

Background: The cysteine desulfurase Nfs1 provides sulfur for Fe-S cluster biogenesis in mitochondria.
Results: Frataxin or a mutant Fe-S scaffold protein (Isu1Sup) with frataxin-bypassing ability stimulates cysteine binding to Nfs1.
Conclusion: Frataxin or Isu1Sup likely induces a conformational change in Nfs1, exposing substrate-binding sites.
Significance: Data presented here may help develop a drug for treating Friedreich ataxia associated with frataxin deficiency.
Keywords: Iron-Sulfur Protein, Metal Homeostasis, Metals, Mitochondria, Mitochondrial Metabolism, Pyridoxal Phosphate, Sulfur, Yeast
Abstract
For iron-sulfur (Fe-S) cluster synthesis in mitochondria, the sulfur is derived from the amino acid cysteine by the cysteine desulfurase activity of Nfs1. The enzyme binds the substrate cysteine in the pyridoxal phosphate-containing site, and a persulfide is formed on the active site cysteine in a manner depending on the accessory protein Isd11. The persulfide is then transferred to the scaffold Isu, where it combines with iron to form the Fe-S cluster intermediate. Frataxin is implicated in the process, although it is unclear where and how, and deficiency causes Friedreich ataxia. Using purified proteins and isolated mitochondria, we show here that the yeast frataxin homolog (Yfh1) directly and specifically stimulates cysteine binding to Nfs1 by exposing substrate-binding sites. This novel function of frataxin does not require iron, Isu1, or Isd11. Once bound to Nfs1, the substrate cysteine is the source of the Nfs1 persulfide, but this step is independent of frataxin and strictly dependent on Isd11. Recently, a point mutation in Isu1 was found to bypass many frataxin functions. The data presented here show that the Isu1 suppressor mimics the frataxin effects on Nfs1, explaining the bypassing activity. We propose a regulatory mechanism for the Nfs1 persulfide-forming activity. Specifically, at least two separate conformational changes must occur in the enzyme for optimum activity as follows: one is mediated by frataxin interaction that exposes the “buried” substrate-binding sites, and the other is mediated by Isd11 interaction that brings the bound substrate cysteine and the active site cysteine in proximity for persulfide formation.
Introduction
Iron-sulfur (Fe-S) clusters are essential cofactors of proteins that perform important functions such as electron transfer, catalysis, DNA repair, tRNA thiolation, ribosome biogenesis, and iron regulation (1–3). Fe-S cluster biogenesis occurs in mitochondria, and disruption of the process is associated with human diseases. For example, Friedreich ataxia, an inherited disease characterized by neurodegeneration, cardiomyopathy, and diabetes, is caused by frataxin deficiency (4). The cellular phenotypes of the disease include Fe-S cluster deficiency and accumulation of toxic iron in mitochondria (5–7). Fe-S cluster biogenesis in mitochondria is conserved from the yeast Saccharomyces cerevisiae to humans. Thus, knock-out of the yeast frataxin homolog (Yfh1)3 models many features of the human disease, including Fe-S cluster deficiency and mitochondrial iron accumulation (8–10). Importantly, frataxin can substitute for Yfh1 in yeast, indicating that the human protein exerts its effects via a pathway similar to that in yeast (11, 12). The exact function of frataxin in Fe-S cluster biogenesis has remained elusive, and roles in controlling iron donation and/or sulfur donation have been proposed (13–15).
Recently, frataxin/Yfh1 was found to be a component of a core mitochondrial Fe-S cluster assembly complex, containing the cysteine desulfurase Nfs1, the accessory protein Isd11, and the scaffold protein Isu (14, 16–18). Fe-S cluster assembly in mitochondria involves formation of an Fe-S cluster intermediate on a scaffold protein and subsequent transfer of the intermediate to apoproteins (2, 3). The frataxin-containing complex has been implicated in the first phase of the process, involving formation of the Fe-S cluster scaffold intermediate (19).
An initial requirement for forming the Fe-S cluster scaffold intermediate in mitochondria is the supply of sulfur, and for this, the Nfs1 cysteine desulfurase is required (20). Cysteine is the only possible source of the sulfur, and other sulfides and sulfates in the cell do not suffice. Sulfur donation by the yeast mitochondrial Nfs1 cysteine desulfurase involves discrete steps (21). The enzyme requires pyridoxal phosphate (PLP) as the cofactor, and the binding of the substrate cysteine occurs in a PLP-dependent manner. Subsequently, the interaction of Nfs1 with Isd11 initiates a conformational change in the enzyme such that the bound substrate and the active site cysteine are brought into close proximity, allowing persulfide formation on the active site cysteine (21). The persulfide sulfur is then donated to Isu1 for the synthesis of Fe-S cluster intermediates. Frataxin in the core assembly complex reconstituted from purified human proteins was shown to stimulate cysteine desulfurase as assessed by continuous sulfide production in the presence of cysteine and DTT (16). However, the effects of frataxin/Yfh1 on each of the steps of the cysteine desulfurase enzymatic cycle are unknown. A second recent finding is that a single amino acid substitution (methionine to isoleucine) at position 107 in Isu1 is able to rescue Fe-S cluster synthesis and other iron-related phenotypic defects in yeast cells/mitochondria lacking frataxin (Δyfh1) to a significant extent (22). Thus, the Isu1 (M107I) suppressor (called Isu1Sup) must bypass at least some of the critical functions of Yfh1. A specific effect of the Isu1Sup in stimulating one or more steps of the cysteine desulfurase, thereby mimicking the frataxin effect, can be considered.
Here, we make use of highly sensitive radioactive assays that allow monitoring of the separate steps of cysteine desulfurase activity-substrate cysteine binding by Nfs1 and subsequent persulfide formation by the Nfs1·Isd11 complex for a single round of the enzymatic cycle. The approach involves experiments entirely with purified proteins as well as experiments with a combination of purified proteins and mitochondrial lysates. We find that frataxin/Yfh1 by itself can specifically enhance substrate binding to Nfs1 and that Isu1Sup mimics this effect in the absence of Yfh1. Evidence is presented that Nfs1 likely undergoes at least two conformational changes. One change is mediated by the frataxin/Yfh1 interaction and promotes substrate binding. A second change is mediated by Isd11 and induces persulfide formation.
EXPERIMENTAL PROCEDURES
Bacterial Expression of Proteins
Upon import into mitochondria, the N-terminal targeting signals of Nfs1, Isu1, or Yfh1 precursor proteins are proteolytically removed by the matrix processing peptidases, generating the corresponding mature form (20, 22–24). Isd11 does not contain a cleavable mitochondrial targeting signal (25, 26). The mature form of Nfs1 with a C-terminal His6 tag (Nfs1-His6) and Isd11 were coexpressed from a polycistronic vector in BL21 (DE3) Codon Plus cells (Stratagene). The proteins formed a soluble complex (Nfs1·Isd11) because Isd11 lacking the His6 tag was copurified with Nfs1-His6 by Ni-NTA-agarose chromatography (21, 27). Wild-type and mutant forms of mature Nfs1, Yfh1, and Isu1 proteins, each with a C-terminal His6 tag, were also separately expressed in bacteria in soluble forms and purified. The Nfs1 (K263A), Yfh1 (W98G), and Isu1 (M107I) substitutions were introduced by QuikChange mutagenesis (Stratagene) of the corresponding open reading frame. These mutant proteins are called Nfs1mut, Yfh1mut, and Isu1Sup, respectively. For purification of Isu1-His6 and Isu1Sup-His6, an additional wash step with 0.5 m NaCl and 6 m urea was performed while the proteins were bound to Ni-NTA-agarose. Isd11 with a C-terminal His6 tag was expressed in bacteria in soluble form and purified as described previously (21). All purified proteins were stored at −80 °C until further use.
Formation of Covalent [35S]Persulfide on Purified Nfs1
In a typical one-step assay, purified proteins were incubated with 5 μCi of [35S]cysteine (1075 Ci/mmol) in HS buffer (20 mm Hepes/KOH, pH 7.5, 0.6 m sorbitol) containing 0.15 m NaCl and 0.15 mm PLP in a final volume of 50 μl. After incubation at 30 °C for 15 min, an equal volume of ice-cold 20% trichloroacetic acid (TCA) was added, and reaction mixtures were left on ice for 1 h. Samples were then centrifuged at 15,000 × g for 30 min at 4 °C, and the protein pellets thus obtained were analyzed by nonreducing SDS-PAGE followed by autoradiography (21). Assays were performed with numerous variations as indicated in the figure legends. Radiolabeled Nfs1 (Nfs1-S-35SH) bands were quantified by densitometric analysis of autoradiographs using the ImageJ software (National Institutes of Health).
Formation of [35S]Persulfide on Nfs1 in Mitochondrial Lysate
The generation of S. cerevisiae YPH499 strains with regulated YFH1 (28) or ISD11 (21) expression from the GAL10 promoter and the conditions for depletion of the corresponding proteins have been described previously. Depletion of Yfh1 or Isd11 was confirmed by immunoblotting using the corresponding antibodies. Mitochondria were isolated from wild-type (WT), Yfh1-depleted (Yfh1▾), and Isd11-depleted (Isd11▾) cells. To deplete endogenous nucleotides, intact mitochondria in HS buffer were incubated at 30 °C for 10 min (29–31). After nucleotide depletion, assays were performed with intact mitochondria (isotonic condition), mitochondrial lysate (hypotonic condition), or a combination of both (isotonic followed by hypotonic conditions) as necessary. For experiments with the lysate, nucleotide-depleted intact mitochondria (200 μg of proteins) were recovered and then resuspended in 20 mm Hepes/KOH, pH 7.5. Mitochondrial membranes were ruptured by freezing the samples at −80 °C followed by thawing and bath sonication for 30 s at 4 °C. The lysis process was repeated three times. The mitochondrial lysate thus obtained was supplemented with 10 μCi of [35S]cysteine, and the volume was adjusted to 100 μl with final concentrations of 20 mm Hepes/KOH, pH 7.5, 0.15 m NaCl, 40 mm KOAc, and 10 mm Mg(OAc)2. After incubation at 30 °C for 15 min, proteins were precipitated with 10% TCA and analyzed by nonreducing SDS-PAGE and autoradiography (21, 27). Variations of the assay are described in the corresponding figure legends.
