

Background: TAP proteins tagged with CFP and YFP were used for FRET spectroscopy measurements.
Results: Conformational changes of the nucleotide binding domains (NBD) were measurable in permeabilized cells.
Conclusion: TAP-specific peptides induce NBD closure, and maximal NBD closure is induced by the combination of a peptide and a non-hydrolysable ATP analog.
Significance: These studies elucidate distinct steps of the TAP transport cycle.
Keywords: ABC Transporter, Antigen Presentation, ATPases, Major Histocompatibility Complex (MHC), Peptide Transport, Transporters, Transporter Associated with Antigen Processing (TAP)
Abstract
The transporter associated with antigen processing (TAP) plays a critical role in the MHC class I antigen presentation pathway. TAP translocates cellular peptides across the endoplasmic reticulum membrane in an ATP hydrolysis-dependent manner. We used FRET spectroscopy in permeabilized cells to delineate different conformational states of TAP in a native subcellular membrane environment. For these studies, we tagged the TAP1 and TAP2 subunits with enhanced cyan fluorescent protein and enhanced yellow fluorescent protein, respectively, C-terminally to their nucleotide binding domains (NBDs), and measured FRET efficiencies under different conditions. Our data indicate that both ATP and ADP enhance the FRET efficiencies but that neither induces a maximally closed NBD conformation. Additionally, peptide binding induces a large and significant increase in NBD proximity with a concentration dependence that is reflective of individual peptide affinities for TAP, revealing the underlying mechanism of peptide-stimulated ATPase activity of TAP. Maximal NBD closure is induced by the combination of peptide and non-hydrolysable ATP analogs. Thus, TAP1-TAP2 NBD dimers are not fully stabilized by nucleotides alone, and substrate binding plays a key role in inducing the transition state conformations of the NBD. Taken together, these findings show that at least three steps are involved in the transport of peptides across the endoplasmic reticulum membrane for antigen presentation, corresponding to three dynamically and structurally distinct conformational states of TAP. Our studies elucidate structural changes in the TAP NBD in response to nucleotides and substrate, providing new insights into the mechanism of ATP-binding cassette transporter function.
Introduction
The transporter associated with antigen processing (TAP)2 plays a crucial role in the processing and presentation of the MHC class I-restricted antigens (1). TAP transports peptides from the cytosol into the endoplasmic reticulum (ER), thereby selecting peptides for binding to MHC class I molecules (2). Upon loading onto MHC class I molecules, the trimeric MHC class I·β2-microglobulin·peptide complex is then transported to the cell surface and presented to CD8+ cytotoxic T cells. Given the importance of TAP in adaptive immune responses, a number of TAP-targeted strategies have emerged as potential therapeutic approaches in different diseases (3). However, the molecular mechanisms of TAP are still not fully understood.
TAP belongs to ATP binding cassette (ABC) transporter superfamily, which is a group of membrane proteins that use the binding and hydrolysis of ATP to power the translocation of a wide variety of substrates across cellular membranes (4). These proteins share a common architecture, comprising two nucleotide-binding domains (NBDs) that bind and hydrolyze ATP and two transmembrane domains (TMDs) that contain the substrate binding sites and form the transport pathway (5). Two major models have been suggested for the ATP hydrolysis mechanism of ABC transporters (6). In the “switch” model, NBD dimerization following ATP binding provides the “power stroke” driving substrate transport, whereas ATP hydrolysis is required to reset the transporter. In the “constant contact” model, the NBD monomers never fully separate, and ATP hydrolysis occurs alternately at each of the two active sites.
As in other ABC transporters, the TAP1-TAP2 complex comprises two nucleotide-binding domains, located on the cytosolic side of the ER membrane (Fig. 1). Two composite ATP-binding active sites are formed by the interaction of the signature motif on each NBD with an ATP molecule bound to the Walker A and B motifs of an opposing NBD (7, 8). Biochemical studies showed that purified TAP1-TAP2 NBDs are unable to form a heterodimer in solution in the presence of either ADP or ATP (8). However, interactions between the NBD of full-length TAP1-TAP2 complexes have not been investigated previously.
