

Background: PTPα has been implicated in breast cancer, but its function remains to be defined.
Results: Suppression of PTPα led to a GRB7-dependent, ErbB2-mediated increase in mammary epithelial cell migration. PTPα dephosphorylated FAK on Tyr-407.
Conclusion: PTPα functions to suppress ErbB2 signaling events that lead to migration of breast cancer cells.
Significance: PTPα may play positive or negative roles in signaling, depending upon context.
Keywords: Breast Cancer, Cell Motility, Oncogene, Phosphatase, Phosphorylation, Protein Kinases, ErbB2, FAK, PTPα, Dephosphorylation
Abstract
We investigated the role of protein-tyrosine phosphatase α (PTPα) in regulating signaling by the ErbB2 oncoprotein in mammary epithelial cells. Using this model, we demonstrated that activation of ErbB2 led to the transient inactivation of PTPα, suggesting that attenuation of PTPα activity may contribute to enhanced ErbB2 signaling. Furthermore, RNAi-induced suppression of PTPα led to increased cell migration in an ErbB2-dependent manner. The ability of ErbB2 to increase cell motility in the absence of PTPα was characterized by prolonged interaction of GRB7 with ErbB2 and increased association of ErbB2 with a β1-integrin-rich complex, which depended on GRB7-SH2 domain interactions. Finally, suppression of PTPα resulted in increased phosphorylation of focal adhesion kinase on Tyr-407, which induced the recruitment of vinculin and the formation of a novel focal adhesion kinase complex in response to ErbB2 activation in mammary epithelial cells. Collectively, these results reveal a new role for PTPα in the regulation of motility of mammary epithelial cells in response to ErbB2 activation.
Introduction
Reversible tyrosine phosphorylation, catalyzed by the synchronized and complementary activity of protein tyrosine kinases (PTKs)6 and protein-tyrosine phosphatases (PTPs), is primarily utilized in multicellular eukaryotes to communicate between and within cells (1). The coordinated activity of the large families of PTKs and PTPs regulates the function of proteins involved in a plethora of cellular processes, and the disruption of this PTK-PTP balance has been linked to the etiology of several diseases, including cancer. Initial studies revealing the critical role of PTKs in promoting oncogenesis (2) led naturally to the concept that PTPs may function as tumor suppressors (3). However, the situation is more complex. Interestingly, PTPs that function as the products of oncogenes have been discovered, such as the SRC homology 2 domain containing phosphatase (SHP2) (3, 4) as well as PTKs with tumor suppressor activity, such as spleen tyrosine kinase (SYK) (5).
Protein-tyrosine phosphatase α (PTPα, encoded by the PTPRA gene) is a receptor-like transmembrane member of the PTP family that catalyzes phosphoryl hydrolysis on proteins through a well defined mechanism (6). These enzymes are characterized by the active-site signature motif HCX5R, in which the cysteine residue is involved in nucleophile attack on the phosphotyrosyl residue of the substrate. PTPα is broadly expressed (7–10) and has been implicated in a variety of biological and pathological processes, including cell cycle arrest (11), neuronal differentiation (12), and tumorigenesis (reviewed in Ref. 13). Of particular significance, PTPα has been implicated in the positive regulation of signaling pathways and is among a small group of receptor-like PTPs, which includes PTPβ (PTPRB), PTPξ (PTPRZ), PTP-LAR (PTPRF), PTPγ (PTPRG), and SAP1 (PTPRH), showing oncogenic potential (3).
The catalytic activity of PTPα resides within a tandem arrangement of cytosolic phosphatase domains (6). The membrane-proximal D1 domain of PTPα is essential and contains most of the catalytic activity. Uniquely to PTPα, the membrane-distal D2 domain is also active, but with lower specific activity than D1. Furthermore, D2 appears to play a role in sensing reactive oxygen species (14, 15) and, following oxidation, may participate in “inside-out” signaling by altering the rotational coupling of PTPα molecules within a receptor dimer (16). There is considerable evidence supporting a role for PTPα in activating SRC and other SRC family kinases (13, 17–19). However, the biological activity of PTPs is highly context-dependent (20), and it is possible that PTPα may recognize other physiological substrates. In fact, p130cas (21), Kv1.2 (22), and the insulin receptor (23) have also been proposed to be substrates of PTPα.
The ability of PTPα to activate SRC family kinases is the mechanism by which this receptor-like phosphatase transforms rat embryo fibroblasts (17). On this basis, it has been assumed that PTPα functions positively to promote tumorigenesis. Consistent with this, PTPα is overexpressed in late-stage colon carcinoma (24), oral squamous carcinoma (25), and gastric carcinoma (26). Nevertheless, the situation is actually more complex. Expression of PTPα varies widely in breast cancer. In some cases, high levels of PTPα are associated with low tumor grade and reduced aggressiveness (27). In addition, metastasizing breast tumors (stage 3) were reported to express low levels of PTPα (27). Consistent with this, ectopic expression of PTPα in ErbB2-positive human mammary tumor cells reduces tumor growth and delays lung metastasis (27). In contrast, experiments in MMTV-ErbB2/PTPα−/− mice suggest that ablation of PTPα does not contribute to ErbB2-induced mammary tumor initiation or metastasis (28). In light of these apparently conflicting observations, this study was designed to address the function of PTPα in ErbB2-induced signaling in human mammary epithelial cells.
EXPERIMENTAL PROCEDURES
Materials
Anti-PTPα and 4G10 antibodies were from Millipore. Anti-PTPα pY789 and anti-FAK antibodies were from Cell Signaling Technology. Anti-GRB7 antibodies were from Abnova, HRP-conjugated anti-HA antibodies were from Roche, and anti-β1-integrin antibodies were from BD Transduction Laboratories. Anti-FLAG, anti-β-actin, PT-66-agarose-conjugated beads, anti-FLAG M2 beads, and anti-HA beads were purchased from Sigma. HRP-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories, Inc. Protease inhibitor mixture tablets were from Roche. Catalase and superoxide dismutase were from Calbiochem. Surfact-Amps Nonidet P-40, zeba desalt spin columns, EZ-Link iodoacetyl-PEG2-biotin, and iodoacetic acid were from Thermo Scientific. G7–18NATE peptide (sequence WFEGYDNTFPC cyclized via a thioether bond) was prepared by S. Pero (29). Peroxyfluor-6 acetoxymethyl ester (PF6-AM) was prepared by B. Dickinson (30). AP1510 was purchased from ARIAD Pharmaceuticals.
Generation of FLAG-tagged PTPα Fusion Proteins
Full-length Human PTPα was cloned into p3XFLAG-CMV-13 mammalian expression vector (Sigma, catalog no. E4776), in which the N-terminal preprotrypsin leader sequence preceding the multiple cloning region was deleted. Using p3XFLAG-CMV13-PTPα (WT) as the template, expression constructs for trapping mutants (PTPα (D1DA) and PTPα (D2EA)) were generated by QuikChange mutagenesis. All these constructs have a C-terminal 3XFLAG tag.
Hydrogen Peroxide Molecular Imaging
Molecular imaging of ErbB2-induced hydrogen peroxide production in 10A.B2 cells was studied using a PerkinElmer Life Sciences Ultraview spinning disk confocal operating on a Nikon Ti microscope with the in vivo scientific chamber, heater, and gas regulator as described previously (30). Images were analyzed using ImageJ (Wayne Rasband, National Institutes of Health).