RESULTS
Isd11 Promotes Persulfide Formation on Nfs1, and Neither Yfh1 nor Isu1Sup Can Substitute for This Function
Nfs1 is the only known cysteine desulfurase in mitochondria, and the enzyme activity is essential for Fe-S cluster biogenesis in mitochondria and hence for cell survival (20). However, too much cysteine desulfurase activity is toxic.4 Thus, Nfs1 activity in mitochondria must be tightly controlled. Nfs1 interacts with a small accessory protein, Isd11, and forms a stable complex, Nfs1·Isd11 (21, 25–27). A higher order complex, consisting of Nfs1, Isd11, Yfh1, and Isu1, has also been described (14, 16–18). We sought to determine the effects of various proteins on Nfs1 activity. For this purpose, Nfs1, Isd11, Yfh1, and Isu1 proteins, each with a C-terminal His6 tag, were expressed in bacteria in soluble forms and purified by Ni-NTA-agarose chromatography. Several mutant proteins were expressed in similar fashion, including inactive forms of Nfs1 (Nfs1mut) and Yfh1 (Yfh1mut). The suppressor form of Isu1 (Isu1Sup) was also expressed and purified. In addition, Nfs1 with a C-terminal His6 tag was coexpressed with Isd11, and the Nfs1·Isd11 complex formed in vivo in bacteria was purified. Proteins were analyzed by SDS-PAGE followed by silver staining. All individually expressed proteins were extremely pure, and no contaminating bacterial proteins were detected (Fig. 1A, lanes 2–8). The Nfs1·Isd11 complex was also more than 90% homogeneous with a small amount of contaminating bacterial proteins (Fig. 1A, lane 1).
FIGURE 1.

Analysis of proteins expressed in bacteria. A, various proteins were expressed in bacteria in soluble forms. These include Nfs1-His6 together with Isd11 (Nfs1·Isd11, lane 1), Nfs1-His6 alone (lane 2), Nfs1 (K263A)-His6 alone (Nfs1mut, lane 3), Isd11-His6 alone (lane 4), Yfh1-His6 (lane 5), Yfh1 (W98G)-His6 (Yfh1mut, lane 6), Isu1-His6 (lane 7), and Isu1 (M107I)-His6 (Isu1Sup, lane 8). These soluble proteins were purified by Ni-NTA-agarose chromatography. Proteins were analyzed on 15% gel by SDS-PAGE under reducing conditions, followed by silver staining of the gel. Std (lane 9) indicates protein standards with their molecular mass in kDa. B, reaction mixture contained the Nfs1·Isd11 complex (100 ng, lane 1), Nfs1 alone (100 ng, lane 2), or Nfs1 (100 ng) plus various proteins (100 ng each) as indicated (lanes 3–8) in HS buffer (20 mm Hepes/KOH, pH 7.5, 0.6 m sorbitol) containing 0.15 m NaCl. Samples were supplemented with 0.1 μm [35S]cysteine (5 μCi) and 0.15 mm PLP in a final volume of 50 μl. After incubation at 30 °C for 15 min, proteins were precipitated with 10% TCA and analyzed on 12% gel by nonreducing SDS-PAGE and autoradiography for covalent [35S]persulfide formation on Nfs1.
The enzymatic cycle of cysteine desulfurase includes formation of a covalent persulfide on the active site cysteine (21, 27, 32, 33). To determine persulfide formation, purified proteins were incubated with [35S]cysteine and analyzed by nonreducing SDS-PAGE followed by autoradiography (Fig. 1B). Nfs1-bound persulfide (Nfs1-S-35SH) was formed by the Nfs1·Isd11 complex (Fig. 1B, lane 1). Treatment with a reducing agent such as DTT released the persulfide sulfur, and accordingly, no radiolabeled Nfs1 was detected as reported earlier (27). Nfs1 alone, incubated with [35S]cysteine prior to resolution on nonreducing SDS gel, was not radiolabeled (Fig. 1B, lane 2). However, addition of Isd11 to Nfs1 resulted in Nfs1-bound persulfide formation (Fig. 1B, lane 8). In the absence of Isd11, no radiolabeled persulfide was detected when Nfs1 was supplemented with Isu1 (Fig. 1B, lane 3), Yfh1 (lane 4), Isu1 plus Yfh1 (lane 5), Isu1Sup (lane 6), or Yfh1mut (lane 7). Thus, Isd11 is required for covalent persulfide formation on Nfs1, and neither Yfh1 nor Isu1Sup can replace this Isd11 function. However, Yfh1, Isu1, and/or Isu1Sup may participate in a different step of the enzymatic cycle involving formation of a noncovalent intermediate that cannot be directly detected by SDS gels. In the following sections, experiments are designed to test this hypothesis.
Yfh1 by Itself Stimulates Covalent Persulfide Formation on Nfs1 by the Nfs1·Isd11 Complex, and Isu1Sup Mimics the Stimulatory Activity of Yfh1
The Nfs1·Isd11 complex was supplemented with Yfh1 and/or Isu1. Reaction mixtures were then incubated with [35S]cysteine, and samples were analyzed by nonreducing SDS-PAGE and autoradiography (Fig. 2A). As expected, Nfs1-bound persulfide (Nfs1-S-35SH) was formed by the Nfs1·Isd11 complex to some extent (Fig. 2A, lane 1). Strikingly, addition of Yfh1 greatly stimulated persulfide formation in a concentration-dependent manner (Fig. 2A, lanes 2–4). Whereas wild-type Isu1 alone exhibited some (∼20%) inhibitory effects (Fig. 2A, lane 5), a combination of Yfh1 and Isu1 was as stimulatory as Yfh1 alone (Fig. 2A, compare lanes 2–4 with lanes 6–8, respectively). This result was unanticipated because a previously published study using mixtures of the human proteins found that only the addition of both frataxin and Isu, and not frataxin alone, was able to stimulate sulfide production by the Nfs1·Isd11 complex (16). Human and yeast proteins might be different in this respect, or the studies might have overlooked the effects of frataxin alone due to lower sensitivity of the methylene blue spectrophotometric assays.
FIGURE 2.

Yfh1 or Isu1Sup stimulates persulfide formation by the purified Nfs1·Isd11 complex. A, as indicated, the Nfs1·Isd11 complex (100 ng) was mixed with Yfh1 (1× = 50 ng) and/or Isu1 (100 ng) in HS buffer containing 0.15 m NaCl. Samples were supplemented with 0.1 μm [35S]cysteine (5 μCi) and 0.15 mm PLP in a final volume of 50 μl. After incubation at 30 °C for 15 min, proteins were precipitated with 10% TCA and analyzed by nonreducing SDS-PAGE and autoradiography. B, Nfs1·Isd11 complex (100 ng) was mixed with Isu1Sup (1× = 50 ng) and/or Isu1 (100 ng) as indicated, and assays were performed as in A. C, Nfs1·Isd11 complex (100 ng) in HS buffer containing 0.15 m NaCl was incubated at 30 °C for 10 min, with or without added Yfh1mut (1× = 50 ng). As indicated, reaction mixtures were then supplemented with Yfh1 or Isu1Sup (1× = 50 ng). Following addition of 0.1 μm [35S]cysteine (5 μCi) and 0.15 mm PLP, samples were incubated at 30 °C for 15 min and analyzed as in A.
Interestingly, persulfide formation by the Nfs1·Isd11 complex was also enhanced by the addition of Isu1Sup alone in a concentration-dependent manner (Fig. 2B, lanes 1–4), and the stimulatory effect of the suppressor was comparable with that of Yfh1 (see Fig. 2C). Furthermore, addition of wild-type Isu1 did not interfere with the suppressor-mediated stimulation of persulfide formation (Fig. 2B, compare lanes 2–4 with lanes 6–8, respectively). Thus, Isu1Sup behaved exactly like Yfh1 in terms of the stimulatory effects on persulfide-forming activity of the Nfs1·Isd11 complex, and this may explain its ability to restore Fe-S cluster biogenesis in Δyfh1 mitochondria to a significant extent (22). Because we did not observe any major effects of Isu1 on Yfh1 or Isu1Sup-mediated stimulation of Nfs1-bound persulfide formation in our assays, experiments with purified proteins in the following sections were performed in the absence of added Isu1.
To further assess the similarities between Yfh1- and Isu1Sup-mediated stimulatory effects on Nfs1 activity, we used a mutant of Yfh1 (called Yfh1mut) in which the tryptophan residue at position 98 of the mature protein was changed to glycine. Unlike wild-type Yfh1, Yfh1mut failed to exhibit any stimulatory effects on persulfide formation by the Nfs1·Isd11 complex (Fig. 2C, compare lanes 1 and 2). These results are consistent with the inability of the mutant protein, expressed from the native YFH1 promoter, to rescue growth and other Fe-S cluster phenotypic defects of the Δyfh1 cells (data not shown). We then performed competition experiments to determine the effects of Yfh1mut on wild-type Yfh1-mediated stimulation of persulfide formation by the Nfs1·Isd11 complex. Briefly, the complex was preincubated without or with increasing concentrations of Yfh1mut for 10 min at 30 °C. A fixed amount of Yfh1 was then added, and reaction mixtures were incubated with [35S]cysteine. In the absence of Yfh1mut, Yfh1 greatly stimulated persulfide formation (Fig. 2C, compare lanes 1 and 3). However, in the presence of Yfh1mut, Yfh1-mediated stimulation was gradually abolished in a manner consistent with the increasing concentrations of the mutant protein (Fig. 2C, lanes 4–6). Most likely, Yfh1mut was able to bind Nfs1 but unable to activate persulfide formation by the Nfs1·Isd11 complex. With the increasing concentrations of Yfh1mut, more and more of the mutant protein bound to Nfs1, thereby inhibiting interaction of wild-type Yfh1 with the enzyme. Thus, Yfh1 must be functionally active to be able to stimulate persulfide formation by the Nfs1·Isd11 complex.
In parallel, a similar experiment was performed to determine the effects of Yfh1mut on Isu1Sup-mediated stimulation of persulfide formation by the Nfs1·Isd11 complex. In the absence of Yfh1mut, Isu1Sup was as effective as Yfh1 in stimulating persulfide formation (Fig. 2C, compare lanes 3 and 7). Strikingly, like in the case for Yfh1, Isu1Sup-mediated stimulation of persulfide formation was also inhibited by Yfh1mut in a concentration-dependent manner (Fig. 2C, lanes 8–10), and the inhibitory patterns for Yfh1 and Isu1Sup were highly comparable (Fig. 2C, compare lanes 4–6 with lanes 8–10, respectively). Thus, Yfh1 and Isu1Sup exhibited the stimulatory effects on the enzyme complex in a very similar, if not identical, fashion.