FIGURE 1.
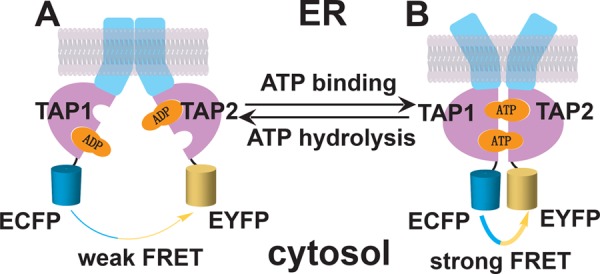
Schematic depiction of the FRET assay for measuring TAP1-TAP2 NBD interactions. TAP comprises two subunits, TAP1 and TAP2, each containing a transmembrane region (blue) and cytosolic NBD (pink). TAP is thought to exist in an NBD-open, inward-facing (cytosol-facing) conformation (A) or an NBD-closed, outward-facing (ER-facing) conformation (B). However, the factors required to drive transitions between the conformations are poorly understood. Prior studies with other ABC transporters have implicated ATP binding and hydrolysis in driving conformational changes. Because of the predicted NBD distance changes during peptide transport, we expected that different TAP conformations could be monitored on the basis of FRET efficiencies between ECFP and EYFP tagged to the NBDs of TAP1 and TAP2.
Although the crystal structures of isolated TAP1 nucleotide-binding domains have been solved (7, 8), high-resolution structural data for full-length TAP are currently lacking. According to the current mechanistic model for ABC transporter function (reviewed in Refs 5, 6), TAP can exist in at least two major conformations during the catalytic cycle: an “inward”-facing conformation, with the peptide binding pocket exposed to the cytoplasmic side and separated NBDs (Fig. 1A), and an “outward”-facing conformation with a low-affinity peptide binding site exposed to the ER and closely interacting NBDs (Fig. 1B). The C termini of the two NBDs are predicted to undergo large distance changes when going from an inward-facing conformation to an outward-facing conformation (9). In this report, we investigated the movement of TAP NBDs during the catalytic cycle using a FRET approach, which allows us to monitor the distance changes between the TAP1-TAP2 NBDs in a native cellular membrane environment under different conditions without the need of biochemical sample preparation.
EXPERIMENTAL PROCEDURES
Baculovirus Constructs
A baculovirus encoding the enhanced cyan fluorescent protein-tagged version of human TAP1 (TAP1-ECFP) was obtained as described previously (10, 11). For construction of the enhanced yellow fluorescent protein-tagged version of human TAP2 (TAP2-EYFP), the TAP2 was excised from pPCRscript (11) with BglII and ligated into pEYFP (Clontech) at the BamHI site to generate the TAP2 fusion with EYFP. TAP2-EYFP was then excised from pEYFP with XbaI, ligated into the vector pVL1393 (BD Pharmingen) at the XbaI site, and used to generate a TAP2-EYFP-encoding baculovirus using the BaculoGold transfection kit (BD Pharmingen). The hydrolysis-deficient TAP2 mutant TAP2-EYFP(E632Q) was made by introducing the E632Q mutation into TAP2-EYFP using the QuikChange site-directed mutagenesis kit. The primers were 5′-GGGTCCTCATTCTAGATCAGGCTACTAGTGCCCTAG-3′ (forward) and 5′-CTAGGGCACTAGTAGCCTGATCTAGAATGAGGACCC-3′ (reverse) (11). All primers were purchased from Invitrogen.
