

Background: PI3K regulates the surface expression of Kv7.1.
Results: Kv7.1 surface expression depends on PI3K activity in polarized epithelial cells. SGK1 is the primary downstream target of PI3K in this process, which involves inhibition of Nedd4-2-dependent endocytosis of the channel.
Conclusion: The surface expression of Kv7.1 is regulated by a PI3K-SGK1-Nedd4-2-mediated pathway.
Significance: This pathway could regulate Kv7.1 cell surface expression levels in epithelial cells and cardiac myocytes.
Keywords: E3 Ubiquitin Ligase, Endocytosis, Ion Channels, PI 3-Kinase (PI3K), Trafficking, KCNQ1
Abstract
Epithelial cell polarization involves several kinase signaling cascades that eventually divide the surface membrane into an apical and a basolateral part. One kinase, which is activated during the polarization process, is phosphoinositide 3-kinase (PI3K). In MDCK cells, the basolateral potassium channel Kv7.1 requires PI3K activity for surface-expression during the polarization process. Here, we demonstrate that Kv7.1 surface expression requires tonic PI3K activity as PI3K inhibition triggers endocytosis of these channels in polarized MDCK. Pharmacological inhibition of SGK1 gave similar results as PI3K inhibition, whereas overexpression of constitutively active SGK1 overruled it, suggesting that SGK1 is the primary downstream target of PI3K in this process. Furthermore, knockdown of the ubiquitin ligase Nedd4-2 overruled PI3K inhibition, whereas a Nedd4-2 interaction-deficient Kv7.1 mutant was resistant to both PI3K and SGK1 inhibition. Altogether, these data suggest that a PI3K-SGK1 pathway stabilizes Kv7.1 surface expression by inhibiting Nedd4-2-dependent endocytosis and thereby demonstrates that Nedd4-2 is a key regulator of Kv7.1 localization and turnover in epithelial cells.
Introduction
The potassium channel Kv7.1 (KCNQ1, KvLQT) plays an important role in a number of tissues where it associates with the auxiliary KCNE β-subunits. In the heart, Kv7.1, together with KCNE1, forms the delayed rectifier potassium current IKs, which is an important contributor to the repolarization of the cardiac action potential (1, 2). Kv7.1 is also present in pancreatic β-cells, where it is thought to be implicated in the regulation of insulin secretion (3, 4). In addition, Kv7.1 is expressed in several epithelia, where it is involved in salt and water transport (5, 6). Most importantly, the channel regulates gastric acid secretion (7, 8) and contributes to the release of potassium to the endolymph in the inner ear (9, 10). Accordingly, Kv7.1 knock-out mice show gastric hyperplasia and are completely deaf (11). Mutations in the KCNQ1 gene are furthermore associated with long QT (LQT)4 syndrome, an inherited form of cardiac arrhythmia that can lead to cardiac arrest (12). In its recessive form, the Jervell and Lange-Nielsen syndrome (13), the disease additionally leads to hearing loss due to disturbances in the flow of potassium in the inner ear. The mechanism underlying the LQT syndrome is reflected in a loss of Kv7.1 function, frequently originating from trafficking disorders, and hence a decrease in number of channels in the plasma membrane (14–16). Nevertheless, the molecular and cellular mechanisms controlling the cell surface expression of Kv7.1 in cardiomyocytes and epithelial cells are still largely unknown.
We recently observed that the basolateral Kv7.1 potassium channel displays a very dynamic localization pattern during Madin-Darby canine kidney (MDCK) cell polarization controlled by a calcium switch (17). We found that initiation of MDCK cell polarization results in removal and degradation of surface-expressed Kv7.1 and subsequent accumulation of newly synthesized channels in the endoplasmic reticulum (ER). Later in the polarization process, Kv7.1 is released from the ER, and surface expression is recovered. While the initial removal of Kv7.1 from the cell surface is mediated by the AMP-activated protein kinase and E3 ubiquitin ligase Nedd4-2 (neuronal precursor cell expressed developmentally down-regulated 4-2) (18), the subsequent recovery of Kv7.1 surface expression depends on PI3K activity (17). PI3K is an important kinase that is implicated in the control of a number of cellular processes including cell proliferation, cell survival, and epithelial cell polarization (19–22). It has in particular received a lot of attention in relation to human cancer as the kinase is one of the most common oncogenes (reviewed in Ref. 23). PI3K is composed of a regulatory subunit and a catalytic subunit that phosphorylates phosphatidylinositol 4,5-bisphosphate into phosphatidylinositol (3,4,5)-trisphosphate. Phosphatidylinositol (3,4,5)-trisphosphate is an important signaling molecule that binds proteins via a pleckstrin homology domain, which is found in e.g. 3-phosphoinositide-dependant-kinase 1 and the Akt kinase (also denoted protein kinase B) (24, 25). In polarizing MDCK cells, PI3K is activated by adherens junction assembly, resulting in Rac1-dependent changes in the actin cytoskeleton (26, 27). In polarized MDCK cells, adherens junctions are enriched in phosphatidylinositol (3,4,5)-trisphosphate suggesting that PI3K remains tonically active at this subcellular location (28). Furthermore, long term inhibition of PI3K reduces MDCK cell height, suggesting that tonic PI3K activity regulates basolateral membrane formation and maintenance (19, 28).
Two well described downstream targets of PI3K are the serum- and glucocorticoid-inducible kinase 1 (SGK1 (29)) and Akt (reviewed in Ref. 30). Both protein kinases have been reported to stimulate Kv7.1-KCNE1 currents in Xenopus oocytes (31, 32) and inhibit the actions of Nedd4-2 (33–35), another well known regulator of Kv7.1 (36). Nedd4-2 is an E3 ubiquitin ligase that ubiquitylates target membrane proteins such as ion channels, thereby increasing the rate of their internalization and degradation (37, 38). SGK1 and Akt can phosphorylate Nedd4-2, thus increasing the binding affinity to 14-3-3 proteins (39). For the epithelial sodium channel ENaC, it has been found that 14-3-3 protein binding to Nedd4-2 prevents Nedd4-2-mediated ubiquitylation and thereby increases surface expression levels of the channel (39, 40). Because the interaction of Nedd4-2 with both ENaC and Kv7.1 is mediated by intrinsic sequences known as PY motifs, it is possible that the interaction of Nedd4-2 with Kv7.1 is inhibited by the same phosphorylation of Nedd4-2. Overall, these observations position SGK1 and Akt as possible downstream targets of the PI3K effect upon Kv7.1 localization during the polarization process.
In this study, we demonstrate that a PI3K pathway stabilizes cell surface expression of Kv7.1 channels in both polarizing and polarized cells. We find that PI3K primarily acts through SGK1 and demonstrate that SGK1 controls Kv7.1 localization by inhibiting Nedd4-2-dependent endocytosis of the channel. Intriguingly, the PY motif Kv7.1 mutant that cannot interact with Nedd4-2 displayed increased expression in the apical membrane, suggesting that Nedd4-2-dependent endocytosis is important to avoid apical trapping of the channel during polarization. Overall, our data suggest that a PI3K-SGK1 pathway promotes cell surface stabilization of Kv7.1 by inhibiting Nedd4-2-dependent endocytosis and places Nedd4-2 as a key regulator of Kv7.1 surface expression and turnover.
EXPERIMENTAL PROCEDURES
Chemical Compounds
The activators and inhibitors used in this study were as follows: 52-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride (LY294002, Sigma-Aldrich); 2-cyclopentyl-4-(5-phenyl-1H-pyrrolo[2,3-b]pyridin-3-yl-benzoic acid (GSK650394, Tocris Bioscience); N4-(7-chloro-4-quinolinyl)-N1,N1-dimethyl-1,4-pentanediamine diphosphate salt (chloroquine, Sigma-Aldrich); Akt inhibitor IV (Santa Cruz Biotechnology); and cycloheximide (Sigma-Aldrich).
Antibodies
The antibodies used in this study were as follows: goat anti-Kv7.1 (1:50, C-20, Santa Cruz Biotechnology, Heidelberg, Germany), rabbit anti-Kv7.1 (Western blots, 1:1000, APC-022, Alomone, Israel), mouse anti-actin (Western blots, 1:500, A-2066, Sigma), mouse anti-ZO-1 (1:25, ZO1-1A12, Invitrogen), mouse anti-desmoplakins (1:200, multiepitope mixture to desmoplakin 1 and 2, Progen, Heidelberg, Germany), mouse anti-EEA1 (1:100, clone 14, BD Transduction Laboratories), mouse anti-LAMP2 (1:100, AC17, Acris Antibodies, Herford, Germany), mouse anti-mannose-6-phosphate receptor (1:100, M6PR clone 2G11, Abcam, Cambridge, UK), rabbit anti-Nedd4-2 (Western blots, 1:1000, no. 4013, Cell Signaling Technology), rabbit anti-Nedd4-2 Ser-448 (Western blots, 1:1000, no. 8063, Cell Signaling Technology), mouse anti-T7 (1:200, Novagene, Madison, WI), mouse anti-E-cadherin (1:20, rr1, Developmental Studies Hybridoma Bank, University of Iowa). The rr1 anti-E-cadherin antibody developed by Barry Gumbiner was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biology (Iowa City, IA).
As secondary antibodies an Alexa Fluor® 488-conjugated donkey anti-mouse IgG (1:200), an Alexa Fluor® 488-conjugated donkey anti-goat IgG (1:200), and an Alexa Fluor® 568-conjugated donkey anti-goat IgG (1:800) were employed. Alexa Fluor® 647 phalloidin (1:200) was used to stain actin filaments, and DAPI (Invitrogen) was used to stain the nucleus. All Alexa Fluor®-coupled reagents were purchased from Invitrogen. HRP-conjugated donkey anti-rabbit and donkey anti-mouse F(ab′)2 fragments were from Jackson ImmunoResearch Laboratories (1:10,000, West Grove, PA).
