

Background: Regulation of c-Myb transcriptional activity and protein level is essential for hematopoietic homeostasis.
Results: p38 MAPKs phosphorylate c-Myb on Thr486, resulting in repression of SUMOylation in stressed cells.
Conclusion: There is negative cross-talk between the p38 MAPK phosphorylation pathway and the SUMO-2/3 conjugation pathway that regulates c-Myb activity in stressed cells.
Significance: The novel mechanism that modulates activity of c-Myb in stressed cells was identified.
Keywords: Apoptosis, p38 MAPK, Phosphorylation, Protein Degradation, Protein Stability, Stress, Sumoylation, Transcription, Myb
Abstract
c-Myb plays an essential role in regulation of properly balanced hematopoiesis through transcriptional regulation of genes directly controlling cellular processes such as proliferation, differentiation, and apoptosis. The transcriptional activity and protein levels of c-Myb are strictly controlled through post-translational modifications such as phosphorylation, acetylation, ubiquitination, and SUMOylation. Conjugation of small ubiquitin-like modifier (SUMO) proteins has been shown to suppress the transcriptional activity of c-Myb. SUMO-1 modifies c-Myb under physiological conditions, whereas SUMO-2/3 conjugation was reported in cells under stress. Because stress also activates several cellular protein kinases, we investigated whether phosphorylation of c-Myb changes in stressed cells and whether a mutual interplay exists between phosphorylation and SUMOylation of c-Myb. Here we show that several types of environmental stress induce a rapid change in c-Myb phosphorylation. Interestingly, the phosphorylation of Thr486, located in close proximity to SUMOylation site Lys499 of c-Myb, is detected preferentially in nonSUMOylated protein and has a negative effect on stress-induced SUMOylation of c-Myb. Stress-activated p38 MAPKs phosphorylate Thr486 in c-Myb, attenuate its SUMOylation, and increase its proteolytic turnover. Stressed cells expressing a phosphorylation-deficient T486A mutant demonstrate decreased expression of c-Myb target genes Bcl-2 and Bcl-xL and accelerated apoptosis because of increased SUMOylation of the mutant protein. These results suggest that phosphorylation-dependent modulation of c-Myb SUMOylation may be important for proper response of cells to stress. In summary, we have identified a novel regulatory interplay between phosphorylation and SUMOylation of c-Myb that regulates its activity in stressed cells.
Introduction
The c-Myb transcription factor plays an essential role during definitive hematopoiesis through transcriptional regulation of genes that encode protein products that directly control cellular processes such as proliferation, differentiation, and apoptosis (1, 2). Targeted disruption of the gene is embryonically lethal, because embryos lacking functional c-Myb die in utero around day 15 because of severe anemia (3). Conditional deletion of the gene in specific hematopoietic compartments has shown specific roles for c-Myb at different stages during T-cell and B-cell development (4–7), in balanced myelopoiesis (8), in erythropoiesis (9, 10), and in maintenance of proper hematopoietic stem cell homeostasis (11, 12). Different protein levels of c-Myb are critical for proper development of specific hematopoietic lineages as demonstrated in experiments with mice carrying hypomorphic c-myb alleles (9). The importance of specific protein levels for development was confirmed in ex vivo differentiation assays using embryonic stem cells from knock-out mice with regulated c-Myb expression (13). Therefore, maintenance of appropriate levels of protein seems to be critical for balanced production of hematopoietic cells.
Oncogenic forms of c-Myb, in animal model systems, are frequently associated with truncations of either amino, carboxyl, or both termini (14, 15). These truncations result in removal of structural regions with a negative impact on proteolytic stability and transactivation capacity (16, 17). In contrast to many animal leukemic models, increased expression of unaltered c-MYB is commonly detected in human leukemia (18, 19). More recently rearrangements of the MYB locus were also reported (20–25). However, these alterations result mostly in increased expression of the full-length, unaltered c-MYB protein (21–23). These examples are similar to an activation of c-myb in murine leukemia, where a retrovirus integrates several kilobases upstream of the c-myb transcription start site and deregulates its expression (26, 27). Nevertheless, these leukemic cells seem to be dependent on the elevated levels of c-Myb, because knocking down the gene expression is detrimental for those cells, whereas normal untransformed progenitors survive similar treatment (28, 29). Thus, these results clearly show that strict regulation of the protein level of c-Myb and/or activity is essential for balanced hematopoietic cell homeostasis, and its deregulation can be leukemogenic.
Post-translational modifications of c-Myb, such as phosphorylation, acetylation, ubiquitination, and SUMOylation are crucial for regulation of the transcriptional activity and steady-state levels of c-Myb (16). c-Myb is a short-lived protein that is rapidly turned over by the ubiquitin/26S proteasome proteolytic system (30). In contrast to ubiquitination, conjugation of the SUMO2 proteins to the lysine residues, located in the negative regulatory domain of c-Myb, results in decreased transcriptional activity and increased proteolytic stability (31–33). Although SUMO-1 conjugates to c-Myb predominantly under normal physiological growth conditions, cellular stress induces rapid inactivation of c-Myb through covalent conjugation of SUMO-2/3 paralogs (34). SUMOylation of target proteins, like conjugation of polyubiquitin chains, can be regulated by phosphorylation (35, 36). Because cellular stresses activate several protein kinases that phosphorylate many downstream targets (37), we decided to investigate whether phosphorylation of c-Myb changes in cells subjected to cellular stress and whether these changes in phosphorylation of c-Myb influence its stress-induced SUMOylation.
Here, we report that the phosphorylation of Thr486 in the negative regulatory domain of c-Myb by p38 MAPKs occurs in cells subjected to several metabolic stresses. Furthermore, we show that phosphorylation of Thr486 negatively regulates conjugation of SUMO-2/3 to c-Myb and leads to modulation of its transcriptional activity in stressed cells.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
Constructs encoding wild-type c-Myb (c-MybWT) and a p5xMRE-A-luc construct were described previously (31). The mutant T486A of c-Myb (cMybT486A) was generated by the QuikChange site-directed mutagenesis kit (Stratagene) and subcloned into pcDNA3.1+(Invitrogen) vector. Plasmid encoding GST-cMyb(464–544aa) was prepared by subcloning of c-Myb(464–544aa), in frame with GST, into the pGEX-KG bacterial expression vector. The base pair substitution and in frame cloning was confirmed by dideoxy sequencing using the BigDyeTM Terminator version 1.0 ready reaction cycle sequencing kit (Applied Biosystems). Retroviral vectors expressing c-MybWT and cMybT486A were constructed by subcloning murine c-Myb cDNAs into BglII and XhoI sites of the retroviral vector pMSCV-IRES-GFP (MIG), kindly provided by Scott Kogan (University of California, San Francisco, CA). Plasmids encoding pcDNA-FLAG-p38 MAPK isoforms were provided by S. Ferrari (University of Zurich, Zurich, Switzerland) with kind permission from J. Han (Scripps Research Institute, La Jolla, CA). pCMV-HA-SUMO-2 (pCMV-HA-SUMO-2-GG, HA-SUMO2) was provided by R. Hay (University of St. Andrews, St. Andrews, United Kingdom).
Cell Cultures and Viruses
COS-7 cells (SV40-transformed monkey kidney) and RAW264.7 macrophage cells (both from American Type Culture Collection) were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Sigma), 2 mm l-glutamine (Invitrogen), and penicillin/streptomycin (100 μg/ml each) (Invitrogen). Murine M1 myeloid cells (30) were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated horse serum (Invitrogen). M1 cells were transduced with MSCV-IRES-GFP-based retroviruses expressing cMybWT or cMybT486A or pool of lentiviruses expressing shRNAs targeting mouse p38 MAPKα (Sigma-Aldrich) in the presence of Polybrene (10 μg/ml; Sigma-Aldrich). Twenty-four hours after retrovirus infection, green fluorescent protein-positive (GFP+) cells were sorted using a FACS. Knockdown of p38 MAPKα was evaluated 72 h after infection by immunoblotting.
