

Background: The reason for the variability in degree of atherosclerosis in Tangier disease remains poorly understood.
Results: Tangier fibroblasts with different molecular defects display marked phenotypic variability.
Conclusion: Complete ABCA1 deficiency is associated with various potentially atherogenic effects and is compensated for by marked oxysterol-mediated down-regulation of cholesterol biosynthesis.
Significance: Elucidation of the link between the degree of ABCA1 deficiency and the in vivo phenotype.
Keywords: ABC Transporter, Cholesterol, Cholesterol Metabolism, High Density Lipoprotein (HDL), Telomerase, Oxysterols
Abstract
We compared the consequences of an ABCA1 mutation that produced an apparent lack of atherosclerosis (Tangier family 1, N935S) with an ABCA1 mutation with functional ABCA1 knockout that was associated with severe atherosclerosis (Tangier family 2, Leu548:Leu575-End), using primary and telomerase-immortalized fibroblasts. Telomerase-immortalized Tangier fibroblasts of family 1 (TT1) showed 30% residual cholesterol efflux capacity in response to apolipoprotein A-I, whereas telomerase-immortalized Tangier fibroblasts of family 2 (TT2) showed only 20%. However, there were a number of secondary differences that were often stronger and may help to explain the more rapid development of atherosclerosis in family 2. First, the total cellular cholesterol content increase was 2–3-fold and 3–5-fold in TT1 and TT2 cells, respectively. The corresponding increase in esterified cholesterol concentration was 10- and 40-fold, respectively. Second, 24-, 25-, and 27-hydroxycholesterol concentrations were moderately increased in TT1 cells, but were increased as much as 200-fold in TT2 cells. Third, cholesterol biosynthesis was moderately decreased in TT1 cells, but was markedly decreased in TT2 cells. Fourth, potentially atheroprotective LXR-dependent SREBP1c signaling was normal in TT1, but was rather suppressed in TT2 cells. Cultivated primary Tangier fibroblasts were characterized by premature aging in culture and were associated with less obvious biochemical differences. In summary, these results may help to understand the differential atherosclerotic susceptibility in Tangier disease and further demonstrate the usefulness of telomerase-immortalized cells in studying this cellular phenotype. The data support the contention that side chain-oxidized oxysterols are strong suppressors of cholesterol biosynthesis under specific pathological conditions in humans.
Introduction
Identification of ATP-binding cassette transporter A1 (ABCA1)3 as a defective protein in the rare human genetic disorder Tangier disease (TD) demonstrated that this ATP-binding cassette transporter protein is responsible for a rate-limiting step in the efflux of cholesterol from peripheral cells and formation of atheroprotective high density lipoprotein (HDL) (1–5). TD is characterized by a nearly complete absence of normal HDL in plasma and by the accumulation of cholesterol esters in many peripheral tissues, including those from the tonsils, spleen, intestinal mucosa, Schwann cells, thymus, skin, and cornea (6). It is estimated that individuals lacking functional ABCA1 have a prevalence of cardiovascular disease at least 6-fold higher than the general population (7). The study of TD and identification of ABCA1 as a defective protein in TD illustrate the importance of the transport of cholesterol from peripheral cells to the liver as protective function of HDL (reverse cholesterol transport).
On the other hand, atherosclerosis in many TD patients occurs at a relatively advanced age and the observed increase in adverse coronary events is, in many individuals, less than predicted for such a severe reduction in HDL (7). It is not understood why the manifestation of atherosclerosis greatly varies from patient to patient and why it is apparently completely lacking in some patients (8, 9) despite near-complete HDL deficiency and virtually zero cholesterol efflux capacity in cultivated TD fibroblast cell lines (10–12). This observation can only in part be explained by the multifactorial origin of atherosclerosis, the long disease process before clinical signs become manifest, and the existence of unknown compensatory mechanisms that may vary from patient to patient.
Another possible explanation for the discrepancies between clinical and cellular phenotype is genetic heterogeneity and differential biochemical and pathophysiological effects of the respective ABCA1 mutation. However, such subtle differences are hard to detect in standard cell culture models. For example, we demonstrated that cholesterol efflux from senescent cells and cultivated Tangier fibroblasts is significantly improved after immortalization with the ectopic expression of the catalytic subunit of human telomerase (hTERT) (5). HTERT is a specialized cellular reverse transcriptase that can compensate for the erosion of telomeres by synthesizing new telomeric DNA (13). As cholesterol efflux is sensitive to aging artifacts in cell culture, the “improved” cholesterol efflux in hTERT immortalized TD cells, relative to primary TD cells, is likely to more closely reflect the true genetic phenotype. In contrast to cells transformed with oncogenes or carcinogens, cells immortalized with telomerase have normal cell cycle controls, functional p53 and pRB checkpoints, are contact inhibited, anchorage dependent, and require growth factors for proliferation and possess a normal karyotype (13). Thus, hTERT immortalization allows, for the first time, study of TD cells at comparable (“nearly youthful”) conditions. Moreover, immortalization of Tangier fibroblasts with telomerase allows analyses that require relatively large amounts of cell culture material that are difficult to perform with primary TD cells, which have growth abnormalities (14) and a limited proliferative life span in culture.
Here we compared primary and hTERT-immortalized cell lines from a TD family expressing an ABCA1 point mutation whose carriers lack atherosclerosis (family 1), with cells from a TD family with a functional knockout of ABCA1 (family 2) that is associated with severe atherosclerosis in three homozygous patients of the family. The goal of this study was to characterize the consequences of partial and complete ABCA1 loss of function on the regulation of cellular cholesterol homeostasis and cellular pathways that govern cholesterol balance. Family 1 is characterized by an asparagine to serine amino acid substitution in exon 19 (AAT/AGT, amino acid position 935 of the primary translation product) resulting in the expression of defective full-length protein (15); Family 2 is characterized by a homozygous 1-bp deletion in exon 1. This deletion introduces a stop codon at position 575, resulting in the omission of the majority of the ABCA1 protein sequence and expression of a truncated protein (2). The asparagine to serine mutation of family 1 is not associated with premature atherosclerosis (8), whereas all three homozygous patients of family 2 suffer from severe premature atherosclerosis (2). HDL cholesterol levels were close to zero in all 5 homozygous patients of both families. Experiments were performed with hTERT-immortalized cell lines TT1 and TT2, the respective primary cell lines (T1 and T2), and immortalized and non-immortalized control fibroblasts.
We demonstrate that TD cells are characterized by marked cellular phenotype variability. In particular, we found marked differences in the cellular content of side chain-oxidized oxysterols, accompanied by differences in the regulation of key regulatory pathways of cholesterol biosynthesis and potentially protective LXR-dependent gene regulation.
EXPERIMENTAL PROCEDURES
Materials
Cholesterol, essentially fatty acid-free bovine serum albumin, and apolipoprotein A-I (apoA-I) were purchased from Sigma. Thin layer chromatography plates (LK6DF Silica Gel 60 A) were purchased from Whatman and [1,2-3H]cholesterol (48 Ci/mmol) from PerkinElmer Life Sciences. Reagents for lipid analysis were of analytical grade and supplied by Sigma, except for HPLC-grade water and organic solvents, which were purchased from Mallinckrodt Baker, Griesheim, Germany. Human lipoprotein-deficient serum (LPDS) was prepared from pooled human plasma from healthy donors by flotation ultracentrifugation (d > 1.215 g/ml) as described in Ref. 16. The plasma was kindly provided by the Institute for Transfusion Medicine in Münster, Germany. The absence of lipoproteins in the LPDS was confirmed by lipoprotein electrophoresis. Antibodies against SREBP2 (SC-8151) and nonimmune control IgG (SC-2027) were purchased from Santa Cruz Biotechnology.
Cell Culture
Human skin fibroblasts cultured from biopsies of adult human hip-skin were isolated as described previously (17). The Tangier cells were initially maintained using 1:3 split ratios; based on this, we estimated that the cell lines had undergone 22 and 25 population doublings. Subsequent doublings were determined by cell counts. For immortalization, cells were infected with retroviral supernatants obtained from packaging cells stably expressing hTERT cloned into the pBabePuro vector (18). Cells were then selected for 2 weeks using puromycin at a concentration of 750 ng/ml. For cholesterol efflux measurements, normal cells between passage levels 20 and 25, and immortalized cells between passage levels 15 and 30 after immortalization were plated at 25,000 cells/15-mm well or 50,000 cells/35-mm well (for radioactive measurements) and grown to confluence in DMEM containing 10% fetal bovine serum. For GC measurements, cells were grown to near-confluence in the presence of 10% lipoprotein-deficient serum or in the presence of fatty acid-free albumin (1 mg/ml) in the presence or absence of 20 μg/ml of free cholesterol.