Insect Cell Infections, Microsome Preparations, and Peptide Translocation
Sf9 cells were cultured in Sf-900TM II serum free media (SFM) (Invitrogen). The cells were grown to confluence and infected with the appropriate baculoviruses at multiplicities of infection of 5–20, depending on the protein expression level of individual baculoviruses. Following these infections, the cells were harvested after 72 h, and microsomal membranes were generated as described (12). Peptide transport was analyzed using the fluorescent peptide RYWANATKFITCSR as described previously (13). The translocation assay was performed by incubating microsomes with 10 μm of FITC-labeled peptide at 37 °C for 15 min in the presence of 5 mm ATP. Microsomes were lysed in 1% Nonidet P-40. The fraction of transported and glycosylated peptide was recovered by overnight incubation with ConA-Sepharose (Pharmacia, Freiburg, Germany) and quantified by plate reader. Data were plotted as the mean ± S.E. of triplicate measurements. To measure TAP-dependent transport in whole cells, insect cells (2 × 106) were semipermeabilized with 0.05% saponin for 10 min on ice (14). After washing, the cells were resuspended in a final volume of 200 μl of buffer A (20 mm HEPES, 150 mm NaCl, 5 mm MgCl2 (pH 7.4)) containing 10 mm ATP. The transport reaction was initiated by adding 1 μm fluorescent peptide (RYWANATKFITCSR) for 15 min at 37 °C. Transported peptides were isolated using ConA beads and quantified as described above.
FRET Measurements
Sf9 cells were infected with recombinant baculoviruses, harvested by scraping 40–60 h post-infection, and washed once with cold buffer A. Sf9 cells were semipermeabilized with 0.05% saponin on ice for 10 min in buffer A (14). Cells were resuspended at a fixed density (1 × 106 cells/ml) for all measurements. FRET spectra were generated by exciting cells at 430 nm (bandpass, 8 nm) and scanning from 450–600 nm (bandpass, 4 nm) on a FluoroMax-4 fluorometer (Horiba Scientific). The apparent FRET efficiency was measured by calculating the ratio of emission of EYFP (527 nm) to ECFP (475 nm) (15), and FRET efficiency changes are shown as (A/D (sample) − A/D (control)) / A/D (control). Results are expressed as mean values ± S.E. of at least three independent experiments, with each experiment analyzed in triplicate. Statistical analysis was carried out using GraphPad Prism 5.0 (Graphad Software Inc.) Statistical significances evaluated using paired Student's t tests are indicated in the figure legends with corresponding p values of *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; and ****, p ≤ 0.0001.
Peptide Binding Analysis
To calculate the dissociation constant KD for the peptides, the FRET efficiency change B was plotted against the peptide concentration [P]. Data were fitted using Equation 1,
and KD values were determined by nonlinear least squares analysis.
Trapping of the TAP Complex by Vanadate
Permeabilized cells were incubated in buffer A containing 1 μm peptide RKL, 100 μm ATP, and 100 μm vanadate for 10 min at 27 °C. Vanadate was prepared as follows. Sodium orthovanadate was dissolved in water, and the pH of the solution was adjusted to 10 using HCl. The solution had a dark orange color and was boiled for 2 min until it turned colorless. Then it was cooled to room temperature. The process of adjusting the pH, boiling, and cooling was repeated two more times. The solution was stored frozen at −80 °C until use.
RESULTS
Expression of Functional FRET Fusions of TAP Proteins in sf9 Insect Cells
To perform FRET assays, the fluorescent proteins ECFP and EYFP were fused to the C-terminal ends of TAP1 (T1C) and TAP2 (T2Y) and used as donor and acceptor, respectively (Fig. 1). Recombinant baculoviruses encoding T1C and T2Y were obtained as described under “Experimental Procedures” (10, 11) and used to coinfect sf9 insect cells. To characterize the optical properties of ECFP and EYFP when attached to TAP1 and TAP2, the fluorescence of cells expressing T1C, T2Y, or T1C/T2Y were detected using a fluorometer, which showed that the fusion of ECFP or EYFP to TAP1 or TAP2 did not alter the spectral properties of the fluorescent proteins (Fig. 2A). When excited at 430 nm or 490 nm, T1C and T2Y displayed typical spectra for ECFP and EYFP, respectively, with emission peaks at 475 and 527 nm. Cells expressing both T1C and T2Y showed a significant FRET signal at 527 nm following excitation at 430 nm, indicating the formation of TAP1-TAP2 heterodimers (Fig. 1A).