Assay of PTP Oxidation
In PTPs, the catalytic cysteinyl residue is present as a thiolate anion in resting cells. After ErbB2 activation by AP1510, the cells were lysed in a degassed buffer at pH 5.5 containing iodoacetic acid. The active-site cysteinyl residue of PTPs that remained in a reduced state was terminally inactivated by alkylation. Conversely, the active-site cysteines of PTPs that were oxidized by second-messenger reactive oxygen species molecules were protected from irreversible alkylation. Iodoacetic acid was then removed from the lysate by size exclusion chromatography, and the reversibly oxidized active-site cysteinyl residues were reduced back to the thiolate ion with Tris(2-carboxyethyl)phosphine (TCEP). PTPs were maintained in pH 5.5 buffers and incubated with a biotinylated sulfhydryl-reactive iodoacetyl-PEG2 probe. After purification by streptavidin pull-down, PTPs that were oxidized in response to ErbB2 signaling were identified by immunoblotting (31).
Generation of Cells Expressing shRNA PTPα
For stable PTPα knockdown in 10A.B2 cells, we expressed a pMLP retroviral vector in a pMSCV backbone (32) using the targeting sequence CAGATGGTGCAAACCGATA incorporated into the sequence of the human microRNA-30 (miR30). The infected cells were selected in medium containing 1–2 μg of puromycin, and EGFP coexpression was verified using a Zeiss Axiovert 200 M microscope.
Immunoprecipitation and Immunoblotting
HA-ErbB2, tyrosine-phosphorylated proteins, and FAK were immunoprecipitated as follows. Cells were grown to 90% confluence in 10-cm plates, serum-starved for 16 h, and stimulated with AP1510 to induce ErbB2 dimerization and activation for the indicated times. After treatment, the cells were washed with cold PBS and extracted in 800 μl of a lysis buffer consisting of 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 5 mm EDTA, 10 mm EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 20 mm β-glycerophosphate, 1 mm Na3VO4, 20 mm NaF, 1 mm PMSF, and protease inhibitor mixture. All subsequent steps were carried out on ice or at 4 °C. Cells were lysed on a rotating wheel at 4 °C for 30 min. Cell debris were centrifuged at 12,000 × g for 10 min, and protein concentrations were determined. An equal amount of protein was diluted in cold lysis buffer and precleared for 60 min with protein A/G-Sepharose. The supernatants were first incubated for 60 min on a rotating wheel with appropriate antibodies, and 10 μl of protein A/G-Sepharose was then added for another 60 min. The immune complexes were pelleted at 3000 × g for 5 min and washed three times with lysis buffer. The beads were resuspended in 20 μl of 4× Laemmli sample buffer and heated at 95 °C for 1 min. Proteins were separated by SDS-PAGE and detected by immunoblotting.
Cell Migration Assays
Cell motility was studied using a Boyden chamber-based migration assay (33) using cell culture inserts (8.0-μm pore size) for 6-well plates (BD Falcon). For siRNA studies, knockdowns were performed with specified siRNAs (siα-1, 5′-CAGAUGGUGCAAACCGAUA dTdT-3′; siα-2, 5′-AAGCUGGGAGCCAUUCCAAUU dTdT-3′) using Lipofectamine as described (34). To quantitate cell motility, 100,000 cells were seeded on the inserts. After 48 h, the cells were washed with 1× PBS and fixed with 5% buffered Formalin solution, stained, and counted. The cells retained inside the insert were removed, and those that had migrated through the pores to the bottom surface of the transwell were counted. For each condition, the number of migrating cells was counted in eight random microscopic fields. The number of migrating cells in the control 10A.B2 cells without stimulation was normalized to 1. Where indicated, AP1510 (1 μm), G7–18NATE-Penetratin (G7–18NATE-P) peptide (GRB7 inhibitory peptide, WFEGYDNTFPC-RQIKIWFQNRRMKWKK) or Penetratin (RQIKIWFQNRRMKWKK) were added to the culture medium at the beginning of the assay. Cell motility was quantitated after 48 h.
In Vitro Phosphatase Assay
HA-tagged PTPα was expressed in HEK293T cells, purified, and washed several times with ice-cold reducing buffer (50 mm HEPES (pH7.4), 100 mm NaCl, 0.1% Triton X-100, 2 mm DTT, and a protease inhibitors tablet) for 10 min on ice to complete the reduction of PTPα. The reduced enzyme was then incubated with phosphorylated FAK at 30 °C for 30 min. The reaction was stopped by addition of 20 μl of 4× Laemmli sample buffer and heated at 95 °C for 1 min. Proteins were separated by SDS-PAGE, and substrate dephosphorylation was visualized by immunoblotting.
RESULTS
Cooperation between PTPα and ErbB2 Signaling in Mammary Epithelial Cells
We tested the effects of suppressing the expression of PTPα on ErbB2-induced signaling in mammary epithelial cells using two independent siRNA sequences. The effect of PTPα suppression on ErbB2-induced cell motility was examined using a Boyden chamber-based migration assay. Dimerization and activation of ErbB2 was induced in MCF10A cells that expressed a well characterized chimeric form of the kinase (10A.B2) the activity of which can be induced by addition of a small molecule dimerizer, AP1510, as described previously (35). Activation of ErbB2 in parental 10A.B2 cells or cells treated with scrambled siRNA resulted in an ∼3.5-fold stimulation of migration. Following treatment with an siRNA sequence shown previously to suppress PTPα (34) and a second siRNA designed using the RNAi Codex program at Cold Spring Harbor Laboratory, we observed that ErbB2 activation resulted in an 6- to 8-fold increase in cell motility compared with the basal migration observed in unstimulated 10A.B2 cells (Fig. 1A). Consistent with this observation, both siRNAs efficiently suppressed PTPα expression, whereas the nonspecific siRNA did not (Fig. 1B).
FIGURE 1.

Suppression of PTPα induced ErbB2-mediated 10A.B2 cell motility. A, 10A.B2 cells, either untransfected or transfected with a nonspecific control siRNA (si-N.S.) or two distinct siRNAs targeting PTPα (siα-1 and siα-2) were seeded in transwell migration chambers. After incubation in the absence (−) or presence (+) of 1 μm AP1510 for 48 h, migration was quantitated as described under “Experimental Procedures.” B, the effect of siRNA on the expression of PTPα assessed by immunoblotting of cell lysates using actin as a loading control. C, 10A.B2 cells were infected with a retroviral vector encoding shRNA targeting PTPα (shPTPα) and selected to create a stable cell line. Lysates from 10A.B2 and 10A.B2-shPTPα cells were immunoblotted with either anti-PTPα rabbit polyclonal antibody or with anti-PTPϵ goat polyclonal antibody. Loading controls were performed by immunoblotting for actin. D, cells (10A.B2 and 10A.B2-shPTPα) were seeded in transwell migration chambers in the absence (−) or presence (+) of 1 μm AP1510 for 48 h, and migration was quantitated as described under “Experimental Procedures.”
A stable cell line expressing the most effective shRNA targeting PTPα was then established, and cells were selected. Following the selection, the depletion of PTPα by shRNA in 10A.B2 cells was estimated to be ∼90%. Specificity in the effect of the shRNA was illustrated by the fact that expression of the closely related PTP family member PTPϵ was not affected (Fig. 1C). Using these shRNA-expressing 10A.B2 cells, the effect of PTPα suppression on ErbB2-induced cell motility was then re-examined. In the presence of AP1510, the migration of parental 10A.B2 cells was increased 3-fold. In contrast, following shRNA-depletion of PTPα, ErbB2 activation resulted in a 5-fold increase in cell motility compared with the basal migration of unstimulated 10A.B2 cells (Fig. 1D). Hence, attenuation of PTPα contributed to increased ErbB2 signaling.