Transient Transfection, Immunoprecipitation, and Immunoblotting
COS-7 cells were transiently transfected with EffecteneTM transfection reagent (Qiagen) as described previously (34). Protein expression was analyzed by immunoblotting of total cell lysates or immunoprecipitates fractionated by SDS-PAGE with the following antibodies: mouse monoclonal antibody anti-cMyb (34), rabbit polyclonal anti-cMyb (34), rabbit polyclonal anti-P-T486pept, anti-T486pept (Rockland Immunochemicals, Inc.), anti-SUMO-2/3 (34), anti-FLAG, anti-HA (Sigma-Aldrich), anti-p38 MAPKα, anti-p38 MAPKβ, anti-p38 MAPKγ, anti-p38 MAPKδ, anti-actin, anti-tubulin, anti-cleaved PARP, anti-Bcl-2, anti-Bcl-xL, and anti-Mcl-1 antibody (Cell Signaling).
Phosphoamino Acid Analysis
Phosphoamino acid analysis was performed as described previously (38). COS7 cells, transiently expressing c-MybWT, were metabolically labeled in phosphate-free DMEM with 0.5 mCi/ml of carrier-free 32Pi orthophosphate (MP Biomedicals) for 4 h. c-Myb was immunoprecipitated, separated by SDS-PAGE, and transferred to an Immobilon-P membrane (Millipore). The membrane immobilized c-Myb was excised, hydrolyzed in 200 μl of 6 n HCl (constant boiling) (Sigma) at 110 °C for 2 h, lyophilized in a SpeedVac (Savant), and resuspended in 10 μl of water containing 0.1 mg/ml of each phosphoamino acid standard, phosphoserine, phosphothreonine, and phosphotyrosine (Sigma). The samples were spotted onto thin layer cellulose plates (Sigma) and separated by electrophoresis (38). The migration of phosphoamino acid standards was detected by staining with ninhydrin (Sigma), and 32P-labeled phosphoamino acids were visualized by direct autoradiography.
LC-MS/MS Analyses to Detect Phosphorylation of c-Myb
c-Myb protein immunoprecipitated from stressed or unstressed cells with anti-c-Myb antibody was separated by SDS-PAGE and stained with a PageBlue protein staining solution (Fermentas). The gel bands were destained, reduced, alkylated, and digested with trypsin in the presence of ProteaseMAXTM Surfactant (Promega) at 50 °C for 1 h according to the manufacturer's protocol. MS was performed in the Laboratory of Proteomics and Analytical Technologies, SAIC-Frederick, Inc., NCI (Frederick, MD). The tryptic peptides were analyzed by nanobore reversed phase LC-MS/MS. nanobore reversed phase LC-MS/MS was performed using an Agilent 1100 nanoflow liquid chromatography system coupled online with a linear ion trap-Fourier transform mass spectrometer (ThermoElectron). The linear ion trap-Fourier transform mass spectrometer was operated in a data-dependent mode where each full MS scan was followed by seven tandem MS scans, and the seven most abundant molecular ions were dynamically selected for collision-induced dissociation using a normalized collision energy of 35%. Tandem mass spectra were searched against a mouse database using SEQUEST software (ThermoElectron).
Luciferase Assay
RAW264.7 cells (2 × 106 cells/ml) were washed in PBS and resuspended in 100 μl of Nucleofector kit V (Lonza) supplemented with 0.5 μg of a cMyb-dependent firefly luciferase reporter construct (p5xMRE-A-Luc), 30 ng of Renilla luciferase pCMV-RL (Promega), a control construct used for normalization, and 1.5 μg of constructs encoding either cMybWT, cMybT486A, or cMybSU. Cells were transfected by electroporation using Nucleofector II (Lonza) in accordance with the manufacturer's instructions. Luciferase activity was assessed using a dual-luciferase reporter system (Promega) and the TurnerTD-20e luminometer (Turner Designs).
RESULTS
Hyperthermic Stress Induces Rapid Changes in Phosphorylation of c-Myb
We have shown previously that cellular stresses induce rapid conjugation of SUMO-2/3 to two lysines (Lys523 and Lys499) located in NRD of c-Myb. Several protein kinases, including p38 MAPK and JNK families, are also rapidly activated in response to cellular stresses and phosphorylate many targets (37). Therefore, we decided to investigate whether cellular stresses affect phosphorylation of c-Myb and whether these changes in c-Myb phosphorylation may regulate stress-induced SUMOylation of c-Myb. To compare the c-Myb phosphorylation status in cells treated (43 °C) or not treated (37 °C) with hyperthermia, we first labeled metabolically cellular proteins with [32P]orthophosphate in COS7 cells transiently transfected with a c-Myb construct. Labeled cells were split in half and either treated or not treated with hyperthermia for another 30 min. The c-Myb protein was immunoprecipitated from cells, digested with trypsin, and analyzed by reverse phase HPLC (Fig. 1A). There are both increases (Fig. 1A, up arrows) and decreases (Fig. 1A, down arrows) in the intensities of 32P-labeled tryptic peptides generated by digestion of c-Myb immunoprecipitated from cells treated with hyperthermia as compared with untreated cells (Fig. 1A). These results suggest that cellular stresses, as exemplified by hyperthermia, induce rapid changes in phosphorylation of c-Myb. Next we decided to assess the phosphoamino acids content in c-Myb isolated from cells treated or not treated with stress. Because we wanted to investigate a possible relation between phosphorylation and SUMOylation, we decided to also compare phosphorylation of unmodified and SUMO-2 modified c-Myb treated with hyperthermia or untreated. To increase the amount of SUMOylation of c-Myb, COS-7 cell were cotransfected with constructs encoding both c-Myb and SUMO-2. Cells were labeled with [32P]orthophosphate and either stressed (43 °C) or not stressed (37 °C) with hyperthermia for another 30 min and lysed. NonSUMOylated and SUMOylated c-Myb forms were immunoprecipitated, separated by SDS-PAGE, transferred to the membrane, and analyzed by direct exposure to x-ray film. As expected, heat stress induced a strong increase in SUMO-2 conjugation to c-Myb (34) (Fig. 1B). Immobilized 32P-labeled c-Myb was hydrolyzed, and phosphoamino acid analysis was performed. There was an apparent increase in the phosphorylation of Thr residues in the nonSUMOylated form of c-Myb isolated from hyperthermia-treated cells as compared with cells growing under physiological conditions (Fig. 1C). Quantitative analysis of the phosphoamino acid content on TLC plates revealed that the amount of phosphorylated Thr in nonSUMOylated c-Myb isolated from stressed cells increased significantly (Thr(P) contents 29%) as compared with that from unstressed cells (Thr(P) 11%) (Fig. 1C). However, when we compared phosphoamino acid composition of nonSUMOylated and SUMOylated c-Myb protein isolated from stressed cells, we detected a decrease in Thr phosphorylation in the SUMOylated form (Thr(P) contents 18%) as compared with the nonSUMOylated form (Thr(P) 28%) of c-Myb (Fig. 1D). Phosphorylation of the Tyr residues was not detected. These results clearly show that cellular stress, exemplified here by hyperthermia, causes rapid changes in the phosphorylation of c-Myb.
FIGURE 1.

Stress induces changes in phosphorylation of c-Myb. A, metabolically labeled c-Myb with [32P]orthophosphate was isolated from cell lysates prepared from unstressed (37 °C) or stressed cells (43 °C) by immunoprecipitation and separated by SDS-PAGE. Tryptic peptides from 32P-labeled c-Myb were separated by reverse phase HPLC. Black arrows pointing up and gray arrows pointing down mark fractions with increased and decreased intensity, respectively. The inset shows 32P-labeled c-Myb immunoprecipitated from COS-7 cells transfected with c-Myb construct and treated with heat stress as indicated. B, [32P]orthophosphate-labeled c-Myb immunoprecipitated from COS-7 cells transfected with constructs encoding c-Myb and HA-SUMO-2 and treated (43 °C) or untreated (37 °C) with hyperthermia was separated by SDS-PAGE, transferred to membrane, and directly exposed to x-ray film. Mobility of unmodified and SUMO-conjugated forms of c-Myb with one or two molecules of SUMO are labeled as c-Myb, S1-c-Myb, and S2-c-Myb, respectively. C, comparative phosphoamino acid analysis of the nonSUMOylated form of c-Myb isolated from cells untreated (c-Myb@37) or treated (c-Myb@43) with hyperthermia. D, comparative phosphoamino acid analysis of the nonSUMOylated (c-Myb@43) and the SUMOylated c-Myb (S21-c-Myb@43) isolated from cells treated with hyperthermia. The positions of the phosphoserine (P-S), phosphothreonine (P-T), and phosphotyrosine (P-Y) markers are visualized by ninhydrin staining and are indicated on the right. Quantitative analysis of radioactive signals was performed on a PhosphorImager 425 using ImageQuant software (Molecular Dynamics) and is shown as a percentage of phosphoserine and phosphothreonine; total phosphorylation of both phosphoserine and phosphothreonine was assigned to 100%.