DNA Constructs
The expression vectors for pXOOM-hKv7.1, pXOOM-hKv7.1-YA, pXOOM-hNedd4-2, and pXOOM-Nedd4-2-CS have been described previously (36, 41, 42). The pDsRed2-ER construct was purchased from Clontech. The pEGFP-N2-hKv7.1 construct was a kind gift from Professor Guiscard Seebohm. pXOOM-T7-SGK1 was subcloned from pcDNA3-T7-SGK1, which was a kind gift from Professor Oliver Staub. The T7-tagged pXOOM-SGK1-S422D mutant was obtained by PCR-based site-directed mutagenesis (Pfu Turbo polymerase, Stratagene/Agilent Technologies, Waldbronn, Germany) using pXOOM-T7-SGK1 as template.
Transient and Stable Expression in MDCK Cells
MDCK (strain II) cells were grown in DMEM (Invitrogen) supplemented with 100 units/ml penicillin, 100 mg/ml streptomycin, and 10% FCS (Sigma-Aldrich; henceforth called normal calcium medium, NCM) at 37 °C in a humidified atmosphere with 5% CO2.
Transient Transfections
MDCK cells were transfected in suspension using Lipofectamine and Plus Reagent (Invitrogen) and plated on glass coverslips (12 mm in diameter, Thermo Fischer Scientific).
Stable Transfections
The monoclonal stable Kv7.1 cell lines MDCK-Kv7.1 and MDCK-Kv7.1-YA as well as the polyclonal stable cell line expressing pEGFP-N2-hKv7.1 have been described previously (18).
Calcium Switch and Inhibitor Experiments
MDCK cells stably expressing hKv7.1, hKv7.1-YA (and when relevant, transiently expressing the ER marker DsRed2-ER) or empty cells (negative control for Western blotting) were plated on glass coverslips or in Petri dishes (for Western blotting), and the calcium switch experiment was performed as described previously (17). Briefly, the cells were allowed to attach to the coverslips for 1.5 h in full DMEM and then washed three times in a low calcium medium (calcium-free MEM, Spinner Modification (Sigma-Aldrich) supplemented with 5% dialyzed fetal bovine serum (Invitrogen), 100 units/ml penicillin, 100 mg/ml streptomycin, 2 mm l-glutamine, and 1.6 μm calcium chloride) and grown for 2 days in low calcium medium until reaching confluency. The medium was then changed to NCM, which started the polarization process, and the cells were allowed to polarize for up to 24 h.
For the calcium switch inhibitor studies, NCM was added for 1 h and 45 min and then changed to NCM supplemented with either 10 μm LY294002, 10 μm Akt inhibitor IV, or 1 μm GSK650394 to inhibit PI3K, Akt, or SGK1, respectively. The polarization process was followed for up to 24 h. For inhibitor studies performed on polarized cells, a 24-h calcium switch was performed followed by a 3-h treatment with NCM supplemented with either 10 μm LY294002, 1 or 10 μm Akt inhibitor IV, or 1 μm GSK650394.
Immunofluorescence
Transiently or stably transfected cells grown on glass coverslips were fixed in 4% paraformaldehyde in PBS for 30 min at room temperature. Blocking and permeabilization was performed by a 30-min incubation with 0.2% fish skin gelatin in PBS supplemented with 0.1% Triton X-100 (PBST). The cells were incubated for 1 h in primary antibodies diluted in PBST. Secondary antibodies were diluted in PBST and applied for 45 min. The coverslips were mounted in Prolong Gold (Invitrogen).
Confocal Microscopy and Imaging
Laser scanning confocal microscopy was performed using either the Leica TCS SP2 system equipped with argon and helium-neon lasers or the Zeiss LSM 780 confocal system. Images were acquired using a 63× water immersion objective, NA of 1.2 (Leica TCS SP2) or a 63× oil immersion objective, numerical aperture of 1.4 (Zeiss LSM 780) with a pinhole size of 1 and a pixel format of 1024 × 1024. Line averaging was used to reduce noise. For double- and triple-labeling experiments, sequential scanning was employed to allow the separation of signals from the individual channels.
Quantifications
Total cell fluorescence and intracellular fluorescent signals were quantified using the Zen 2010 confocal software and the submembranous actin cytoskeleton to define the localization of the plasma membrane. Specifically, the exterior and interior of plasma membrane-associated Alexa Fluor® 647 phalloidin staining was used to define the regions quantified. Signals were subtracted for background fluorescence (the mean intensity of the background was obtained from an area of the nucleus as none of the proteins quantified would be expressed there.) For data analysis, signals originating from the surface membrane were obtained by subtracting the intracellular signal from the total cell fluorescent signal. The surface-associated signal was subsequently expressed as a percentage of the total cell fluorescence. n > 20 cells from three independent experiments were quantified. Data analysis was carried out using one-way analysis of variance test or t test and the Prism statistical software package. Data are presented as S.E.
Live Cell Imaging
MDCK cells stably expressing pEGFP-N2-hKv7.1 were subjected to a 24-h calcium switch before being transferred to an imaging chamber. The medium was changed from NCM to Hanks' balanced salt solution supplemented with 10 μm LY294002, 10 mm HEPES, pH 7.3. The experiment was performed at 37 °C. Cells were visualized by confocal laser scanning microscopy using the CSU-X1 spinning disk module on a Zeiss CellObserver (Carl Zeiss microimaging GmbH, Jena, Germany) with a 63× oil immersion objective (numerical aperture of 1.2).
Time Lapse
The first z-stack was acquired 30 min after addition of Hanks' balanced salt solution supplemented with 10 μm LY294002, 10 mm HEPES, pH 7.3, and thereafter, z-stacks were acquired every 3 min with an exposure time of 350 ms using the AxioVision software (version 4.8). The three-dimensional reconstruction of the obtained time lapse was created by using the Imaris software (version 7.2). Data are presented as a QuickTime movie.
Cell Lysates
MDCK cells stably expressing Kv7.1 or Kv7.1-YA were plated in 25 cm2 flasks and subjected to a 24-h calcium switch followed by a 3-h incubation in NCM supplemented with 40 μg/ml cycloheximide. Cells were collected and solubilized in solubilization buffer (50 mm Tris, pH 7.4, 10 mm NaCl, 10 mm KCl, 10 mm NaF, 1% Triton X-100 (Sigma Aldrich) and 0.5% sodium deoxycholate (Sigma Aldrich), 50 μl of protease inhibitor mixture (dilution of 1:200, Sigma Aldrich), and a phosphatase inhibitor tablet (Roche Diagnostics)) for 3 h at 4 °C. The samples were centrifuged at 15,000 rpm for 10 min, and the supernatant was collected. The protein concentration was determined by performing Bradford assay (Bio-Rad) according to manufacturer's instructions and calibrated to 2 μg/μl.
Western Blotting
Cell lysates were loaded in equal amounts and separated on gradient (5–15%) SDS-PAGE gels. Proteins were transferred onto a Hybond-P PVDF transfer membrane (Amersham Biosciences, 0.45 μm) in 25 mm Tris base, 200 mm glycine, 20% methanol using a mini transblot (Hercules, CA). After transfer, the membranes were incubated for 1 h in blocking buffer (PBS containing 4% lowfat milk powder or 5% bovine serum albumin). The membrane was incubated overnight at room temperature in blocking buffer containing anti-Kv7.1, anti-Nedd4-2, anti-Nedd4-2 Ser-468, or anti-actin antibodies. After washing, bound antibody was revealed with HRP-conjugated F(ab′)2 fragment donkey anti-rabbit or donkey anti-mouse IgG (1/10,000, Jackson ImmunoResearch Laboratories) in blocking buffer for 30 min. For visualization, blots were incubated for 5 min in a 1:1 mixture of the following: 1) 100 mm Tris, pH 8.5, containing 2.5 mm luminol (5-amino-2,3-dihydro 1,4-phthalazinedione) and 0.4 mm p-coumaric acid and 2) 100 mm Tris, pH 8.5, containing 0.018% hydrogen peroxide. The solutions were prepared and mixed just before use. Immunoblots were exposed on hyperfilm ECL (Amersham Biosciences).
Nedd4-2 Knockdown in MDCK Cells
A double-stranded 5′-GGGAAGAGAAGGUGGACAA-3′ siRNA targeting canine Nedd4-2 was employed as it has previously been demonstrated to result in robust knockdown of Nedd4-2 in MDCK cells (43). Non-targeting siRNA (5′-CCAUCCUGAUGUCGCAAUA-3′) was used as a negative control. Both siRNA oligonucleotides were purchased from Eurogentec. MDCK cells stably expressing Kv7.1 were transfected with 20 nm total siRNA oligonucleotides using siLentFectTM (Bio-Rad) according to manufacturer's instructions. In addition, eGFP (eGFP-pcDNA3) was co-transfected serving as a marker for siRNA-transfected cells. The cells were plated on glass coverslips and grown for 2 days before being treated with 10 μm LY294002 for 90 min prior to fixation.
cRNA Generation and Oocyte Injection
cRNA was generated from linearized pXOOM plasmids carrying the gene of interest using the Ambion T7 m-Message Machine kit (Ambion, Austin, TX) according to the manufacturer's instructions. cRNA concentrations were determined using the ND-1000 NanoDrop UV spectrophotometer, and its integrity was confirmed by gel electrophoresis. cRNAs were stored at −80 °C until injection. Xenopus laevis oocytes were obtained from EcoCyte Bioscience (Castrop-Rauxel, Germany). Oocytes were kept at 19 °C in Kulori solution consisting of 4 mm KCl, 90 mm NaCl, 1 mm MgCl2, 1 mm CaCl2, 5 mm HEPES, pH 7.4, for a minimum 24 h before injection. Injection of 50 nl of cRNA (1 ng of all Kv7.1, SGK1, and Akt variants, and 0.2 ng of Nedd4-2/Nedd4-2 CS) was accomplished using a Nanoject microinjector from Drummond (Drummond Scientific, Broomall, PA). Injected oocytes were kept at 19 °C for 2 days prior to measurements.