Cell Donors
Two patients of family 1, each homozygous for an asparagine to serine amino acid substitution in exon 19 (AATAGT; amino acid position 935 of the primary translation product), were used: patient 1 was a 63-year-old, female; patient 2 was a 66-year-old male and the brother of patient 1. The molecular defect (15) and clinical and biochemical manifestations including HDL deficiency, splenomegaly, lipid storage in reticuloendothelial tissues, and lack of severe atherosclerosis have been described in detail in previous reports (5, 6, 8, 10, 12, 14, 15, 17, 20). The third patient, from a different family (family 2), was a 60-year-old male characterized by a homozygous 1-bp deletion in exon 13 (dG 1764:Leu548:Leu575-End). This deletion introduces a stop codon at position 575, resulting in deletion of the majority of the ABCA1 protein sequence. The virtually complete absence of HDL in three homozygous patients with this mutation is associated with atherosclerosis and coronary heart disease (2). Control fibroblasts included primary and hTERT immortalized fibroblasts from healthy volunteers that have been described elsewhere (19) as well as another primary (C2) and hTERT-immortalized (CT2) cell line from a 58-year-old healthy donor, and an apparently healthy 45-year-old male individual (C3 and CT3); all of these individuals had normal plasma lipid values.
Cholesterol Efflux Measurements
Cholesterol efflux measurements were performed as described previously (20). At ∼50% confluence, 0.5 or 1 μCi/ml of [3H]cholesterol was added to the cell medium. When the cells reached confluence, they were washed three times in phosphate-buffered saline containing 1 mg/ml of bovine serum albumin, whereupon the medium was replaced for 24 h with DMEM containing 1 mg/ml of fatty acid-free albumin (FAFA) and 20 μg/ml of free non-lipoprotein cholesterol, added from a 10 mg/ml stock in an ethanol solution. Cellular cholesterol pools were allowed to equilibrate for another 24 h in DMEM containing 1 mg/ml of FAFA. Efflux studies (0–24 h) were then performed in DMEM containing 1 mg/ml of FAFA and different concentrations of apoA-I.
At the end of the efflux time, the medium was centrifuged for 10 min at 14,000 × g to remove floating cells. Cellular [3H]cholesterol content was measured by dissolving cells in 500 μl of 0.2% SDS (15 min of shaking at room temperature) and scintillation counting of a 100-μl aliquot. Percent cellular cholesterol efflux was calculated as (3H dpm in medium/(3H dpm in medium + 3H dpm in cells)) × 100. Cell protein content was determined with the DC Bio-Rad assay by using bovine serum albumin as a standard.
Oxysterol Preparation
Oxysterols were prepared and analyzed as recently described (21). For oxysterol measurement as an internal standard and antioxidant reagent, a methanolic solution of 1 ng/μl of 22S-HC with 20 ng/μl of 3,5-di-tert-butyl-4-hydroxytoluene was used. Four hundred microliters of this solution were added to 1 ml of cell homogenate and subjected to saponification with 2 ml of 20% tetramethyl-ammoniumhydroxide/2-propanol (w/v, 30 min, 80 °C). Three milliliters of water were added to the cold sample followed by extraction twice with 4 ml of n-hexane. The upper layers were combined in a silanized glass tube and the organic solvent was removed under a nitrogen stream. The residue was resolved in 100 μl of HPLC eluent (solvent system 2-propanol/n-heptane/acetonitrile, 35:12:52, v/v/v). To remove cholesterol, the sample was subjected to HPLC fractionation (as in the conditions described previously, but with a 100-μl loop injector). Oxysterols were collected in silanized glass tubes within the 3rd and 6th min after HPLC injection (fractionating apparatus: Advantec SF2120), derivatized (22), and subjected to further analysis by gas chromatography-mass spectrometry (GC-MS) (22, 23).
Gas Chromatography-Mass Spectrometry
GC-MS analysis of oxysterols was performed on a GCQ system (Thermo-Finnigan, Bremen, Germany) equipped with an ion trap mass analyzer and a HT-5 fused silica capillary column (SGE, Darmstadt, Germany; 25 m, inner diameter 0.22 mm, film thickness 0.1 μm) as previously described (21). Twenty microliters of the sample were injected in a large volume of solvent split mode at 55 °C and vaporized splitlessly at 280 °C. Helium was used as carrier gas at a constant velocity of 30 cm/s. The temperature of the column oven was maintained at 50 °C for 4 min, then increased at 25 °C/min to 200 °C and finally increased at 10 °C/min to 280 °C. The parameters for the ion trap mass analyzer were as follows: transfer line at 280 °C, positive electron ionization mode (70 eV), ion source at 180 °C, full scan on masses 50–650 Da. Oxysterol compound concentrations were quantified by comparing the ion current response of the sum of characteristic quantitation masses with the peak area response obtained for the known amount of the added internal standard. Amounts of oxysterol were normalized to the protein content. Duplicate or triplicate samples were analyzed.
Gas-liquid Chromatography
The concentrations of free and total cholesterol in the cells were determined by GLC of the trimethylsilyl ether derivatives as previously described (22, 23). Cells were harvested and homogenized as described above. Ten micrograms of 5β-cholestan-3a-ol were added to 100 μl of cell lysate as an internal standard. Lipid extraction was performed using the method of Klansek et al. (24). Derivatization was performed according to standard methods (25) and GLC was carried out using a Dani 8521a gas chromatograph (Monza, Italy) equipped with a programmable injector (PTV), a CP-Wax 57CB fused silica capillary column (25 m × 0.32-mm inner diameter, 0.2-μm film thickness, Chromspec, Bridgewater, NJ), a flame ionization detector, and a chromatogram data processor MT2 (Kontron, Neufahrn, Germany).
Quantitiative Real Time-PCR
Quantitiative real time-PCR was performed using an Applied Biosystems Prism 7900HT sequence detection system. Total RNA was treated with DNase I (RNase-free, Qiagen), and reverse-transcribed with oligo(dT) primer using Moloney murine leukemia virus SuperScript III (Invitrogen) to generate cDNA. Primers for each gene were designed using Primer Express Software (PerkinElmer Life Sciences) and validated by analysis of template titration and dissociation curves. Each quantitative RT-PCR contained (final volume of 12 μl) 25 ng of reverse-transcribed RNA, each primer at 400 nm, and 6 μl of 2× SYBR Green PCR Master Mix (Applied Biosystems). Each sample was analyzed in duplicate. Results were evaluated by the comparative CT method (user Bulletin No. 2, PerkinElmer Life Sciences) using cyclophilin as the invariant control gene. RNA levels are expressed relative to those obtained for the controls and reflect the average ± S.D. for n = 3–4 independent experiments. The following PCR primers were used for amplification of mouse and human RNAs: ABCA1, 5′-ATTGCCAGACGGAGTCGGA-3′ (forward) and 5′-GTGGCACGATCAGGCTGAAC-3′ (reverse); SREBP2, 5′-AGGCAGGCTTTGAAGACGAA-3′ (forward) and 5′-GAACAGGCGGATCCTGCAG-3′ (reverse); SREBP1c, 5′-GGCGGGCGCAGATC-3′ (forward) and 5′-TTGTTGATAAGCTGAAGCATGTCT-3′ (reverse); HMG-CoA reductase, 5′-TTGGCAGCAGGACATCTTGTC-3′ (forward) and 5′-TCTTGGTGCAAGCTCCTTGG-3′ (reverse); LDL receptor, 5′-ATGAGGTCCACATTTGCCACA-3′ (forward) and 5′-ATGTTCACGCCACGTCATCC-3′ (reverse); fatty acid synthase, 5′-GCAAATTCGACCTTTCTCAGAA-3′ (forward) and 5′-GTAGGACCCCGTGGAATGTC-3′ (reverse); cyclophilin, 5′-TTTCATCTGCACTGCCAAGA-3′ (forward) and 5′-TTGCCAAACACCACATGCT-3′ (reverse); SCD I, GCCCACCTCTTCGGATATCG-3′ (forward) and TGATGTGCCAGCGGTACTCA-3′ (reverse). Results were normalized to cyclophilin.
Western Analysis
Whole cell extracts were prepared as described in Ref. 26. Fibroblasts were solubilized in Laemmli buffer by aspirating 20 times through a 0.90 × 120-mm TLK-Supra syringe. The protein concentration was determined with the Pierce BCA Protein Assay Kit protein assay. 15 μg of whole cell lysate protein was loaded in each lane. PageRuler protein ladder (Fermentas, SM0671) was used as molecular weight standard. Cell lysates were transferred electrophoretically to a polyvinylidene difluoride membrane (30 V, 14 h). Proteins were visualized by sequential treatment with specific antibodies, horseradish peroxidase-conjugated secondary antibodies, and an ECL kit (Amersham Biosciences/GE Healthcare). In detail, the blotted membrane was fixed in methanol (15 s), dried for cutting or storage at 4 °C, briefly (15s) activated in methanol, rinsed with PBS, blocked 1–1.5 h or overnight in 5% milk powder in 1× PBS, 0.5% Tween. The blot was then incubated for 1.5 h with the first antibody (anti-SREBP-2 monoclonal antibody, BD Bioscience) in 5% milk in 1× PBS, 0.1% Tween. This antibody recognizes the C terminus of human SREBP-2, the cleaved mature form having an apparent molecular mass of 60–70 kDa in SDS gel electrophoresis. The blot was then washed in 1× PBS, 0.1% Tween (1 × 15 min, 3 × 5 min) and subsequently incubated for 1 h with the second antibody (horseradish peroxidase-conjugated goat anti-mouse, dilution 1:5000, Bio-Rad) in 5% milk powder dissolved in 1× PBS, 0.1% Tween. After washing in 1× PBS, 0.1% Tween (1 × 15 min, 3 × 5 min) the blot was developed (1 min) in ECL solution (Amersham Biosciences), as indicated by the manufacturer.