Identification of Phosphorylated Residues in c-Myb by Mass Spectrometry
Reversed phase liquid chromatography-tandem mass spectrometry was used to identify phosphorylated residues in c-Myb isolated from stressed or unstressed cells. As shown in Fig. 2A, the c-Myb-specific peptide coverage that was detected ranged from 42–44%. Phosphorylation of Ser528, described previously (39, 40), and Thr462 and Thr464 were detected in nonSUMOylated c-Myb isolated from both stressed and unstressed cells. Phosphorylation of Thr208, Ser444, and Thr486 was detected in cMyb isolated from stressed cells, but not in c-Myb isolated from cells growing under physiological conditions (Fig. 2A). Because Thr486 is located closely to Lys499, which is an acceptor of the SUMO-2 molecule and conjugation of SUMO-2 to Lys499 is strongly modified in response to stress (34), we decided to investigate whether phosphorylation of Thr486 may regulate stress-induced SUMOylation of c-Myb. High conservation of the NRD region of c-Myb surrounding Thr486 among species (Fig. 2B) also suggests an important function. Interestingly, however, phosphorylation of Thr486 was detected only in the nonSUMOylated form of c-Myb isolated from cells treated with hyperthermia (Fig. 2A). This observation is also in line with the phosphoamino acid experiment, where decreased Thr phosphorylation was detected in the SUMO-2-modified form of c-Myb (Fig. 1D).
FIGURE 2.
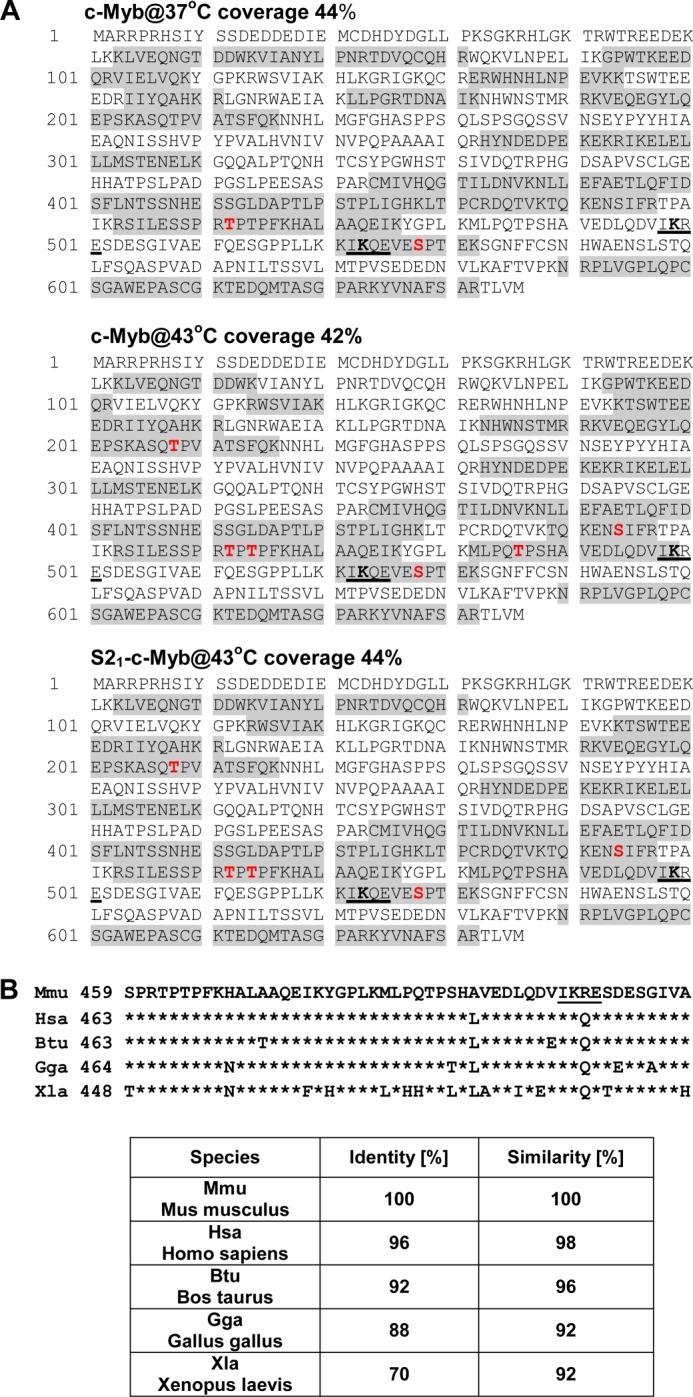
Identification of phosphorylation sites in c-Myb isolated from stressed cells by mass spectrometry. A, protein sequence of mouse c-Myb protein. The shaded residues were identified in the tryptic peptides from the nonSUMOylated form of c-Myb isolated from untreated cells (c-Myb@37°C) and nonSUMOylated c-Myb (c-Myb@43°C) and SUMOylated c-Myb (S21-c-Myb@43°C) from cells treated with hyperthermia. Total protein coverage is shown for every sample analyzed. Phosphorylation sites identified in analyzed samples are marked in red. Three new phosphorylation sites Thr208, Ser444, and Thr486 were identified in c-Myb isolated from stressed cells. SUMOylation motifs 498IKRE501 and 522IKQE525 are underlined. B, conservation of c-Myb protein sequences (459–509 amino acids, murine numbering). Phosphorylation site Thr486 is located amino-terminally to SUMOylation site Lys499. The SUMOylation motif IKRE is underlined. Identity and similarity of the c-Myb sequences between mouse and other species are presented in the table.
Phosphorylation of Thr486 Negatively Regulates Stress-induced Conjugation of SUMO-2/3
To further confirm the results from mass spectrometry, we decided to prepare an antibody that specifically recognizes the phosphorylated Thr486 residue in murine c-Myb. In collaboration with Rockland Immunochemicals Inc., rabbit polyclonal antibody that recognizes either phosphorylated Thr486 (α-P-T486pept) or nonphosphorylated region in murine c-Myb (amino acid residues 480–494, α-T486pept) were prepared and tested in COS7 cells transfected with c-Myb. Strong induction of Thr486 phosphorylation in c-Myb (Fig. 3A, P-T486-cMyb) was detected in cells treated with hyperthermia using Thr(P)486-specific antibody, whereas no difference in the c-Myb level was detected using antibody raised against the nonphosphorylated peptide (Fig. 3A). To further investigate phosphorylation of Thr486 in SUMOylated and nonSUMOylated forms of c-Myb, we transfected COS-7 cells with constructs encoding c-MybWT, HA-SUMO-2 and treated them with hyperthermia for 15 min. Strong conjugation of SUMO-2 protein was detected by immunoblotting using anti-c-Myb monoclonal antibody (Fig. 3B). c-Myb was immunoprecipitated from cell lysates using rabbit antibodies that recognize specifically either phosphorylated (α-P-T486pept) or nonphosphorylated Thr486 (α-T486pept) in murine c-Myb and analyzed by immunoblotting. Anti-HA antibody detected SUMO-2 conjugated to c-Myb, whereas anti-c-Myb monoclonal antibody recognized both SUMOylated and nonSUMOylated forms of c-Myb. Antibodies specific for phosphorylated Thr486 (α-P-T486pept) precipitated largely nonSUMOylated c-Myb, whereas antibody specific for nonphosphorylated c-Myb (α-T486pept) immunoprecipitated efficiently both SUMOylated (S21-c-Myb and S22-c-Myb) and nonSUMOylated (c-Myb) protein (Fig. 3C). These results confirmed mass spectrometry data and suggested that phosphorylation of Thr486 may negatively regulate stress-induced conjugation of SUMO-2 to c-Myb. To further investigate this possibility, we mutated Thr486 to Ala (cMybT486A) and assessed its stress-induced SUMOylation. Immunoblotting analyses of whole cell lysates and c-Myb immunoprecipitates showed that mutation of T486A resulted in enhanced stress-induced SUMOylation of c-Myb as compared with wild-type protein specifically in stressed cells (Fig. 3D).