Transient Oxidation and Inactivation of PTPα in Response to ErbB2 Signaling in Mammary Epithelial Cells
Hydrogen peroxide (H2O2) has been shown to inactivate protein-tyrosine phosphatases and, thereby, to promote protein-tyrosine phosphorylation-dependent signal transduction (36). Suppressing the expression of a particular PTP effectively reproduces oxidation-mediated inactivation and increases the phosphorylation of sites that are targeted by that PTP to promote downstream events in the signaling cascade (37). To determine whether PTPα was reversibly oxidized in the context of ErbB2 signaling, first we measured H2O2 production using molecular imaging with a specific fluorescence indicator, PF6-AM (30). This probe features an aryl boronate chemical switch that allows for selective detection of H2O2 over other reactive oxygen species molecules (38–41). Following serum withdrawal, 10A.B2 cells were loaded with PF6-AM and treated with AP1510 (Fig. 2A). ErbB2 activation caused a rapid and time-dependent increase in intracellular fluorescence. Considering the known selectivity of PF6-AM, our data indicate that, following addition of AP1510 and ErbB2 dimerization in 10A.B2 mammary epithelial cells, endogenous generation of H2O2 occurred in less than 2 min and peaked at 5 min. In addition, preincubation of 10A.B2 cells with a chemical inhibitor of NADPH oxidases, diphenyleneiodonium (DPI), prevented ErbB2-mediated H2O2 production and intracellular fluorescence of PF6-AM.
FIGURE 2.

ErbB2-mediated H2O2 production enhanced phospho-tyrosine signaling and PTPα-reversible oxidation in 10A. B2 cells. A, ErbB2-induced H2O2 production was assessed by molecular imaging using PF6-AM. Serum-starved 10A.B2 cells were loaded with 5 μm PF6-AM for 20 min and stimulated with 1 μm AP1510 for the indicated times (t) (in minutes) and then imaged. Alternatively, 100 μm H2O2 was added to the medium for 5 min. For diphenyleneiodonium (DPI) treatment, cells were preincubated in medium containing 10 μm DPI for 30 min prior to AP1510 stimulation for 5 min. Scale bars = 50 μm. B, DPI inhibition of ErbB2-induced tyrosine phosphorylation. Serum-starved 10A.B2 cells were stimulated with 1 μm AP1510 in the presence or absence of DPI (10 μm, 30 min) for the indicated times. Tyrosine-phosphorylated proteins were immunoprecipitated (IP) from 200 μg of cell lysate using PT-66 and immunoblotted using 4G10 anti-pTyr antibodies. ErbB2 was detected using anti-HA antibodies. Loading controls were performed by immunoblotting lysates for actin. C, schematic of the cysteinyl labeling assay (31). IAA, iodoacetic acid; IAP, iodoacetyl-PEG2. D, serum-deprived 10A.B2 cells were incubated with AP1510 (1 μm) for the indicated times and subjected to the cysteinyl labeling assay. Biotinylated proteins were purified on streptavidin-Sepharose (s-S) beads, resolved on SDS-PAGE, and immunoblotted for PTPα. Lysates were also probed for PTPα as controls. PD, pull down.
We tested whether DPI compromised signaling downstream of ErbB2, as reported previously for other receptor tyrosine kinases (31, 42). Serum-starved cells were stimulated with AP1510 in the presence or absence of DPI. Cellular extracts were prepared, and tyrosine phosphorylated proteins were visualized by anti-pTyr immunoprecipitation and immunoblotting. Interestingly, inhibition of H2O2 production by DPI led to decreased tyrosine phosphorylation of proteins of ∼180, 75, 55, and 45 kDa (Fig. 2B). In addition, blotting with anti-HA revealed that the ∼180-kDa band comigrated with ErbB2. Hence, decreasing the acute generation of H2O2, which would be expected to increase the level of active PTPs, compromised tyrosine phosphorylation and activation of ErbB2 and its downstream targets in mammary epithelial cells.
We have shown previously that PTPα is reversibly oxidized upon EGF receptor activation in IMR90 fibroblasts (43). Hence, we used a modified cysteinyl-labeling assay (43) to ascertain whether PTPα activity was also regulated by H2O2 generated upon ErbB2 activation in 10A.B2 cells. This is a three-step method in which the reversibly oxidized invariant catalytic cysteine residue is specifically biotinylated by a thiolate anion-directed probe (Fig. 2C). Serum-starved 10A.B2 cells were incubated with AP1510 and lysed in an anaerobic work station that was purged and constantly supplied with ultrapure argon gas to prevent post-lysis oxidation of PTPs. We detected only minimal biotin labeling of PTPα in untreated cells. However, the biotinylation pattern from the cysteinyl labeling assay revealed that the reversible oxidation of PTPα occurred in a biphasic manner upon ErbB2 activation (Fig. 2D). Thus, ErbB2 activation attenuated PTPα activity by reversible oxidation in mammary epithelial cells.
Identification of Focal Adhesion Kinase as a Substrate of PTPα
To identify substrates of PTPα that were components of ErbB2-induced signaling pathways, we treated 10A.B2 cells with AP1510 to activate ErbB2 and pervanadate to amplify the signal of potential phosphotyrosine-containing substrates for analysis. Substrate-trapping mutant forms of PTPα, PTPαD1DA, and PTPαD2EA (Fig. 3A) were then utilized to identify potential physiological substrates of the PTP, as described previously (34). Proteins involved in ErbB2 signaling and cell migration were tested as potential interacting partners of PTPα(WT), PTPα(D1DA), or PTPα(D2EA). Interestingly, the PTK FAK was enriched with PTPα-trapping mutants (Fig. 3B). Considering previous studies implicating FAK in ErbB2-induced cell migration, invasion, and focal adhesion turnover (44, 45), we investigated this novel PTPα-FAK interaction further.
FIGURE 3.

Identification of focal adhesion kinase as a PTPα substrate. A, schematic of the domain composition of PTPα. B, immunoblot analysis of FAK associated with substrate-trapping mutants of PTPα. 10A.B2 cells were treated with 1 μm AP1510 for 5 min and 50 μm pervanadate for an additional 30 min prior to lysis. FLAG-PTPα(WT), FLAG-PTPα(D1DA) (DA), or FLAG-PTPα(D2EA) (EA) was incubated with cell lysate, immunoprecipiated (IP) with anti-FLAG, and then protein complexes were analyzed by SDS-PAGE and immunoblotting with anti-FAK or anti-FLAG antibodies. C, the effects of sodium orthovanadate on FAK-PTPαD1DA interaction. Purified PTPα(D1DA) and PTPα(WT) were incubated with cell lysates as in B, with or without the indicated concentration of Na3VO4. Protein complexes were immunoprecipitated using anti-FLAG antibodies, analyzed by SDS-PAGE, and immunoblotted with anti-FAK. AP, AP1510; PV, pervanadate. D, immunoblot analysis of the catalytic activity of PTPα on FAK in vitro. 10A.B2 cellular lysates were prepared as described in B. Phospho-FAK was immunoprecipitated from 100 μm lysate using 1 μg anti-FAK antibody and incubated, or not incubated, with PTPα for 30 min. Proteins were separated by SDS-PAGE and immunoblotted using specific anti-phospho FAK antibodies for pTyr-397, pTyr-407, pTyr-576, pTyr-861, and pTyr-965. Blots were stripped and reprobed for total FAK. E, 10A.B2 and shPTPα cells were incubated with AP1510 for the indicated times. Lysates were prepared, separated by SDS-PAGE, and immunoblotted with either anti-phospho FAK Tyr-407, anti-PTPα, or anti-actin antibodies.
