (15). Thermodynamic studies for N239Y, however, showed that this suppressor mutation increases the stability of the p53 core domain (24). This result is further supported by recent crystallographic studies of a superstable quadruple p53 mutant that included N239Y (32). The identification of N235K as a crucial suppressor further supports a rescue mechanism that is based on stabilization of the core domain, because 235 is part of the β-strand S8 of the β-sandwich (29). Crystallographic studies have begun to determine the exact nature of the suppressor mechanism.
Understanding the rescue mechanism of the 235-239-240 suppressor motif on a structural level will provide the basis to pursue the ultimate challenge, designing small molecules able to restore integrity to mutant p53 core domains in a similar fashion. This task requires a multidisciplinary approach that combines the expertise of chemists, structural biologists, computer scientists, and molecular biologists.
Supplementary Material
Acknowledgments
We thank Carrie B. Brachmann for critically reading the manuscript. This work was supported by National Institutes of Health Grant CA81511 (to R.K.B.).
Abbreviations: DBS, DNA-binding site; IARC, International Agency for Research on Cancer.
References
- 1.Vousden, K. H. (2000) Cell 103, 691-694. [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein, B., Lane, D. & Levine, A. J. (2000) Nature 408, 307-310. [DOI] [PubMed] [Google Scholar]
- 3.Prives, C. & Hall, P. A. (1999) J. Pathol. 187, 112-126. [DOI] [PubMed] [Google Scholar]
- 4.Beroud, C. & Soussi, T. (2003) Hum. Mutat. 21, 176-181. [DOI] [PubMed] [Google Scholar]
- 5.Olivier, M., Eeles, R., Hollstein, M., Khan, M. A., Harris, C. C. & Hainaut, P. (2002) Hum. Mutat. 19, 607-614. [DOI] [PubMed] [Google Scholar]
- 6.Gallagher, W. M. & Brown, R. (1999) Ann. Oncol. 10, 139-150. [DOI] [PubMed] [Google Scholar]
- 7.Lane, D. P. & Lain, S. (2002) Trends Mol. Med. 8, S38-S42. [DOI] [PubMed] [Google Scholar]
- 8.Bullock, A. N. & Fersht, A. R. (2001) Nat. Rev. Cancer 1, 68-76. [DOI] [PubMed] [Google Scholar]
- 9.Ferlay, J., Bray, F., Pisani, P. & Parkin, D. M. (2001) GLOBOCAN 2000: Cancer Incidence, Mortality and Prevalence Worldwide, Version 1.0. IARC CancerBase No. 5 (IARC Press, Lyon, France).
- 10.Harris, C. C. (1996) Carcinogenesis 17, 1187-1198. [DOI] [PubMed] [Google Scholar]
- 11.Maurici, D., Monti, P., Campomenosi, P., North, S., Frebourg, T., Fronza, G. & Hainaut, P. (2001) Oncogene 20, 3533-3540. [DOI] [PubMed] [Google Scholar]
- 12.Peng, Y., Li, C., Chen, L., Sebti, S. & Chen, J. (2003) Oncogene 22, 4478-4487. [DOI] [PubMed] [Google Scholar]
- 13.Foster, B. A., Coffey, H. A., Morin, M. J. & Rastinejad, F. (1999) Science 286, 2507-2510. [DOI] [PubMed] [Google Scholar]
- 14.Bykov, V. J. N., Issaeva, N., Shilov, A., Hultcrantz, M., Pugacheva, E., Chumakov, P., Bergman, J., Wiman, K. G. & Selivanova, G. (2002) Nat. Med. 8, 282-288. [DOI] [PubMed] [Google Scholar]
- 15.Brachmann, R. K., Yu, K., Eby, Y., Pavletich, N. P. & Boeke, J. D. (1998) EMBO J. 17, 1847-1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin-Goerke, J. L., Robbins, D. J. & Burczak, J. D. (1997) BioTechniques 23, 409-412. [DOI] [PubMed] [Google Scholar]
- 17.Svetlov, V. & Cooper, T. G. (1998) Yeast 14, 89-91. [DOI] [PubMed] [Google Scholar]
- 18.Vidal, M., Brachmann, R. K., Fattaey, A., Harlow, E. & Boeke, J. D. (1996) Proc. Natl. Acad. Sci. USA 93, 10315-10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brachmann, R. K., Vidal, M. & Boeke, J. D. (1996) Proc. Natl. Acad. Sci. USA 93, 4091-4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian, H., Wang, T., Naumovski, L., Lopez, C. D. & Brachmann, R. K. (2002) Oncogene 21, 7901-7911. [DOI] [PubMed] [Google Scholar]
- 21.Wang, T., Kobayashi, T., Takimoto, R., Denes, A. E., Snyder, E. L., el-Deiry, W. S. & Brachmann, R. K. (2001) EMBO J. 20, 6404-6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.el-Deiry, W. S., Tokino, T., Velculescu, V. E., Levy, D. B., Parsons, R., Trent, J. M., Lin, D., Mercer, W. E., Kinzler, K. W. & Vogelstein, B. (1993) Cell 75, 817-825. [DOI] [PubMed] [Google Scholar]
- 23.Zauberman, A., Lupo, A. & Oren, M. (1995) Oncogene 10, 2361-2366. [PubMed] [Google Scholar]
- 24.Nikolova, P. V., Wong, K. B., DeDecker, B., Henckel, J. & Fersht, A. R. (2000) EMBO J. 19, 370-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inga, A. & Resnick, M. A. (2001) Oncogene 20, 3409-3419. [DOI] [PubMed] [Google Scholar]
- 26.Wieczorek, A. M., Waterman, J. L., Waterman, M. J. & Halazonetis, T. D. (1996) Nat. Med. 2, 1143-1146. [DOI] [PubMed] [Google Scholar]
- 27.Furuhata, T., Tokino, T., Urano, T. & Nakamura, Y. (1996) Oncogene 13, 1965-1970. [PubMed] [Google Scholar]
- 28.Takimoto, R. & el-Deiry, W. S. (2000) Oncogene 19, 1735-1743. [DOI] [PubMed] [Google Scholar]
- 29.Cho, Y., Gorina, S., Jeffrey, P. D. & Pavletich, N. P. (1994) Science 265, 346-355. [DOI] [PubMed] [Google Scholar]
- 30.Shortle, D. & Lin, B. (1985) Genetics 110, 539-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bullock, A. N., Henckel, J. & Fersht, A. R. (2000) Oncogene 19, 1245-1256. [DOI] [PubMed] [Google Scholar]
- 32.Joerger, A. C., Allen, M. D. & Fersht, A. R. (2004) J. Biol. Chem. 279, 1291-1296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}