Filipin Staining
Cells were grown on coverslips and fixed with 3% paraformaldehyde in PBS for 20 min, washed three times with PBS, and stained with filipin as described in Ref. 27, with a slight modification. Cells were incubated in PBS with 1.5 mg/ml of glycine for 10 min, washed three times with PBS, and stained with 50 μg/ml of filipin in PBS for 30 min. Coverslips were mounted with ProLong Antifade reagent (Molecular Probes), and filipin fluorescence was detected by fluorescence microscopy using a Zeiss Axiovert epifluorescence microscope. The following filter sets (Chroma) were used for filipin, excitation filter 360/40 nm, beam splitter 400 nm, and emission filter 460/50 nm.
Measurement of de Novo Cholesterol Synthesis
Metabolic labeling of de novo synthesized cholesterol was performed as described (28). On day 0, cells were seeded in triplicate in DMEM, 10% FCS. On day 1, the cells were washed three times with PBS and re-fed DMEM, 10% LPDS. On day 2, the cells were washed three times with PBS and re-fed DMEM, 10% LPDS with [3H]acetate (40 μCi/ml). After a 24-h incubation, the cells were washed three times with Tris-buffered saline at 4 °C, and lipids were extracted by a hexane/isopropyl alcohol (3:2) solution. A chromatography recovery standard was added (20 μg of cholesterol), the samples were dried under a stream of nitrogen (dissolved in 50 μl of hexane/isopropyl alcohol) and separated by TLC using a hexane/diethyl ether/acetic acid (130:30:1.5) solution as the solvent. [3H]Cholesterol was quantified by scintillation counting.
Statistics
Quantitative data were expressed as mean ± S.D. Statistical significance of the data were evaluated by analysis of variance or Student's t test. p values less than 0.05 were considered significant. Results were representative from at least three independent experiments.
Ethics
The study including skin biopsies was reviewed and approved by the ethics committee of the University of Münster, Germany, and informed consent was obtained from all patients.
RESULTS
Cholesterol Efflux from Primary and hTERT-immortalized Tangier Fibroblasts
In various studies, apolipoprotein-mediated cholesterol efflux has previously been shown to be markedly reduced in fibroblasts from Tangier patients (10–12). In accordance with these data, we found a marked reduction of cholesterol efflux in T1 and T2 fibroblasts, compared with fibroblasts from healthy control donors (Fig. 1). As published elsewhere (5), the typical characteristics of Tangier cells (reduced cholesterol efflux and cellular cholesterol accumulation) were also visible in both hTERT-immortalized TT1 and TT2 cells. However, the lipid efflux capacity was significantly improved in both hTERT-infected Tangier cells, compared with the cholesterol efflux in the respective primary cells (T1 versus TT1 and T2 versus TT2 in Fig. 1; p < 0.01). The overall improvement in cholesterol efflux to 30% of normal values remained higher in TT1 cells, compared with 20% in TT2 cells (TT1 versus TT2, p < 0.05, as indicated in Fig. 1). In fibroblasts from healthy control donors, by contrast, the cholesterol efflux was not significantly different in the primary and respective hTERT-immortalized cells (5).
FIGURE 1.

Promotion of apoA-I-inducible cholesterol efflux from primary and hTERT-infected control and TD fibroblasts. Cholesterol-loaded normal (C1, C2), TD (T1, T2), and the respective hTERT-infected fibroblasts (CT1, CT2, TT1, TT2) were radiolabeled with [3H]cholesterol and loaded with non-lipoprotein cholesterol (20 μg/ml) for 24 h, as described under “Experimental Procedures.” Cells were then incubated for 4 h in the presence of 1 mg/ml of FAFA and 20 mg/ml of apoA-I plus 1 mg/ml of FAFA. Values are expressed as % of total [3H]cholesterol (cells plus medium) appearing in the medium and were then compared with control 1, which was set as 100% value. Cholesterol efflux in the presence of FAFA alone varied between 1.3 and 1.7%. The values represent the mean ± S.D. and are representative of three experiments with similar results. p < 0.05 for differences of TT1 versus TT2, and p < 0.01 for the differences of T1 versus TT1 and T2 versus TT2, as indicated.
The most likely reason for this improvement in cholesterol efflux subsequent to immortalization of Tangier fibroblasts is the higher stress sensitivity of sick cells under artificial culture conditions. We hypothesized that the improved apoA-I-inducible cholesterol efflux in hTERT-TD cells relative to primary TD cells more closely reflects the true genetic phenotype separated from secondary changes due to replicative aging (5). This interpretation was further supported by other findings from aged cells in culture and the observation that the primary cells from TD patients have growth abnormalities and a limited proliferative life span in culture (14). As shown in Fig. 2, primary Tangier fibroblasts develop a flat senescence-like appearance at relatively low cell passages, accompanied by loss of feeder cell growth and damage-specific alterations in mitochondria (indicated by the arrow). These abnormalities, which also occur in healthy fibroblasts at higher population doublings, can be completely reversed by the immortalization procedure and likely do not represent the “normal” in vivo state, but rather represent an accelerated development of senescence in TD cells under unphysiological culture conditions. Next, we performed a detailed analysis of cellular cholesterol metabolism in primary and hTERT-immortalized TD fibroblasts. If telomerase-immortalized cells more closely reflect the normal phenotype, such cells should be suitable for elucidation of the phenotype variability in TD.
FIGURE 2.

Filipin staining and electron microscopy of primary and hTERT-infected control and Tangier disease fibroblasts. A, primary and hTERT-infected control (C1) and TD fibroblasts (T1/T2) were grown on coverslips and stained with 50 μg/ml of filipin in PBS for 30 min as described under ”Experimental Procedures.“ Filipin fluorescence was detected by fluorescence microscopy on a Zeiss Axiovert epifluorescence microscope. The following filter sets (Chroma) were used: for filipin, excitation filter 360/40 nm, beam splitter 400 nm, emission filter 460/50 nm. B, electron microscopy of T1 fibroblasts before and after hTERT immortalization. The arrow indicates abnormalities in mitochondrial cristae in primary T1 cells.
Cholesterol Accumulation in Tangier Disease Fibroblasts
Cellular cholesterol concentrations were compared in primary and hTERT-immortalized healthy control and TD fibroblasts using gas liquid chromatography. Total cellular cholesterol concentration was increased ∼3.7- and 4.1-fold in primary T1 and T2 cells, respectively. Total cellular cholesterol concentration increased 2.7- and 4.3-fold in hTERT-immortalized TT1 and TT2 cells, respectively. In three different control cell lines and in T2/TT2 cells, we did not find significant differences between the primary and hTERT-immortalized cells (Table 1). Immortalization of T1 reduced cellular cholesterol accumulation by 30–40% (Table 1).
TABLE 1.
Cholesterol concentrations in primary and hTERT-immortalized fibroblasts
n = 3; NS, not significant; * representative for homozygous patients from TD1 and TD2 kindred.