FIGURE 2.
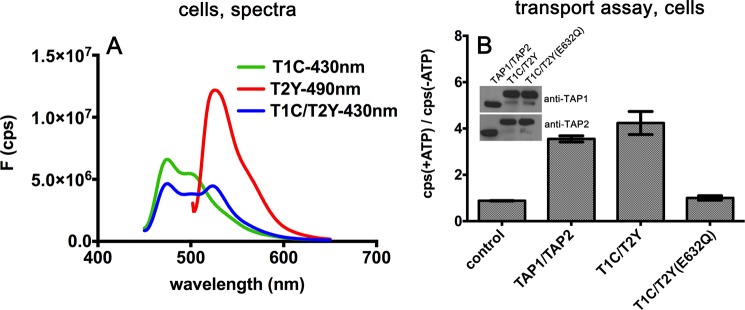
Functional characterization of the TAP1-ECFP and TAP2-EYFP complexes. A, fluorescence spectra of sf9 cells infected with TAP1-ECFP (T1C), TAP2-EYFP (T2Y), and TAP1-ECFP/TAP2-EYFP (T1C/T2Y). The excitation wavelength is indicated. On the y axis, F indicates the fluorescence values in counts/s (cps). B, ATP-dependent peptide transport in whole cells. Peptide translocation was performed as described under “Experimental Procedures.” Representative data are shown from one of two experiments, each performed in triplicate. Inset, immunoblot analyses indicating the expression levels of TAP1 and TAP2 in cells used for the peptide transport assays.
To ensure that attachment of fluorescent proteins to the TAP subunits did not interfere with the formation of a functional heterodimeric complex, peptide translocation experiments were next performed. A fluorophore-labeled TAP substrate (RYWANATKFITCSR) was incubated with control cells, cells expressing wild-type TAP complexes, T1C-T2Y, or an ATPase-deficient mutant complex, T1C-T2Y(E632Q) (11). The glutamate-to-glutamine mutation at the residue C-terminal to the Walker B motif, as in TAP2(E632Q), impairs ATP hydrolysis and NBD dimer separation in many ABC transporters (8, 16, 17). Transport of the peptide in whole cells was assessed in the presence or absence of ATP using previously described transport assays (14). Cells lacking TAP were used as the negative control. Similar to untagged wild-type TAP complexes, peptide fluorescence intensities from cells expressing T1C-T2Y were about 4-fold higher in the presence of ATP (+ATP) compared with the corresponding signals observed in the absence of ATP (-ATP) (Fig. 2B), indicating that the T1C-T2Y complex is competent for translocation and, thus, folded and assembled similarly to the wild-type TAP complex. The ATPase-deficient mutant complex T1C/T2Y(E632Q) did not transport peptides, as expected from previous measurements with untagged mutant complexes (11) (Fig. 2B). Thus, the attachment of ECFP or EYFP at the C terminus does not affect the function of TAP, which allowed us to use the tagged TAP1-TAP2 complexes to study NBD interactions in permeabilized cells.