Yfh1 or Isu1Sup Enhances Substrate Binding to Purified Nfs1
The ability of Yfh1 or Isu1Sup to stimulate the Nfs1 persulfide-forming activity raises the question of how the effect is mediated. We developed a two-step assay in which the substrate-binding step of the cysteine desulfurase can be separated from the persulfide-forming step. In this assay, purified Nfs1 by itself is able to bind the substrate [35S]cysteine, and the enzyme·substrate complex can be recovered by ammonium sulfate precipitation. The bound substrate cannot be detected by SDS gels because SDS releases the radioactive substrate from the enzyme. Only when Isd11 is added during a second step is a covalently bound Nfs1-S-35SH persulfide formed and detected by the nonreducing SDS gel system (21). We took advantage of this assay to evaluate the effects of Yfh1 or Isu1Sup on substrate binding or persulfide formation by Nfs1.
A fixed concentration of Nfs1 was incubated with [35S]cysteine in the presence of increasing concentrations of Yfh1 or Isu1Sup. Proteins were precipitated with ammonium sulfate, removing unbound and excess [35S]cysteine (first step). The precipitates containing Nfs1 with bound [35S]cysteine were solubilized with buffer, and radioactivity was determined by scintillation counting. Increasing 35S signal was detected with increasing concentrations of Yfh1 or Isu1Sup used in the assay, indicating stimulatory effects on Nfs1 for [35S]cysteine binding (Fig. 3A). In parallel, Nfs1 with bound [35S]cysteine was incubated with purified Isd11 (second step), and samples were analyzed by nonreducing SDS-PAGE and autoradiography (Fig. 3B). Again, the signals for [35S]persulfide on Nfs1 were enhanced with increasing concentrations of Yfh1 or Isu1Sup used in the substrate-binding step (first step) of the assay.
FIGURE 3.
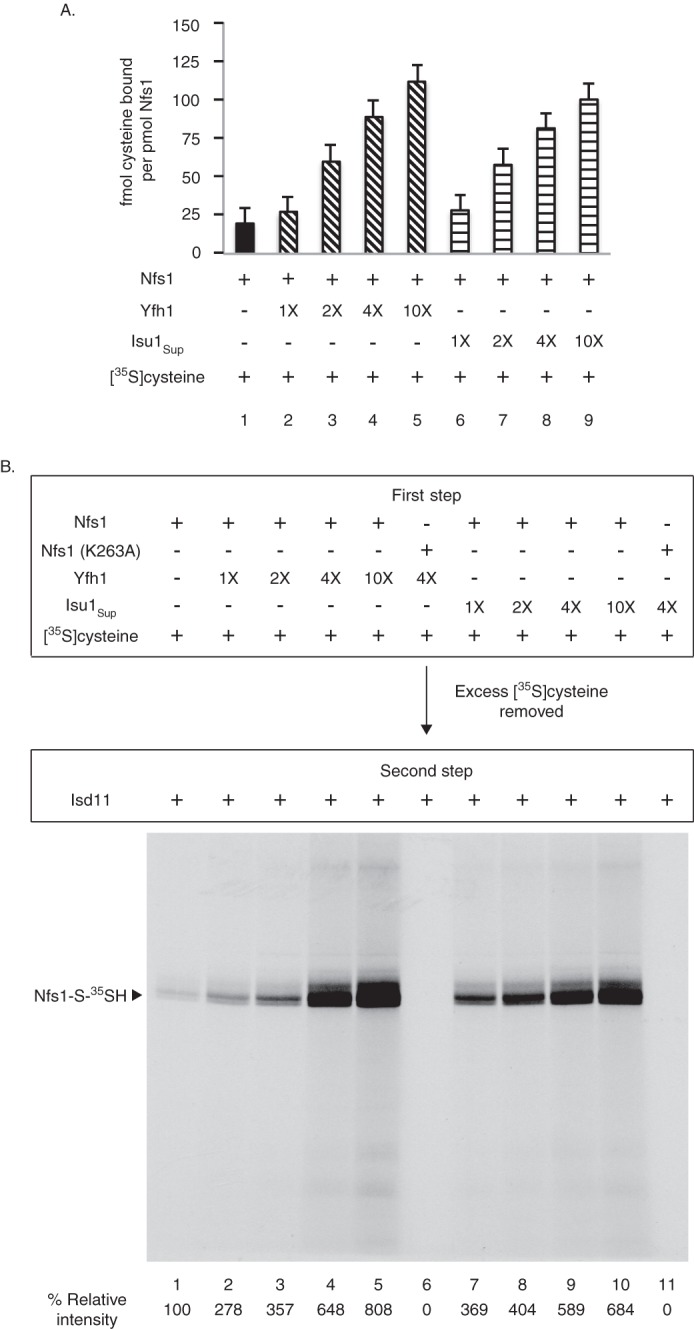
Yfh1 or Isu1Sup stimulates cysteine binding to purified Nfs1. A, as indicated, Nfs1 (100 ng) was mixed with Yfh1 or Isu1Sup (1× = 50 ng) in HS buffer containing 0.15 m NaCl. Samples were then supplemented with [35S]cysteine (0.1 μm; 5 μCi) and 0.15 mm PLP in a final volume of 50 μl. After incubation at 30 °C for 15 min, proteins were precipitated with ice-cold ammonium sulfate (final 67%) and centrifuged to remove excess and unbound [35S]cysteine. Protein-bound radioactivity was determined by scintillation counting. Identical samples in which wild-type Nfs1 was replaced with the Nfs1 (K263A) mutant served as respective background controls. Background counts were subtracted, and data are from three separate experiments. B, Nfs1 (100 ng) was incubated at 30 °C for 15 min with increasing amounts of Yfh1 or Isu1Sup (1× = 50 ng) in HS buffer containing NaCl (0.15 m), [35S]cysteine (0.1 μm; 5 μCi) and PLP (0.15 mm) (first step). Nfs1 (K263A) served as negative control. Proteins were precipitated with ammonium sulfate and centrifuged. The protein pellets were solubilized with HS buffer containing 0.15 m NaCl and 0.15 mm PLP and incubated with Isd11 (100 ng) at 30 °C for 15 min (second step). Samples were analyzed by nonreducing SDS-PAGE and autoradiography.
These data can be interpreted as follows. During the first step, a small portion of the added [35S]cysteine bound to Nfs1 through interactions with PLP in the substrate-binding site. With the addition of Yfh1, [35S]cysteine binding to Nfs1 was greatly enhanced in a manner that was dependent on the concentration of added Yfh1. Isu1Sup behaved exactly like Yfh1, enhancing [35S]cysteine binding to Nfs1 (Fig. 3A). The Nfs1·substrate complexes formed in the first step were converted to persulfide intermediates by addition of Isd11 in the second step, and these were then visible on the SDS gel (Fig. 3B). A mutant of Nfs1 (substituting alanine for lysine at position 263) was constructed, with an altered substrate-binding site such that it cannot form the cysteine-PLP adduct. The Nfs1 (K263A) mutant was used as a negative control for cysteine-binding experiments (Fig. 3, A and B).
An experiment was then performed to evaluate the effect of Yfh1 or Isu1Sup specifically on the persulfide-forming step of the enzyme. Nfs1 was incubated with [35S]cysteine (first step). After removal of excess and unbound [35S]cysteine, Nfs1 with bound [35S]cysteine was incubated with Isd11 and increasing concentrations of Yfh1 or Isu1Sup (second step). There were no significant changes in persulfide formation (Fig. 4). Thus, the stimulatory effect of Yfh1 or Isu1Sup on the enzyme can be attributed to enhanced substrate binding by Nfs1 and not to increased persulfide formation from already bound cysteine.
FIGURE 4.
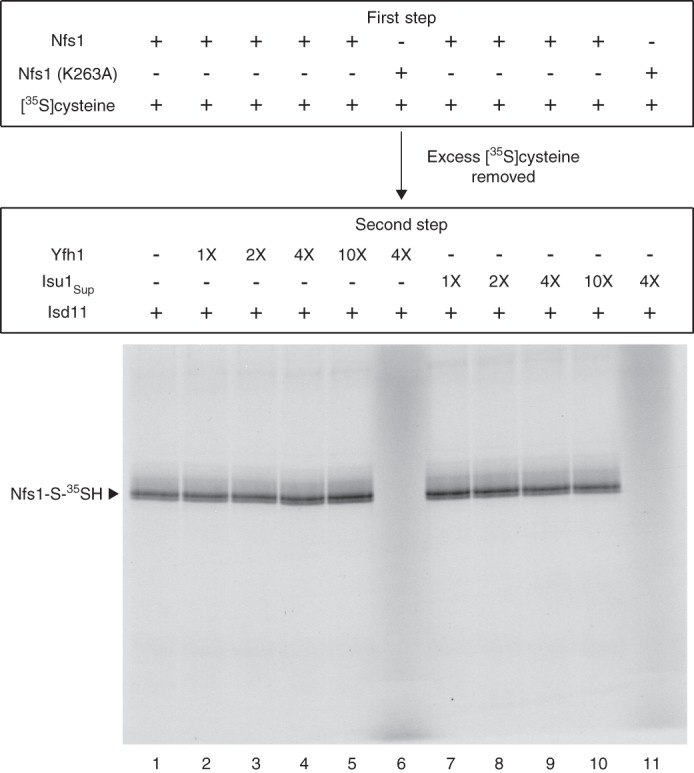
Persulfide formation by purified Nfs1 from already bound cysteine: no significant effect of added Yfh1 or Isu1Sup. Nfs1 (100 ng) or the Nfs1 (K263A) mutant (100 ng) was incubated with [35S]cysteine (0.1 μm; 5 μCi) and PLP (0.15 mm) in HS buffer containing 0.15 m NaCl at 30 °C for 15 min (first step). Proteins were precipitated with ammonium sulfate and centrifuged to remove excess and unbound [35S]cysteine. The protein pellet containing Nfs1 with bound [35S]cysteine was dissolved in HS buffer plus 0.15 m NaCl and 0.15 mm PLP. As indicated, reaction mixtures were supplemented with increasing amounts of Yfh1 or Isu1Sup (1× = 50 ng). Following addition of Isd11 (100 ng), samples were incubated at 30 °C for 15 min (second step) and subsequently analyzed by nonreducing SDS-PAGE and autoradiography.