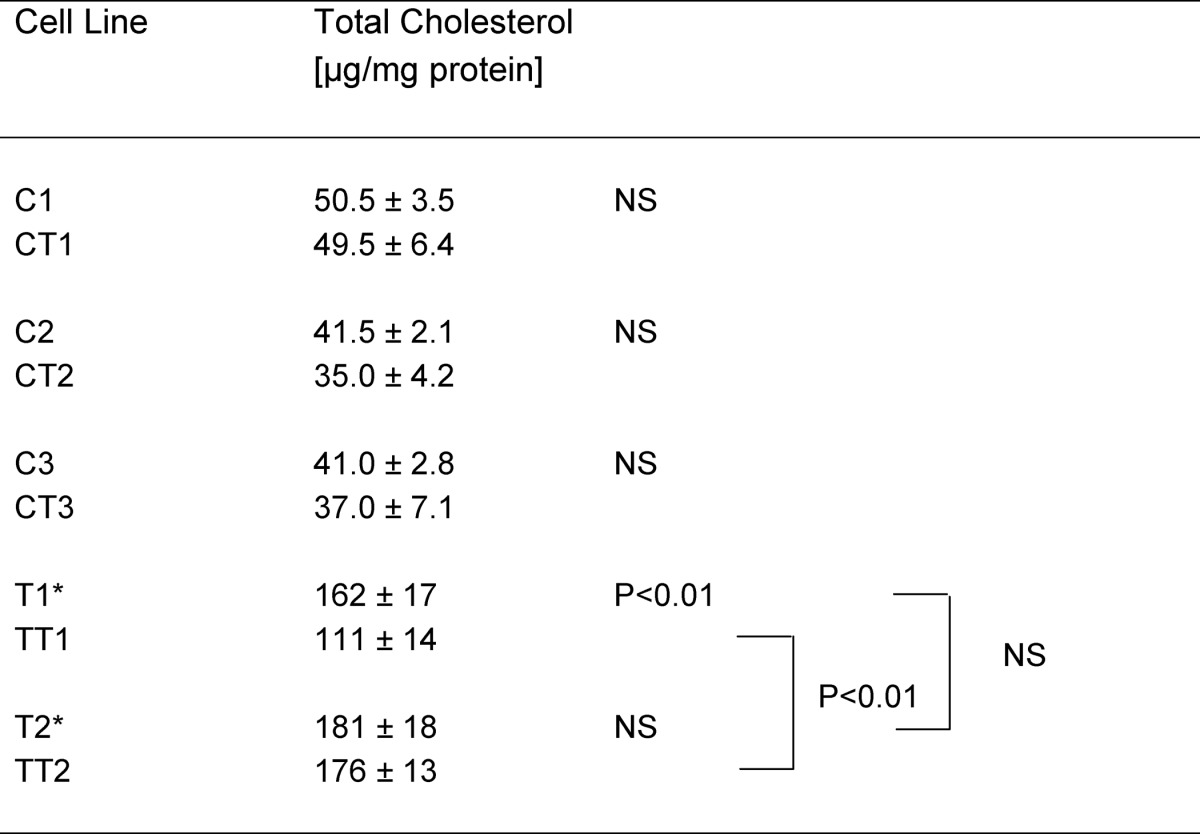
Using the hTERT-immortalized cells, we performed a more detailed analysis at different loading stages. As shown in Fig. 3, the total cellular cholesterol content was increased 2–3-fold in TT1 cells and increased 3–5-fold in TT2 cells in LPDS, compared with healthy controls. Cholesterol loading (20 μg/ml) did not further increase these values. The vast majority of cellular cholesterol was attributable to cellular cholesterol esters, whose levels were found to be increased up to 10-fold in TT1 and up to 30-fold in TT2 fibroblasts. Additional loading with free cholesterol (20 μg/ml) moderately increased cellular cholesterol ester content in control cells, but had almost no effect in TD cells. The free cholesterol content, by contrast, was only slightly increased in TT2 cells (120% of control values), but was moderately increased in TT1 cells (up to 170% of control values). Cholesterol loading did not further increase these values. Lower cholesterol levels were observed under serum starvation (−LPDS) with similar differences between normal and Tangier cells, as compared with normal growth conditions (+LPDS).
FIGURE 3.

Cellular concentration of total, free, and esterified cholesterol in hTERT-infected control and Tangier fibroblasts. The concentrations of free and total cholesterol were determined in cells incubated in LPDS in the presence or absence of 20 μg/ml of free cholesterol. Total and free cholesterol were determined by GLC of the trimethylsilyl ether derivatives, as described under ”Experimental Procedure.“ Ten micrograms of 5β-cholestan-3α-ol was added to 100 μl of cell lysate as an internal standard. *, p < 0.05 for comparison to control. TC, total cholesterol; FC, free cholesterol; EC, esterified cholesterol.
Filipin forms a fluorescent complex with mainly free membrane cholesterol and is primarily used to screen patients for Niemann-Pick disease type C. In these cells, the classic phenotype exhibits an intense, punctate, perinuclear fluorescence that may indicate cholesterol storage in lysosomes and in the perinuclear Golgi region. As shown in Fig. 2, filipin also labels cholesterol storage compartments in both primary and hTERT-immortalized TD cells. Fluorescence distribution closely resembled that of Niemann Pick type C cells. There was little intracellular filipin fluorescence in normal fibroblasts (C1), but intense fluorescence in both T1 and T2 cells. The filipin labeling was mainly observed in the perinuclear area, at the plasma membrane and as vesicular staining throughout the cytoplasm. The intensity of filipin staining was similar in immortalized and primary cells (with a tendency to higher values in the primary cells at identical microscope settings). The staining appeared slightly more intense in TT1 cells, in accordance with higher concentrations of free cholesterol in this cell type.
Oxysterol Accumulation in Tangier Disease Fibroblasts
Side chain-oxidized oxysterols are important intermediates in a number of hepatic and extrahepatic catabolic pathways. In the liver, water-soluble bile acids are produced as final products. On the one hand, oxysterols are atherogenic; on the other hand, formation of side chain-oxidized oxysterols is also a mechanism by which peripheral cells, such as macrophages and fibroblasts, may eliminate excess cholesterol (29, 30). Oxysterols have been proposed to be important regulators of gene expression. Under in vitro conditions oxysterols are strong suppressors of cholesterol biosynthesis, even more potent than cholesterol itself (31). To test whether the cellular oxysterol concentration is increased in TT1 and TT2 cells, we measured the concentrations of 24-, 25-, and 27-hydroxycholesterol from lipid extracts of control and TT1 and TT2 fibroblasts using gas chromatography and mass spectrometry, as shown in Fig. 4, and Tables 2 and 3. All these oxysterols are known to be potent suppressors of hydroxymethylglutaryl-CoA reductase (32–34), and they are known ligands for liver X receptor transcription factors (35, 36). 25-Hydroxycholesterol and 27-hydroxycholesterol have previously been found in cultivated fibroblasts (32).
FIGURE 4.

Chromatograms and mass spectra of lipid extracts from control, TT1 and TT2 fibroblasts (A) and of the respective sera of the patients (B) showing the hydroxylated cholesterol derivatives. The panel shows the chromatograms of hydroxycholesterol standards, CT1, TT1, and TT2 cell extracts and sera with the relative intensities of 24-, 25-, and 27-hydroxycholesterol, using three characteristic mass fragments as indicated.
TABLE 2.
Hydroxysterol concentrations in Tangier disease fibroblasts (experiment 1)
| 24-Hydroxycholesterol | 25-Hydroxycholesterol | 27-Hydroxycholesterol | |
|---|---|---|---|
| ng/mg cells | |||
| Con 2, FAFA | 1.70 ± 0.14 | 4.35 ± 0.49 | 1.60 ± 0.57 |
| Con 2, FAFA + Chol | 2.25 ± 0.21 | 5.65 ± 0.21a | 1.75 ± 0.07 |
| Con 2, LPDS | 1.60 ± 0.25 | 6.75 ± 0.78 | 1.40 ± 0.57 |
| Con 2, LPDS + Chol | 1.85 ± 0.07 | 6.05 ± 0.07 | 1.45 ± 0.07 |
| Con 3, FAFA | 1.95 ± 0.21 | 5.90 ± 0.85 | 1.45 ± 0.07 |
| Con 3, FAFA + Chol | 3.35 ± 0.78 | 7.15 ± 0.35 | 3.40 ± 1.56 |
| Con 3, LPDS | 1.20 ± 0.50 | 8.10 ± 0.57 | 0.70 ± 0.10 |
| Con 3, LPDS + Chol | 2.10 ± 0.45 | 9.50 ± 0.42 | 2.65 ± 0.07a |
| TT 1, FAFA | 6.90 ± 0.14b | 2.30 ± 1.06 | 2.30 ± 3.25 |
| TT 1, FAFA + Chol | 8.00 ± 1.70 | 2.60 ± 0.38c | 3.20 ± 1.20 |
| TT 1, LPDS | 4.15 ± 0.78 | 4.15 ± 0.78c | 3.42 ± 0.70c |
| TT 1, LPDS + Chol | 4.65 ± 0.07b | 4.65 ± 0.07 | 4.86 ± 0.79c |
| TT 2, FAFA | 66.95 ± 3.89b | 53.05 ± 4.17b | 288.60 ± 88.11c |
| TT 2, FAFA + Chol | 56.05 ± 4.60b | 55.40 ± 4.81b | 317.45 ± 52.68b |
| TT 2, LPDS | 54.20 ± 0.85d | 30.55 ± 1.91b | 313.40 ± 3.54d |
| TT 2, LPDS + Chol | 52.70 ± 3.58b | 33.90 ± 1.27d | 299.15 ± 18.88b |
a p < 0.05, Chol− versus Chol+.
b p < 0.01 for T1/T2 versus controls.
c p < 0.05 for T1/T2 versus controls.
d p < 0.001 for T1/T2 versus controls.
TABLE 3.