To determine whether the interaction of FAK with the PTPα-trapping mutant occurred via the PTP active site, the trapping experiment was performed in the presence of sodium orthovanadate, a competitive inhibitor and transition state analog of phosphate that prevents substrate binding (46). The interaction of FAK with PTPα(D1DA) was inhibited by vanadate, indicating the involvement of the active site and suggesting that FAK may represent a direct substrate of PTPα in cells (Fig. 3C). Under the conditions of our substrate-trapping experiments, it is possible that multiprotein complexes containing the mutant PTP may be isolated. Therefore, we investigated whether PTPα could dephosphorylate FAK directly in vitro. FAK was immunoprecipitated from lysates of 10A.B2 cells treated with AP1510 and pervanadate. Under these conditions, FAK was phosphorylated on Tyr-397, Tyr-407, Tyr-576, Tyr-861, and Tyr-925 (Fig. 3D). The immunoprecipitates of phosphorylated FAK were then incubated with wild-type active PTPα, and dephosphorylation was measured using phospho-specific anti-pTyr antibodies. Immunoblot analyses revealed that PTPα could dephosphorylate phospho-Tyr-407 of FAK specifically in vitro, whereas none of the other Tyr residues was dephosphorylated following incubation with the phosphatase. A role of PTPα in regulating the phosphorylation status of FAK Tyr-407 was verified in parental 10A.B2 cells and in 10A.B2 cells stably expressing shRNA for PTPα. By immunoblotting using a phosphosite-specific antibody, we observed transient phosphorylation of Tyr-407 of FAK 30–45 min following ErbB2 activation with AP1510 (Fig. 3E). In contrast, FAK Tyr-407 phosphorylation was sustained from 15–60 min following ErbB2 activation when PTPα expression was compromised. Collectively, these results suggest a novel role for PTPα in regulating phosphorylation of FAK on Tyr-407.
Interaction of FAK pTyr-407 with Vinculin
Tyrosine phosphorylation of FAK by SRC and PYK2 (proline-rich tyrosine kinase 2) leads to its activation and association with several SH2 domain-containing proteins as well as with focal adhesion proteins such as paxillin and vinculin. Previous studies in human ventricular endothelial cells have shown that phosphorylation of FAK on tyrosine 407 by PYK2 led to the recruitment of FAK to vinculin and paxillin (47, 48). Hence, to understand the role of PTPα in dephosphorylating FAK, we tested whether FAK pTyr-407 behaved similarly in mammary epithelial cells. FAK was immunoprecipitated from 10A.B2 cells that were incubated with AP1510 for 30 min. We observed FAK interaction with paxillin and vinculin in 10A.B2 cells when PTPα was suppressed. However, vinculin was not coimmunoprecipitated with FAK in parental 10A.B2 cells (Fig. 4). In addition, because FAK interacts directly with GRB7 (growth factor receptor bound 7) to promote cell migration (49), we also tested for the presence of GRB7 and its interacting partner ErbB2 (50) in FAK complexes. Both GRB7 and ErbB2 were detected in FAK complexes upon ErbB2 activation, independently of PTPα expression. This shows that the hyperphosphorylation of FAK Tyr-407 observed in the absence of PTPα contributed to the recruitment of vinculin to FAK in a multiprotein complex.
FIGURE 4.
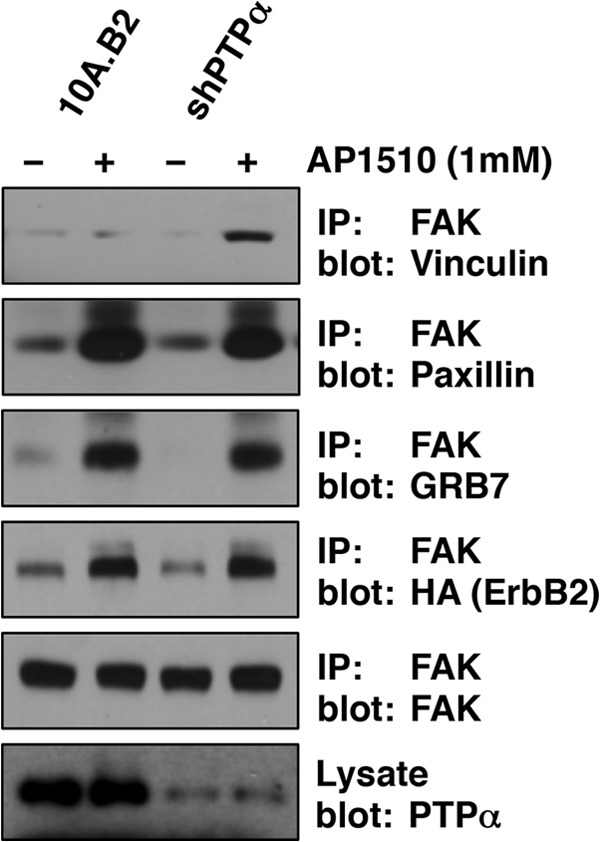
Effect of suppression of PTPα on the recruitment of focal adhesion proteins to FAK. FAK was immunoprecipitated (IP) from serum-deprived 10A.B2 or 10A.B2-shPTPα cells plated on fibronectin (25 μg/ml), incubated or not incubated with AP1510 (1 μm) for 30 min, and lysed. Proteins were separated by SDS-PAGE and immunoblotted using anti-vinculin, anti-paxillin, anti-GRB7, anti-HA, or anti-FAK antibodies. Lysates were probed for PTPα expression.
Increased Association of β1-Integrin and GRB7 with ErbB2 upon Suppression of PTPα
A significant body of evidence indicates that the presence of vinculin in focal adhesions is critical for integrin-mediated cell adhesion and migration (reviewed in Ref. 51). β1-integrin is required for proliferation, survival, and invasiveness of human breast cancer cell lines (52). Integrins associate with the EGF receptor (53, 54), and ErbB2 transactivation is impaired in β1-integrin-deficient breast tumors (55). Considering that an ErbB2-vinculin complex was detected in the absence of PTPα and that vinculin is recruited to the cytoplasmic tails of β-integrins (56), we tested whether the increased ErbB2-mediated migration observed in the absence of PTPα coincided with a change in the formation of an ErbB2-β1-integrin complex. We monitored the association of β1-integrin and GRB7 with ErbB2. ErbB2 was immunoprecipitated following activation in intact 10A.B2 cells, or in cells in which PTPα expression was compromised, and immunoblotted for interacting proteins (Fig. 5). The interaction of PTPα, GRB7, and β1-integrin with ErbB2 was transient and peaked between 30–45 min in parental 10A.B2 cells, whereas suppression of PTPα levels resulted in a rapid and sustained association of β1-integrin with ErbB2. Hence, ErbB2-dependent migration in the absence of PTPα is likely to coincide with enhanced signaling of the receptor at β1-integrin-rich focal adhesions.
FIGURE 5.
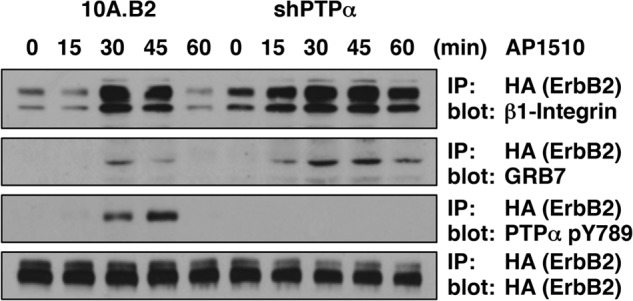
Effect of PTPα suppression on ErbB2 interaction with a β1-integrin complex. Serum-deprived 10A.B2 and 10A.B2-shPTPα cells plated on fibronectin (25 μg/ml) were treated with 1 μm AP1510 for the indicated times. HA-ErbB2 was immunoprecipitated (IP) from 100 μg of cell lysate using anti-HA and immunoblotted using anti-β1-integrin, anti-HA, or anti-GRB7 antibodies. ErbB2 was detected using anti-HA antibody.