Electrophysiological Recordings on X. laevis Oocytes
Kv7.1 currents were recorded using a two-electrode voltage-clamp amplifier (Dagan CA-1B, Minneapolis, MN). Electrodes were pulled from borosilicate glass capillaries on a horizontal patch electrode puller (DMZ universal puller, Zeitz Instruments, München, Germany) and had tip resistance between 0.5 and 2.0 megohms when filled with 2 m KCl. The recordings were done at room temperature under continuous superfusion with Kulori solution. Kv7.1 currents were analyzed by applying a standard step protocol with pulses from −60 to +40 mV (5 s) in 20-mV increments from a holding potential of −80 mV. The tail currents were measured at −30 mV. The condition of each single oocyte was tested before measurements by recording membrane potentials. Only oocytes with a membrane potential more negative than −40 mV were used in the experiments. The results were analyzed by GraphPad Prism software (GraphPad Software, San Diego, CA).
Data Analysis
Data analysis and drawings were performed using IGOR software (version 4, WaveMetrics, Lake Oswego, OR) or GraphPad Prism software (version 4, GraphPad Software, San Diego, CA). All deviations of calculated mean values are given as S.E. values. Statistical significance was determined by t test or one-way analysis of variance for more than two groups of data. The quantitative analysis of Western blot experiments was done using Odyssey Imaging Software (LI-COR Biosciences, Lincoln, NE). Confocal images were adjusted by the use of either Leica Confocal Software (Leica Microsystems, Mannheim, Germany) or Zen 2009 (Carl Zeiss) and Adobe Photoshop CS5.
RESULTS
Kv7.1 Surface Expression Requires Continuous PI3K Activity in Polarized MDCK Cells
Epithelial cell polarization, a process dependent on the calcium concentration in the extracellular medium, can be controlled by the calcium switch assay (44). We have previously reported that Kv7.1 is retained intracellularly during the early stages of epithelial cell polarization induced by the calcium switch but is redistributed to the basolateral cell surface in a PI3K-dependent manner at a later stage (17). As PI3K activity not only regulates the formation of the basolateral membrane but also appears to be required for its preservation (28), we speculated whether tonic PI3K activity would be required to maintain Kv7.1 expression at the basolateral plasma membrane of polarized MDCK cells. To test this, MDCK cells were subjected to a 24-h calcium switch and then incubated in NCM containing 10 μm of the PI3K inhibitor LY294002 for an additional 3 h. As illustrated in Fig. 1A, LY294002 treatment resulted in a disappearance of Kv7.1 from the basolateral cell surface and an intracellular accumulation of the channel (27 h, LY294002). Kv7.1 was primarily observed in vesicular structures and in the ER, as revealed by a partial co-localization with the ER marker, DsRed-ER. LY294002-treated cells displayed a 60% decrease in Kv7.1 signal at the cell surface compared with control cells (see Fig. 8B). Thus, PI3K activity appears to be required for maintaining basolateral surface expression of Kv7.1 in polarized MDCK cells. This effect was not due to changes in the overall polarity of the cells as the localizations of the tight junction protein ZO-1, the adherens junction protein E-cadherin, and the desmosome component desmoplakin all remained unaffected by a 3-h treatment with LY294002 (Fig. 1, C and D).
FIGURE 1.
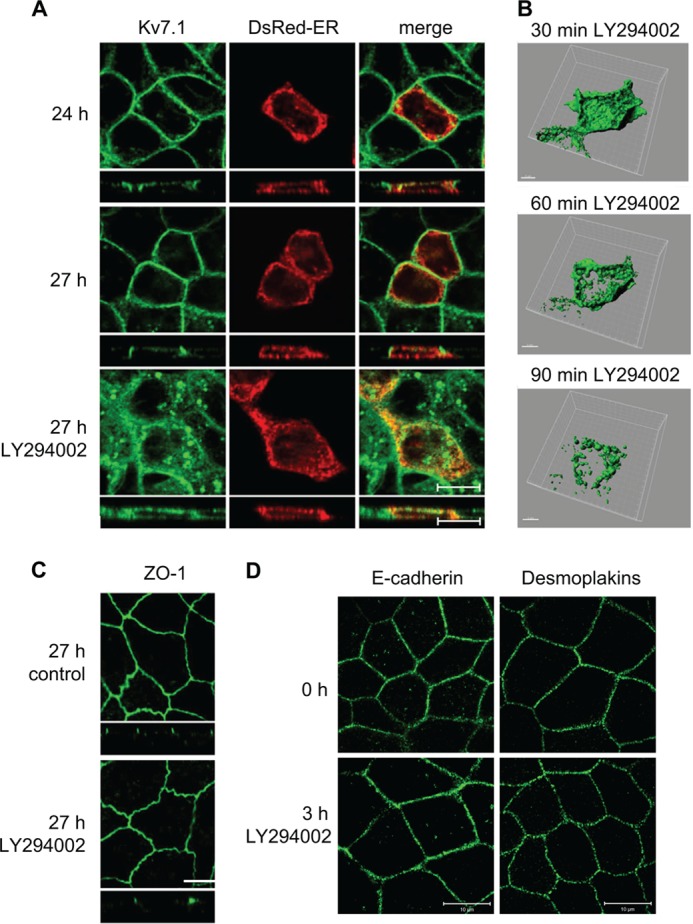
Kv7.1 changes localization upon PI3K inhibition in polarized MDCK cells. A, MDCK cells stably expressing Kv7.1 were transiently transfected with pDsRed-ER and subjected to a calcium switch assay for 24 h (24 h). At this point, the medium was changed to NCM containing 10 μm of the PI3K inhibitor LY294002 (27 h LY294002) or just NCM (control, 27 h) for 3 h. The cells were fixed and stained for Kv7.1. Scale bar, 10 μm. B, MDCK cells stably expressing Kv7.1-GFP were subjected to a calcium switch assay for 24 h and thereafter treated with 10 μm LY294002 for up to 1 h and 30 min. Z-stacks were obtained on living cells every 3 min during the experiment and used for three-dimensional reconstruction. Representative cell are illustrated after 30, 60, and 90 min of PI3K inhibitor treatment. Scale bar, 5 μm. C, MDCK-Kv7.1 cells stained for the tight junction protein ZO-1. The cells were subjected to the calcium switch for 24 h and thereafter treated with the LY294002 (10 μm) for 3 h (27 h LY294002). Control cells (27 h control) were not treated with the inhibitor. As illustrated, cell polarization, indicated by the presence of the tight junctions (stained by ZO-1), was unaffected by PI3K inhibition. Scale bar, 10 μm. D, polarized MDCK cells were treated for 3 h with 10 μm LY294002 and then fixed and stained for the adherens junction protein E-cadherin or desmosome components desmoplakins. As shown, 3 h of PI3K inhibition did not affect the localizations of E-cadherin or desmoplakins compared with untreated control cells. Scale bar, 10 μm.
FIGURE 8.
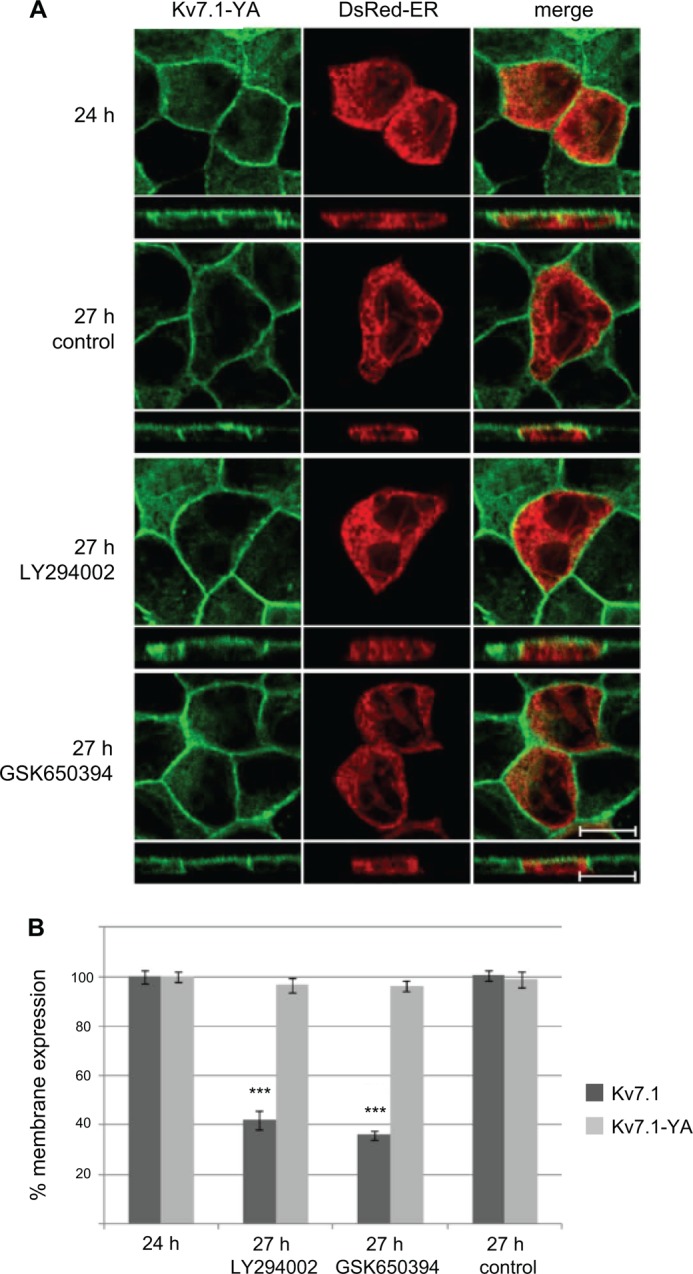
Kv7.1-YA mutant does not change localization upon inhibition of SGK1 and PI3K in polarized cells. A, MDCK cells stably expressing Kv7.1-YA were transiently transfected with DsRed-ER cDNA and subjected to a calcium switch for 24 h. The cells were then incubated in NCM containing SGK1 inhibitor GSK650394 (1 μm) or the PI3K inhibitor LY294002 (10 μm) for 3 h (27 h) and fixed and stained for Kv7.1. Illustrated are untreated cells (27 h control) and polarized cells treated for 3 h with the inhibitors (27 h GSK650394, 27 h LY294002). Scale bar, 10 μm. B, quantification of the cell surface-associated signal of Kv7.1 (dark gray bars) and Kv7.1-YA (light gray bars) in response to PI3K and SGK1 inhibition. The signal obtained 24 h into the calcium switch was set to 100% and the membrane-associated signals 3 h after incubation in NCM without (27 h, control) or with application of PI3K and SGK1 inhibitors (27 h, LY294002 and 27 h, GSK650394) were expressed as a percentage of the 24 h signal. 20–30 cells were quantified per condition. Quantification was performed as described under “Experimental Procedures”; ***, p < 0.001. Bars represent means of each group ± S.E.