Hydroxysterol concentrations in Tangier fibroblasts (experiment 2)
| 24-Hydroxycholesterol | 25-Hydroxycholesterol | 27-Hydroxycholesterol | |
|---|---|---|---|
| ng/mg cells | |||
| Con 1, LPDS | 2.0 ± 1.0 | 5.0 ± 1.0 | 1.0 ± 0.5 |
| TT 1, LPDS | 8.0 ± 3.0 | 11.0 ± 3.0 | 9.0 ± 4.0 |
| TT 2, LPDS | 24.0 ± 2.0a | 19.0 ± 4.0b | 213.0 ± 63.0b |
a p < 0.01, TT versus control.
b p < 0.05, TT versus control.
The cellular concentrations of 24-, 25-, and 27-hydroxycholesterol were increased ∼10–40-, 4–10-, and 150–200-fold, respectively, in TT2 fibroblasts (compared with healthy controls). In contrast the increase of these oxysterols was more moderate in TT1 cells (2–3-fold for 24- and 25- hydroxycholesterol; and 3–9-fold for 27-hydroxycholesterol). Additional loading with cholesterol (30 μg/ml) had no or only moderate influence on cellular hydroxysterol concentration (Table 2). We observed a relatively high interassay variability in hydroxycholesterol measurements (for comparison, see data from two independent experiments in Tables 2 and 3), which can at least in part be explained by a dependence of intracellular sterol concentration on subtle differences in the cell density. However, the hydroxycholesterol concentrations were, in all experiments, markedly increased in TT2 compared with TT1 cells, with the highest relative and absolute difference in 27-hydroxycholesterol. Oxysterol measurements in primary control fibroblasts versus hTERT immortalized control fibroblasts showed similar oxysterol concentrations. In primary Tangier fibroblasts these measurements could not be performed due to lack of sufficient cell material.
It was hypothesized that oxysterols are key regulators of cellular cholesterol balance and that one or more oxysterol(s) are sensors for the cellular cholesterol concentration. We were therefore interested in investigating a correlation between oxysterol concentrations and the cellular total, free, and esterified cholesterol concentrations in the different Tangier, and control cells, and all under different loading conditions (with or without cholesterol in the presence or absence of LPDS). We were, however, unable to demonstrate any correlation between any of the investigated oxysterols and cellular cholesterol concentrations (data not shown). Stress induction by serum starvation (absence of LPDS) slightly increased the concentration of 24-hydroxycholesterol in all cells and 25-hydroxycholesterol in TT2 cells (Table 2).
Oxysterol Accumulation in Tangier Disease Serum
Mean plasma concentrations of 24-, 25-, and 27-hydroxycholesterol were not significantly increased in plasma from TD1 patients, with a tendency to slightly elevated levels of 25- and 27-hydroxycholesterol. By contrast, in TD2 plasma 24-, 25-, and 27-hydroxycholesterol concentrations were markedly increased up to 8.5-, 42-, and 11-fold, respectively, relative to plasma from healthy controls (Table 4). The highest absolute increases were observed for 27- hydroxycholesterol, followed by 24- and 25-hydroxycholesterol.
TABLE 4.
Hydroxysterol concentrations in Tangier serum
| 24-Hydroxycholesterol | 25-Hydroxycholesterol | 27-Hydroxycholesterol | |
|---|---|---|---|
| ng/ml | |||
| Con | 64 ± 20 | 3.0 ± 2.0 | 135 ± 46 |
| Tangier 1 | 55 ± 6.0 | 5.0 ± 2.0 | 185 ± 16 |
| Tangier 2 | 546 ± 61a | 124 ± 19a | 1473 ± 225a |
a p < 0.01, TT versus control.
Regulation of Cholesterol Biosynthesis in Tangier Disease Fibroblasts
Only 0.1–2% of total cellular cholesterol is present in the endoplasmic reticulum. Nevertheless, endoplasmic reticulum cholesterol levels can fluctuate widely and the local endoplasmic reticulum cholesterol pool controls cellular cholesterol content through regulated proteolysis of SREBPs (37). A high cellular concentration of cholesterol leads to suppression of cholesterol biosynthesis. Side chain-oxidized oxysterols are also potent suppressors of cholesterol biosynthesis (33). Because TT1 fibroblasts have only a moderately increased cellular free cholesterol and oxysterol concentration, whereas TT2 fibroblasts have normal free cholesterol, but markedly increased oxysterol concentration, we were interested to compare the degree of suppression of de novo synthesis of cellular cholesterol in TT1 and TT2.
To analyze newly synthesized lipids, cells were labeled with [3H]acetate for 24 h before harvest, after which lipids were extracted, hydrolyzed, and analyzed by thin layer chromatography. Cholesterol was visualized by lipid staining on TLC sheets and quantified by measurement of radioactivity. As shown in Fig. 5, cholesterol biosynthesis was slightly reduced (by ∼30%) in TT1 cells and was markedly reduced (by ∼60%) in TT2 fibroblasts. In control fibroblasts, no significant difference was observed in cholesterol biosynthesis before and after hTERT immortalization. By contrast, in Tangier fibroblasts, suppression of cholesterol biosynthesis was more pronounced in the respective primary cells, and particularly in T1 cells.
FIGURE 5.

A, cholesterol biosynthesis in control, TT1, and TT2 fibroblasts. B, Western blot of SREBP2. A, primary (white columns) and hTERT immortalized (black columns) fibroblasts were pulsed with [3H]acetate, as described under ”Experimental Procedures.“ On day 0, cells were seeded in triplicate in DMEM, 10% FCS. On day 1, the cells were washed three times with PBS and re-fed DMEM, 10% LPDS. On day 2, the cells were washed three times with PBS and re-fed DMEM, 10% LPDS with [3H]acetate (40 μCi/ml). After a 24-h incubation lipids were extracted and analyzed by TLC, and incorporation of [3H]acetate into [3H]cholesterol was quantified. Results are mean values ± S.D. from triplicate dishes from one representative experiment. *, p < 0.05; **, p < 0.01 for comparison to C1. B, activation of SREBPs: cholesterol depletion (Chol−) results in a significant (at least 10-fold) increase in the active cleavage product in Western blot analysis in control cells compared with cholesterol-loaded cells (Chol+). In TT1 cells, the protein signal at both conditions (Chol− and Chol+) was markedly reduced, compared with controls. In TT2 cells, SREBP2 cleavage product was almost non-detectable, or only a very faint band was visible. The arrow denotes the mature (cleaved) nuclear form of SREBP2 at 55–65 kDa.
Under basal conditions, SREBP is bound to endoplasmic reticulum membranes as a glycosylated precursor protein. Upon cholesterol depletion, the protein is cleaved to its active forms (about 50–68 kDa) and translocated into the nucleus to stimulate transcription of genes involved in the uptake and synthesis of cholesterol. As shown in Fig. 5B, cholesterol depletion (Chol−) results in a significant (at least 10-fold) increase in the active cleavage product in Western blot analysis in control cells compared with cholesterol-loaded cells (Chol+). In TT1 cells, the protein signal in both conditions (Chol− and Chol+) was markedly reduced compared with controls. In TT2 cells, SREBP2 cleavage product was almost non-detectable, or a very faint band was visible at the expected molecular weight of the SREBP2 cleavage product.
Diminished cleavage of SREBP2 can be expected to result in lower expression levels of the SREBP2-dependent target genes low density lipoprotein (LDL) receptor and 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase. Indeed, as shown in Fig. 6, the mRNA synthesis of LDL receptor was markedly decreased in TT2 fibroblasts and was slightly reduced in TT1 cells. HMG-CoA reductase mRNA levels were reduced in TT2, but were not significantly altered in TT1 cells.
FIGURE 6.

MRNA expression of SREBP2, LDL receptor and HMG-CoA reductase. mRNA concentrations in control and TD fibroblasts. Results are the ratio of the gene of interest relative to cyclophilin A. Values are expressed relative to control and were set at 100% in control CT1. Values represent the mean ± S.D. of data from three or four independent experiments. *, p < 0.05 for comparison to CT1.
It has previously been shown in CHO cells that cholesterol balance is not only post-transcriptionally but can also be transcriptionally regulated at the SREBP2 level (38). In accordance with these data, as shown in Fig. 6, the SREBP2 mRNA level was slightly reduced in TT1 and was markedly reduced in TT2 under the experimental conditions described here. Altogether these data show moderate suppression of cholesterol biosynthesis, SREBP2 cleavage, and SREBP2 expression in TT1 cells, but marked suppression of these factors in TT1 cells.
Regulation of SREBP1c and Downstream Genes
The nuclear receptors liver X receptors, α and β (LXRα and LXRβ), bind with high affinity to side chain-oxidized oxysterols, which are believed to be possible physiological activators of these receptors. A potentially protective mechanism by which LXR agonists stimulate lipogenesis (and may thus compensate for toxicity of free sterols) appears to be through direct activation of the SREBP-1c promoter. Free cholesterol is highly toxic to cells and esterification to fatty acids is an important mechanism for buffering free cholesterol levels. We therefore measured mRNA of the sterol regulatory element-binding protein 1c (SREBP-1c) and downstream genes of SREBP1c, fatty acid synthase and steroyl-coenzyme A desaturase 1.