Decreased PTPα Expression Led to Enhanced ErbB2-GRB7 Interaction and GRB7-dependent Cell Migration
The GRB7 gene, encoding the SH2-containing adaptor protein GRB7, is part of the ERBB2 amplicon, an ∼86,000-bp region that includes six genes (TCAP, PNMT, PERLD1, HER2, C17orf37/C35, and GRB7) that is amplified in breast cancer (57). It has been reported that GRB7 is present at focal adhesions (58) regulating motility and tumorigenesis in cancer cells (59). GRB7 was present in FAK and ErbB2 immunoprecipitates following the activation of ErbB2 by AP1510. Hence, the presence of GRB7 in the ErbB2-FAK-β1-integrin complexes prompted us to investigate the relationship between PTPα, GRB7, and ErbB2 as part of a potential mechanism leading to increased motility of 10A.B2 cells. To this effect, ErbB2 was activated, immunoprecipitated from 10A.B2 cells expressing shRNA for PTPα or from parental 10A.B2 cells, and then the immunoprecipitates were probed for the presence of the GRB7 adaptor. Consistent with previous observations (50), GRB7 associated with ErbB2 (Fig. 6A). In 10A.B2 cells, there was a low basal level of association, with gradual GRB7 recruitment to ErbB2 upon receptor activation. In contrast, following PTPα knockdown, the basal level of interaction was increased, and ErbB2-induced association of GRB7 to ErbB2 occurred more rapidly (Fig. 6A).
FIGURE 6.

Effects of suppressing PTPα on GRB7-ErbB2 signaling in 10A.B2 cells. A, association of GRB7 with ErbB2. Serum-deprived 10A.B2 and 10A.B2-shPTPα cells were treated with AP1510 (1 μm) for the indicated times. HA-ErbB2 was immunoprecipitated (IP) from 100 μg of cell lysate using anti-HA and immunoblotted using anti-GRB7 antibody. ErbB2 was detected using anti-HA antibody, and lysates were probed for actin and PTPα for loading and PTPα expression. B, sequence of GRB7 SH2-domain inhibitor peptide G7–18NATE. C, the effects of GRB7 inhibitor peptide on ErbB2-induced migration in 10A.B2-shPTPα cells. 10A.B2 and 10A.B2-shPTPα cells were seeded in transwell migration chambers in the absence (−) or presence (+) of 1 μm AP1510, 10 μm G7–18NATE-Penetratin (WFEGYDNTFPC-RQIKIWFQNRRMKWKK), or 10 μm penetratin peptide (RQIKIWFQNRRMKWKK), incubated for 48 h, and then migration was quantitated as described under “Experimental Procedures.”
To examine the importance of the ErbB2-GRB7 interaction on cell motility observed in the absence of PTPα, we tested the effects of a GRB7 inhibitor on ErbB2-induced migration. We used a non-phosphorylated inhibitor peptide specific for the GRB7 SH2 domain, G7–18NATE (GRB7-peptide18-No Arms Thioether, Fig. 6B) bound to a penetratin peptide (G7–18NATE-P) (29) that has been shown previously to attenuate the migration of pancreatic cancer cells (60). The effect of inhibiting GRB7 on ErbB2-induced cell motility in 10A.B2 cells stably expressing shRNA for PTPα was examined using a Boyden chamber-based migration assay. ErbB2 activation resulted in a 5-fold increase in cell migration compared with the basal migration observed in unstimulated 10A.B2 cells. However, treatment of these PTPα-depleted cells with AP1510 in the presence of the GRB7 inhibitor G7–18NATE-P abolished ErbB2-induced cell motility, whereas incubation with the penetratin peptide alone had no effect (Fig. 6C). This suggests that the increased interaction between GRB7 and ErbB2 observed in the absence of PTPα led to a GRB7-dependent increase in 10A.B2 cell migration.
DISCUSSION
Although PTPα has the capacity to display oncogenic properties, its biological role in mammary epithelial cells and breast cancer is unclear (27, 34, 61 and reviewed in Ref. 4). It has been shown that PTPα expression levels vary widely among breast tumors. Furthermore, it is unclear whether PTPα plays a positive or negative role in signaling in breast cancer (27). In this study, we examined the role of PTPα in ErbB2 signaling using a chimeric form of the kinase that could be induced by addition of a small-molecule dimerizing agent, AP1510, in human mammary epithelial 10A.B2 cells. We found that PTPα is a negative regulator of ErbB2-dependent 10A.B2 cell motility. In addressing the function of PTPα in ErbB2 signaling, we uncovered a novel function of the phosphatase in regulating the phosphorylation of FAK on tyrosine 407, regulating FAK binding to vinculin and prolonging the association of ErbB2 with GRB7 and β1-integrins. In addition, PTPα-mediated, ErbB2-dependent cell motility was also dependent upon GRB7 acting as an ErbB2-interacting protein. The consequences of RNAi-induced suppression of PTPα suggest an important role for this receptor protein-tyrosine phosphatase in controlling ErbB2 signal transduction, leading to migration of human mammary cancer cells.
We have shown previously that PTPα is reversibly oxidized following EGF receptor activation (43). Others have also observed a role of PTPα in EGF receptor signaling in diverse mechanisms, such as aging (62), as well as in cell-substratum adhesion (63). However, this is the first study implicating PTPα in the regulation of ErbB2-mediated cell motility. We utilized siRNA targeting human PTPα, designed using the RNAi Codex program at Cold Spring Harbor Laboratory, and confirmed the migration phenotype with another siRNA sequence shown previously to be a potent suppressor of PTPα expression in the Shalloway laboratory (34). By repressing PTPα expression, which would mimic the oxidation-mediated reversible inactivation of the enzyme that occurs in signaling in cells, we increased the phosphorylation of sites targeted by PTPα. This suggested that the transient inactivation of PTPα may control the phosphorylation of FAK and the formation of GRB7 complexes involved in the migratory phenotype. Reversible oxidation of the catalytic cysteine of the D1 domain would be expected to inactivate its function directly. Furthermore, reversible oxidation of the D2 domain of PTPα has been shown to cause the formation of a disulfide bond with the catalytic cysteine of the counterpart D2 domain in the dimer, thereby inducing a conformational change and inhibition of the D1 domain (reviewed in Ref. 6). Hence, the reversible oxidation of either the D1 or D2 cysteine of PTPα, as detected by the cysteinyl labeling assay, is a measure of the inactivation of PTPα occurring following the rapid increase in intracellular hydrogen peroxide that takes place upon acute ErbB2 activation.
The highly dynamic process of cell migration, regulated by tyrosine phosphorylation within focal complexes, involves modulation of cell-substrate adhesion and recruitment of over 50 structural proteins to the cytoplasmic segments of α- and β-integrins (64). FAK is a central regulator of focal complexes. It has been implicated in cancer cell motility in vitro in addition to being an important contributor to tumor invasion, metastasis, and malignancy (65–67). There have been reports indicating that, under certain circumstances, PTPα, acting via stimulation of SRC, may promote phosphorylation of Tyr-397 in FAK (68). In this study, analysis by RNAi-induced suppression of PTPα, application of substrate-trapping mutant forms of the enzyme, and a direct phosphatase activity assay all illustrate dephosphorylation of Tyr-407 of FAK by PTPα. We did not observe significant changes in any other sites of tyrosine phosphorylation in FAK, highlighting the specificity of the phosphatase. Moreover, we found that FAK Tyr-407 phosphorylation was prolonged upon ErbB2 activation when PTPα expression was compromised. FAK Tyr-407 has been shown previously to be phosphorylated by PYK2 in response to VEGF stimulation (48) as well as by SRC (69). In addition to its function as a kinase, phosphorylation of FAK is known to promote its function as a scaffold protein (49). We have observed that FAKpTyr-407 displayed preferential recruitment to vinculin in addition to being associated with ErbB2, GRB7, and paxillin. This pTyr-407-dependent interaction between FAK and vinculin has also been observed by others (47). However, the significance of pTyr-407 phosphorylation is still unclear. Previous groups have shown that FAK Tyr-407 phosphorylation occurs at focal adhesions and at the periphery of migrating cells (48, 70), in tumor cell differentiation (71), and in epithelial mesenchymal transdifferentiation (70). Interestingly, it has also been proposed to be a FAK regulatory site (72). Thus, because ErbB2-induced cell migration, invasion, and focal adhesion turnover is dependent on FAK signaling (44, 45), identifying the SH2-containing signaling protein bound to phosphoTyr-407 in this context may yield further insight into the role of PTPα in the ErbB2-dependent migration of human mammary epithelial cells.