To follow the effects of PI3K inhibition upon the localization of Kv7.1 more dynamically, live-cell imaging was performed on MDCK stably expressing EGFP-tagged hKv7.1. Z-stacks were acquired every 3 min for 1 h (starting at 30 min (24 h, 30 min) after addition of LY294002, Fig. 1B). The obtained images were then presented as a three-dimensional reconstruction (see supplemental Movie 1 or Fig. 1B). The live-cell imaging experiments confirmed our observations from fixed cells as LY294002 treatment resulted in loss of the Kv7.1-GFP membrane-associated signal and an accumulation of the channel in vesicular structures.
To reveal the identity of the intracellular structures in which Kv7.1 resided, polarized MDCK cells treated with PI3K inhibitor for up to 1 h were co-stained for Kv7.1 and endosomal markers. The vesicular structures containing Kv7.1 partly co-localized with markers of the early (EEA1) and late (M6PR) endosomes as well as lysosomes (LAMP2, Fig. 2). This observation suggests that PI3K inhibition initiates endocytosis and subsequent degradation of surface-expressed Kv7.1 channels.
FIGURE 2.
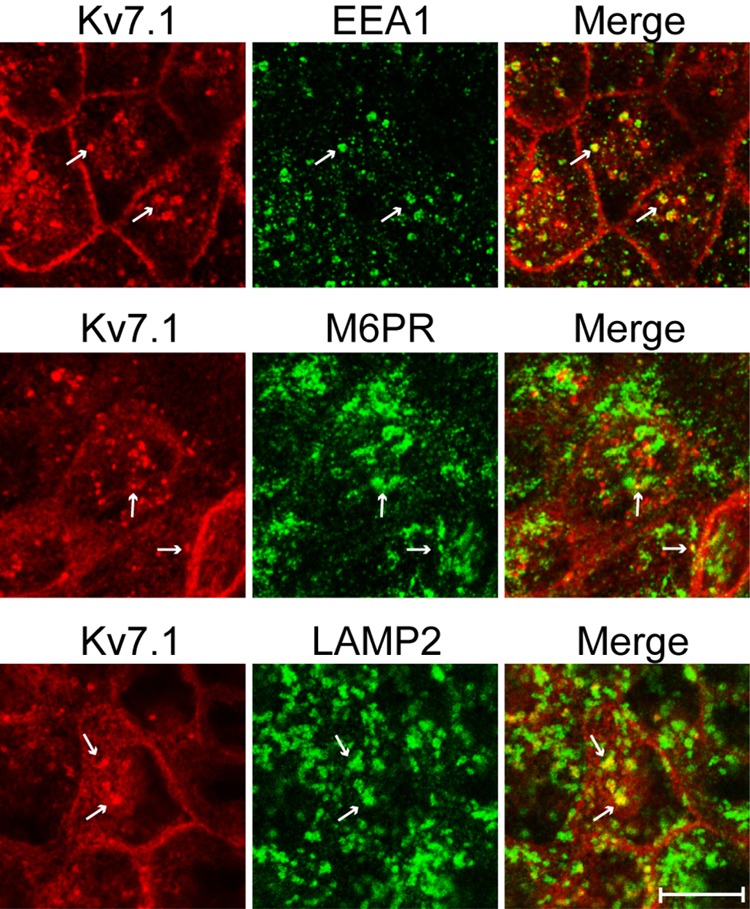
Kv7.1 is endocytosed upon inhibition of PI3K. MDCK cells stably expressing Kv7.1 were subjected to a 24-h calcium switch assay and thereafter treated with 10 μm PI3K inhibitor LY294002 for up to 1 h. The cells were then fixed and co-stained for Kv7.1 and markers of the early endosomes (EEA1), late endosomes (M6PR), and lysosomes (LAMP2). As illustrated, Kv7.1-containing vesicular structures were found to partly co-localize with the three endosomal markers (white arrows). Scale bar, 10 μm.
SGK1 Inhibition Mimics the Effects of PI3K Inhibition
As mentioned above, interesting downstream partners of PI3K are the SGK1 and Akt kinases, which have been reported to up-regulate Kv7.1 currents (31, 32, 45–47). To test for a possible involvement of the two kinases upon Kv7.1 localization, the effects of the inhibitors GSK650394 (SGK1) and Akt inhibitor IV (Akt) were examined. In polarized MDCK cells, addition of 1 μm of GSK650394 resulted in an intracellular accumulation of Kv7.1 (Fig. 3). In contrast, 1 μm Akt inhibitor IV was without effect (Fig. 3). At higher concentrations (10 μm), Akt inhibitor IV also caused intracellular accumulation of Kv7.1 (Fig. 3). However, at this concentration, Akt inhibitor IV can also target SGK1 (48), thus causing uncertainty in regards to the specificity of the effect. Quantifications of the Kv7.1 fluorescent signals revealed a decrease of ∼60% in the level of Kv7.1 at the plasma membrane when inhibiting SGK1 (see Fig. 8B). Similar to the observations in polarized cells, GSK650394 prevented accumulation of Kv7.1 at the basolateral cell surface when applied during the calcium switch (Fig. 4). These observations suggest that SGK1, similarly to PI3K, regulates the basolateral surface expression of Kv7.1 in both polarizing and polarized MDCK cells.
FIGURE 3.

Inhibition of SGK1 leads to intracellular accumulation of Kv7.1 in polarized MDCK cells. MDCK cells stably expressing Kv7.1 were subjected to a calcium switch for 24 h. The cells were then incubated in NCM containing 1 μm SGK1 inhibitor GSK650394 or 1–10 μm Akt IV inhibitor for 3 h, after which they were fixed and stained for Kv7.1. Illustrated are confocal horizontal and vertical scans of MDCK-Kv7.1 cells before addition of the inhibitor (control), 3 h after addition of the Sgk1 inhibitor (1 μm GSK650394 3 h) or Akt inhibitor (1–10 μm Akt IV inhibitor 3 h), and control cells not treated with the inhibitor (27 h, control). Actin was stained by phalloidin, and DAPI (blue) was used to stain the nucleus (shown in merged pictures). Scale bars, 10 μm.
FIGURE 4.

SGK1 inhibition prevents surface expression of Kv7. 1 in polarizing MDCK cells. MDCK cells stably expressing Kv7.1 were transiently transfected with DsRed-ER cDNA and subjected to a calcium switch in the presence of the SGK1 inhibitor GSK650394 (1 μm). GSK650394 was added 1 h and 45 min after initiation of the switch, and cells were fixed at different time points after initiation of the switch. Illustrated are confocal horizontal and vertical scans of MDCK-Kv7.1 cells before addition of the inhibitor (t = 1 h, 45 min), 24 h after addition of the inhibitor (24 h GSK), and control cells not treated with the inhibitor (24 h control). Scale bar, 10 μm.
Constitutively Active SGK1 Prevents LY294002-induced Internalization of Kv7.1
To examine whether SGK1 could be the primary mediator of the PI3K effects on Kv7.1 localization, we tested the impact of PI3K inhibition in MDCK cells overexpressing a constitutively active SGK1 mutant, SGK1-S422D. We transfected MDCK cells stably expressing Kv7.1 with this mutant and applied LY294002 for 1–3 h to the cells at the polarized state. In support of SGK1 being the primary target of PI3K, SGK1-S422D overexpression prevented LY294002-induced removal of Kv7.1 from the basolateral cell surface (Fig. 5). To further establish the important role of SGK1, we examined the localization of Kv7.1 during the calcium switch in cells co-expressing SGK1-S422D (Fig. 6). SGK1-S422D expression prevented the clearance of Kv7.1 from the cell surface, which is normally observed upon switch initiation (17). This observation supports an important role for SGK1 in regulating Kv7.1 cell surface expression.
FIGURE 5.

Overexpression of constitutively active SGK1 prevents internalization of Kv7. 1 in response to PI3K inhibition. A, MDCK cells stably expressing Kv7.1 were transfected with SGK1-S422D, subjected to a 24-h calcium switch, and then treated with the PI3K inhibitor (LY294002; 10 μm) for 1 and 3 h. The cells were then fixed and stained for Kv7.1, SGK1, and actin. Illustrated are untreated cells (24 h) and polarized cells treated for 1 and 3 h with the inhibitor (25 h LY294002 and 27 h LY294002). Scale bar, 10 μm. B, quantification of the cell surface-associated signal of Kv7.1 in control cells (dark gray bars) and cells expressing the constitutively active SGK1-S422D (light gray bars) in response to PI3K inhibition. The signal obtained 24 h into the calcium switch was set to 100%, and the membrane-associated signals 1 and 3 h after application of PI3K inhibitor (25 h LY294002 and 27 h LY294002) were expressed as a percentage of the signal obtained at 24 h. 9–14 cells were quantified per condition. Quantifications was performed as described under “Experimental Procedures”; ***, p < 0.001. Bars represent means of each group ± S.E.
FIGURE 6.