FIGURE 3.

Phosphorylation of Thr486 negatively regulates stress-induced SUMOylation of c-Myb. A, COS7 cells were transfected with a construct encoding c-MybWT and treated with hyperthermia as indicated. Immunoblotting results are of total cell lysates analyzed with rabbit polyclonal antibody specific for phosphorylated Thr486 (P-cMybT486, top panel), nonphosphorylated Thr486 in murine c-Myb (c-Myb, middle panel), or anti-actin antibody (bottom panel). B, cell lysates from COS7 cells transfected with construct encoding c-MybWT and HA-SUMO-2 and treated with hyperthermia were analyzed by immunoblotting with monoclonal anti-c-Myb (top panel), or anti-actin antibody (bottom panel). The mobility of unmodified (c-Myb) and SUMOylated forms (HAS21-cMyb and HAS22-cMyb) are marked. C, immunoprecipitation of c-Myb using rabbit polyclonal antibody specific for phosphorylated (α-P-T486pept) or nonphosphorylated Thr486 (α-T486pept) were analyzed by immunoblotting with monoclonal anti-HA (α-HA, left panel) or anti-c-Myb antibody (α-cMyb, right panel). D, COS7 cells transfected with HA-SUMO-2, c-MybWT, and c-MybT486A and treated with hyperthermia as indicated. Cell lysates analyzed by immunoblotting with monoclonal anti-c-Myb (α-cMyb, right panel). c-Myb was immunoprecipitated with rabbit anti-c-Myb polyclonal antibody and analyzed by immunoblotting with anti-HA (α-HA, middle panel) or anti-c-Myb (α-cMyb, right panel) monoclonal antibodies. The mobility of c-Myb or SUMOylated forms of c-Myb are indicated on the right, and molecular mass markers (in kDa) are indicated on the left. IP, immunoprecipitation; IB, immunoblotting.
Activation of p38 MAPKs Negatively Regulates SUMOylation of c-Myb
Several stress-induced protein kinases are rapidly activated in response to different types of cellular stresses (37). Hyperthermia and other types of metabolic stresses rapidly induce activation of p38 MAPKs in M1 myeloid cells (34) (Fig. 4, A and B). It was previously reported that p38 MAPKs regulate steady-state levels of c-Myb (41). Furthermore, several Ser and Thr residues in the NRD, including Thr486, were implicated in p38 MAPK-dependent destabilization of c-Myb (41). However, the authors did not investigate whether any of these residues were indeed phosphorylated by p38 MAPKs. To examine the possibility that the p38 MAPKs phosphorylate Thr486 in response to stress, we immunoprecipitated activated forms of endogenous p38 MAPKs from M1 cells treated with hyperthermia using phospho-specific p38 MAPK antibody. The p38 MAPK kinase assay with GST-cMyb(464–544aa) fusion protein as a substrate was performed in vitro, and products were analyzed by immunoblotting using anti-P-T486cMyb phospho-specific antibody. Increased phosphorylation of Thr486 was detected in GST-cMyb(464–544aa) after in vitro incubation with activated p38 MAPKs (Fig. 4C), suggesting that at least in vitro the p38 MAPKs may phosphorylate Thr486 in c-Myb. Next, we investigated the efficiency of four different p38 MAPKs isoforms to suppress SUMOylation of c-Myb in COS-7 cells. All four p38 MAPK isoforms suppressed conjugation of HA-SUMO-2 to c-Myb in anisomycin-pretreated cells. However, there was an obvious difference in the suppressive activities of the different p38 MAPKs isoforms, with the strongest repressive effect detected with p38 MAPKγ and p38 MAPKσ (Fig. 4D).
FIGURE 4.

Activation of p38 MAPKs represses stress-induced SUMOylation of c-Myb. A and B, M1 cells treated with hyperthermia (43 °C) for the indicated times or anisomycin (5 μg/ml) (A) or with other metabolic stresses (0.7 m NaCl, 100 μm H2O2 peroxide, 7% EtOH) for 15 min (B). Total cell lysates were analyzed by immunoblotting with phospho-specific p38 MAPK and total p38 MAPK antibodies. Levels of activated (P-p38MAPK) and total p38 MAPK are shown. C, activated forms of p38 MAPKs immunoprecipitated from M1 cells treated with hyperthermia and used in an in vitro p38 MAPK kinase assay following the manufacturer's instructions (Cell Signaling) with GST-cMyb(464–544aa) protein as a substrate. Phosphorylation of Thr486 in GST-cMyb(464–544aa) was evaluated by immunoblotting with anti-P-T486cMyb phospho-specific antibody, and loading of GST-cMyb(464–544aa) was evaluated with anti-c-Myb antibody. D, COS7 cells transfected with constructs encoding c-MybWT, HA-SUMO-2, and four different FLAG-tagged p38 MAPK isoforms were pretreated with anisomycin (5 μg/ml; upper panel) or not treated with anisomycin (lower panel) and stressed by heat for another 30 min. SUMOylation of c-Myb was assessed by immunoblotting with anti-c-Myb monoclonal antibody. Expression of p38 MAPK isoforms was evaluated with anti-FLAG monoclonal antibody and protein loading with anti-actin antibody.
p38 MAPKs Negatively Regulate SUMOylation of Endogenous c-Myb in Myeloid Cells
Immunoprecipitation of c-Myb from anisomycin-treated myeloid M1 cells with anti-Thr(P)486-cMyb-specific serum confirmed phosphorylation of Thr486 in c-Myb at the endogenous level (Fig. 5A). Similar to our previous experiments in COS7 cells ectopically expressing c-Myb, preactivation of p38 MAPKs in myeloid M1 cells resulted in a substantial decrease of stress-induced conjugation of SUMO-2/3 to c-Myb at the endogenous level (Fig. 5B). In contrast, treatment of M1 cells with an inhibitor of p38 MAPK activity SB203580 resulted in an increase of SUMOylated c-Myb (Fig. 5C). The identity of slower migrating c-Myb-immunoreactive bands as SUMO-2/3-modified forms of c-Myb was confirmed by immunoprecipitation with anti-c-Myb-specific antibody and immunoblotting with anti-SUMO-2/3 antibody (Fig. 5D). Immunoblotting analyses revealed that M1 cells express high levels of the p38 MAPKα isoform and low levels of the p38 MAPKγ isoform (Fig. 5E). However, expression of p38 MAPKγ is rapidly increased upon IL-6-induced monocytic differentiation of M1 cells. Interestingly, induction of p38 MAPKγ expression is accompanied by a rapid reduction of c-Myb in M1 cells (Fig. 5E) that can be reversed by treatment with MG132, an inhibitor of 26S proteasome (Fig. 5F). Knockdown of p38 MAPKα by shRNA (Fig. 5G) resulted in accumulation of total and SUMOylated forms of c-Myb in M1 cells (Fig. 5G). These results, along with experiments employing the T486A mutant, show that there is negative cross-talk between stress-induced phosphorylation of Thr486 and conjugation of SUMO-2/3 to c-Myb in stressed cells.
FIGURE 5.