Our study, demonstrating that the SH2 domain peptide inhibitor of GRB7 (G7–18NATE-P) completely abolished ErbB2-mediated 10A.B2 cell migration following suppression of PTPα, stressed the important scaffolding role of GRB7. GRB7 was initially characterized as an interacting partner of ErbB2 at the tyrosine 1139 site (50) and has been implicated in the regulation of focal adhesion function and cell migration (58). Our studies also illustrate that disruption of PTPα expression regulates the interaction between GRB7 and ErbB2 and suggest a potential role for the GRB7 adaptor protein in the effects of PTPα. It has been reported that GRB7 can form dimers (73), suggesting the possibility that these may provide anchorage points for proteins at focal adhesion complexes. It has been shown that phosphorylation of PTPα at Tyr-789, previously identified as a binding site for GRB2 (74), was critical in targeting PTPα to focal adhesions (75, 76). It would be interesting to investigate whether GRB7 is a candidate SH2-containing protein that mediates the recruitment of PTPα to focal adhesions. A phosphotyrosine displacement mechanism has been proposed to facilitate the activation of SRC by PTPα in which pTyr-789 of PTPα engages the SH2 domain of SRC, thereby exposing the C-terminal pTyr for dephosphorylation and activation of the kinase (77). Perhaps pTyr-789, functioning as a GRB7 docking site on PTPα, could provide a competing phospho site to tyrosine 1139 on ErbB2. Therefore, the transient localization of ErbB2 at β1-integrin complexes may be regulated in a similar manner to that observed for the activation of SRC by PTPα, in that engagement of pTyr-789 on the phosphatase by GRB7 may expose other sites for dephosphorylation. The presence of GRB7 together with β1-integrins and FAK following ErbB2 activation may also be significant because it has been reported previously that the β1-integrin-FAK axis controls the initial proliferation of micrometastases of mammary carcinoma cells in the lung (78).
Overall, we have shown for the first time that the suppression of PTPα expression leads to a GRB7-dependent increase in migration of human mammary epithelial cells in response to ErbB2 activation. Our data support a novel role for PTPα in regulating the phosphorylation of FAK at tyrosine 407, thereby promoting its association with vinculin at β1-integrin focal adhesion complexes. These novel aspects of PTPα signaling reveal an important role of the phosphatase in the regulation of a key mediator of focal adhesions and cell migration and of ErbB2-mediated mammary cancer cell migration.
Acknowledgments
We thank the members of the Tonks laboratory, D. Shalloway, and S. Muthuswamy for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants CA53840 and GM55989 (to N. K. T.) and GM79465 (to C. J. C.). This work was also supported by Cold Spring Harbor Laboratory Cancer Centre Support Grant CA45508 and by The Gladowsky Breast Cancer Foundation, The Don Monti Memorial Research Foundation, the Hansen Memorial Foundation, the West Islip Breast Cancer Coalition for Long Island, Glen Cove CARES, Find a Cure Today (FACT), Constance Silveri, the Robertson Research Fund, and the Masthead Cove Yacht Club Carol Marcincuk Fund (to N. K. T.).
- PTK
- protein-tyrosine kinase
- PTP
- protein-tyrosine phosphatase
- PF6-AM
- peroxyfluor acetoxymethyl ester
- FAK
- focal adhesion kinase
- DPI
- diphenyleneiodonium
- TCEP
- tris(2-carboxyethyl)phosphine.
REFERENCES
- 1. Hunter T. (2009) Tyrosine phosphorylation. Thirty years and counting. Curr. Opin. Cell Biol. 21, 140–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blume-Jensen P., Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365 [DOI] [PubMed] [Google Scholar]
- 3. Julien S. G., Dubé N., Hardy S., Tremblay M. L. (2011) Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 11, 35–49 [DOI] [PubMed] [Google Scholar]
- 4. Ostman A., Hellberg C., Böhmer F. D. (2006) Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer 6, 307–320 [DOI] [PubMed] [Google Scholar]
- 5. Mócsai A., Ruland J., Tybulewicz V. L. (2010) The SYK tyrosine kinase. A crucial player in diverse biological functions. Nat. Rev. Immunol. 10, 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tonks N. K. (2006) Protein tyrosine phosphatases. From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846 [DOI] [PubMed] [Google Scholar]
- 7. Sap J., D'Eustachio P., Givol D., Schlessinger J. (1990) Cloning and expression of a widely expressed receptor tyrosine phosphatase. Proc. Natl. Acad. Sci. U.S.A. 87, 6112–6116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaplan R., Morse B., Huebner K., Croce C., Howk R., Ravera M., Ricca G., Jaye M., Schlessinger J. (1990) Cloning of three human tyrosine phosphatases reveals a multigene family of receptor-linked protein-tyrosine-phosphatases expressed in brain. Proc. Natl. Acad. Sci. U.S.A. 87, 7000–7004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krueger N. X., Streuli M., Saito H. (1990) Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J. 9, 3241–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Matthews R. J., Cahir E. D., Thomas M. L. (1990) Identification of an additional member of the protein-tyrosine-phosphatase family. Evidence for alternative splicing in the tyrosine phosphatase domain. Proc. Natl. Acad. Sci. U.S.A. 87, 4444–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng X. M., Shalloway D. (2001) Two mechanisms activate PTPα during mitosis. EMBO J. 20, 6037–6049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. den Hertog J., Pals C. E., Peppelenbosch M. P., Tertoolen L. G., de Laat S. W., Kruijer W. (1993) Receptor protein tyrosine phosphatase α activates pp60c-src and is involved in neuronal differentiation. EMBO J. 12, 3789–3798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pallen C. J. (2003) Protein tyrosine phosphatase α (PTPα). A Src family kinase activator and mediator of multiple biological effects. Curr. Top. Med. Chem. 3, 821–835 [DOI] [PubMed] [Google Scholar]
- 14. Persson C., Sjöblom T., Groen A., Kappert K., Engström U., Hellman U., Heldin C. H., den Hertog J., Ostman A. (2004) Preferential oxidation of the second phosphatase domain of receptor-like PTP-α revealed by an antibody against oxidized protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 101, 1886–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang J., Groen A., Lemeer S., Jans A., Slijper M., Roe S. M., den Hertog J., Barford D. (2007) Reversible oxidation of the membrane distal domain of receptor PTPα is mediated by a cyclic sulfenamide. Biochemistry 46, 709–719 [DOI] [PubMed] [Google Scholar]
- 16. van der Wijk T., Blanchetot C., Overvoorde J., den Hertog J. (2003) Redox-regulated rotational coupling of receptor protein-tyrosine phosphatase α dimers. J. Biol. Chem. 278, 13968–13974 [DOI] [PubMed] [Google Scholar]
- 17. Zheng X. M., Wang Y., Pallen C. J. (1992) Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature 359, 336–339 [DOI] [PubMed] [Google Scholar]