Stimulatory Effect of Yfh1 or Isu1Sup on Substrate Binding to Nfs1 Is Independent of Iron Availability
Yfh1 is an iron-binding protein (34), prompting us to test if iron is required for the stimulatory effect of Yfh1 on substrate binding to Nfs1. The physiological source of iron for Fe-S cluster synthesis in mitochondria is unknown. However, addition of iron as ferrous ascorbate greatly stimulates Fe-S cluster synthesis in isolated mitochondria (29, 31). Conversely, treatment of isolated mitochondria with a membrane-permeable iron chelator blocks Fe-S cluster synthesis (27, 31). We therefore used ferrous ascorbate for iron supplementation in our assays. Likewise, assays were performed in the presence of EDTA expected to remove protein-bound iron and chelate contaminating free iron, if any, of the reaction mixtures. Briefly, assays were performed in two steps. During the first step, Nfs1 was incubated with [35S]cysteine and PLP in the absence or presence of Yfh1 (or Isu1Sup), with or without added increasing concentrations of ferrous ascorbate or EDTA. Following removal of excess and free [35S]cysteine, PLP, and iron (or EDTA), Nfs1 with bound [35S]cysteine was incubated with Isd11 to allow covalent persulfide formation (second step). Samples were analyzed by SDS-PAGE followed by autoradiography (Fig. 5). The [35S]persulfide on Nfs1 was greatly enhanced in the presence of Yfh1 (Fig. 5, compare lanes 1 and 2) or Isu1Sup (compare lanes 1 and 7) to a comparable extent. However, neither iron supplementation (Fig. 5, compare lane 2 with lanes 3 and 4) nor iron chelation (compare lane 2 with lanes 5 and 6) during the first step had any significant effects on Yfh1-mediated stimulation of substrate binding to Nfs1 as judged by subsequent Nfs1-bound persulfide formation. Likewise, Isu1Sup-mediated stimulation of persulfide formation on Nfs1 was also not affected by the presence or absence of available iron (Fig. 5, lanes 8–11). Thus, the stimulatory effect of Yfh1 or Isu1Sup on substrate binding to Nfs1 does not require iron, reinforcing similarities between the two proteins.
FIGURE 5.
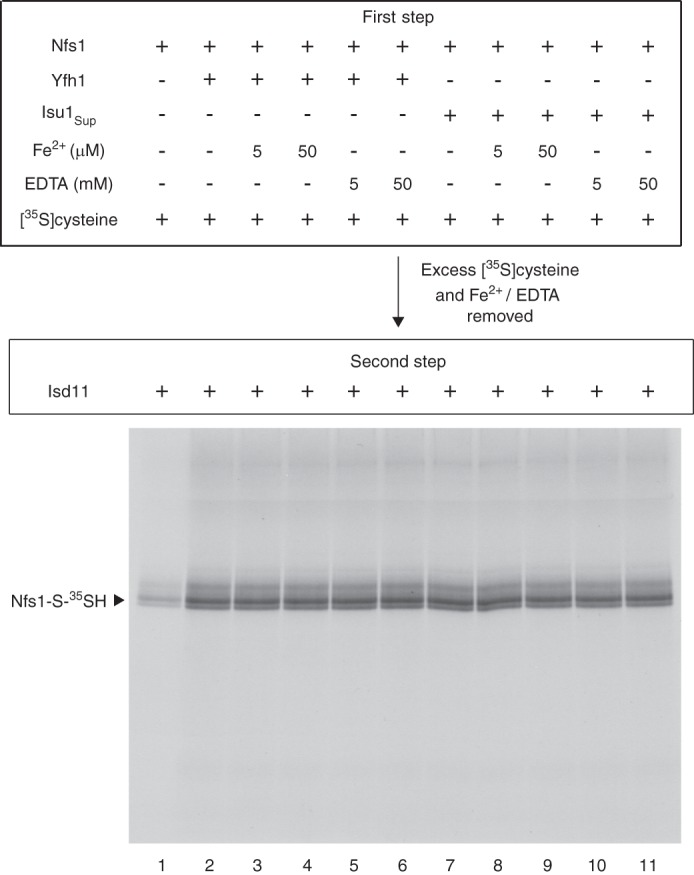
Yfh1 or Isu1Sup does not require iron for stimulating cysteine binding to purified Nfs1. Nfs1 (100 ng) was mixed with [35S]cysteine (0.1 μm; 5 μCi) and 0.15 mm PLP in the absence or presence of Yfh1 (100 ng) or Isu1Sup (100 ng) in 50 μl of HS buffer containing 0.15 m NaCl. As indicated, reaction mixtures were supplemented with ferrous ascorbate (5–50 μm) or EDTA (5–50 mm). After incubation at 30 °C for 15 min (first step), proteins were precipitated with ammonium sulfate and centrifuged to remove excess and unbound [35S]cysteine, PLP, and ferrous ascorbate (or EDTA). The protein pellets were solubilized with HS buffer containing 0.15 m NaCl and 0.15 mm PLP. After addition of Isd11 (100 ng), reaction mixtures were incubated at 30 °C for 15 min (second step). Samples were analyzed by nonreducing SDS-PAGE and autoradiography.
Yfh1 or Isu1Sup-mediated Stimulation of the Nfs1 Cysteine Desulfurase Is Not Because of Increased PLP Binding
Cysteine desulfurases require PLP as the cofactor. For Nfs1, a conserved lysine residue (Lys263) forms a Schiff base with PLP, which in turn interacts with the substrate cysteine. The cysteine-PLP ketimine adduct thus formed is the key intermediate for persulfide formation. Nfs1 purified from the bacterial expression system contains small amounts of bound PLP and exhibits low cysteine desulfurase activity. Addition of PLP during the enzyme assays results in stimulation of activity (Fig. 6A, compare lanes 1 and 2). This observation prompted us to test whether the stimulation of substrate binding to Nfs1 by Yfh1 or Isu1Sup is mediated via enhancement of PLP binding.
FIGURE 6.

Cysteine interaction with purified Nfs1-bound cofactor: stimulatory effects of added Yfh1 or Isu1Sup. A, Nfs1 (100 ng) in HS buffer containing 0.15 m NaCl was incubated at 30 °C for 10 min, with or without PLP (0.15 mm) (first step). Proteins were precipitated with ammonium sulfate and centrifuged to remove excess and unbound PLP. The protein pellets were dissolved in HS buffer plus 0.15 m NaCl. Samples were then supplemented with increasing amounts of Yfh1 as indicated (1× = 50 ng). Following addition of [35S]cysteine (0.1 μm; 5 μCi) and Isd11 (100 ng), reaction mixtures were incubated at 30 °C for 15 min in the absence or presence of added PLP (0.15 mm) (second step). Samples were analyzed by nonreducing SDS-PAGE and autoradiography. B, same as in A except Yfh1 was replaced with Isu1Sup during the second step of the assay.
Assays were performed in two steps, separating PLP binding from the substrate binding. During the first step, Nfs1 was incubated with a saturating concentration of PLP (0.15 mm). Excess PLP was removed by ammonium sulfate precipitation of Nfs1. During the second step of the assay, Nfs1 with bound PLP was supplemented with [35S]cysteine and increasing concentrations of Yfh1. Following addition of Isd11, samples were incubated without or with added PLP and analyzed by nonreducing SDS-PAGE and autoradiography (Fig. 6A). The [35S]persulfide signal was greatly increased when Nfs1 with prebound PLP (generated in the first step) was incubated with Yfh1 (second step) (Fig. 6A, compare lane 2 with lanes 5 and 6). The stimulation was dependent on concentrations of added Yfh1 and occurred completely independent of added PLP during the second step (Fig. 6A, compare lanes 3 and 4 with lanes 5 and 6, respectively). Thus, Yfh1-mediated stimulation of the persulfide-forming activity of Nfs1 was not due to enhanced PLP binding. An identical experiment was then performed with Isu1Sup, replacing Yfh1 in the second step of the assay (Fig. 6B). Results show that Isu1Sup behaved just like Yfh1. Like Yfh1, Isu1Sup also promoted cysteine interaction with PLP already bound to the enzyme as judged by enhanced persulfide formation. Thus, Yfh1 and Isu1Sup appear to act in a very similar manner to enhance substrate binding by the enzyme.
Yfh1 or Isu1Sup Induces Accessibility of the Substrate-binding Sites of Purified Nfs1 for Efficient Interaction with Cysteine
For the experiments described above, we used a low concentration of [35S]cysteine (0.1 μm). The enhanced substrate binding to Nfs1 in the presence of Yfh1 or Isu1Sup under these conditions could be mediated by enhanced affinity of Nfs1 for cysteine or by exposure of new binding sites. To distinguish between these possibilities, any exposed cysteine-binding sites on Nfs1 were first blocked by incubation with unlabeled cysteine at 100–1000-fold higher concentrations (10–100 μm) (first step). The protein was recovered by ammonium sulfate precipitation, removing excess unlabeled cysteine. When this treated Nfs1 was incubated with [35S]cysteine (0.1 μm) and Isd11 (second step), as expected, no radiolabeled persulfide was detected because all of the exposed cysteine-binding sites were blocked (Fig. 7, lane 2). Surprisingly, radiolabeled persulfide formation was again detected when Yfh1 (Fig. 7, lanes 3, 4, 7, and 8) or Isu1Sup (lanes 5, 6, 9, and 10) was included in the second step, and the signal was enhanced with increasing concentrations of Yfh1 or Isu1Sup.
FIGURE 7.