Regulation of endogenous c-Myb SUMOylation by p38 MAPKs in myeloid cells. A, M1 cells expressing endogenous c-Myb and SUMO-2/3 were treated with anisomycin for 20 min as indicated, and c-Myb was immunoprecipitated with rabbit polyclonal antibody specific for phosphorylated (IP:α-P-T486cMyb, left panel) or nonphosphorylated Thr486 (IP:α-T486cMyb, right panel). Immunoprecipitates were analyzed by immunoblotting with monoclonal anti-c-Myb antibody (IB:α-cMyb). B and C, M1 cells were pretreated with anisomycin (B) or SB203580 (10 μm, an inhibitor of p38 MAPKs) (C) for 20 min followed by heat stress for another 30 min. B–D, expression of c-Myb and its SUMOylated forms was evaluated by immunoblotting with anti-c-Myb antibody (B and C) or by immunoprecipitation with anti-c-Myb antibody and by immunoblotting with anti-SUMO-2/3 antibody (D). E, expression of c-Myb and p38 MAPK isoforms in M1 myeloid cells treated with IL-6 (100 ng/ml) for the indicated times. F, inhibition of 26 S proteasome by MG132 (10 μm) prevents increased proteolytic processing of c-Myb in M1 cells treated by IL-6. G, expression of c-Myb and its SUMOylated forms in control M1 cells and M1 cells with p38 MAPKα knocked down (shRNA-p38 α). Protein loadings were evaluated with anti-actin or anti-tubulin antibodies. The positions of SUMO-2/3 modified c-Myb (S2/31-c-Myb, S2/32-c-Myb) and unmodified c-Myb (c-Myb) are marked on the right. The migrations of molecular mass markers are shown on the left. IP, immunoprecipitation; IB, immunoblotting.
Mutation of T486A Modulates Proteolytic Stability and Transcriptional Activity of c-Myb
A previous report showed that activation of p38 MAPKs may regulate the proteolytic stability of murine c-Myb. Through mutational analyses, the authors implicated several Ser and Thr phosphorylation sites located in the NRD of c-Myb in destabilization of the protein (41). We also reported previously that conjugation of SUMO to c-Myb increases its half-life (31). Because T486A mutation results in an enhanced SUMOylation under stress (Fig. 3D), we compared protein turnovers of c-MybWT and cMybT486A in COS7 cells. There was no significant difference in the proteolytic processing of wild-type and mutant c-Myb in cells without activation of p38 MAPKs (data not shown). However, activation of p38 MAPKs by anisomycin treatment resulted in a slower protein turnover of cMybT486A as compared with cMybWT (Fig. 6, A and B). We hypothesized that the observed partial resistance of the cMybT486A mutant to proteolytic degradation in cells with chronic activation of p38 MAPKs can be explained, at least in part, by increased conjugation of SUMO-2/3 proteins to c-MybT486A, as compared with wild-type protein. To test this, we constructed the c-MybSU protein, resistant to a cleavage by SUMO-specific proteases, by in frame fusion of SUMO (2–93 amino acids) to the NRD of c-Myb (1–519aa). A significantly increased proteolytic stability of c-MybSU, as compared with both cMybWT and cMybT486A, in cells treated with anisomycin (Fig. 6, A, B, and C) provided additional evidence that SUMOylation of c-Myb increases its resistance to proteolytic degradation. Interestingly, we detected phosphorylation of Thr486 in c-MybSU after treatment of cells with anisomycin (data not shown), suggesting that conjugation of SUMO stabilizes the cMyb transcription factor independently of Thr486 phosphorylation.
FIGURE 6.

Mutation T486A affects proteolytic stability and transactivation activity of c-Myb in stressed cells. A, COS7 cells transfected with constructs encoding c-MybWT, c-MybT486A, or c-MybSU and treated with anisomycin (5 μg/ml) and cycloheximide (CHX, 10 μg/ml) for the indicated times and analyzed by immunoblotting with anti-c-Myb and anti-actin antibody. The migrations of molecular mass markers are shown on the left. B, scanned images were quantitated using ImageJ software. The relative amount of c-Myb protein during the cycloheximide treatment was normalized to actin, and the level of c-Myb at 0 min was assigned to value 1 (normalized intensity). Each point on the graph represents an average ± S.E. (error bars) from two independent experiments. C, bar graph presents half-lives of cMybWT, cMybT486A, and cMybSU. Data are shown as averages ± S.E. (error bars) from three independent experiments, calculated as described previously (30). *, p < 0.05; **, p < 0.01. D, Raw264.7 cells were transfected by electroporation in triplicate with c-Myb-responsive reporter construct p5xMRE-A-luc (5xMRE), and constructs encoding cMybWT, cMybT486, and cMybSU as indicated. Plasmid pRL-TK (encoding Renilla luciferase gene) was also transfected to control for transfection efficiency. Twenty-four hours after transfection, cells were left untreated (gray bars) or pretreated with anisomycin (5 μg/ml) for 30 min, followed by hyperthermic stress for another 60 min (black bars). Firefly luciferase activity was normalized using Renilla luciferase values. The results show the mean ± S.D. (error bars) for two independent experiments performed in triplicate. The unpaired two-tailed Student's t test was used to calculate p values. *, p < 0.05; RLU, relative light units.
To investigate the transactivation activity of c-MybWT and cMybT486A, we used macrophage cell line RAW264.7 that does not express endogenous c-Myb (data not shown), but it expresses many cofactors that modulate the transcriptional activity of c-Myb. Transactivation activity of the wild-type and the T486A mutant was evaluated in cells transfected with a c-Myb-responsive firefly luciferase reporter construct (p5xMRE-A-luc) and constructs encoding either cMybWT or cMybT486A as indicated. We did not detect any significant difference in transcriptional activity between c-MybWT and c-MybT486A in unstressed cells. However, pretreatment of cells with anisomycin for 30 min followed by heat stress for 60 min resulted in stronger suppression of transcriptional activity by cMybT486A than cMybWT (Fig. 6D). Additionally, transcriptional activity of the cMybSU protein was very low and confirmed a strong suppressive activity of SUMO conjugation to c-Myb (Fig. 6D).
Ectopic Expression of cMybT486A Results in Accelerated Apoptosis of Myeloid Cells
To evaluate the physiological consequences of c-Myb Thr486 phosphorylation under more normal conditions, we prepared myeloid M1 cell lines that ectopically express either wild-type (M1/cMybWT) or T486A mutant of c-Myb (M1/cMybT486A). Both M1/cMybWT and M1/cMybT486A cells proliferated with a similar doubling time as parental M1 cells (data not shown). Activation of p38 MAPKs by anisomycin treatment resulted in a rapid decrease in cell viability with virtually no viable cells in parental M1 cells or cell lines ectopically expressing either cMybWT or cMybT486A after 24 h of treatment (data not shown). Interestingly, however, at early time points, acceleration of cell death was detected in a cell line ectopically expressing the mutant form of c-Myb (cMybT486A) (Fig. 7A). Enhanced apoptotic cell death in M1/c-MybT486A was confirmed by higher levels of the cleaved PARP protein in cells expressing mutant protein (Fig. 7B) and by increased degradation of genomic DNA (Fig. 7C). Decreased transcriptional activity of cMybT486A detected in stressed cells suggests that some of the transcriptional targets of c-Myb that are involved in regulation of apoptosis may be misregulated. Therefore, we analyzed expression of two important anti-apoptotic regulators, Bcl-2 and Bcl-xL that were shown to be transcriptionally activated by c-Myb (42–44). Indeed, we detected lower levels of Bcl-2 and Bcl-xL in stressed M1/cMybT486A as compared with M1/cMybWT, whereas the level of another anti-apoptotic protein Mcl-1 was not considerably changed (Fig. 7, D and E). Together, these results suggest that phosphorylation of Thr486 modulates the transcriptional activity of c-Myb through suppression of SUMOylation in cells under stress and can lead to stress-triggered apoptotic cell death.
FIGURE 7.