- 18. Ponniah S., Wang D. Z., Lim K. L., Pallen C. J. (1999) Targeted disruption of the tyrosine phosphatase PTPα leads to constitutive downregulation of the kinases Src and Fyn. Curr. Biol. 9, 535–538 [DOI] [PubMed] [Google Scholar]
- 19. Su J., Muranjan M., Sap J. (1999) Receptor protein tyrosine phosphatase α activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr. Biol. 9, 505–511 [DOI] [PubMed] [Google Scholar]
- 20. Tonks N. K., Muthuswamy S. K. (2007) A brake becomes an accelerator. PTP1B. A new therapeutic target for breast cancer. Cancer Cell 11, 214–216 [DOI] [PubMed] [Google Scholar]
- 21. Buist A., Blanchetot C., Tertoolen L. G., den Hertog J. (2000) Identification of p130cas as an in vivo substrate of receptor protein-tyrosine phosphatase α. J. Biol. Chem. 275, 20754–20761 [DOI] [PubMed] [Google Scholar]
- 22. Tsai W., Morielli A. D., Cachero T. G., Peralta E. G. (1999) Receptor protein tyrosine phosphatase α participates in the m1 muscarinic acetylcholine receptor-dependent regulation of Kv1.2 channel activity. EMBO J. 18, 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Møller N. P., Møller K. B., Lammers R., Kharitonenkov A., Hoppe E., Wiberg F. C., Sures I., Ullrich A. (1995) Selective down-regulation of the insulin receptor signal by protein-tyrosine phosphatases α and ϵ. J. Biol. Chem. 270, 23126–23131 [DOI] [PubMed] [Google Scholar]
- 24. Tabiti K., Smith D. R., Goh H. S., Pallen C. J. (1995) Increased mRNA expression of the receptor-like protein tyrosine phosphatase α in late stage colon carcinomas. Cancer Lett. 93, 239–248 [DOI] [PubMed] [Google Scholar]
- 25. Berndt A., Luo X., Böhmer F. D., Kosmehl H. (1999) Expression of the transmembrane protein tyrosine phosphatase RPTPα in human oral squamous cell carcinoma. Histochem. Cell Biol. 111, 399–403 [DOI] [PubMed] [Google Scholar]
- 26. Wu C. W., Kao H. L., Li A. F., Chi C. W., Lin W. C. (2006) Protein tyrosine-phosphatase expression profiling in gastric cancer tissues. Cancer Lett. 242, 95–103 [DOI] [PubMed] [Google Scholar]
- 27. Ardini E., Agresti R., Tagliabue E., Greco M., Aiello P., Yang L. T., Ménard S., Sap J. (2000) Expression of protein tyrosine phosphatase α (RPTPα) in human breast cancer correlates with low tumor grade, and inhibits tumor cell growth in vitro and in vivo. Oncogene 19, 4979–4987 [DOI] [PubMed] [Google Scholar]
- 28. Meyer D. S., Aceto N., Sausgruber N., Brinkhaus H., Müller U., Pallen C. J., Bentires-Alj M. (2013) Tyrosine phosphatase PTPα contributes to HER2-evoked breast tumor initiation and maintenance. Oncogene, in press [DOI] [PubMed] [Google Scholar]
- 29. Pero S. C., Oligino L., Daly R. J., Soden A. L., Liu C., Roller P. P., Li P., Krag D. N. (2002) Identification of novel non-phosphorylated ligands, which bind selectively to the SH2 domain of Grb7. J. Biol. Chem. 277, 11918–11926 [DOI] [PubMed] [Google Scholar]
- 30. Dickinson B. C., Peltier J., Stone D., Schaffer D. V., Chang C. J. (2011) Nox2 redox signaling maintains essential cell populations in the brain. Nat. Chem. Biol. 7, 106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boivin B., Tonks N. K. (2010) Analysis of the redox regulation of protein tyrosine phosphatase superfamily members utilizing a cysteinyl-labeling assay. Methods Enzymol. 474, 35–50 [DOI] [PubMed] [Google Scholar]
- 32. Dickins R. A., Hemann M. T., Zilfou J. T., Simpson D. R., Ibarra I., Hannon G. J., Lowe S. W. (2005) Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat. Genet. 37, 1289–1295 [DOI] [PubMed] [Google Scholar]
- 33. Lin G., Aranda V., Muthuswamy S. K., Tonks N. K. (2011) Identification of PTPN23 as a novel regulator of cell invasion in mammary epithelial cells from a loss-of-function screen of the “PTP-ome.” Genes Dev. 25, 1412–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng X., Resnick R. J., Shalloway D. (2008) Apoptosis of estrogen-receptor negative breast cancer and colon cancer cell lines by PTP α and Src RNAi. Int. J. Cancer 122, 1999–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Muthuswamy S. K., Li D., Lelievre S., Bissell M. J., Brugge J. S. (2001) ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol. 3, 785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chiarugi P., Cirri P. (2003) Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem. Sci. 28, 509–514 [DOI] [PubMed] [Google Scholar]
- 37. Meng T. C., Buckley D. A., Galic S., Tiganis T., Tonks N. K. (2004) Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J. Biol. Chem. 279, 37716–37725 [DOI] [PubMed] [Google Scholar]
- 38. Miller E. W., Chang C. J. (2007) Fluorescent probes for nitric oxide and hydrogen peroxide in cell signaling. Curr. Opin, Chem. Biol. 11, 620–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dickinson B. C., Chang C. J. (2011) Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 7, 504–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lippert A. R., Van de Bittner G. C., Chang C. J. (2011) Boronate oxidation as a bioorthogonal reaction approach for studying the chemistry of hydrogen peroxide in living systems. Acc. Chem. Res. 44, 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin V. S., Dickinson B. C., Chang C. J. (2013) Boronate-based fluorescent probes. Imaging hydrogen peroxide in living systems. Methods Enzymol. 526, 19–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Finkel T. (2011) Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boivin B., Yang M., Tonks N. K. (2010) Targeting the reversibly oxidized protein tyrosine phosphatase superfamily. Sci. Signal. 3, pl2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Benlimame N., He Q., Jie S., Xiao D., Xu Y. J., Loignon M., Schlaepfer D. D., Alaoui-Jamali M. A. (2005) FAK signaling is critical for ErbB-2/ErbB-3 receptor cooperation for oncogenic transformation and invasion. J. Cell Biol. 171, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu Y., Benlimame N., Su J., He Q., Alaoui-Jamali M. A. (2009) Regulation of focal adhesion turnover by ErbB signalling in invasive breast cancer cells. Br. J. Cancer 100, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huyer G., Liu S., Kelly J., Moffat J., Payette P., Kennedy B., Tsaprailis G., Gresser M. J., Ramachandran C. (1997) Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 272, 843–851 [DOI] [PubMed] [Google Scholar]
- 47. Le Boeuf F., Houle F., Huot J. (2004) Regulation of vascular endothelial growth factor receptor 2-mediated phosphorylation of focal adhesion kinase by heat shock protein 90 and Src kinase activities. J. Biol. Chem. 279, 39175–39185 [DOI] [PubMed] [Google Scholar]