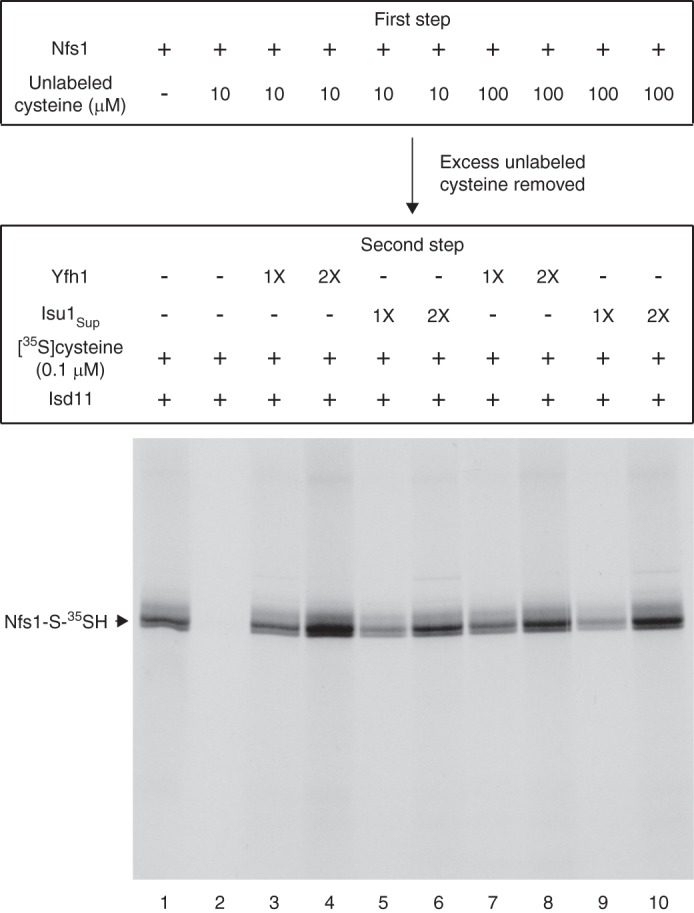
Yfh1 or Isu1Sup exposes the substrate-binding sites of purified Nfs1 for effective interaction with cysteine. Nfs1 (100 ng) was incubated at 30 °C for 15 min with unlabeled cysteine (10 or 100 μm) in HS buffer containing NaCl (0.15 m) and PLP (0.15 mm) (first step). After ammonium sulfate precipitation, the protein pellets were solubilized with HS buffer plus 0.15 m NaCl, and Yfh1 or Isu1Sup (1× = 50 ng) was added as indicated. Samples were incubated with [35S]cysteine (0.1 μm; 5 μCi), PLP (0.15 mm), and Isd11 (100 ng) at 30 °C for 15 min (second step) and then analyzed by nonreducing SDS-PAGE and autoradiography.
We conclude that the substrate-binding site of purified Nfs1 is not readily accessible for interaction with the substrate cysteine and that an interaction of Yfh1 or Isu1Sup with Nfs1 exposes the “buried” substrate-binding sites for efficient interaction with the substrate. A conformational change in the enzyme induced by Yfh1 or Isu1Sup interaction would be consistent with such a notion. In the absence of Yfh1 or Isu1Sup, unlabeled cysteine even at the 100 μm physiological concentration (16) did not have access to the buried sites and hence failed to occupy these sites. The buried sites became exposed only after addition of Yfh1 (or Isu1Sup), and [35S]cysteine at a concentration as low as 0.1 μm was then able to interact with these newly exposed sites. These conclusions were further substantiated by additional experiments as follows.
Nfs1 was incubated with unlabeled cysteine (10 μm) and PLP in the presence of Yfh1 (first step). Under these conditions, all of the Nfs1 substrate-binding sites, including the buried sites, should be exposed, and consequently, all sites should be occupied by unlabeled cysteine. Proteins were precipitated with ammonium sulfate to remove free unlabeled cysteine and PLP. The protein pellets containing Nfs1 with bound unlabeled cysteine and Yfh1 were then dissolved in buffer, and the reaction mixtures were again supplemented with fresh Yfh1. Samples were then incubated with increasing concentrations of [35S]cysteine to allow for any possible exchange between Nfs1-bound (unlabeled) and newly added free (radiolabeled) substrates that might be mediated by Yfh1 (second step). However, no Nfs1-S-35SH was detected when these samples were incubated with Isd11 (Fig. 8A, lanes 5–7). Thus, Yfh1 failed to mediate any exchange between Nfs1-bound (unlabeled) and unbound (radiolabeled) substrates.
FIGURE 8.

Yfh1 or Isu1Sup does not promote exchange of Nfs1-bound substrate with free substrate. A, Nfs1 (100 ng) was supplemented with unlabeled cysteine (10 μm) and/or Yfh1 (100 ng) as indicated. All reaction mixtures contained PLP (0.15 mm). Samples were incubated at 30 °C for 15 min (first step). After ammonium sulfate precipitation, the protein pellets were solubilized with HS buffer plus 0.15 m NaCl, and Yfh1 (100 ng) was added to some samples as indicated. Following addition of [35S]cysteine (1× = 0.1 μm; 5 μCi), reaction mixtures were incubated at 30 °C for 10 min prior to addition of Isd11 (100 ng). The incubation at 30 °C was continued for another 15 min (second step). Following TCA precipitation, proteins were analyzed by nonreducing SDS-PAGE and autoradiography. B, same as in A except Yfh1 was replaced with Isu1Sup in both first and second steps of the assay.
For a direct comparison side by side with the data presented in Fig. 7, Nfs1 was incubated with unlabeled cysteine in the absence of Yfh1 to block only the exposed, and not the buried, substrate-binding sites (first step). Following removal of excess unlabeled cysteine, samples were incubated with [35S]cysteine and Isd11, in the absence or presence of Yfh1 (second step). Nfs1-S-35SH was detected only when Yfh1 was included in the second step (Fig. 8A, compare lanes 3 and 4). As discussed above, this radiolabeled persulfide formation cannot be due to exchange of Nfs1-bound unlabeled substrate with newly added free [35S]cysteine. Most likely, in the presence of Yfh1 during the second step, new substrate-binding sites became available to interact with [35S]cysteine. The radiolabeled substrate thus bound to newly exposed sites was subsequently utilized for Nfs1-S-35SH formation.
As an additional control, Nfs1 was incubated with or without Yfh1, and unlabeled cysteine was omitted (first step). Following ammonium sulfate precipitation, the protein pellets containing either Nfs1 alone (Fig. 8A, lane 1) or both Nfs1 and Yfh1 (lane 2) were dissolved in buffer and incubated with [35S]cysteine and Isd11 (second step). As expected, the presence of Yfh1 greatly stimulated persulfide formation (Fig. 8A, compare lanes 1 and 2). Importantly, very similar results were obtained when the same experiment was performed with Isu1Sup, replacing Yfh1 in the corresponding samples (Fig. 8B). Together, these results strongly suggest that an interaction of Yfh1 with Nfs1 induces the accessibility of the enzyme's substrate-binding sites, thereby promoting enhanced substrate binding that can be productively used for subsequent persulfide formation. In the context of stimulating substrate binding to purified Nfs1, Isu1Sup behaves just like Yfh1.
Yfh1 Stimulates Cysteine Interaction with Nfs1 in Mitochondrial Lysate
Very recently, we identified the Nfs1-bound persulfide in isolated mitochondria that represents a genuine intermediate en route to Fe-S cluster synthesis (21, 27). Fe-S cluster synthesis in isolated mitochondria requires addition of nucleotides such as ATP, GTP, and NADH (29–31). However, these nucleotides are not needed for persulfide formation by the purified Nfs1·Isd11 complex (e.g. see Fig. 1B) or by the enzyme complex in mitochondria (21, 27). We therefore performed experiments with nucleotide-depleted mitochondria to minimize transfer of the persulfide sulfur from Nfs1 to downstream recipients and to “trap” the persulfide on the enzyme. To optimize conditions, initial experiments were done with mitochondria isolated from wild-type (WT) and an nfs1 mutant (MA14) yeast strains. MA14 retained Nfs1 expression comparable with wild-type levels (27) and yet lacked Fe-S clusters because of greatly compromised Nfs1 activity resulting from the hypomorphic mutation (20). Following nucleotide depletion, mitochondrial membranes were disrupted by freeze/thaw and mild bath sonication. The mitochondrial lysates thus generated were incubated with [35S]cysteine and analyzed by nonreducing SDS-PAGE and autoradiography (Fig. 9A). As reported previously (27), a major radiolabeled protein (51 kDa) corresponding to Nfs1-S-35SH was detected in wild-type (WT) and not in MA14 mitochondria. Treatment with DTT prior to SDS-PAGE released the persulfide sulfur, and no radiolabeled Nfs1 was detected in WT mitochondria. Several other radiolabeled proteins were detected in both WT and MA14 mitochondria. The patterns were very similar even though MA14 mitochondria had very little Nfs1 activity, and hence these Nfs1-independent radiolabeled bands were considered to be background bands. The Nfs1-independent radiolabeling of the background bands in mitochondria could be due to cysteinylation or H2S formation and protein S-sulfhydration of these proteins (35–37). This notion is supported by the observation that the background radiolabeling was completely abolished when mitochondria were preincubated with unlabeled cysteine, washed, and then incubated with [35S]cysteine (see Fig. 10). In any case, results show that Nfs1-S-35SH generated by mitochondrial lysates can be identified with certainty.
FIGURE 9.

Addition of purified Yfh1 enhances cysteine binding to Nfs1 in mitochondrial lysate. A, mitochondria were isolated from a wild-type (WT) and a hypomorphic nfs1 mutant (MA14) yeast strains. Intact mitochondria (200 μg of proteins) in HS buffer were incubated at 30 °C for 10 min to deplete endogenous nucleotides. Mitochondria were recovered by centrifugation and resuspended in 20 mm Hepes/KOH, pH 7.5. Mitochondrial membranes were ruptured, and the lysate thus obtained was supplemented with [35S]cysteine (0.1 μm; 10 μCi), NaCl (0.15 m), KOAc (40 mm), and Mg(OAc)2 (10 mm). The final volume was 100 μl. After incubation at 30 °C for 15 min, proteins were precipitated with 10% TCA and analyzed by nonreducing SDS-PAGE and autoradiography. B, mitochondria were isolated from the Gal-Isd11 strain after a 9 h depletion of Isd11 in galactose-free medium. Intact Isd11-depleted mitochondria (Isd11▾) in HS buffer were incubated at 30 °C for 10 min and recovered, and then membranes were ruptured under hypotonic conditions. The lysate was supplemented with Yfh1 (1× = 50 ng) as indicated, and [35S]cysteine (0.1 μm; 10 μCi) was added. The reaction mixture was adjusted to 100 μl with final concentrations of 20 mm Hepes/KOH, pH 7.5, 0.15 m NaCl, 40 mm KOAc, and 10 mm Mg(OAc)2 and incubated at 30 °C for 15 min (first step). Proteins were precipitated with ammonium sulfate (final 67%) and centrifuged to remove free [35S]cysteine. Protein pellets with bound [35S]cysteine were resuspended in 20 mm Hepes/KOH, pH 7.5, 0.15 m NaCl, 40 mm KOAc, and 10 mm Mg(OAc)2. Isd11 (100 ng) was added, and samples were incubated at 30 °C for 15 min, with or without added Yfh1 (second step). Proteins were precipitated with 10% TCA and analyzed by nonreducing SDS-PAGE and autoradiography.