Accelerated apoptosis of M1 cells ectopically expressing cMybT486A. A, M1 cell lines ectopically expressing either wild-type (cMybWT) or T486A mutant of c-Myb (cMybT486A) treated with anisomycin (5 μg/ml) for indicated time and evaluated for viability by trypan blue exclusion assay. Data (percentages of dead cells in cell cultures) are shown as averages ± S.E. (error bars) from three independent experiments. *, p < 0.05. The inset shows expression of c-Myb and tubulin in M1/cMybWT and M1/cMybT486A cells. B, cleavage of PARP protein (cl-PARP) in cells treated with anisomycin evaluated by immunoblotting. The relative intensities of cl-PARP in M1/cMybWT and M1/cMybT486A normalized to levels of tubulin (loading control) are shown below immunoblot. C, apoptotic DNA ladder in M1 cells ectopically expressing cMybWT and cMybT486A untreated (left panel) or treated with anisomycin for 6 h (right panel). D, expression of Bcl-2, Bcl-xL, Mcl-1, and actin in M1/cMybWT and M1/cMybT486A cells untreated (left panel) or treated with anisomycin for 6 h (right panel). E, bar graph presents intensities of Bcl-2, Bcl-xL, and Mcl-1 in anisomycin-treated M1 cells normalized to actin.
DISCUSSION
In the present work, we have shown for the first time that environmental stresses, in addition to rapid covalent conjugation of SUMO-2/3 proteins (34), induce changes in the phosphorylation of c-Myb. Although stress-induced phosphorylation of Thr208 and Ser444 was identified in both SUMOylated and nonSUMOylated forms of c-Myb, the phosphorylation of Thr486, located in close proximity to SUMOylation site Lys499 of c-Myb, was detected preferentially in the nonSUMOylated form. Furthermore, we have shown that a T486A mutation results in increased SUMOylation of c-Myb in cells treated with hyperthermia, suggesting a repressive effect Thr486 phosphorylation on stress-induced conjugation of SUMO-2/3 to c-Myb. p38 MAPKs were identified as protein kinase candidates that regulate the activity of c-Myb through phosphorylation-dependent repression of SUMOylation in stressed cells. Thus, we have uncovered a novel regulatory interplay between phosphorylation and SUMOylation of c-Myb that regulates its activity in stressed cells.
SUMOylation dramatically modulates transcriptional activity, as well as proteolytic turnover of c-Myb (31–33). Phosphorylation-dependent stimulation of SUMOylation of target proteins is frequently detected when modified Ser or Thr residues are located within the specific bipartite phosphorylation-dependent SUMOylation motif composed of a SUMO consensus site and an adjacent proline-directed phosphorylation site (ψIKXEXX(S/T)P) (35). Although the major SUMOylation site Lys523 and constitutively phosphorylated Ser528 (39) are both located within the phosphorylation-dependent SUMOylation motif in c-Myb, no significant regulatory effect of Ser528 phosphorylation on SUMOylation of c-Myb has been detected (31, 34). It has been reported that suppression of SUMOylation through phosphorylation can also occur when phosphorylated residues are located outside of the phosphorylation-dependent SUMOylation motif in target proteins (45). In this regard, we have identified phosphorylation of Thr486 in c-Myb isolated from stressed cells but largely in the nonSUMOylated form of c-Myb, suggesting a negative role for Thr486 phosphorylation in stress-induced SUMOylation of c-Myb. Accordingly, we observed a significant increase in SUMOylation of a cMybT486A mutant as compared with wild-type c-Myb (Fig. 3). Although a detailed mechanism through which phosphorylation of Thr486 suppresses SUMOylation of c-Myb is not known, we can speculate that phosphorylation changes the conformation of the NRD of c-Myb, and these changes result in decreased affinity of phosphorylated c-Myb for SUMO-conjugating enzymes necessary for efficient SUMOylation. Indeed, conformational changes in the NRD of c-Myb in cells treated with okadaic acid, an inhibitor of Ser/Thr protein phosphatases that induces strong induction of Thr phosphorylation, were described previously (38, 46). Alternatively, activation of p38 MAPKs can also stimulate specific SUMO-2/3 proteases that in turn affect SUMOylation and stability of c-Myb in stressed cells.
Our results also indicate that the cMybT486A mutant, in stressed cells, has a decreased proteolytic turnover as compared with wild-type c-Myb. Several protein kinases and phosphatases were reported to destabilize c-Myb through increased phosphorylation of its NRD, but identification of a critical residue previously remained largely elusive (38, 41, 47, 48). More recently, Kitagawa et al. (49) reported that phosphorylation of Thr572 by GSK3 protein kinase in murine c-Myb is recognized by Fbw7b, a protein component of SCF-ubiquitin ligase complex. Fbw7b binds to phosphorylated Thr572 and catalyzes polyubiquitin chain conjugation that triggers degradation of c-Myb by the 26S proteasome. Regulation of c-Myb turnover through phosphorylation of Thr572 seems to be specific for murine c-Myb, because this residue is not evolutionary conserved. Furthermore, both positive and negative roles for GSK3 protein kinase in regulation of human c-Myb were reported for different cell lines (50, 51). Here, we show that phosphorylation of Thr486 by p38 MAPKs in stressed cells destabilizes c-Myb. Our results are in agreement with a previous report where the authors showed increased proteolytic processing of c-Myb in cells upon activation of p38 MAPKs by anisomycin. This report also suggested that several Ser and Thr residues located in the NRD of c-Myb, including Thr486, can be phosphorylated by p38 MAPKs and involved in proteolytic degradation of c-Myb (41). We confirmed this observation, and by using a newly developed anti-phospho-Thr486 cMyb-specific antibody, we confirmed that Thr486 in murine c-Myb is phosphorylated by activated p38 MAPKs (Fig. 4C). Furthermore, we showed that overexpression of all four different isoforms (α, β, γ, and δ) attenuated stress-induced SUMOylation of c-Myb, with the strongest effect observed for p38 MAPKγ and p38 MAPKδ (Fig. 4D). Whether phosphorylated Thr486 is recognized by a specific ubiquitin ligase that catalyzes conjugation of polyubiquitin chains that targets c-Myb for degradation or whether decreased SUMOylation in phosphorylated protein increases proteolytic turnover of c-Myb is not clear at present. However, increased proteolytic stability of the cMybT486A mutant can be at least partially explained by augmented conjugation of SUMO to c-Myb in stressed cells. We have previously reported that conjugation of SUMO increases the half-life of modified c-Myb (31). Here we provide additional evidence based upon the observation that a c-Myb-SUMO fusion protein (cMybSU), resistant to SUMO-specific proteases, has dramatically increased proteolytic stability in cells (Fig. 6, A–C).
Stress-induced SUMOylation of targets proteins can have diverse effects on their activities. Although numerous proteins involved in DNA repair processes are strongly activated by stress-induced SUMOylation (52), transcription factors with a positive role in regulation of cellular proliferation are repressed by conjugation of SUMO proteins (53). Rapid inactivation of positive regulators of cell proliferation may represent a mechanism through which cells adapt to stress by ceasing proliferation, thus providing time for the enzymatic repair machinery to assess and restore damages imposed to cells by stress. c-Myb regulates transcription of diverse genes directly involved in broad set of cellular processes such as proliferation, differentiation, and programmed cells death. Regulated expression of different target genes by a differentially modified transcriptional regulator may represent a mechanism responsible for a proper cellular adaptation response to different growth conditions. Accordingly, attenuation of stress-induced SUMOylation of c-Myb through phosphorylation of Thr486 may maintain expression of anti-apoptotic genes, resulting in modulation of apoptotic processes in stressed cells. Indeed, in myeloid M1 cells ectopically expressing the T486A mutant (M1/cMybT486A), we detected lower levels of two anti-apoptotic proteins Bcl-2 and Bcl-xL that are encoded by genes directly regulated by c-Myb (42–44). Consequently, M1/cMybT486A cells displayed accelerated induction of programmed cell death in response to stress (Fig. 7). We speculate that a fraction of c-Myb protein that regulates genes directly involved in cellular proliferation is rapidly inactivated by conjugation of SUMO in stressed cells. Inactivation of the second fraction of c-Myb by SUMOylation can be attenuated through phosphorylation of Thr486 and possibly other residues and may still positively regulate a different set of genes important for survival and adaptation to stress. Whether differently modified populations of c-Myb exist in cells and regulate diverse target genes is not clear at present. However, there is experimental evidence that in a single cell, several differently modified histone molecules serve diverse functions in regulation of gene transcription (54).