Nucleotide Binding Induces Temperature-dependent Conformational Changes
Nucleotides are important cofactors that regulate the conformation of ABC transporters. The crystal structures of NBDs of ABC proteins and biochemical studies support the concept of an ATP binding-driven dimerization and ATP hydrolysis-promoted separation of NBDs (18, 19). Purified TAP1-TAP2 NBDs are unable to form a heterodimer in solution in the presence of either ADP or ATP (8). We investigated the effect of nucleotides on NBD interaction in the context of full-length TAP1 and TAP2 by using fluorescent protein-based FRET spectroscopy. Comparing the acceptor to donor (A/D) ratio between spectra is an established method of estimating relative distance changes between two fluorophores (20), and applications of this method to examine averaged conformational changes in intact cells have been described recently (for example, in Ref. 15). An increase in the A/D ratio indicates an increase in FRET efficiency and a decrease of distance between the donor and acceptor molecules. A decrease in the A/D ratio, on the other hand, would indicate an increase in the distance.
To exclude the influence of endogenous ATP, we incubated the cells with saponin to permeabilize the cells, and intracellular ATP was rinsed out with buffer lacking ATP (21). Fig. 3, A and B, shows FRET efficiency changes, normalized to the apo state, obtained for the wild-type T1C-T2Y complex in the presence of ADP, ATP, and a non-hydrolysable ATP analog, AMP-PNP (adenosine 5′-(β,γ-imido)triphosphate). Previously, it had been shown that AMP-PNP can bind ABC transporters and induce a “closed” NBD conformation (19, 22, 23). At 4 °C, neither ADP, ATP, nor AMP-PNP induced a significant FRET efficiency enhancement (Fig. 3A). However, at 27 °C, all three nucleotides enhanced FRET efficiency (Fig. 3B). ADP has been shown to be less effective than ATP in inducing NBD dimerization of other ABC transporters (24, 25). However, in the case of full-length TAP complexes in native cell membranes, there was no consistent difference between the FRET signals induced by ATP and ADP or even the non-hydrolysable AMP-PNP.
FIGURE 3.

Effects of nucleotides on FRET efficiency changes in permeabilized cells expressing the indicated TAP complexes. FRET assays were performed at 4 °C or 27 °C as indicated. Nucleotides were used at 100 μm unless indicated otherwise. The ratios of measured fluorescence intensities at 527 and 475 nm (A/D) were used to characterize the FRET efficiencies, which were then normalized relative to the observed FRET efficiencies in the apo state. Error bars represent the S.E. from at least three experiments, each performed in triplicate. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. N.S., not significant.
Stable nucleotide-bound dimers of ABC transporter NBDs have been obtained previously by preventing ATP hydrolysis by mutagenesis (16). As can be seen in Fig. 3, C and D, with the ATPase-deficient TAP mutant complex T1C/T2Y(E632Q), the addition of ATP, but not ADP, markedly increased the FRET signal even at 4 °C (Fig. 3C). At 27 °C, both ADP and ATP enhanced FRET efficiency significantly, but the effect of ATP is stronger than that of ADP (Fig. 3D). Thus, the mutant complex is more sensitive to the effect of ATP compared with the wild-type TAP counterpart. Additionally, nucleotides have different effects on TAP NBD closure in the context of the ATPase-deficient TAP mutant complex compared with the wild-type complex.
NBD Closure Following Peptide Binding Is the Key Step in Peptide-stimulated ATP Hydrolysis
To test the effects of peptides on NBD interactions, we chose two peptides substrates, RRYQKSTEL (RKL) and RRYNASTEL (RAL), with a high and intermediate affinity for TAP, respectively (26). Additionally, because peptide acetylation at the N terminus is expected to impair binding to TAP (27, 28), the acetylated peptide GILGCVFTL (Acetyl-GL) was used as a control. As shown in Fig. 4, A and B, addition to the T1C-T2Y complex of the TAP-specific peptides RKL and RAL, but not the control peptide Acetyl-GL, leads to a significant increase in FRET efficiency, suggesting that the binding of substrate peptide alone can induce a conformational change in the NBDs, which enhances NBD proximity. To exclude the influence of residual endogenous ATP, we used apyrase-treated microsomes prepared from T1C- and T2Y-infected insect cells in buffers lacking nucleotides. Similar to the cell-based FRET study (Fig. 4B), peptide binding altered the distances between the two NBDs, even under the ATP-depleted microsome condition (Fig. 4C), confirming that peptide binding alone in the absence of bound nucleotides can induce conformational changes of the NBD.