FIGURE 10.
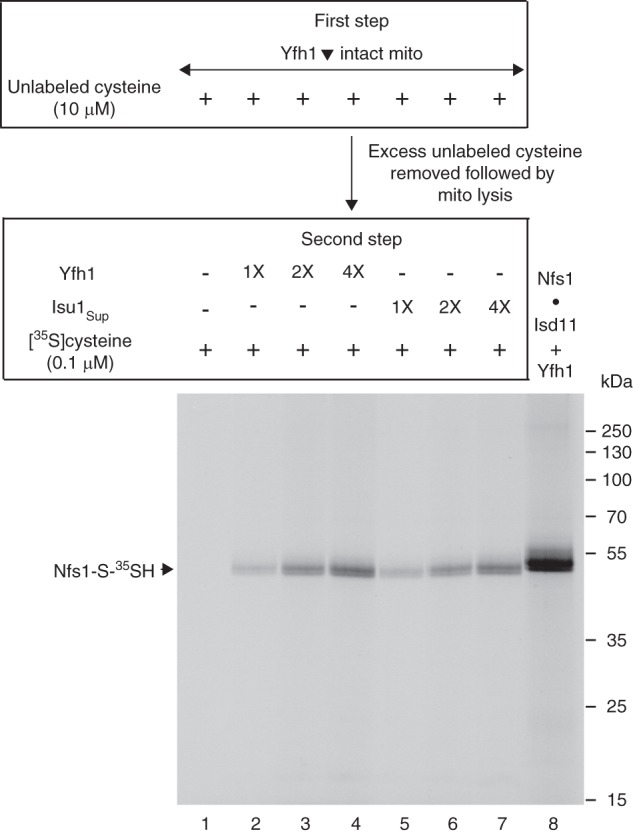
Addition of purified Yfh1 or Isu1Sup exposes the substrate-binding sites of Nfs1 for effective interaction with cysteine in mitochondrial lysate. Mitochondria were isolated from the Gal-Yfh1 strain after an 18 h depletion of Yfh1 in galactose-free medium. Intact Yfh1-depleted mitochondria (Yfh1▾; 200 μg of proteins) in HS buffer were incubated at 30 °C for 10 min in the presence of unlabeled cysteine (10 μm) (first step). After dilution with HS buffer, mitochondria were recovered by centrifugation, removing excess and free cysteine. The mitochondrial pellet was resuspended in 20 mm Hepes/KOH, pH 7.5, and membranes were ruptured. The lysate thus obtained was supplemented with purified Yfh1 or Isu1Sup (1× = 50 ng) as indicated. The reaction mixtures were adjusted to 100 μl with final concentrations of 20 mm Hepes/KOH, pH 7.5, 0.1 μm [35S]cysteine (10 μCi), 0.15 m NaCl, 40 mm KOAc, and 10 mm Mg(OAc)2, and samples were incubated at 30 °C for 15 min (second step) (lanes 1–7). As control (lane 8), a mixture of purified Nfs1·Isd11 complex (100 ng) and Yfh1 (100 ng) was incubated with 0.1 μm [35S]cysteine (5 μCi) and 0.15 mm PLP in a final volume of 50 μl. Proteins were precipitated with 10% TCA and analyzed by nonreducing SDS-PAGE followed by autoradiography.
To evaluate cysteine binding to Nfs1 independent of persulfide formation in mitochondria, the persulfide-forming activity had to be blocked. This was accomplished by in vivo depletion of Isd11. The Gal-Isd11 strain was shifted from an inducing carbon source (galactose) to a noninducing carbon source (raffinose). After 9 h, Isd11 was found to be severely depleted with only a marginal decrease in Nfs1 protein level (21). Mitochondria were isolated from wild-type (WT) and Isd11-depleted (Isd11▾) cells. After depletion of endogenous nucleotides, mitochondrial membranes were ruptured as described above. Assays were performed in two steps. In the first step, the mitochondrial lysate was incubated with [35S]cysteine in the absence or presence of added Yfh1. Proteins were precipitated with ammonium sulfate and centrifuged, removing excess and free [35S]cysteine. The protein pellets were resuspended/solubilized in buffer and incubated with Isd11 in the absence or presence of added Yfh1, with no further addition of [35S]cysteine. Samples were analyzed by nonreducing SDS-PAGE and autoradiography (Fig. 9B). When purified Yfh1 was added in the first step, the Nfs1-S-35SH signal was increased in a manner that was dependent on concentrations of added Yfh1 (Fig. 9B, lanes 3–5). The stimulatory effect was highly specific for Nfs1, and there was practically no change in radiolabeling of background bands. Furthermore, Yfh1 added during the second step had no effect on the Nfs1-bound persulfide signal (Fig. 9B, compare lane 2 with lanes 6–8). As expected, persulfide formation was strictly dependent on Isd11 addition in the second step, and the Nfs1-S-35SH signal was observed only when Isd11 was included (Fig. 9B, compare lanes 1 and 2). In the absence of added Isd11, no radiolabeled Nfs1 was detected, but nonspecific radiolabeling of background bands persisted (Fig. 9B, lane 1) as in MA14 nfs1 mutant mitochondria (Fig. 9A, lane 1).
The results presented in Fig. 9B can be interpreted as follows. In the presence of increasing concentrations of Yfh1 added during the first step, increasing amounts of [35S]cysteine bound to Nfs1 through interactions with PLP in the substrate-binding site. In Isd11-depleted mitochondrial extract, the bound substrate could not be used for persulfide formation. Nfs1 with noncovalently bound [35S]cysteine was not detected by SDS gels because SDS released the radioactive substrate from the enzyme. Addition of Isd11 to the lysate during the second step induced Nfs1 with prebound [35S]cysteine to form the covalent Nfs1-S-35SH persulfide that was detected by nonreducing SDS gels. The persulfide signal was proportional to the amount of prebound [35S]cysteine. Yfh1 included in the first step promoted [35S]cysteine binding to Nfs1, and hence these samples exhibited enhanced persulfide signal. Yfh1 added during the second step (free [35S]cysteine absent) had no effects on persulfide formation. We conclude that Yfh1 acts specifically on the Nfs1 substrate-binding step and not on the subsequent persulfide-forming step in mitochondria.
Yfh1 or Isu1Sup Induces Accessibility of the Substrate-binding Sites of Nfs1 for Efficient Interaction with Cysteine in Mitochondrial Lysate
The experiment (Fig. 9B) described above was performed with purified Yfh1 and Isd11-depleted mitochondria. However, endogenous Yfh1 was still present in Isd11-depleted mitochondria. To more thoroughly ascertain the effects of Yfh1 on Nfs1 in mitochondria, experiments must therefore be performed with mitochondria lacking endogenous Yfh1. For this purpose, the Gal-Yfh1 strain was grown under noninducing conditions for 18 h (28). Under these conditions, there was no detectable Yfh1 as judged by immunoblotting. Mitochondria were isolated from these Yfh1-depleted (Yfh1▾) cells and used in a two-step assay as follows. Following depletion of endogenous nucleotides, intact mitochondria were incubated with unlabeled cysteine (10 μm) to block any exposed cysteine-binding sites on Nfs1 (first step). After dilution with isotonic buffer, mitochondria were recovered by centrifugation, removing excess cysteine. Mitochondrial membranes were ruptured, and the lysate was incubated with [35S]cysteine (0.1 μm) in the absence or presence of Yfh1 or Isu1Sup. Proteins were precipitated and analyzed by nonreducing SDS-PAGE and autoradiography (Fig. 10). When mitochondria were pretreated with unlabeled cysteine during the first step, all of the exposed cysteine-binding sites of Nfs1 were blocked. Consequently, these mitochondria completely failed to generate Nfs1-S-35SH when incubated with [35S]cysteine in the second step (Fig. 10, lane 1). However, [35S]persulfide formation on Nfs1 occurred when Yfh1 (Fig. 10, lanes 2–4) or Isu1Sup (lanes 5–7) was included in the second step. The signal was enhanced with increasing concentrations of Yfh1 or Isu1Sup, and the effects were comparable. As indicated above (see Fig. 9), incubation of mitochondria with [35S]cysteine gave rise to nonspecific labeling independent of Nfs1 activity. These background bands were completely abolished by preincubation with unlabeled cysteine during the first step of this assay (Fig. 10, lanes 1–7). With the addition of Yfh1 or Isu1Sup during the second step, [35S]persulfide formation on Nfs1 occurred, but practically no background radiolabeling was observed. Thus, the observed stimulatory effect of Yfh1 or Isu1Sup on Nfs1 is quite specific even in total mitochondrial lysate. The purified Nfs1·Isd11 complex incubated with [35S]cysteine in the presence of Yfh1 served as positive control (Fig. 10, lane 8). The Nfs1-S-35SH generated by the purified complex contained the His6 tag and migrated close to but slightly slower than the radiolabeled Nfs1 containing no His6 tag that was generated in mitochondria.
We conclude that as in the case for purified Nfs1, Yfh1 plays a critical role in Nfs1 activity in mitochondria as well. An interaction of Yfh1 with Nfs1 makes the substrate-binding sites of the enzyme available for efficient binding of cysteine. Isu1Sup mimics the effects of Yfh1 on Nfs1 substrate binding consistent with its Yfh1-bypassing activity in cells and mitochondria.
DISCUSSION
Fe-S cluster biogenesis in mitochondria is essential for cell survival, and the process is conserved from yeast to humans (2). During the biosynthetic process, Fe-S cluster intermediates are first formed on the Isu scaffold and then transferred to recipient apoproteins, forming holoproteins. Key components required for the synthesis of Fe-S cluster intermediates on Isu include the Nfs1 cysteine desulfurase, the accessory protein Isd11, and frataxin. The enzyme Nfs1 binds the substrate cysteine through the cofactor PLP, subsequently removing sulfur from the substrate and forming a covalent persulfide on the active site cysteine. The persulfide sulfur is then transferred to the Isu scaffold for Fe-S cluster synthesis. Here, we show for the first time that yeast frataxin (Yfh1) by itself directly and specifically stimulates binding of the substrate cysteine to Nfs1. An interaction of Yfh1 with Nfs1 induces exposure of substrate-binding sites, which are otherwise mostly buried. The lack of this effect may explain, at least in part, deficiency of Fe-S clusters in mitochondria/cells associated with frataxin/Yfh1 deficiency.