In summary, we have identified phosphorylation of Thr486 in c-Myb as a new stress-induced post-translational modification that regulates c-Myb function. Moreover, discovery of negative cross-talk between the stress-activated p38 MAPKs and SUMOylation pathways add further complexity to post-translational regulation of the NRD of c-Myb, which affects transactivation activity and proteolytic stability of c-Myb. Strict regulation of c-Myb during hematopoiesis is critical for hematopoietic homeostasis, and deregulation of even wild-type c-Myb protein can be leukemogenic (21–23). Therefore, it is essential to identify novel post-translational modifications and signal transduction pathways that mutually cross-talk and modulate c-Myb activity because they may represent important targets for pharmacological intervention in diseases where deregulated activity of c-Myb plays important roles.
This work was supported, in whole or in part, by the Intramural Research Program of the National Cancer Institute, Center for Cancer Research, National Institutes of Health.
- SUMO
- small ubiquitin-like modifier
- HA
- hemagglutinin
- NRD
- negative regulatory domain
- MRE
- Myb-responsive element.
REFERENCES
- 1. Oh I. H., Reddy E. P. (1999) The myb gene family in cell growth, differentiation and apoptosis. Oncogene 18, 3017–3033 [DOI] [PubMed] [Google Scholar]
- 2. Ramsay R. G., Gonda T. J. (2008) MYB function in normal and cancer cells. Nat. Rev. Cancer 8, 523–534 [DOI] [PubMed] [Google Scholar]
- 3. Mucenski M. L., McLain K., Kier A. B., Swerdlow S. H., Schreiner C. M., Miller T. A., Pietryga D. W., Scott W. J., Jr., Potter S. S. (1991) A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell 65, 677–689 [DOI] [PubMed] [Google Scholar]
- 4. Allen R. D., 3rd, Bender T. P., Siu G. (1999) c-Myb is essential for early T cell development. Genes Dev. 13, 1073–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomas M. D., Kremer C. S., Ravichandran K. S., Rajewsky K., Bender T. P. (2005) c-Myb is critical for B cell development and maintenance of follicular B cells. Immunity 23, 275–286 [DOI] [PubMed] [Google Scholar]
- 6. Pearson R., Weston K. (2000) c-Myb regulates the proliferation of immature thymocytes following beta-selection. EMBO J. 19, 6112–6120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lieu Y. K., Kumar A., Pajerowski A. G., Rogers T. J., Reddy E. P. (2004) Requirement of c-myb in T cell development and in mature T cell function. Proc. Natl. Acad. Sci. U.S.A. 101, 14853–14858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sumner R., Crawford A., Mucenski M., Frampton J. (2000) Initiation of adult myelopoiesis can occur in the absence of c-Myb whereas subsequent development is strictly dependent on the transcription factor. Oncogene 19, 3335–3342 [DOI] [PubMed] [Google Scholar]
- 9. Emambokus N., Vegiopoulos A., Harman B., Jenkinson E., Anderson G., Frampton J. (2003) Progression through key stages of haemopoiesis is dependent on distinct threshold levels of c-Myb. EMBO J. 22, 4478–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vegiopoulos A., García P., Emambokus N., Frampton J. (2006) Coordination of erythropoiesis by the transcription factor c-Myb. Blood 107, 4703–4710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lieu Y. K., Reddy E. P. (2009) Conditional c-myb knockout in adult hematopoietic stem cells leads to loss of self-renewal due to impaired proliferation and accelerated differentiation. Proc. Natl. Acad. Sci. U.S.A. 106, 21689–21694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sandberg M. L., Sutton S. E., Pletcher M. T., Wiltshire T., Tarantino L. M., Hogenesch J. B., Cooke M. P. (2005) c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev. Cell 8, 153–166 [DOI] [PubMed] [Google Scholar]
- 13. Sakamoto H., Dai G., Tsujino K., Hashimoto K., Huang X., Fujimoto T., Mucenski M., Frampton J., Ogawa M. (2006) Proper levels of c-Myb are discretely defined at distinct steps of hematopoietic cell development. Blood 108, 896–903 [DOI] [PubMed] [Google Scholar]
- 14. Wolff L. (1996) Myb-induced transformation. Crit. Rev. Oncogenesis 7, 245–260 [DOI] [PubMed] [Google Scholar]
- 15. Lipsick J. S., Wang D. M. (1999) Transformation by v-Myb. Oncogene 18, 3047–3055 [DOI] [PubMed] [Google Scholar]
- 16. Zhou Y., Ness S. A. (2011) Myb proteins. Angels and demons in normal and transformed cells. Front. Biosci. 16, 1109–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bies J., Nazarov V., Wolff L. (1999) Identification of protein instability determinants in the carboxy-terminal region of c-Myb removed as a result of retroviral integration in murine monocytic leukemias. J. Virol. 73, 2038–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barletta C., Pelicci P. G., Kenyon L. C., Smith S. D., Dalla-Favera R. (1987) Relationship between the c-myb locus and the 6q-chromosomal aberration in leukemias and lymphomas. Science 235, 1064–1067 [DOI] [PubMed] [Google Scholar]
- 19. Pelicci P. G., Lanfrancone L., Brathwaite M. D., Wolman S. R., Dalla-Favera R. (1984) Amplification of the c-myb oncogene in a case of human acute myelogenous leukemia. Science 224, 1117–1121 [DOI] [PubMed] [Google Scholar]
- 20. Tomita A., Watanabe T., Kosugi H., Ohashi H., Uchida T., Kinoshita T., Mizutani S., Hotta T., Murate T., Seto M., Saito H. (1998) Truncated c-Myb expression in the human leukemia cell line TK-6. Leukemia 12, 1422–1429 [DOI] [PubMed] [Google Scholar]
- 21. Clappier E., Cuccuini W., Kalota A., Crinquette A., Cayuela J. M., Dik W. A., Langerak A. W., Montpellier B., Nadel B., Walrafen P., Delattre O., Aurias A., Leblanc T., Dombret H., Gewirtz A. M., Baruchel A., Sigaux F., Soulier J. (2007) The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood 110, 1251–1261 [DOI] [PubMed] [Google Scholar]
- 22. Lahortiga I., De Keersmaecker K., Van Vlierberghe P., Graux C., Cauwelier B., Lambert F., Mentens N., Beverloo H. B., Pieters R., Speleman F., Odero M. D., Bauters M., Froyen G., Marynen P., Vandenberghe P., Wlodarska I., Meijerink J. P., Cools J. (2007) Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat. Genet. 39, 593–595 [DOI] [PubMed] [Google Scholar]
- 23. O'Neil J., Tchinda J., Gutierrez A., Moreau L., Maser R. S., Wong K. K., Li W., McKenna K., Liu X. S., Feng B., Neuberg D., Silverman L., DeAngelo D. J., Kutok J. L., Rothstein R., DePinho R. A., Chin L., Lee C., Look A. T. (2007) Alu elements mediate MYB gene tandem duplication in human T-ALL. J. Exp. Med. 204, 3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murati A., Gervais C., Carbuccia N., Finetti P., Cervera N., Adélaïde J., Struski S., Lippert E., Mugneret F., Tigaud I., Penther D., Bastard C., Poppe B., Speleman F., Baranger L., Luquet I., Cornillet-Lefebvre P., Nadal N., Nguyen-Khac F., Pérot C., Olschwang S., Bertucci F., Chaffanet M., Lessard M., Mozziconacci M. J., Birnbaum D. (2009) Genome profiling of acute myelomonocytic leukemia. Alteration of the MYB locus in MYST3-linked cases. Leukemia 23, 85–94 [DOI] [PubMed] [Google Scholar]