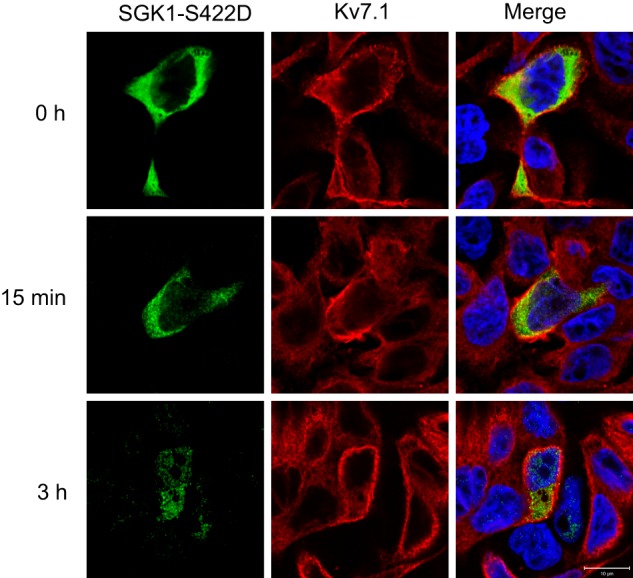
Overexpression of constitutively active SGK1 prevents internalization of Kv7.1 in response to initiation of a calcium switch. MDCK cells stably expressing Kv7.1 were transiently transfected with SGK1-S422D and subjected to a calcium switch. Cells were fixed before initiation of the switch (t = 0 h), 15 min into the switch (15 min), as well as 3 h after the switch was initiated (3 h). As shown, overexpression of SGK1-S422D prevents the reduction in Kv7.1 cell surface expression observed after 15 min as well as 3 h. Scale bar, 10 μm.
SGK1 Counteracts Nedd4-2-mediated Down-regulation of Kv7.1
As mentioned, SGK1 can inhibit Nedd4-2 (33, 34), a ubiquitin ligase that down-regulates Kv7.1 currents (36). We therefore hypothesized that SGK1 promotes Kv7.1 surface expression by inhibiting Nedd4-2. To address this, we performed two-electrode voltage-clamp experiments on oocytes co-expressing Kv7.1, Nedd4-2, and SGK1, as well as catalytically inactive forms of Nedd4-2 and SGK1 (Nedd4-2 CS and SGK1 KA; Fig. 7A). In line with previous observations (32, 36), SGK1 co-expression significantly increased Kv7.1 current levels, whereas Nedd4-2 co-expression significantly decreased them (Fig. 7A). Interestingly, SGK1 rescued the effect of Nedd4-2 co-expression. Although Nedd4-2 decreased the Kv7.1 current by approximately half, the additional co-expression of SGK1 caused a significant increase in the current amplitude, suggesting that SGK1 inhibits Nedd4-2 and thereby recovers the Kv7.1 current (Fig. 7A). This relationship was not observed for the catalytically inactive SGK1 KA mutant. In contrast to SGK1, Akt co-expression did not antagonize the Nedd4-2-mediated down-regulation of the Kv7.1 current. Kv7.1 current levels in oocytes co-expressing Kv7.1, Nedd4-2, and Akt were not statistically different from the current levels obtained in oocytes co-expressing Kv7.1, Nedd4-2, and catalytically inactive Akt 3A (Fig. 7B). Thus, SGK1 appears to be the primary regulator of Kv7.1, and the kinase could be acting through inhibition of Nedd4-2 activity.
FIGURE 7.

SGK1, but not Akt, counteracts Nedd4-2-mediated down-regulation of Kv7.1 currents in X. laevis oocytes. A, the effect of SGK1 on the Kv7.1 current in X. laevis oocytes. Two-electrode voltage clamp measurements of Xenopus oocytes are shown. The obtained current levels were measured in the end of the +40-mV step. Presented is a summary of three individual two-electrode voltage clamp experiments (n ≥ 6 in each) in oocytes expressing Kv7.1 alone (normalized to 100) or in combination with Nedd4-2/Nedd4-2-CS and/or SGK/SGK KA, where Nedd4-2-CS and SGK KA are catalytically inactive versions of the respective enzymes. The Kv7.1 current measured in oocytes co-expressing Kv7.1, Nedd4-2, and SGK1 was significantly increased compared with oocytes co-expressing Kv7.1, Nedd4-2, and the catalytically inactive version of SGK1 (SGK1 KA). The difference in Kv7.1 current level between oocytes co-expressing Kv7.1 and Nedd4-2 and those additionally expressing SGK1 KA, was not significant. In all of the conditions where active SGK1 was co-expressed, an increased Kv7.1 current, as compared with Kv7.1 alone, was observed; ***, p < 0.001. Bars represent means of each group ± S.E. B, the effect of Akt on the Kv7.1 current in X. laevis oocytes. Two-electrode voltage clamp measurements of Xenopus oocytes are shown. The obtained current levels were measured in the end of the +40-mV step. Presented is a summary of three individual two-electrode voltage clamp experiments (n ≥ 6 in each) in oocytes expressing Kv7.1 alone (normalized to 100) or in combination with Nedd4-2/Nedd4-2-CS and/or Akt/Akt 3A, where Nedd4-2-CS and Akt 3A are catalytically inactive versions of the respective enzymes. Akt has very little or no effect on Nedd4-2-mediated decrease of Kv7.1 current. There was no difference in Kv7.1 current level in oocytes co-expressing Kv7.1, Nedd4-2, and Akt as compared with Kv7.1, Nedd4-2 and catalytically inactive Akt 3A. Bars represent means of each group ± S.E. ns, not significant.
The PI3K-SGK1 Pathway Promotes Kv7.1 Surface Expression by Inhibiting Nedd4-2-dependent Endocytosis
To further examine whether PI3K and SGK1 act on Kv7.1 through a Nedd4-2-dependent mechanism, we tested the effects of PI3K and SGK1 inhibition upon the localization of a Nedd4-2 interaction-deficient Kv7.1 mutant (Kv7.1-YA). As speculated, the localization of Kv7.1-YA was unaffected by inhibition of PI3K and SGK1 in polarized MDCK cells, suggesting that the two kinases act through Nedd4-2 (Fig. 8). Moreover, the involvement of Nedd4-2 was additionally supported by the observation that inhibition of both PI3K and SGK1 were without effect on the membrane localization of Kv7.1-YA in polarizing cells, i.e. during the calcium switch (data not shown). Interestingly, the Kv7.1-YA mutant also displayed localization in the apical membrane (side scans in Fig. 8A, 27 h control, versus Fig. 1A, 27 h), indicating that Nedd4-2-dependent endocytosis is important to avoid apical trapping of the channel upon cell polarization.
To confirm that Nedd4-2 acts downstream of PI3K-SGK1, we reduced the expression of Nedd4-2 in MDCK cells stably expressing Kv7.1 by using a previously employed Nedd4-2 siRNA oligonucleotide (43). First, we verified that the siRNA oligonucleotide indeed decreased Nedd4-2 expression levels. As illustrated in Fig. 9C, we observed a reduction in the protein level of Nedd4-2 in cells transfected with the Nedd4-2-specific siRNA compared with control cells transfected with a non targeting control siRNA. By quantifying the signal intensity of the Nedd4-2 band and the corresponding actin band, we calculated an ∼30% reduction in Nedd4-2 expression (data not shown). As our transfection efficiencies rarely exceed 25–30%, it indicates that cells transfected with the Nedd4-2-specific siRNA have a very robust reduction in Nedd4-2 protein expression. This is in agreement with previous results using this specific Nedd4-2 siRNA in MDCK cells (43). Therefore, MDCK cells were next transfected with Nedd4-2 siRNA followed by treatment of the cells with LY294002. Nedd4-2 silencing prevented LY294002-induced internalization of Kv7.1, which was not the case when non-targeting siRNA was used as a control (Fig. 9A). Quantifications revealed that LY294002-treated cells transfected with Nedd4-2 siRNA had significantly higher membrane to intracellular Kv7.1 signal ratio than the cells transfected with control siRNA (Fig. 9B), suggesting that Nedd4-2 silencing protects Kv7.1 from internalization upon PI3K inhibition.
FIGURE 9.

Nedd4-2 knockdown protects Kv7.1 from internalization upon PI3K inhibition. A, MDCK cells stably expressing Kv7.1 were transiently co-transfected with eGFP and siRNA targeting Nedd4-2 or a non-coding siRNA, which was used as a negative control. Polarized cells were treated with 10 μm PI3K inhibitor LY294002 for 90 min. The localization of Kv7.1 was examined in transfected cells expressing eGFP (GFP) as these were expected also to be transfected with the siRNA. As demonstrated, Kv7.1 was internalized in cells expressing the non-targeting control siRNA. This internalization was not observed in cells expressing siRNA targeting Nedd-2 (Nedd4-2 siRNA). Scale bar, 10 μm. B, quantification of membrane and intracellular Kv7.1 signals from cells expressing either non-targeting control siRNA or siRNA targeting Nedd4-2 (Nedd4-2 siRNA). The two bars demonstrate the ratios between membrane and intracellular signals in cells expressing the two siRNAs. The data are from four experiments with a total n > 17. The cells transfected with Nedd4-2 siRNA have a significantly higher membrane to intracellular signal ratio than the cells transfected with control siRNA; ***, p < 0.001. Bars represent means of each group ± S.E. C, Western blot of MDCK cells transfected with control siRNA (lane 1) or Nedd4-2 siRNA (lane 2). A reduction in the signal intensity of the band at ∼120 kDa, which correspond to Nedd4-2, is observed in lane 2 compared with the lane 1. Actin was used as a loading control. D, Western blots of cell lysates from polarized, non-treated control MDCK cells (control) and cells treated with 10 μm LY294002 (LY294002) for 2 h. The blots were probed for total Nedd4-2 (left blot) or with Ser-468 phospho-specific Nedd4-2 antibody (right blot). The graph to the right displays band intensity quantifications from n = 3 experiments. The amount of phospho-Nedd4-2 relative to the total amount of Nedd4-2 is shown. The control condition was set to 1. A significant reduction in the relative signal for phospho-Nedd4-2 was observed in response to LY294002 treatment. *, p < 0.05. Actin was used as a loading control.