Several important findings on the regulatory effects of frataxin/Yfh1 on Nfs1 as presented here follow. 1) Purified Yfh1 by itself stimulates the persulfide-forming activity of the purified Nfs1·Isd11 complex (Fig. 2A). 2) Yfh1 specifically enhances substrate binding to Nfs1 (Fig. 3) rather than persulfide formation by the enzyme from already bound substrate (Fig. 4). The stimulatory effect of Yfh1 on substrate binding to Nfs1 is independent of Isu1 or Isd11. 3) Although Yfh1 is an iron-binding protein (34), its stimulatory role on Nfs1 substrate binding does not require iron (Fig. 5). 4) The Yfh1-mediated stimulation of cysteine interaction with Nfs1 is not due to enhanced PLP binding by the enzyme. Nfs1 by itself is capable of acquiring PLP in the absence of Yfh1 or any other proteins. However, efficient interaction of cysteine with Nfs1-bound PLP requires the presence of Yfh1 (Fig. 6A). 5) Yfh1 promotes accessibility of the buried substrate-binding sites of Nfs1 for efficient interaction with cysteine (Fig. 7). When these newly exposed sites are saturated by cysteine, Yfh1 does not facilitate exchange of Nfs1-bound cysteine with free cysteine (Fig. 8A). 6) Until now, a direct role of frataxin/Yfh1 in Nfs1 activity in mitochondria has never been demonstrated. We find that as in the case for purified Nfs1, Yfh1 also exhibits similar specific effects on Nfs1 in mitochondria. These include enhanced cysteine binding to Nfs1 (Fig. 9), likely resulting from increased accessibility of the substrate-binding sites induced by Yfh1 (Fig. 10). 7) Finally, Isu1Sup mimics the stimulatory effects of Yfh1 on Nfs1 (Figs. 2B, 3, 5, 6B, 7, 8B, and 10) consistent with its ability to restore Fe-S cluster biogenesis in Δyfh1 mitochondria to a significant extent (22). The Yfh1-bypassing activity of Isu1Sup may therefore be attributed, at least in part, to its stimulatory effects on Nfs1 activity required for Fe-S cluster synthesis.
The stoichiometry of Nfs1 and Yfh1 interaction under physiological conditions in mitochondria is unknown. In some of our assays using purified proteins, Yfh1 was present in molar excess over Nfs1, and increasing the Yfh1/Nfs1 molar ratio enhanced substrate binding to Nfs1 (e.g. Fig. 3). A possible explanation for this finding is that some of the purified Yfh1 may have become inactivated, due to misfolding/aggregation over time. Previous studies have shown that Yfh1 binds two atoms of ferrous iron per molecule of the protein with low affinity (∼3 μm) (34). The bacterially expressed Yfh1 was purified in apo-form, i.e. without metal bound, as indicated by a colorimetric assay using the chelator, bathophenanthroline disulfonic acid (38) (data not shown). Thus, this purified apo-Yfh1 may have been more susceptible to inactivation, as has been described (34). In any case, the stimulatory effect of Yfh1 on substrate binding to Nfs1 does not depend on iron availability (Fig. 5). In addition to stimulating the substrate binding to Nfs1 (iron-independent role), Yfh1 may have iron-requiring functions as well. For example, iron-loaded Yfh1, and not apo-Yfh1, may interact with the Isu1 scaffold and such a process may allow transfer of iron from Yfh1 to Isu1 for Fe-S cluster synthesis (39).
In a previous study using the methylene blue colorimetric assay, addition of frataxin to a mixture of Nfs1, Isd11, Isu, and iron was found to stimulate the cysteine desulfurase activity (16). The assay measures continuous sulfide production from unlabeled cysteine in the presence of DTT. In this assay, frataxin by itself did not exhibit any effects on the basal cysteine desulfurase activity of the Nfs1·Isd11 complex in the absence of Isu. Thus, a direct role of frataxin on cysteine desulfuration could not be ascertained. Likewise, the cysteine binding cannot be separated from the subsequent persulfide formation and/or release of persulfide sulfur in these assays. In addition, these assays are too insensitive and cannot be used to determine the cysteine desulfurase activity in mitochondrial extracts (21, 27). For example, mitochondria almost entirely lacking Nfs1 exhibit strong sulfide-forming activity. The use of highly sensitive radioactive assays as described here overcomes all of these inherent problems associated with the spectrophotometric assays, allowing us to identify a novel role of frataxin/Yfh1 in initiating the cysteine desulfurase activity of Nfs1.
The best explanation for the data presented here is that an interaction of frataxin/Yfh1 with Nfs1 induces a conformational change in the enzyme, exposing the buried substrate-binding sites for interaction with cysteine (Fig. 11). In the structure of a bacterial cysteine desulfurase complex (40), the PLP in the substrate-binding site is found buried deep inside a tunnel at some distance from the hidden entry point for substrate access. One can imagine a conformational change induced by frataxin binding that leads to opening and exposure of this tunnel. There is as yet no solved structure for any eukaryotic cysteine desulfurase or cysteine desulfurase complex. However, the binding sites for Yfh1 and Isu1 on Nfs1 do not appear to overlap, because both can join in higher order complex formation (16). Yfh1 and Isu1Sup may interact with Nfs1 at different sites, and yet they are able to exert similar stimulatory effects on the enzyme. Surprisingly, Yfh1mut blocked not only Yfh1-mediated stimulation but also Isu1Sup-mediated stimulation of persulfide formation on Nfs1 (Fig. 2C). Furthermore, addition of Isu1 did not seem to interfere with the stimulatory effects of Isu1Sup on persulfide formation by the Nfs1·Isd11 complex (Fig. 2B). It is possible that unlike the wild-type Isu1, Isu1Sup interacts with Nfs1 at or close to the Yfh1-binding site of the enzyme, thereby inducing conformational changes required for substrate binding just like Yfh1. Alternatively, Isu1Sup may bind Nfs1 at the usual Isu1 site, leading to propagation of a conformational change in the Yfh1 interaction site. The Yfh1 interaction site in effect may be acting as a downstream trigger for the Isu1Sup effect. Thus, if this site on Nfs1 is blocked by Yfh1mut, activation by Isu1Sup will not occur. Much work and new structural information regarding the protein complexes will be needed to determine whether the conformational change induced by frataxin/Yfh1 or Isu1Sup is triggered by distinct or same/close interaction sites on Nfs1. Finally, the changes induced by frataxin or Isu1Sup binding do not result in persulfide formation. An additional conformational change of Nfs1, induced by Isd11 binding, is required to bring the bound substrate in proximity to the active site cysteine for persulfide formation (Fig. 11) (21). The persulfide sulfur can then be utilized for Fe-S cluster synthesis, tRNA thiolation, and other processes.
FIGURE 11.
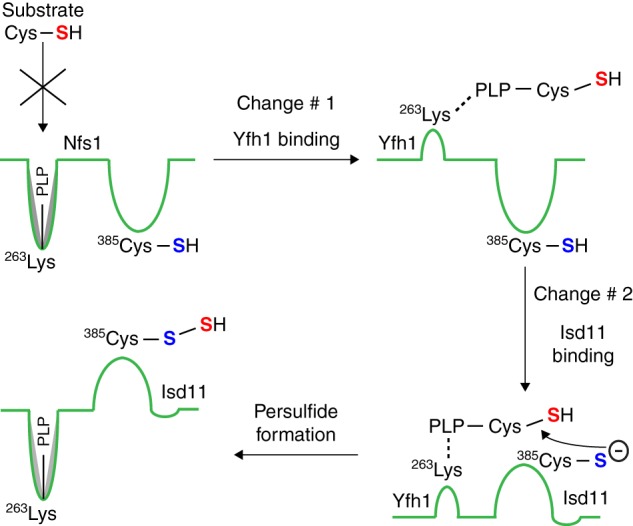
Model for two-tier regulation of persulfide formation by Nfs1. The enzyme Nfs1 is shown by a green line. The Lys263 residue of the Nfs1 substrate-binding site forms a Schiff base with the cofactor PLP. Nfs1 by itself inefficiently binds the substrate cysteine (Cys-SH, sulfur indicated in red). Frataxin/Yfh1 interacts with Nfs1, inducing a conformational change in the enzyme (Change #1) and exposing new sites with Lys263-PLP, now able to efficiently bind the substrate cysteine. Isd11 then interacts with Nfs1 with substrate cysteine already bound, inducing a second conformational change (Change #2), and bringing the substrate cysteine (red sulfur) into proximity to the Cys385 (sulfur indicated in blue) in the peptide backbone of the Nfs1 active site loop. A nucleophilic attack by the thiolate anion of the active site Cys385 extracts the –SH group of the substrate, forming a covalent persulfide (Nfs1-S-SH). The substrate cysteine bound to PLP after Change #1 is shown as an external aldimine. However, this has not been shown experimentally, and it is possible that at this stage the substrate cysteine interacts with PLP still bound to the Lys263 residue of Nfs1 as an internal aldimine.
Friedreich ataxia is a neurodegenerative disease associated with frataxin deficiency (4). Currently, there is no cure for the disease, and some of the developing therapeutic approaches rely on increasing frataxin protein level or decreasing iron toxicity (41). In principle, the data presented here may lead to a new therapeutic approach. For example, a small molecule or an Isu1Sup mimic capable of exposing the buried cysteine-binding sites of the cysteine desulfurase perhaps could be developed into a drug for treating Friedreich ataxia.
Acknowledgments
We thank Elise R. Lyver for generating some of the constructs used for bacterial expression of proteins and Heeyong Yoon for evaluating iron content of purified Yfh1.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1AG030504 from NIA (to D. P.), RO1DK068139 (to T. L. S.), and R37DK053953 (to A. D.).
This article was selected as a Paper of the Week.