SREBP1c mRNA showed a high variability from experiment to experiment, with higher values in overconfluent cells. In few experiments, we observed a moderate up-regulation of SREBP1c in TT1. However, as shown in Fig. 7, the mean mRNA expression of SREBP1c, steroyl-coenzyme A desaturase 1, and fatty acid synthase was not significantly altered in TT1 cells. By contrast, we always observed a strong suppression of SREBP1c and SREBP1c target genes in TT2 cells.
FIGURE 7.

MRNA expression of SREBP1c, fatty acid synthase, and SCD1 in control and Tangier fibroblasts. Results are the ratio of the gene of interest relative to cyclophilin A, and values are expressed relative to control and were set at 100% in control CT1. Values represent the mean ± S.D. of data from three or four independent experiments. * indicates p < 0.05 for a significant increase in comparison to CT1.
DISCUSSION
The most important result of this study is the marked phenotype variability in hTERT-immortalized TD fibroblasts with different molecular defects, and the observation that the differences were less obvious in the respective primary cells. In hTERT-immortalized TD cells we found various biochemical differences between cells with complete ABCA1 knockout (TT2) and cells with resting ABCA1 activity (TT1). Most but not all of these alterations are consistent with an increased atherogenic susceptibility in ABCA1 knockout TD patients (Table 5).
TABLE 5.
Potentially atherogenic and potentially protective effects
| Presumed effect | TT1 cells |
TT2 cells |
||
|---|---|---|---|---|
| Atherogenic | Protective | Atherogenic | Protective | |
| Cholesterol efflux | ↓ | ↓↓ | ||
| CE accumulation | ↑ | ↑↑ | ||
| FC accumulation | ↑ | |||
| OS accumulation | ↑ | ↑↑ | ||
| FC biosynthesis | ↓ | ↓↓ | ||
| “LXR protection”a | (↑) | ↓ | ||
a LXR-dependent target gene activation of SREBP1c, FAS, SCD1; CE, cholesterol ester; FC, free cholesterol; OS, oxysterols.
Sterol Efflux and Cellular Accumulation
Apolipoprotein-inducible cholesterol efflux was reduced in both cell lines. However, there was a measurable resting efflux capacity in both cell lines, which was higher in TT1 cells compared with TT2 cells. Resting apolipoprotein-inducible cholesterol efflux capacity reached almost 30% of normal values, which may contribute to the milder phenotype in TT1 cells. Moreover, the inducible cholesterol efflux in TT2 cells (with complete functional knockout) must be caused by ABCA1-independent mechanisms, and it is currently not known if ABCA1-dependent and ABCA1-independent cholesterol efflux mechanisms are equally effective in atheroprotection.
Cholesterol ester accumulation was 4-fold higher in TT2 compared with TT1 cells. Remarkably, the cellular oxysterol concentration was markedly increased in TT2 cells, but was only moderately increased in TT1 cells. Free cholesterol, by contrast, was only moderately increased in TT1 cells. The cholesterol ester, and particularly the extreme oxysterol accumulation in TT2 cells (upto 200-fold of controls), may point to an increased atherogenic susceptibility in TD2. The extremely high concentration of 24-, 25-, and particularly 27-hydroxycholesterol may have proatherogenic and direct cytotoxic effects (39, 40).
Mitochondrial 24-, 25-, and 27-hydroxylation of cholesterol by a 27-hydroxylase preparation has previously been described in liver cells (41). Parallel to the experiments described here, Lange and co-workers (42) found production of 24-, 25-, and 27-hydroxycholesterol in wild-type fibroblasts, but could not detect 24- and 27-hydroxycholesterol production in cerebrotendinous xanthomatosis fibroblasts, which are deficient in Cyp27A1. Altogether these and our findings suggest that human fibroblasts produce all major side chain-oxidized hydroxysterols and that 24- and 27-hydroxycholesterol are almost completely formed via Cyp27.
The accumulation of oxysterols in the plasma of the Tangier patient 2 is an indication for the physiological significance of a diffusion-like sterol efflux mechanism that may at least in part compensate for the cellular sterol accumulation. As 27-hydroxycholesterol can more easily transverse the plasma membrane than cholesterol this mechanism is potentially atheroprotective (29, 30). Thus, the described data present a human disease model that can support this previously formed hypothesis (29, 30). On the other hand, this potentially protective efflux pathway can apparently not overcome the defect, but might have prevented an even more severe phenotype. We were, however, unable to demonstrate any correlation between one of the investigated oxysterols and cellular cholesterol concentrations. These findings do not support the concept that total cellular 24-, 25-, and 27-hydroxycholesterol concentration is an indicator for cellular cholesterol concentration.
Regulation of Cholesterol Biosynthesis
Cholesterol biosynthesis was markedly suppressed in TT2 cells but was only moderately reduced in TT1 cells. This may reflect the more severe phenotype of TT2, but is also an effective protective mechanism that may compensate for the marked cholesterol efflux defect in TT2 cells.
Interestingly, the cellular concentration of free cholesterol was only slightly increased in TT1 cells, but was normal in TT2 cells. Apparently, the massive oxysterol accumulation in TT2 cells (in the absence of free cholesterol increase) was a stronger stimulus to suppress cholesterol biosynthesis than the moderate accumulation of free cholesterol and hydroxysterols in TT1 cells. Tangier fibroblasts thus present a cell model for oxysterol-mediated suppression of cholesterol biosynthesis. In this context, it is interesting to note that Wang and colleagues (43) have recently shown that 24-, 25-, and 27-hydroxycholesterol can suppress cholesterol biosynthesis by directly silencing the expression of two cholesterologenic enzymes (lanosterol 14a-demethylase (CYP51A1) and squalene synthase (farnesyl diphosphate farnesyl transferase 1)) via binding to novel negative LXR DNA response elements. Altogether, these studies suggest a dual regulation of cholesterol biosynthesis via SREBP2 and LXR.
Regulation of SREBP1c-dependent Gene Regulation
Surprisingly, not only SREBP2, but also SREBP1c regulation was suppressed in TT2 cells, but was not significantly altered in TT1 cells. In accordance with this finding, a similar pattern of expression was observed in downstream genes of SREBP1c (fatty acid synthase and steroyl-coenzyme A desaturase 1). SREBP1c, as interplayer between cholesterol and triglyceride metabolism, may counteract cholesterol toxicity by enhancing fatty acid synthesis and cholesterol esterification, with oleoyl-CoA as the preferred substrate for acyl-CoA:cholesterol O-acyltransferase-mediated cholesterol esterification (44–46). Insofar the suppression of SREPB1c and downstream genes would further aggravate the severe phenotype in TT2 cells (Table 5).
The reason for the difference in the regulation of SREBP1c between TT1 and TT2 is unclear. Regulation of SREBP1c is complex. On the one hand, SREBP1c is under the influence of LXR, and one would expect activation of SREBP1c-inducible genes when cellular (oxy)sterol concentrations are high. On the other hand, SREBP-1c can also negatively be regulated by a sensing mechanism for intracellular cholesterol in addition to, and distinct from, the LXR pathway (47). The extent of counter-regulation, however, was surprising and the possible consequences are of physiological significance: coordinate regulation of SREBP2 and SREBP1c under these extreme conditions may accelerate the development of atherosclerosis. In other words, the price for marked suppression of cholesterol biosynthesis in TT2 would be the parallel down-regulation of the (potentially anti-atherogenic) SREBP1c pathway.
Primary Versus Telomerase-immortalized Tangier Fibroblast
The biochemical differences between TT1 and TT2 cells were less obvious in the respective primary cells. In primary fibroblasts, T1 and T2, apolipoprotein-inducible cholesterol efflux was almost identical: close to zero. Apolipoprotein A-I-inducible cholesterol efflux was significantly higher in hTERT-immortalized cells, as previously shown (5). In accordance with these data, cholesterol accumulation and suppression of cholesterol biosynthesis was more pronounced in primary cells, and particularly in T1 cells, which complicated phenotype discrimination in respective primary cells. Studies on premature aging and genomic instability syndromes also indicated that it is often difficult to distinguish effects intrinsic to a genetic defect from those caused by cellular senescence in vitro (13). Artifacts caused by genetic instability and premature senescence, which are amplified by the high oxygen concentrations in routine incubators, complicate interpretation of findings from sick cells in culture, especially when ambient oxygen tension is used (48). This may be more important for sick cells resulting in loss of subtle biochemical differences. In particular HDL metabolism is likely sensitive to these artificial alterations due to various inter-relationships between HDL metabolism, aging, and atherosclerosis (49). Altogether, these data are in accordance with previously described data showing advantages of hTERT immortalization over primary cells (5) and over oncogene-immortalized fibroblasts (50). The results further suggest that the phenotype in hTERT-immortalized Tangier cells more closely reflects the true genetic phenotype.