- 48. Le Boeuf F., Houle F., Sussman M., Huot J. (2006) Phosphorylation of focal adhesion kinase (FAK) on Ser-732 is induced by rho-dependent kinase and is essential for proline-rich tyrosine kinase-2-mediated phosphorylation of FAK on Tyr-407 in response to vascular endothelial growth factor. Mol. Biol. Cell 17, 3508–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Luo M., Guan J. L. (2010) Focal adhesion kinase. A prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 289, 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stein D., Wu J., Fuqua S. A., Roonprapunt C., Yajnik V., D'Eustachio P., Moskow J. J., Buchberg A. M., Osborne C. K., Margolis B. (1994) The SH2 domain protein GRB-7 is co-amplified, overexpressed and in a tight complex with HER2 in breast cancer. EMBO J. 13, 1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peng X., Nelson E. S., Maiers J. L., DeMali K. A. (2011) New insights into vinculin function and regulation. Int. Rev. Cell Mol. Biol. 287, 191–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lahlou H., Muller W. J. (2011) β1-integrins signaling and mammary tumor progression in transgenic mouse models. Implications for human breast cancer. Breast Cancer Res. 13, 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Giancotti F. G., Ruoslahti E. (1999) Integrin signaling. Science 285, 1028–1032 [DOI] [PubMed] [Google Scholar]
- 54. Miyamoto S., Teramoto H., Gutkind J. S., Yamada K. M. (1996) Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation. Roles of integrin aggregation and occupancy of receptors. J. Cell Biol. 135, 1633–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huck L., Pontier S. M., Zuo D. M., Muller W. J. (2010) β1-Integrin is dispensable for the induction of ErbB2 mammary tumors but plays a critical role in the metastatic phase of tumor progression. Proc. Natl. Acad. Sci. U.S.A. 107, 15559–15564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mangeat P., Burridge K. (1984) Actin-membrane interaction in fibroblasts. What proteins are involved in this association? J. Cell Biol. 99, 95s-103s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Staaf J., Jönsson G., Ringnér M., Vallon-Christersson J., Grabau D., Arason A., Gunnarsson H., Agnarsson B. A., Malmström P. O., Johannsson O. T., Loman N., Barkardottir R. B., Borg A. (2010) High-resolution genomic and expression analyses of copy number alterations in HER2-amplified breast cancer. Breast Cancer Res. 12, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shen T. L., Guan J. L. (2004) Grb7 in intracellular signaling and its role in cell regulation. Front. Biosci. 9, 192–200 [DOI] [PubMed] [Google Scholar]
- 59. Chu P. Y., Huang L. Y., Hsu C. H., Liang C. C., Guan J. L., Hung T. H., Shen T. L. (2009) Tyrosine phosphorylation of growth factor receptor-bound protein-7 by focal adhesion kinase in the regulation of cell migration, proliferation, and tumorigenesis. J. Biol. Chem. 284, 20215–20226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tanaka S., Pero S. C., Taguchi K., Shimada M., Mori M., Krag D. N., Arii S. (2006) Specific peptide ligand for Grb7 signal transduction protein and pancreatic cancer metastasis. J. Natl. Cancer Inst. 98, 491–498 [DOI] [PubMed] [Google Scholar]
- 61. Huang J., Yao L., Xu R., Wu H., Wang M., White B. S., Shalloway D., Zheng X. (2011) Activation of Src and transformation by an RPTPα splice mutant found in human tumours. EMBO J. 30, 3200–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tran K. T., Rusu S. D., Satish L., Wells A. (2003) Aging-related attenuation of EGF receptor signaling is mediated in part by increased protein tyrosine phosphatase activity. Exp. Cell Res. 289, 359–367 [DOI] [PubMed] [Google Scholar]
- 63. Harder K. W., Moller N. P., Peacock J. W., Jirik F. R. (1998) Protein-tyrosine phosphatase α regulates Src family kinases and alters cell-substratum adhesion. J. Biol. Chem. 273, 31890–31900 [DOI] [PubMed] [Google Scholar]
- 64. Carragher N. O., Frame M. C. (2004) Focal adhesion and actin dynamics. A place where kinases and proteases meet to promote invasion. Trends Cell Biol. 14, 241–249 [DOI] [PubMed] [Google Scholar]
- 65. Sieg D. J., Hauck C. R., Ilic D., Klingbeil C. K., Schaefer E., Damsky C. H., Schlaepfer D. D. (2000) FAK integrates growth-factor and integrin signals to promote cell migration. Nature Cell Biol. 2, 249–256 [DOI] [PubMed] [Google Scholar]
- 66. Hauck C. R., Hsia D. A., Ilic D., Schlaepfer D. D. (2002) v-Src SH3-enhanced interaction with focal adhesion kinase at β 1 integrin-containing invadopodia promotes cell invasion. J. Biol. Chem. 277, 12487–12490 [DOI] [PubMed] [Google Scholar]
- 67. Hauck C. R., Hsia D. A., Schlaepfer D. D. (2002) The focal adhesion kinase. A regulator of cell migration and invasion. IUBMB Life 53, 115–119 [DOI] [PubMed] [Google Scholar]
- 68. Zeng L., Si X., Yu W. P., Le H. T., Ng K. P., Teng R. M., Ryan K., Wang D. Z., Ponniah S., Pallen C. J. (2003) PTP α regulates integrin-stimulated FAK autophosphorylation and cytoskeletal rearrangement in cell spreading and migration. J. Cell Biol. 160, 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cox B. D., Natarajan M., Stettner M. R., Gladson C. L. (2006) New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J. Cell Biochem. 99, 35–52 [DOI] [PubMed] [Google Scholar]
- 70. Nakamura K., Yano H., Schaefer E., Sabe H. (2001) Different modes and qualities of tyrosine phosphorylation of Fak and Pyk2 during epithelial-mesenchymal transdifferentiation and cell migration. Analysis of specific phosphorylation events using site-directed antibodies. Oncogene 20, 2626–2635 [DOI] [PubMed] [Google Scholar]
- 71. Matkowskyj K. A., Keller K., Glover S., Kornberg L., Tran-Son-Tay R., Benya R. V. (2003) Expression of GRP and its receptor in well-differentiated colon cancer cells correlates with the presence of focal adhesion kinase phosphorylated at tyrosines 397 and 407. J. Histochem. Cytochem. 51, 1041–1048 [DOI] [PubMed] [Google Scholar]
- 72. Lim Y., Park H., Jeon J., Han I., Kim J., Jho E. H., Oh E. S. (2007) Focal adhesion kinase is negatively regulated by phosphorylation at tyrosine 407. J. Biol. Chem. 282, 10398–10404 [DOI] [PubMed] [Google Scholar]
- 73. Porter C. J., Wilce M. C., Mackay J. P., Leedman P., Wilce J. A. (2005) Grb7-SH2 domain dimerisation is affected by a single point mutation. Eur. Biophys. J. 34, 454–460 [DOI] [PubMed] [Google Scholar]
- 74. den Hertog J., Tracy S., Hunter T. (1994) Phosphorylation of receptor protein-tyrosine phosphatase α on Tyr789, a binding site for the SH3-SH2-SH3 adaptor protein GRB-2 in vivo. EMBO J. 13, 3020–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lammers R., Lerch M. M., Ullrich A. (2000) The carboxyl-terminal tyrosine residue of protein-tyrosine phosphatase alpha mediates association with focal adhesion plaques. J. Biol. Chem. 275, 3391–3396 [DOI] [PubMed] [Google Scholar]
- 76. Helmke S., Lohse K., Mikule K., Wood M. R., Pfenninger K. H. (1998) SRC binding to the cytoskeleton, triggered by growth cone attachment to laminin, is protein tyrosine phosphatase-dependent. J. Cell Sci. 111, 2465–2475 [DOI] [PubMed] [Google Scholar]
- 77. Zheng X. M., Resnick R. J., Shalloway D. (2000) A phosphotyrosine displacement mechanism for activation of Src by PTPα. EMBO J. 19, 964–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shibue T., Weinberg R. A. (2009) Integrin β1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. U.S.A. 106, 10290–10295 [DOI] [PMC free article] [PubMed] [Google Scholar]