Nedd4-2 is a direct substrate of SGK1 and contains three well described SGK1 phosphorylation sites (33, 35). Phosphorylation of these sites increases interaction with proteins of the 14-3-3 family, keeping Nedd4-2 in an inactive phosphorylated state (40). To determine whether LY294002 treatment of polarized MDCK cells directly affected the phosphorylation state of Nedd4-2, we examined the degree of phosphorylation on one of these key sites, Ser-468 in human Nedd4-2, after treatment. As shown in Fig. 9D, exposure to 10 μm LY294002 for 2 h significantly reduced the fraction of Nedd4-2 phosphorylated at this site by 28%. This observation suggests a direct impact of the PI3K-SGK1 pathway upon Nedd4-2 phosphorylation state.
Kv7.1 Turnover Is Primarily Mediated by a Nedd4-2-related Mechanism in Polarized MDCK Cells
The central involvement of Nedd4-2 in the regulation of Kv7.1 endocytosis suggests that this ubiquitin ligase is a key regulator of Kv7.1 cell surface expression. To examine this further, we investigated the turnover of Kv7.1 and Kv7.1-YA in polarized MDCK cells. MDCK cells stably expressing Kv7.1 or Kv7.1-YA were subjected to a 24-h calcium switch, and protein synthesis was inhibited with cycloheximide for 3 h. Total cell lysates were harvested at time points 24 and 27 h and analyzed by Western blotting. As illustrated in Fig. 10A, Kv7.1 protein levels were reduced by ∼50% after a 3-h cycloheximide treatment, indicating a fast turnover of the channel. In contrast, Kv7.1-YA protein levels were unaffected by the 3-h treatment with cycloheximide (Fig. 10B), demonstrating that the turnover of this mutant is significantly slower. This suggests that the normal turnover of Kv7.1 is primarily mediated by a Nedd4-2-dependent mechanism.
FIGURE 10.
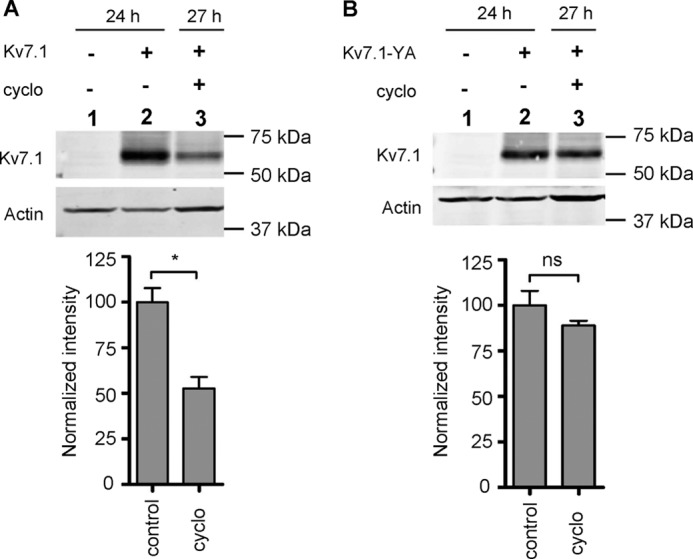
Kv7.1-YA displays a slower turnover than WT Kv7.1. MDCK cells stably expressing Kv7.1 (A) or Kv7.1-YA (B) were subjected to a 24-h calcium switch and thereafter treated with 40 μg/ml cycloheximide (cyclo) for 3 h to inhibit protein synthesis. Cells were harvested at t = 24 h or t = 27 h, and the resulting protein samples were analyzed by Western blotting. Representative blots illustrating the protein levels of Kv7.1 (A) and Kv7.1-YA (B) are presented. The bottom graphs represent band intensity quantification of n = 3 experiments, where each column corresponds to the lane above. Actin was used as a loading control in each experiment. The protein level was normalized to the control (lane 2 in Fig. 7, A and B, set to 100); *, p < 0.01. Bars represent means of each group ± S.E. ns, not significant.
Insulin Counteracts Nedd4-2-mediated Reduction in Kv7.1 Current
As a final step, we wanted to investigate whether activation of PI3K in Xenopus oocytes could rescue the reduction in Kv7.1 current observed upon co-expressing Nedd4-2. Insulin, an activator of PI3K, has previously been demonstrated to stimulate Kv1.5 currents in an SGK1-dependent manner when the channel was expressed in oocytes (49) and was therefore used to activate the proposed pathway. Oocytes expressing Kv7.1 alone or together with Nedd4-2 were incubated in Kulori buffer with and without insulin for 6 h prior to current measurements. Insulin treatment significantly rescued (p < 0.001) the Kv7.1 current reduction caused by Nedd4-2 co-expression (Fig. 11B), thereby indicating that activation of PI3K also can lead to inhibition of Nedd4-2 in Xenopus oocytes.
FIGURE 11.

Insulin rescues Nedd4-2-dependent decrease of Kv7.1 current. A, representative current traces of Kv7.1, Kv7.1 + insulin, Kv7.1 + Nedd4.2, and Nedd4.2 + Kv7.1 + insulin. B, two-electrode voltage clamp experiment on Xenopus oocytes expressing Kv7.1 alone or together with Nedd4-2. The oocytes were incubated in Kulori ± 1 μm insulin for 6 h before measurements. The data are a summary of three individual experiments with a total of 21 < n < 44 oocytes for each group. Insulin treatment significantly increased the current in oocytes co-expressing Kv7.1 and Nedd4-2; ***, p < 0.001. Bars represent means of each group ± S.E.
DISCUSSION
Epithelial cell polarization involves a cascade of processes that ultimately divide the membrane into two distinct parts: an apical and a basolateral. To ensure vectorial transport of ions and solutes, these two membrane domains express different ion channels and transporters. In this study, the mechanisms regulating cell surface expression of the basolateral Kv7.1 channel were studied in MDCK cells both during epithelial cell polarization and in polarized cells. We previously reported that the activity of PI3K is required for recovering Kv7.1 cell surface expression during the polarization process (17). Here, we demonstrate that tonic PI3K activity stabilizes cell surface expression of Kv7.1 in polarized cells, which positions PI3K as an important regulator of Kv7.1 localization and turnover.
To uncover the molecular mechanisms underlying the PI3K-mediated promotion of Kv7.1 cell surface expression, we looked at two well described downstream targets of PI3K, the homologous kinases SGK1 and Akt. We found that pharmacological inhibition of SGK1 in polarized MDCK cells resulted in disappearance of Kv7.1 channels from the surface membrane and intracellular accumulation of the channel. We observed similar effects by using Akt inhibitor IV but only at concentrations where it can also suppress SGK1 (48), suggesting that the observed effect could be due to inhibition of SGK1. In support of this, SGK1, and not Akt, could counteract Nedd4-2-mediated down-regulation of Kv7.1 currents in Xenopus oocytes. Furthermore, overexpression of constitutively active SGK1 overruled the endocytotic response of Kv7.1 to PI3K inhibition, which is supportive of SGK1 being the primary downstream target of PI3K.
SGK1 and Akt are both known inhibitors of Nedd4-2 (33, 34, 50), and several observations in our study support that the PI3K-SGK1 signaling pathway promotes Kv7.1 cell surface expression by inhibiting Nedd4-2. First, SGK1 can antagonize Nedd4-2-mediated down-regulation of Kv7.1 currents in Xenopus oocytes. Second, insulin, a known activator of PI3K, also antagonizes Nedd4-2-mediated reductions in Kv7.1 currents. In addition, the Nedd4-2-interaction deficient Kv7.1-YA mutant did not respond to PI3K and SGK1 inhibition and Nedd4-2 knockdown in MDCK cells prevented endocytosis of the channel in response to PI3K inhibition. Nedd4-2 inhibition by the PI3K-SGK1 pathway is further supported by the observation that the LY294002 treatment results in decreased phosphorylation of Ser-468 in Nedd4-2, a previously described SGK1 phosphorylation site, which plays a critical role in mediating interaction with the inhibitory 14-3-3 proteins (40). Overall, these observations suggest that Nedd4-2 is the primary effector of the PI3K-SGK1 pathway controlling Kv7.1 cell surface expression and positions Nedd4-2 as an important regulator of Kv7.1. Indeed, the Kv7.1-YA mutant displayed a much slower turnover than the wild-type channel, indicating that Nedd4-2 is a key regulator of the Kv7.1 degradation process. In support of this, AMP-activated protein kinase-mediated endocytosis of Kv7.1 in the beginning of the calcium switch is also Nedd4-2-dependent (18). Interestingly, AMP-activated protein kinase activation in polarized MDCK cells is without effect upon Kv7.1 surface expression, which suggests that the Nedd4-2 activation is overruled by other pathways at the more polarized state (18). The PI3K-SGK1-mediated inhibition of Nedd4-2-dependent endocytosis described in this study could explain this observation.
The PI3K-SGK1 pathway possibly promotes Kv7.1 surface expression by additional mechanisms. SGK1 was reported previously to increase the surface expression of the Kv7.1-KCNE1 channel complex by promoting RAB11-mediated channel recycling (32). In line with this observation, we also observed an increase in Kv7.1 current levels upon SGK1 co-expression. Interestingly, co-expression of SGK1 and Nedd4-2 did not increase the current level to that of the control but fully restored it to the same level as SGK1-expressing oocytes. This indicates that there are possibly two different SGK1 pathways that work simultaneously to promote the surface expression of Kv7.1. The Nedd4-2 pathway decreases internalization of surface-expressed channels, whereas the RAB11 pathway enhances forward trafficking and recycling. Interestingly, a similar two-way mechanism appears to regulate the surface expression of the epithelial sodium channel ENaC. ENaC is regulated by Nedd4-2, and SGK1 promotes ENaC surface expression by inhibiting the interaction between Nedd4-2 and ENaC. However, SGK1 also increases the surface expression of ENaC channels carrying PY motif mutations, suggesting that an additional pathway exists (51). RAB11 has recently been shown to enhance the surface expression of ENaC, which raises the possibility that the mechanism is similar to that reported for Kv7.1 (52, 53).