A weakness of this study is the availability of a limited number of cell lines from only two families. On the other hand, the cell lines used originate from two of the best characterized Tangier families with no signs of premature atherosclerosis in two homozygous patients of TD1 kindred and severe premature atherosclerosis in three homozygous patients of the TD2 kindred. The results described here may, for the first time, explain the different atherosclerotic susceptibilities in Tangier families.
In summary, potentially atheroprotective regulatory pathways overweigh in TT1 compared with TT2 cells (Table 5). TD fibroblasts with nonfunctional ABCA1 (TT2) seem to compensate for the defect primarily by down-regulation of de novo cholesterol biosynthesis and efflux of side chain-oxidized oxysterols by diffusion-like mechanisms. This compensation is only partially effective. Massive overall (oxy)sterol accumulation and the limited ability to up-regulate SREBP-1c may contribute to the more severe atherosclerosis in cases of complete ABCA1 knockout. By contrast, fibroblasts with resting ABCA1 activity (TT1) only moderately accumulate oxysterols, and have a higher tone of the potentially protective SREBP1c pathway. SREBP-1c is apparently negatively regulated by a sensing mechanism for intracellular cholesterol in addition to, and distinct from, the LXR pathway. The regulatory importance of side chain-oxidized oxysterols under in vivo conditions has been difficult to demonstrate with use of different mouse models (51). The results of the present work support the contention that such oxysterols are strong suppressors of cholesterol synthesis when accumulating in high concentrations under pathological conditions in humans.
Acknowledgments
We greatly appreciate the gift of hTERT vector and packaging cells from Dr. Jerry Shay and Dr. Woodring Wright (Southwestern Medical Center at Dallas). We also thank Marianne Jansen-Rust and Margit Käse for outstanding technical assistance. We thank Dr. Peter N. Robinson for manuscript editing.
The work was supported by grants from the “Landesversicherungsanstalt Rheinprovinz (LVA)” (to M. W.), the Foundation for Pathobiochemistry and Molecular Diagnostics, German Society for Clinical Chemistry and Laboratory Medicine e.V. (DGKL; Bonn, Germany) (to M. W.), and a grant from the Swedish Research Council (to I. B.).
- ABCA1
- ATP-binding cassette transporter A1
- apoA-I
- apolipoprotein A-I
- FAFA
- fatty acid-free albumin
- HMG-CoA reductase
- 3-hydroxy-3-methylglutaryl-coenzyme A-reductase
- hTERT
- human telomerase reverse transcriptase
- LPDS
- lipoprotein-deficient serum
- LXRα
- liver X receptor α
- LXRβ
- liver X receptor β
- SREBP2
- sterol regulatory element-binding protein 2
- SREBP1c
- sterol regulatory element-binding protein 1c
- TD
- Tangier disease.
REFERENCES
- 1. Rust S., Walter M., Funke H., von Eckardstein A., Cullen P., Kroes H. Y., Hordijk R., Geisel J., Kastelein J., Molhuizen H. O., Schreiner M., Mischke A., Hahmann H. W., Assmann G. (1998) Assignment of Tangier disease to chromosome 9q31 by a graphical linkage exclusion strategy. Nat. Genet. 20, 96–98 [DOI] [PubMed] [Google Scholar]
- 2. Rust S., Rosier M., Funke H., Real J., Amoura Z., Piette J. C., Deleuze J. F., Brewer H. B., Duverger N., Denèfle P., Assmann G. (1999) Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet. 22, 352–355 [DOI] [PubMed] [Google Scholar]
- 3. Bodzioch M., Orsó E., Klucken J., Langmann T., Böttcher A., Diederich W., Drobnik W., Barlage S., Büchler C., Porsch-Ozcürümez M., Kaminski W. E., Hahmann H. W., Oette K., Rothe G., Aslanidis C., Lackner K. J., Schmitz G. (1999) The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 22, 347–351 [DOI] [PubMed] [Google Scholar]
- 4. Brooks-Wilson A., Marcil M., Clee S. M., Zhang L. H., Roomp K., van Dam M., Yu L., Brewer C., Collins J. A., Molhuizen H. O., Loubser O., Ouelette B. F., Fichter K., Ashbourne-Excoffon K. J., Sensen C. W., Scherer S., Mott S., Denis M., Martindale D., Frohlich J., Morgan K., Koop B., Pimstone S., Kastelein J. J., Genest J., Jr., Hayden M. R. (1999) Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 22, 336–345 [DOI] [PubMed] [Google Scholar]
- 5. Walter M., Forsyth N. R., Wright W. E., Shay J. W., Roth M. G. (2004) The establishment of telomerase-immortalized Tangier disease cell lines indicates the existence of an apolipoprotein A-I-inducible but ABCA1-independent cholesterol efflux pathway. J. Biol. Chem. 279, 20866–20873 [DOI] [PubMed] [Google Scholar]
- 6. Assmann G., von Eckardstein A., Brewer H. B., Jr. (1995) in The Metabolic and Molecular Bases of Inherited Disease (Scriver C. A., Beaudet A. L., Sly W. S., Valle D., Eds) 7th Ed., pp. 2053–2072, McGraw-Hill, New York [Google Scholar]
- 7. Serfaty-Lacrosniere C., Civeira F., Lanzberg A., Isaia P., Berg J., Janus E. D., Smith M. P., Jr., Pritchard P. H., Frohlich J., Lees R. S. (1994) Homozygous Tangier disease and cardiovascular disease. Atherosclerosis 107, 85–98 [DOI] [PubMed] [Google Scholar]
- 8. Walter M., Kerber S., Fechtrup C., Seedorf U., Breithardt G., Assmann G. (1994) Characterization of atherosclerosis in a patient with familial high-density lipoprotein deficiency. Atherosclerosis 110, 203–208 [DOI] [PubMed] [Google Scholar]
- 9. Rader D. J., Ikewaki K., Duverger N., Feuerstein I., Zech L., Connor W., Brewer H. B., Jr. (1993) Very low high-density lipoproteins without coronary atherosclerosis. Lancet 342, 1455–1458 [DOI] [PubMed] [Google Scholar]
- 10. Walter M., Gerdes U., Seedorf U., Assmann G. (1994) The high density lipoprotein- and apolipoprotein A-I-induced mobilization of cellular cholesterol is impaired in fibroblasts from Tangier disease subjects. Biochem. Biophys. Res. Commun. 205, 850–856 [DOI] [PubMed] [Google Scholar]
- 11. Francis G. A., Knopp R. H., Oram J. F. (1995) Defective removal of cellular cholesterol and phospholipids by apolipoprotein A-I in Tangier disease. J. Clin. Invest. 96, 78–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rogler G., Trümbach B., Klima B., Lackner K. J., Schmitz G. (1995) HDL-mediated efflux of intracellular cholesterol is impaired in fibroblasts from Tangier disease patients. Arterioscler. Thromb. Vasc. Biol. 15, 683–690 [DOI] [PubMed] [Google Scholar]
- 13. Ouellette M. M., McDaniel L. D., Wright W. E., Shay J. W., Schultz R. A. (2000) The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum. Mol. Genet. 9, 403–411 [DOI] [PubMed] [Google Scholar]
- 14. Drobnik W., Liebisch G., Biederer C., Trumbach B., Rogler G., Müller P., Schmitz G. (1999) Growth and cell cycle abnormalities of fibroblasts from Tangier disease patients. Arterioscler. Thromb. Vasc. Biol. 19, 28–38 [DOI] [PubMed] [Google Scholar]
- 15. Utech M., Höbbel G., Rust S., Reinecke H., Assmann G., Walter M. (2001) Accumulation of RhoA, RhoB, RhoG, and Rac1 in fibroblasts from Tangier disease subjects suggests a regulatory role of Rho family proteins in cholesterol efflux. Biochem. Biophys. Res. Commun. 280, 229–236 [DOI] [PubMed] [Google Scholar]
- 16. Havel R. J., Eder H. A., Bragdon J. H. (1955) The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Invest. 34, 1345–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walter M., Reinecke H., Gerdes U., Nofer J.-R., Höbbel G., Seedorf U., Assmann G. (1996) Defective regulation of phosphatidylcholine-specific phospholipases C and D in a kindred with Tangier disease. Evidence for the involvement of phosphatidylcholine breakdown in HDL-mediated cholesterol efflux mechanisms. J. Clin. Invest. 98, 2315–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morgenstern J. P., Land H. (1990) Advanced mammalian gene transfer. High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18, 3587–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Steinert S., Shay J. W., Wright W. E. (2000) Transient expression of human telomerase extends the life span of normal human fibroblasts. Biochem. Biophys. Res. Commun. 273, 1095–1098 [DOI] [PubMed] [Google Scholar]