- 25. Belloni E., Shing D., Tapinassi C., Viale A., Mancuso P., Malazzi O., Gerbino E., Dall'Olio V., Egurbide I., Odero M. D., Bertolini F., Pelicci P. G. (2011) In vivo expression of an aberrant MYB-GATA1 fusion induces leukemia in the presence of GATA1 reduced levels. Leukemia 25, 733–736 [DOI] [PubMed] [Google Scholar]
- 26. Koller R., Krall M., Mock B., Bies J., Nazarov V., Wolff L. (1996) Mml1, a new common integration site in murine leukemia virus-induced promonocytic leukemias maps to mouse chromosome 10. Virology 224, 224–234 [DOI] [PubMed] [Google Scholar]
- 27. Zhang J., Markus J., Bies J., Paul T., Wolff L. (2012) Three murine leukemia virus integration regions within 100 kilobases upstream of c-myb are proximal to the 5′ regulatory region of the gene through DNA looping. J. Virol. 86, 10524–10532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anfossi G., Gewirtz A. M., Calabretta B. (1989) An oligomer complementary to c-myb-encoded mRNA inhibits proliferation of human myeloid leukemia cell lines. Proc. Natl. Acad. Sci. U.S.A. 86, 3379–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Calabretta B., Sims R. B., Valtieri M., Caracciolo D., Szczylik C., Venturelli D., Ratajczak M., Beran M., Gewirtz A. M. (1991) Normal and leukemic hematopoietic cells manifest differential sensitivity to inhibitory effects of c-myb antisense oligodeoxynucleotides. An in vitro study relevant to bone marrow purging. Proc. Natl. Acad. Sci. U.S.A. 88, 2351–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bies J., Wolff L. (1997) Oncogenic activation of c-Myb by carboxyl-terminal truncation leads to decreased proteolysis by the ubiquitin-26S proteasome pathway. Oncogene 14, 203–212 [DOI] [PubMed] [Google Scholar]
- 31. Bies J., Markus J., Wolff L. (2002) Covalent attachment of the SUMO-1 protein to the negative regulatory domain of the c-Myb transcription factor modifies its stability and transactivation capacity. J. Biol. Chem. 277, 8999–9009 [DOI] [PubMed] [Google Scholar]
- 32. Dahle O., Andersen T. O., Nordgård O., Matre V., Del Sal G., Gabrielsen O. S. (2003) Transactivation properties of c-Myb are critically dependent on two SUMO-1 acceptor sites that are conjugated in a PIASy enhanced manner. Eur. J. Biochem. 270, 1338–1348 [DOI] [PubMed] [Google Scholar]
- 33. Molvaersmyr A. K., Saether T., Gilfillan S., Lorenzo P. I., Kvaloy H., Matre V., Gabrielsen O. S. (2010) A SUMO-regulated activation function controls synergy of c-Myb through a repressor-activator switch leading to differential p300 recruitment. Nucleic Acids Res. 38, 4970–4984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sramko M., Markus J., Kabát J., Wolff L., Bies J. (2006) Stress-induced inactivation of the c-Myb transcription factor through conjugation of SUMO-2/3 proteins. J. Biol. Chem. 281, 40065–40075 [DOI] [PubMed] [Google Scholar]
- 35. Hietakangas V., Anckar J., Blomster H. A., Fujimoto M., Palvimo J. J., Nakai A., Sistonen L. (2006) PDSM, a motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. U.S.A. 103, 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang S. H., Jaffray E., Senthinathan B., Hay R. T., Sharrocks A. D. (2003) SUMO and transcriptional repression. Dynamic interactions between the MAP kinase and SUMO pathways. Cell Cycle 2, 528–530 [DOI] [PubMed] [Google Scholar]
- 37. Cargnello M., Roux P. P. (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bies J., Feiková S., Bottaro D. P., Wolff L. (2000) Hyperphosphorylation and increased proteolytic breakdown of c-Myb induced by the inhibition of Ser/Thr protein phosphatases. Oncogene 19, 2846–2854 [DOI] [PubMed] [Google Scholar]
- 39. Aziz N., Miglarese M. R., Hendrickson R. C., Shabanowitz J., Sturgill T. W., Hunt D. F., Bender T. P. (1995) Modulation of c-Myb-induced transcription activation by a phosphorylation site near the negative regulatory domain. Proc. Natl. Acad. Sci. U.S.A. 92, 6429–6433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miglarese M. R., Richardson A. F., Aziz N., Bender T. P. (1996) Differential regulation of c-Myb-induced transcription activation by a phosphorylation site in the negative regulatory domain. J. Biol. Chem. 271, 22697–22705 [DOI] [PubMed] [Google Scholar]
- 41. Pani E., Ferrari S. (2008) p38MAPKδ controls c-Myb degradation in response to stress. Blood Cells Mol. Dis. 40, 388–394 [DOI] [PubMed] [Google Scholar]
- 42. Taylor D., Badiani P., Weston K. (1996) A dominant interfering Myb mutant causes apoptosis in T cells. Genes Dev. 10, 2732–2744 [DOI] [PubMed] [Google Scholar]
- 43. Frampton J., Ramqvist T., Graf T. (1996) v-Myb of E26 leukemia virus up-regulates bcl-2 and suppresses apoptosis in myeloid cells. Genes Dev. 10, 2720–2731 [DOI] [PubMed] [Google Scholar]
- 44. Yuan J., Crittenden R. B., Bender T. P. (2010) c-Myb promotes the survival of CD4+CD8+ double-positive thymocytes through upregulation of Bcl-xL. J. Immunol. 184, 2793–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bossis G., Malnou C. E., Farras R., Andermarcher E., Hipskind R., Rodriguez M., Schmidt D., Muller S., Jariel-Encontre I., Piechaczyk M. (2005) Down-regulation of c-Fos/c-Jun AP-1 dimer activity by sumoylation. Mol. Cell Biol. 25, 6964–6979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bies J., Feiková S., Markus J., Wolff L. (2001) Phosphorylation-dependent conformation and proteolytic stability of c-Myb. Blood Cells Mol. Dis. 27, 422–428 [DOI] [PubMed] [Google Scholar]
- 47. Kanei-Ishii C., Ninomiya-Tsuji J., Tanikawa J., Nomura T., Ishitani T., Kishida S., Kokura K., Kurahashi T., Ichikawa-Iwata E., Kim Y., Matsumoto K., Ishii S. (2004) Wnt-1 signal induces phosphorylation and degradation of c-Myb protein via TAK1, HIPK2, and NLK. Genes Dev. 18, 816–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Corradini F., Cesi V., Bartella V., Pani E., Bussolari R., Candini O., Calabretta B. (2005) Enhanced proliferative potential of hematopoietic cells expressing degradation-resistant c-Myb mutants. J. Biol. Chem. 280, 30254–30262 [DOI] [PubMed] [Google Scholar]
- 49. Kitagawa K., Hiramatsu Y., Uchida C., Isobe T., Hattori T., Oda T., Shibata K., Nakamura S., Kikuchi A., Kitagawa M. (2009) Fbw7 promotes ubiquitin-dependent degradation of c-Myb. Involvement of GSK3-mediated phosphorylation of Thr-572 in mouse c-Myb. Oncogene 28, 2393–2405 [DOI] [PubMed] [Google Scholar]
- 50. Kitagawa K., Kotake Y., Hiramatsu Y., Liu N., Suzuki S., Nakamura S., Kikuchi A., Kitagawa M. (2010) GSK3 regulates the expressions of human and mouse c-Myb via different mechanisms. Cell Div 5, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou F., Zhang L., van Laar T., van Dam H., Ten Dijke P. (2011) GSK3beta inactivation induces apoptosis of leukemia cells by repressing the function of c-Myb. Mol. Biol. Cell 22, 3533–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jackson S. P., Durocher D. (2013) Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807 [DOI] [PubMed] [Google Scholar]
- 53. Ouyang J., Gill G. (2009) SUMO engages multiple corepressors to regulate chromatin structure and transcription. Epigenetics 4, 440–444 [DOI] [PubMed] [Google Scholar]
- 54. Zentner G. E., Henikoff S. (2013) Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 20, 259–266 [DOI] [PubMed] [Google Scholar]