One explanation for activating and inhibiting Nedd4-2-dependent endocytosis during polarization could be to avoid localization of Kv7.1 channels in the apical membrane after the establishment of tight junctions. By promoting degradation of surface-expressed Kv7.1 and not allowing the channel to restore cell surface expression before proper protein trafficking pathways are established, the cell can prevent trapping of Kv7.1 in the apical membrane. This hypothesis is supported by the increased localization of the Nedd4-2-interaction-deficient Kv7.1 mutant in the apical membrane, which could result from hindered endocytosis during the early stages of polarization.
The PI3K-SGK1-Nedd4-2 pathway described here could also regulate Kv7.1 surface expression levels under other conditions than cell polarization. As mentioned, Kv7.1 regulates acid secretion in the stomach. Interestingly, glucocorticoids have been reported to increase gastric acid secretion and up-regulate Kv7.1 by an SGK1-dependent mechanism (54). SGK1-mediated inhibition of Nedd4-2 could possibly explain this effect. Furthermore, the pathway could be important in other cell types and not only limited to epithelial cell physiology. As mentioned, Kv7.1, together with KCNE1, forms the delayed rectifier potassium current IKs, which is one of the main contributors to the repolarization of the cardiac tissue (1, 2). Mutations leading to loss of channel function causes the most common type of congenital LQT syndrome, type 1 (LQT1). It results in a reduction of IKs and thus a prolongation of action potential duration and the QT interval (see Ref. 55), which often causes dangerous arrhythmias. A recent study reported a mechanism for drug-induced QT interval prolongation that involves changes in several ion currents caused by a decrease in PI3K signaling (56). Although PI3K inhibition of canine cardiac myocytes caused an increase in action potential duration, this could be reversed by intracellular infusion of PIP3. This increase in action potential duration was caused by a decrease in IKs as well as a number of other action potential-shaping currents. Interestingly, the effects of PI3K inhibition were only observed after prolonged exposure suggesting that the effect was not due to direct channel modulation. As Nedd4-2 is also strongly expressed in heart (36), the identified PI3K-SGK1-Nedd4-2 pathway could well explain how PI3K inhibition can cause the observed decrease in IKs.
In summary, the present study positions Nedd4-2 as a key regulator of Kv7.1 cell surface expression. In our previous work, we found that an AMP-activated protein kinase pathway inhibits Kv7.1 surface expression through activation of Nedd4-2 (18). Here, we report a PI3K-SGK1 mechanism with the opposing effect on Kv7.1, namely, stabilizing its surface expression, through inhibition of the ubiquitin ligase Nedd4-2.
This work was supported by The Danish National Research Foundation, The Velux Foundation, The Danish Council for Independent Research-Medical Sciences (to S.-P. O.), the Desirée and Niels Yde Foundation (to T. J.), The Novo Nordisk Foundation (to S.-P. O. and T. J.), the Lundbeck Foundation (to T. J.), the Aase og Ejnar Danielsens Fond (to H. B. R. and T. J.), the Danish Cardiovascular Research Academy (to K. K.), as well as the Swiss National Science Foundation (Grant 310030B_135693; to H. A.).

This article contains supplemental Movie 1.
- LQT
- long QT
- MDCK
- Madin-Darby canine kidney
- ER
- endoplasmic reticulum
- SGK1
- serum- and glucocorticoid-inducible kinase 1
- NCM
- normal calcium media
- EGFP
- enhanced GFP.
REFERENCES
- 1. Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G. (1996) K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384, 78–80 [DOI] [PubMed] [Google Scholar]
- 2. Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., Keating M. T. (1996) Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83 [DOI] [PubMed] [Google Scholar]
- 3. Ullrich S., Su J., Ranta F., Wittekindt O. H., Ris F., Rösler M., Gerlach U., Heitzmann D., Warth R., Lang F. (2005) Effects of I(Ks) channel inhibitors in insulin-secreting INS-1 cells. Pflugers. Arch. 451, 428–436 [DOI] [PubMed] [Google Scholar]
- 4. Yamagata K., Senokuchi T., Lu M., Takemoto M., Fazlul Karim M., Go C., Sato Y., Hatta M., Yoshizawa T., Araki E., Miyazaki J., Song W. J. (2011) Voltage-gated K+ channel KCNQ1 regulates insulin secretion in MIN6 β-cell line. Biochem. Biophys. Res. Commun. 407, 620–625 [DOI] [PubMed] [Google Scholar]
- 5. Dedek K., Waldegger S. (2001) Colocalization of KCNQ1/KCNE channel subunits in the mouse gastrointestinal tract. Pflugers. Arch. 442, 896–902 [DOI] [PubMed] [Google Scholar]
- 6. Vallon V., Grahammer F., Volkl H., Sandu C. D., Richter K., Rexhepaj R., Gerlach U., Rong Q., Pfeifer K., Lang F. (2005) KCNQ1-dependent transport in renal and gastrointestinal epithelia. Proc. Natl. Acad. Sci. U.S.A. 102, 17864–17869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grahammer F., Herling A. W., Lang H. J., Schmitt-Gräff A., Wittekindt O. H., Nitschke R., Bleich M., Barhanin J., Warth R. (2001) The cardiac K+ channel KCNQ1 is essential for gastric acid secretion. Gastroenterology 120, 1363–1371 [DOI] [PubMed] [Google Scholar]
- 8. Heitzmann D., Grahammer F., von Hahn T., Schmitt-Gräff A., Romeo E., Nitschke R., Gerlach U., Lang H. J., Verrey F., Barhanin J., Warth R. (2004) Heteromeric KCNE2/KCNQ1 potassium channels in the luminal membrane of gastric parietal cells. J. Physiol. 561, 547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sunose H., Liu J., Marcus D. C. (1997) cAMP increases K+ secretion via activation of apical IsK/KvLQT1 channels in strial marginal cells. Hear. Res. 114, 107–116 [DOI] [PubMed] [Google Scholar]
- 10. Wangemann P., Liu J., Marcus D. C. (1995) Ion transport mechanisms responsible for K+ secretion and the transepithelial voltage across marginal cells of stria vascularis in vitro. Hear. Res. 84, 19–29 [DOI] [PubMed] [Google Scholar]
- 11. Lee M. P., Ravenel J. D., Hu R. J., Lustig L. R., Tomaselli G., Berger R. D., Brandenburg S. A., Litzi T. J., Bunton T. E., Limb C., Francis H., Gorelikow M., Gu H., Washington K., Argani P., Goldenring J. R., Coffey R. J., Feinberg A. P. (2000) Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J. Clin. Invest. 106, 1447–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang Q., Curran M. E., Splawski I., Burn T. C., Millholland J. M., VanRaay T. J., Shen J., Timothy K. W., Vincent G. M., de Jager T., Schwartz P. J., Toubin J. A., Moss A. J., Atkinson D. L., Landes G. M., Connors T. D., Keating M. T. (1996) Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 12, 17–23 [DOI] [PubMed] [Google Scholar]
- 13. Neyroud N., Tesson F., Denjoy I., Leibovici M., Donger C., Barhanin J., Fauré S., Gary F., Coumel P., Petit C., Schwartz K., Guicheney P. (1997) A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat. Genet. 15, 186–189 [DOI] [PubMed] [Google Scholar]
- 14. Dahimène S., Alcoléa S., Naud P., Jourdon P., Escande D., Brasseur R., Thomas A., Baró I., Mérot J. (2006) The N-terminal juxtamembranous domain of KCNQ1 is critical for channel surface expression: implications in the Romano-Ward LQT1 syndrome. Circ. Res. 99, 1076–1083 [DOI] [PubMed] [Google Scholar]
- 15. Schmitt N., Calloe K., Nielsen N. H., Buschmann M., Speckmann E. J., Schulze-Bahr E., Schwarz M. (2007) The novel C-terminal KCNQ1 mutation M520R alters protein trafficking. Biochem. Biophys. Res. Commun. 358, 304–310 [DOI] [PubMed] [Google Scholar]
- 16. Wilson A. J., Quinn K. V., Graves F. M., Bitner-Glindzicz M., Tinker A. (2005) Abnormal KCNQ1 trafficking influences disease pathogenesis in hereditary long QT syndromes (LQT1). Cardiovasc. Res. 67, 476–486 [DOI] [PubMed] [Google Scholar]
- 17. Andersen M. N., Olesen S. P., Rasmussen H. B. (2011) Kv7.1 surface expression is regulated by epithelial cell polarization. Am. J. Physiol. Cell Physiol. 300, C814-C824 [DOI] [PubMed] [Google Scholar]
- 18. Andersen M. N., Krzystanek K., Jespersen T., Olesen S. P., Rasmussen H. B. (2012) AMP-activated protein kinase downregulates Kv7.1 cell surface expression. Traffic 13, 143–156 [DOI] [PubMed] [Google Scholar]
- 19. Gassama-Diagne A., Yu W., ter Beest M., Martin-Belmonte F., Kierbel A., Engel J., Mostov K. (2006) Phosphatidylinositol-3,4,5-trisphosphate regulates the formation of the basolateral plasma membrane in epithelial cells. Nat. Cell Biol. 8, 963–970 [DOI] [PubMed] [Google Scholar]
- 20. Kuwahara K., Saito Y., Kishimoto I., Miyamoto Y., Harada M., Ogawa E., Hamanaka I., Kajiyama N., Takahashi N., Izumi T., Kawakami R., Nakao K. (2000) Cardiotrophin-1 phosphorylates akt and BAD, and prolongs cell survival via a PI3K-dependent pathway in cardiac myocytes. J. Mol. Cell Cardiol. 32, 1385–1394 [DOI] [PubMed] [Google Scholar]