A. Dancis, unpublished observations.
- Yfh1
- yeast frataxin homolog
- Isu1Sup
- Isu1 suppressor that contains a single amino acid substitution (methionine to isoleucine) at position 107 in the mature form of the protein
- Nfs1·Isd11
- a complex of Nfs1-His6 and Isd11 formed in vivo in bacteria
- PLP
- pyridoxal phosphate
- Ni-NTA
- nickel-nitrilotriacetic acid.
REFERENCES
- 1. Stemmler T. L., Lesuisse E., Pain D., Dancis A. (2010) Frataxin and mitochondrial Fe-S cluster biogenesis. J. Biol. Chem. 285, 26737–26743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rouault T. A. (2012) Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis. Model. Mech. 5, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lill R., Hoffmann B., Molik S., Pierik A. J., Rietzschel N., Stehling O., Uzarska M. A., Webert H., Wilbrecht C., Mühlenhoff U. (2012) The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 1823, 1491–1508 [DOI] [PubMed] [Google Scholar]
- 4. Campuzano V., Montermini L., Moltò M. D., Pianese L., Cossée M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A., Zara F., Cañizares J., Koutnikova H., Bidichandani S. I., Gellera C., Brice A., Trouillas P., De Michele G., Filla A., De Frutos R., Palau F., Patel P. I., Di Donato S., Mandel J. L., Cocozza S., Koenig M., Pandolfo M. (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427 [DOI] [PubMed] [Google Scholar]
- 5. Rötig A., de Lonlay P., Chretien D., Foury F., Koenig M., Sidi D., Munnich A., Rustin P. (1997) Aconitase and mitochondrial iron-sulfur protein deficiency in Friedreich ataxia. Nat. Genet. 17, 215–217 [DOI] [PubMed] [Google Scholar]
- 6. Sanchez-Casis G., Cote M., Barbeau A. (1976) Pathology of the heart in Friedreich's ataxia: review of the literature and report of one case. Can. J. Neurol. Sci. 3, 349–354 [DOI] [PubMed] [Google Scholar]
- 7. Whitnall M., Suryo Rahmanto Y., Huang M. L., Saletta F., Lok H. C., Gutiérrez L., Lázaro F. J., Fleming A. J., St Pierre T. G., Mikhael M. R., Ponka P., Richardson D. R. (2012) Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc. Natl. Acad. Sci. U.S.A. 109, 20590–20595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Babcock M., de Silva D., Oaks R., Davis-Kaplan S., Jiralerspong S., Montermini L., Pandolfo M., Kaplan J. (1997) Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 276, 1709–1712 [DOI] [PubMed] [Google Scholar]
- 9. Foury F. (1999) Low iron concentration and aconitase deficiency in a yeast frataxin homologue deficient strain. FEBS Lett. 456, 281–284 [DOI] [PubMed] [Google Scholar]
- 10. Mühlenhoff U., Richhardt N., Ristow M., Kispal G., Lill R. (2002) The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum. Mol. Genet. 11, 2025–2036 [DOI] [PubMed] [Google Scholar]
- 11. Wilson R. B., Roof D. M. (1997) Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue. Nat. Genet. 16, 352–357 [DOI] [PubMed] [Google Scholar]
- 12. Cavadini P., Gellera C., Patel P. I., Isaya G. (2000) Human frataxin maintains mitochondrial iron homeostasis in Saccharomyces cerevisiae. Hum. Mol. Genet. 9, 2523–2530 [DOI] [PubMed] [Google Scholar]
- 13. Cook J. D., Kondapalli K. C., Rawat S., Childs W. C., Murugesan Y., Dancis A., Stemmler T. L. (2010) Molecular details of the yeast frataxin-Isu1 interaction during mitochondrial Fe-S cluster assembly. Biochemistry 49, 8756–8765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Colin F., Martelli A., Clémancey M., Latour J. M., Gambarelli S., Zeppieri L., Birck C., Page A., Puccio H., Ollagnier de Choudens S. (2013) Mammalian frataxin controls sulfur production and iron entry during de novo Fe(4)S(4) cluster assembly. J. Am. Chem. Soc. 135, 733–740 [DOI] [PubMed] [Google Scholar]
- 15. Vaubel R. A., Isaya G. (2013) Iron-sulfur cluster synthesis, iron homeostasis, and oxidative stress in Friedreich ataxia. Mol. Cell. Neurosci. 55, 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsai C.-L., Barondeau D. P. (2010) Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry 49, 9132–9139 [DOI] [PubMed] [Google Scholar]
- 17. Schmucker S., Martelli A., Colin F., Page A., Wattenhofer-Donzé M., Reutenauer L., Puccio H. (2011) Mammalian frataxin: An essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS One 6, e16199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang T., Craig E. A. (2008) Binding of yeast frataxin to the scaffold for Fe-S cluster biogenesis, Isu. J. Biol. Chem. 283, 12674–12679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gerber J., Mühlenhoff U., Lill R. (2003) An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S-cluster synthesis on Isu1. EMBO Rep. 4, 906–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li J., Kogan M., Knight S. A., Pain D., Dancis A. (1999) Yeast mitochondrial protein, Nfs1p, coordinately regulates iron-sulfur cluster proteins, cellular iron uptake, and iron distribution. J. Biol. Chem. 274, 33025–33034 [DOI] [PubMed] [Google Scholar]
- 21. Pandey A., Golla R., Yoon H., Dancis A., Pain D. (2012) Persulfide formation on mitochondrial cysteine desulfurase: Enzyme activation by a eukaryote-specific interacting protein and Fe-S cluster synthesis. Biochem. J. 448, 171–187 [DOI] [PubMed] [Google Scholar]
- 22. Yoon H., Golla R., Lesuisse E., Pain J., Donald J. E., Lyver E. R., Pain D., Dancis A. (2012) Mutation in the Fe-S scaffold protein Isu bypasses frataxin deletion. Biochem. J. 441, 473–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Naamati A., Regev-Rudzki N., Galperin S., Lill R., Pines O. (2009) Dual targeting of Nfs1 and discovery of its novel processing enzyme, Icp55. J. Biol. Chem. 284, 30200–30208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gordon D. M., Kogan M., Knight S. A., Dancis A., Pain D. (2001) Distinct roles for two N-terminal cleaved domains in mitochondrial import of the yeast frataxin homolog, Yfh1p. Hum. Mol. Genet. 10, 259–269 [DOI] [PubMed] [Google Scholar]
- 25. Wiedemann N., Urzica E., Guiard B., Müller H., Lohaus C., Meyer H. E., Ryan M. T., Meisinger C., Mühlenhoff U., Lill R., Pfanner N. (2006) Essential role of Isd11 in mitochondrial iron-sulfur cluster synthesis on Isu scaffold proteins. EMBO J. 25, 184–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adam A. C., Bornhövd C., Prokisch H., Neupert W., Hell K. (2006) The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. EMBO J. 25, 174–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pandey A., Yoon H., Lyver E. R., Dancis A., Pain D. (2012) Identification of a Nfs1p-bound persulfide intermediate in Fe-S cluster synthesis by intact mitochondria. Mitochondrion 12, 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lesuisse E., Santos R., Matzanke B. F., Knight S. A., Camadro J.-M., Dancis A. (2003) Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1). Hum. Mol. Genet. 12, 879–889 [DOI] [PubMed] [Google Scholar]
- 29. Pain J., Balamurali M. M., Dancis A., Pain D. (2010) Mitochondrial NADH kinase, Pos5p, is required for efficient iron-sulfur cluster biogenesis in Saccharomyces cerevisiae. J. Biol. Chem. 285, 39409–39424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Amutha B., Gordon D. M., Dancis A., Pain D. (2009) Nucleotide-dependent iron-sulfur cluster biogenesis of endogenous and imported apoproteins in isolated intact mitochondria. Methods Enzymol. 456, 247–266 [DOI] [PubMed] [Google Scholar]
- 31. Amutha B., Gordon D. M., Gu Y., Lyver E. R., Dancis A., Pain D. (2008) GTP is required for iron-sulfur cluster biogenesis in mitochondria. J. Biol. Chem. 283, 1362–1371 [DOI] [PubMed] [Google Scholar]
- 32. Johnson D. C., Dean D. R., Smith A. D., Johnson M. K. (2005) Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 74, 247–281 [DOI] [PubMed] [Google Scholar]
- 33. Zheng L., White R. H., Cash V. L., Dean D. R. (1994) Mechanism for the desulfurization of l-cysteine catalyzed by the nifS gene product. Biochemistry 33, 4714–4720 [DOI] [PubMed] [Google Scholar]
- 34. Cook J. D., Bencze K. Z., Jankovic A. D., Crater A. K., Busch C. N., Bradley P. B., Stemmler A. J., Spaller M. R., Stemmler T. L. (2006) Monomeric yeast frataxin is an iron-binding protein. Biochemistry 45, 7767–7777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chouchani E. T., James A. M., Fearnley I. M., Lilley K. S., Murphy M. P. (2011) Proteomic approaches to the characterization of protein thiol modification. Curr. Opin. Chem. Biol. 15, 120–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hochgräfe F., Mostertz J., Pöther D.-C., Becher D., Helmann J. D., Hecker M. (2007) S-Cysteinylation is a general mechanism for thiol protection of Bacillus subtilis proteins after oxidative stress. J. Biol. Chem. 282, 25981–25985 [DOI] [PubMed] [Google Scholar]
- 37. Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., Snyder S. H. (2009) H2S signals through protein S-sulfhydration. Sci. Signal. 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lesuisse E., Lyver E. R., Knight S. A., Dancis A. (2004) Role of YHM1, encoding a mitochondrial carrier protein, in iron distribution of yeast. Biochem. J. 378, 599–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kondapalli K. C., Kok N. M., Dancis A., Stemmler T. L. (2008) Drosophila frataxin: An iron chaperone during cellular Fe-S cluster bioassembly. Biochemistry 47, 6917–6927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marinoni E. N., de Oliveira J. S., Nicolet Y., Raulfs E. C., Amara P., Dean D. R., Fontecilla-Camps J. C. (2012) (IscS-IscU)2 complex structures provide insights into Fe2S2 biogenesis and transfer. Angew. Chem. Int. Ed. Engl. 51, 5439–5442 [DOI] [PubMed] [Google Scholar]
- 41. Tsou A. Y., Friedman L. S., Wilson R. B., Lynch D. R. (2009) Pharmacotherapy for Friedreich ataxia. CNS Drugs 23, 213–223 [DOI] [PubMed] [Google Scholar]