- 20. Walter M., Reinecke H., Nofer J.-R., Seedorf U., Assmann G. (1995) HDL3 stimulates multiple signaling pathways in human skin fibroblasts. Arterioscler. Thromb. Vasc. Biol. 15, 1975–1986 [DOI] [PubMed] [Google Scholar]
- 21. Engel T., Kannenberg F., Fobker M., Nofer J. R., Bode G., Lueken A., Assmann G., Seedorf U. (2007) Expression of ATP binding cassette-transporter ABCG1 prevents cell death by transporting cytotoxic 7β-hydroxycholesterol. FEBS Lett. 581, 1673–1680 [DOI] [PubMed] [Google Scholar]
- 22. Cullen P., Tegelkamp K., Fobker M., Kannenberg F., Assmann G. (1997) Measuring cholesterol in macrophages. Comparison of high-performance liquid chromatography and gas-liquid chromatography with enzymatic fluorometry. Anal. Biochem. 251, 39–44 [DOI] [PubMed] [Google Scholar]
- 23. Cullen P., Fobker M., Tegelkamp K., Meyer K., Kannenberg F., Cignarella A., Benninghoven A., Assmann G. (1997) An improved method for quantification of cholesterol and cholesteryl esters in human monocyte-derived macrophages by high performance liquid chromatography with identification of unassigned cholesteryl ester species by means of secondary ion mass spectrometry. J. Lipid Res. 38, 401–409 [PubMed] [Google Scholar]
- 24. Klansek J. J., Yancey P., St. Clair R. W., Fischer R. T., Johnson W. J., Glick J. M. (1995) Cholesterol quantitation by GLC. Artifactual formation of short-chain steryl esters. J. Lipid Res. 36, 2261–2266 [PubMed] [Google Scholar]
- 25. Tint G. S., Irons M., Elias E. R., Batta A. K., Frieden R., Chen T. S., Salen G. (1994) Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 330, 107–113 [DOI] [PubMed] [Google Scholar]
- 26. Arakawa R., Yokoyama S. (2002) Helical apolipoproteins stabilize ATP-binding cassette transporter A1 by protecting it from thiol protease-mediated degradation. J. Biol. Chem. 277, 22426–22429 [DOI] [PubMed] [Google Scholar]
- 27. Frolov A., Zielinski S. E., Crowley J. R., Dudley-Rucker N., Schaffer J. E., Ory D. S. (2003) NPC1 and NPC2 regulate cellular cholesterol homeostasis through generation of low density lipoprotein cholesterol-derived oxysterols. J. Biol. Chem. 278, 25517–25525 [DOI] [PubMed] [Google Scholar]
- 28. Millard E. E., Srivastava K., Traub L. M., Schaffer J. E., Ory D. S. (2000) Niemann-pick type C1 (NPC1) overexpression alters cellular cholesterol homeostasis. J. Biol. Chem. 275, 38445–38451 [DOI] [PubMed] [Google Scholar]
- 29. Björkhem I., Diczfalusy U., Lütjohann D. (1999) Removal of cholesterol from extrahepatic sources by oxidative mechanisms. Curr. Opin. Lipidol. 10, 161–165 [DOI] [PubMed] [Google Scholar]
- 30. Lund E., Andersson O., Zhang J., Babiker A., Ahlborg G., Diczfalusy U., Einarsson K., Sjövall J., Björkhem I. (1996) Importance of a novel oxidative mechanism for elimination of intracellular cholesterol in humans. Arterioscler. Thromb. Vasc. Biol. 16, 208–212 [DOI] [PubMed] [Google Scholar]
- 31. Björkhem I. (2002) Do oxysterols control cholesterol homeostasis? J. Clin. Invest. 110, 725–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Axelson M., Larsson O. (1995) Low density lipoprotein (LDL) cholesterol is converted to 27-hydroxycholesterol in human fibroblasts. Evidence that 27-hydroxycholesterol can be an important intracellular mediator between LDL and the suppression of cholesterol production. J. Biol. Chem. 270, 15102–15110 [DOI] [PubMed] [Google Scholar]
- 33. Kandutsch A. A., Chen H. W. (1975) Regulation of sterol synthesis in cultured cells by oxygenated derivatives of cholesterol. J. Cell Physiol. 85, 415–424 [DOI] [PubMed] [Google Scholar]
- 34. Esterman A. L., Baum H., Javitt N. B., Darlington G. J. (1983) 26-Hydroxycholesterol. Regulation of hydroxymethylglutaryl-CoA reductase activity in Chinese hamster ovary cell culture. J. Lipid Res. 24, 1304–1309 [PubMed] [Google Scholar]
- 35. Fu X., Menke J. G., Chen Y., Zhou G., MacNaul K. L., Wright S. D., Sparrow C. P., Lund E. G. (2001) 27-Hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells. J. Biol. Chem. 276, 38378–38387 [DOI] [PubMed] [Google Scholar]
- 36. Janowski B. A., Grogan M. J., Jones S. A., Wisely G. B., Kliewer S. A., Corey E. J., Mangelsdorf D. J. (1999) Structural requirements of ligands for the oxysterol liver X receptors LXRα and LXRβ. Proc. Natl. Acad. Sci. 96, 266–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang T. Y., Chang C. C., Ohgami N., Yamauchi Y. (2006) Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 22, 129–157 [DOI] [PubMed] [Google Scholar]
- 38. Sato R., Inoue J., Kawabe Y., Kodama T., Takano T., Maeda M. (1996) Sterol-dependent transcriptional regulation of sterol regulatory element-binding protein-2. J. Biol. Chem. 271, 26461–26464 [DOI] [PubMed] [Google Scholar]
- 39. Brown A. J., Jessup W. (1999) Oxysterols and atherosclerosis. Atherosclerosis 142, 1–28 [DOI] [PubMed] [Google Scholar]
- 40. Tabas I. (2002) Cholesterol in health and disease. J. Clin. Invest. 110, 583–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lund E., Björkhem I., Furster C., Wikvall K. (1993) 24-, 25-, and 27-hydroxylation of cholesterol by a purified preparation of 27-hydroxylase from pig liver. Biochim. Biophys. Acta 1166, 177–182 [DOI] [PubMed] [Google Scholar]
- 42. Lange Y., Ory D. S., Ye J., Lanier M. H., Hsu F. F., Steck T. L. (2008) Effectors of rapid homeostatic responses of endoplasmic reticulum cholesterol and 3-hydroxy-3-methylglutaryl-CoA reductase. J. Biol. Chem. 283, 1445–1455 [DOI] [PubMed] [Google Scholar]
- 43. Wang Y., Rogers P. M., Su C., Varga G., Stayrook K. R., Burris T. P. (2008) Regulation of cholesterologenesis by the oxysterol receptor, LXRα. J. Biol. Chem. 283, 26332–26339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Landau J. M., Sekowski A., Hamm M. W. (1997) Dietary cholesterol and the activity of stearoyl CoA desaturase in rats. Evidence for an indirect regulatory effect. Biochim. Biophys. Acta 1345, 349–357 [DOI] [PubMed] [Google Scholar]
- 45. Repa J. J., Turley S. D., Lobaccaro J. A., Medina J., Li L., Lustig K., Shan B., Heyman R. A., Dietschy J. M., Mangelsdorf D. J. (2000) Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 289, 1524–1529 [DOI] [PubMed] [Google Scholar]
- 46. Repa J. J., Liang G., Ou J., Bashmakov Y., Lobaccaro J. M., Shimomura I., Shan B., Brown M. S., Goldstein J. L., Mangelsdorf D. J. (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev. 14, 2819–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Amemiya-Kudo M., Shimano H., Yoshikawa T., Yahagi N., Hasty A. H., Okazaki H., Tamura Y., Shionoiri F., Iizuka Y., Ohashi K., Osuga J., Harada K., Gotoda T., Sato R., Kimura S., Ishibashi S., Yamada N. (2000) Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. J. Biol. Chem. 275, 31078–31085 [DOI] [PubMed] [Google Scholar]
- 48. Forsyth N. R., Evans A. P., Shay J. W., Wright W. E. (2003) Developmental differences in the immortalization of lung fibroblasts by telomerase. Aging Cell 2, 235–243 [DOI] [PubMed] [Google Scholar]
- 49. Walter M. (2009) Interrelationships among HDL metabolism, aging, and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 29, 1244–1250 [DOI] [PubMed] [Google Scholar]
- 50. Oram J. F., Mendez A. J., Lymp J., Kavanagh T. J., Halbert C. L. (1999) Reduction in apolipoprotein-mediated removal of cellular lipids by immortalization of human fibroblasts and its reversion by cAMP. Lack of effect with Tangier disease cells. J. Lipid Res. 40, 1769–1781 [PubMed] [Google Scholar]
- 51. Björkhem I. (2009) Are side-chain oxidized oxysterols regulators also in vivo? J. Lipid Res. 50, (suppl.) S213–S218 [DOI] [PMC free article] [PubMed] [Google Scholar]