- 21. Li Y., Tennekoon G. I., Birnbaum M., Marchionni M. A., Rutkowski J. L. (2001) Neuregulin signaling through a PI3K/Akt/Bad pathway in Schwann cell survival. Mol. Cell Neurosci. 17, 761–767 [DOI] [PubMed] [Google Scholar]
- 22. Nelson C. M., Chen C. S. (2002) Cell-cell signaling by direct contact increases cell proliferation via a PI3K-dependent signal. FEBS Lett. 514, 238–242 [DOI] [PubMed] [Google Scholar]
- 23. Samuels Y., Ericson K. (2006) Oncogenic PI3K and its role in cancer. Curr. Opin. Oncol. 18, 77–82 [DOI] [PubMed] [Google Scholar]
- 24. Datta K., Franke T. F., Chan T. O., Makris A., Yang S. I., Kaplan D. R., Morrison D. K., Golemis E. A., Tsichlis P. N. (1995) AH/PH domain-mediated interaction between Akt molecules and its potential role in Akt regulation. Mol. Cell Biol. 15, 2304–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McManus E. J., Collins B. J., Ashby P. R., Prescott A. R., Murray-Tait V., Armit L. J., Arthur J. S., Alessi D. R. (2004) The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J. 23, 2071–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakagawa M., Fukata M., Yamaga M., Itoh N., Kaibuchi K. (2001) Recruitment and activation of Rac1 by the formation of E-cadherin-mediated cell-cell adhesion sites. J. Cell Sci. 114, 1829–1838 [DOI] [PubMed] [Google Scholar]
- 27. Pece S., Chiariello M., Murga C., Gutkind J. S. (1999) Activation of the protein kinase Akt/PKB by the formation of E-cadherin-mediated cell-cell junctions. Evidence for the association of phosphatidylinositol 3-kinase with the E-cadherin adhesion complex. J. Biol. Chem. 274, 19347–19351 [DOI] [PubMed] [Google Scholar]
- 28. Jeanes A., Smutny M., Leerberg J. M., Yap A. S. (2009) Phosphatidylinositol 3′-kinase signalling supports cell height in established epithelial monolayers. J. Mol. Histol. 40, 395–405 [DOI] [PubMed] [Google Scholar]
- 29. Lang F., Böhmer C., Palmada M., Seebohm G., Strutz-Seebohm N., Vallon V. (2006) (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol. Rev. 86, 1151–1178 [DOI] [PubMed] [Google Scholar]
- 30. Franke T. F. (2008) PI3K/Akt: getting it right matters. Oncogene. 27, 6473–6488 [DOI] [PubMed] [Google Scholar]
- 31. Embark H. M., Böhmer C., Vallon V., Luft F., Lang F. (2003) Regulation of KCNE1-dependent K+ current by the serum and glucocorticoid-inducible kinase (SGK) isoforms. Pflugers. Arch. 445, 601–606 [DOI] [PubMed] [Google Scholar]
- 32. Seebohm G., Strutz-Seebohm N., Birkin R., Dell G., Bucci C., Spinosa M. R., Baltaev R., Mack A. F., Korniychuk G., Choudhury A., Marks D., Pagano R. E., Attali B., Pfeufer A., Kass R. S., Sanguinetti M. C., Tavare J. M., Lang F. (2007) Regulation of endocytic recycling of KCNQ1/KCNE1 potassium channels. Circ. Res. 100, 686–692 [DOI] [PubMed] [Google Scholar]
- 33. Debonneville C., Flores S. Y., Kamynina E., Plant P. J., Tauxe C., Thomas M. A., Münster C., Chraïbi A., Pratt J. H., Horisberger J. D., Pearce D., Loffing J., Staub O. (2001) Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J. 20, 7052–7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee I. H., Dinudom A., Sanchez-Perez A., Kumar S., Cook D. I. (2007) Akt mediates the effect of insulin on epithelial sodium channels by inhibiting Nedd4-2. J. Biol. Chem. 282, 29866–29873 [DOI] [PubMed] [Google Scholar]
- 35. Snyder P. M., Olson D. R., Thomas B. C. (2002) Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J. Biol. Chem. 277, 5–8 [DOI] [PubMed] [Google Scholar]
- 36. Jespersen T., Membrez M., Nicolas C. S., Pitard B., Staub O., Olesen S. P., Baró I., Abriel H. (2007) The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc. Res. 74, 64–74 [DOI] [PubMed] [Google Scholar]
- 37. Abriel H., Staub O. (2005) Ubiquitylation of ion channels. Physiology 20, 398–407 [DOI] [PubMed] [Google Scholar]
- 38. Andersen M. N., Rasmussen H. B. (2012) AMPK: A regulator of ion channels. Commun. Integr. Biol. 5, 480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bhalla V., Daidié D., Li H., Pao A. C., LaGrange L. P., Wang J., Vandewalle A., Stockand J. D., Staub O., Pearce D. (2005) Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol. Endocrinol. 19, 3073–3084 [DOI] [PubMed] [Google Scholar]
- 40. Ichimura T., Yamamura H., Sasamoto K., Tominaga Y., Taoka M., Kakiuchi K., Shinkawa T., Takahashi N., Shimada S., Isobe T. (2005) 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J. Biol. Chem. 280, 13187–13194 [DOI] [PubMed] [Google Scholar]
- 41. Jespersen T., Rasmussen H. B., Grunnet M., Jensen H. S., Angelo K., Dupuis D. S., Vogel L. K., Jorgensen N. K., Klaerke D. A., Olesen S. P. (2004) Basolateral localisation of KCNQ1 potassium channels in MDCK cells: molecular identification of an N-terminal targeting motif. J. Cell Sci. 117, 4517–4526 [DOI] [PubMed] [Google Scholar]
- 42. Krzystanek K., Rasmussen H. B., Grunnet M., Staub O., Olesen S. P., Abriel H., Jespersen T. (2012) Deubiquitylating enzyme USP2 counteracts Nedd4-2-mediated downregulation of KCNQ1 potassium channels. Heart. Rhythm. 9, 440–448 [DOI] [PubMed] [Google Scholar]
- 43. Van Campenhout C. A., Eitelhuber A., Gloeckner C. J., Giallonardo P., Gegg M., Oller H., Grant S. G., Krappmann D., Ueffing M., Lickert H. (2011) Dlg3 trafficking and apical tight junction formation is regulated by nedd4 and nedd4–2 e3 ubiquitin ligases. Dev. Cell 21, 479–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gumbiner B., Simons K. (1986) A functional assay for proteins involved in establishing an epithelial occluding barrier: identification of a uvomorulin-like polypeptide. J. Cell Biol. 102, 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Busjahn A., Seebohm G., Maier G., Toliat M. R., Nürnberg P., Aydin A., Luft F. C., Lang F. (2004) Association of the serum and glucocorticoid regulated kinase (sgk1) gene with QT interval. Cell Physiol. Biochem. 14, 135–142 [DOI] [PubMed] [Google Scholar]
- 46. Sandu C., Artunc F., Grahammer F., Rotte A., Boini K. M., Friedrich B., Sandulache D., Metzger M., Just L., Mack A., Skutella T., Rexhepaj R., Risler T., Wulff P., Kuhl D., Lang F. (2007) Role of the serum and glucocorticoid inducible kinase SGK1 in glucocorticoid stimulation of gastric acid secretion. Pflugers. Arch. 455, 493–503 [DOI] [PubMed] [Google Scholar]
- 47. Strutz-Seebohm N., Henrion U., Steinke K., Tapken D., Lang F., Seebohm G. (2009) Serum- and glucocorticoid-inducible kinases (SGK) regulate KCNQ1/KCNE potassium channels. Channels 3, 88–90 [DOI] [PubMed] [Google Scholar]
- 48. Dunn E. F., Fearns R., Connor J. H. (2009) Akt inhibitor Akt-IV blocks virus replication through an Akt-independent mechanism. J. Virol. 83, 11665–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boehmer C., Laufer J., Jeyaraj S., Klaus F., Lindner R., Lang F., Palmada M. (2008) Modulation of the voltage-gated potassium channel Kv1.5 by the SGK1 protein kinase involves inhibition of channel ubiquitination. Cell Physiol. Biochem. 22, 591–600 [DOI] [PubMed] [Google Scholar]
- 50. Snyder P. M., Olson D. R., Kabra R., Zhou R., Steines J. C. (2004) cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na+ channel through convergent phosphorylation of Nedd4-2. J. Biol. Chem. 279, 45753–45758 [DOI] [PubMed] [Google Scholar]
- 51. Chen S. Y., Bhargava A., Mastroberardino L., Meijer O. C., Wang J., Buse P., Firestone G. L., Verrey F., Pearce D. (1999) Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc. Natl. Acad. Sci. U.S.A. 96, 2514–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Butterworth M. B., Edinger R. S., Silvis M. R., Gallo L. I., Liang X., Apodaca G., Frizzell R. A., Fizzell R. A., Johnson J. P. (2012) Rab11b regulates the trafficking and recycling of the epithelial sodium channel (ENaC). Am. J. Physiol. Renal Physiol. 302, F581-F590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karpushev A. V., Levchenko V., Pavlov T. S., Lam V. Y., Vinnakota K. C., Vandewalle A., Wakatsuki T., Staruschenko A. (2008) Regulation of ENaC expression at the cell surface by Rab11. Biochem. Biophys. Res. Commun. 377, 521–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rotte A., Bhandaru M., Föller M., Biswas R., Mack A. F., Friedrich B., Rexhepaj R., Nasir O., Ackermann T. F., Boini K. M., Kunzelmann K., Behrens J., Lang F. (2009) APC sensitive gastric acid secretion. Cell Physiol. Biochem. 23, 133–142 [DOI] [PubMed] [Google Scholar]
- 55. Goldenberg I., Zareba W., Moss A. J. (2008) Long QT Syndrome. Curr. Probl. Cardiol. 33, 629–694 [DOI] [PubMed] [Google Scholar]
- 56. Lu Z., Wu C. Y., Jiang Y. P., Ballou L. M., Clausen C., Cohen I. S., Lin R. Z. (2012) Suppression of phosphoinositide 3-kinase signaling and alteration of multiple ion currents in drug-induced long QT syndrome. Sci. Transl. Med. 4, 131ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]