FIGURE 4.

Effects of peptides on FRET efficiency changes. A, fluorescence spectra from one representative experiment with T1C-T2Y in response to the peptides RKL, RAL, and Acetyl-GL (1 μm). For ease of viewing, the spectra have been normalized relative to the corresponding donor peaks. On the y axis, F indicates fluorescence values in counts/s (cps). B, in permeabilized cells expressing T1C-T2Y, RKL, and RAL, but not Acetyl-GL (1 μm), showed a significant enhancement of FRET efficiency. N.S., not significant. C, microsomes were prepared from cells expressing T1C-T2Y, and FRET efficiency changes in response to the peptide RKL (1 μm) were measured. Microsomes were prepared in buffer lacking nucleotides, and samples were pretreated with apyrase for 30 min and then incubated with the peptide RKL for 10 min on ice. D, FRET efficiency changes in permeabilized cells expressing T1C-T2Y in the presence of the peptides RKL, RAL, and Acetyl-GL at different concentrations. The peptide dissociation constants KD for TAP binding were fitted by Equation 1 and determined as described under “Experimental Procedures.” RKL, RRYQKSTEL; RAL, RRYNASTEL; Acetyl, Acetyl-GILGCVFTL. Acetyl-GL was used as a control peptide. The FRET efficiencies in the presence of the indicated peptides were normalized relative to the FRET efficiencies measured in the apo state. Error bars (B and D) represent the S.E. from at least three independent experiments, each analyzed in triplicate. **, p < 0.01; ***, p < 0.001.
We next analyzed the FRET efficiency as a function of peptide concentration (Fig. 4D). There was a dependence of FRET efficiency on the peptide concentration for both the high- and intermediate-affinity peptides, but not for the control peptide, which did not enhance the FRET signal, even at a high concentration. The KD values calculated on the basis of the FRET efficiency changes are 68.92 ± 17.42 nm and 239.1 ± 156.2 nm for RKL and RAL, respectively. These values correlate directly with the peptide binding affinity constants reported by saturation binding assays (154 ± 9 and 503 ± 54 nm, respectively), and the half-maximal concentrations for ATPase stimulation for these two substrates (161 ± 15 and 574 ± 134 nm, respectively) (26). It is noteworthy that similar maximal FRET enhancements were achieved with both peptides. Thus, the saturation of all substrate-binding pockets leads to a similar NBD closure. From this correlation between peptide-induced NBD conformational changes, peptide binding affinity, and ATPase activity, we deduce that peptide binding-induced NBD closure is the key step underlying previous findings of peptide-stimulated ATP hydrolysis (26, 29).
Synergistic Effects of Peptides and Nucleotides on NBD Conformations
As shown above, binding of both peptide and ATP induced a significant increase in FRET efficiency. The measured changes indicate that both peptides and ATP independently induce increased proximity of the TAP1 and TAP2 NBD. We next asked whether the conformation of TAP complexes induced by the combination of peptides and nucleotides displayed a further increase in NBD proximity. For these analyses, we used the non-hydrolysable ATP analog AMP-PNP and orthovanadate, a phosphate analog, to inhibit the transport cycle and stabilize the NBD closed conformation. Previously, AMP-PNP has been shown to bind ABC transporters and stabilize their outward-facing conformations (19, 22, 23). Correspondingly, AMP-PNP is unable to transport TAP substrate (Fig. 5A), even though AMP-PNP binding is measurable in the FRET assay (Fig. 3B, compared with control). Additionally, vanadate, which has been shown previously to inhibit peptide-stimulated ATPase activity (26, 29), also inhibits subsequent peptide translocation (Fig. 5A) by trapping ADP at the catalytic sites.
FIGURE 5.
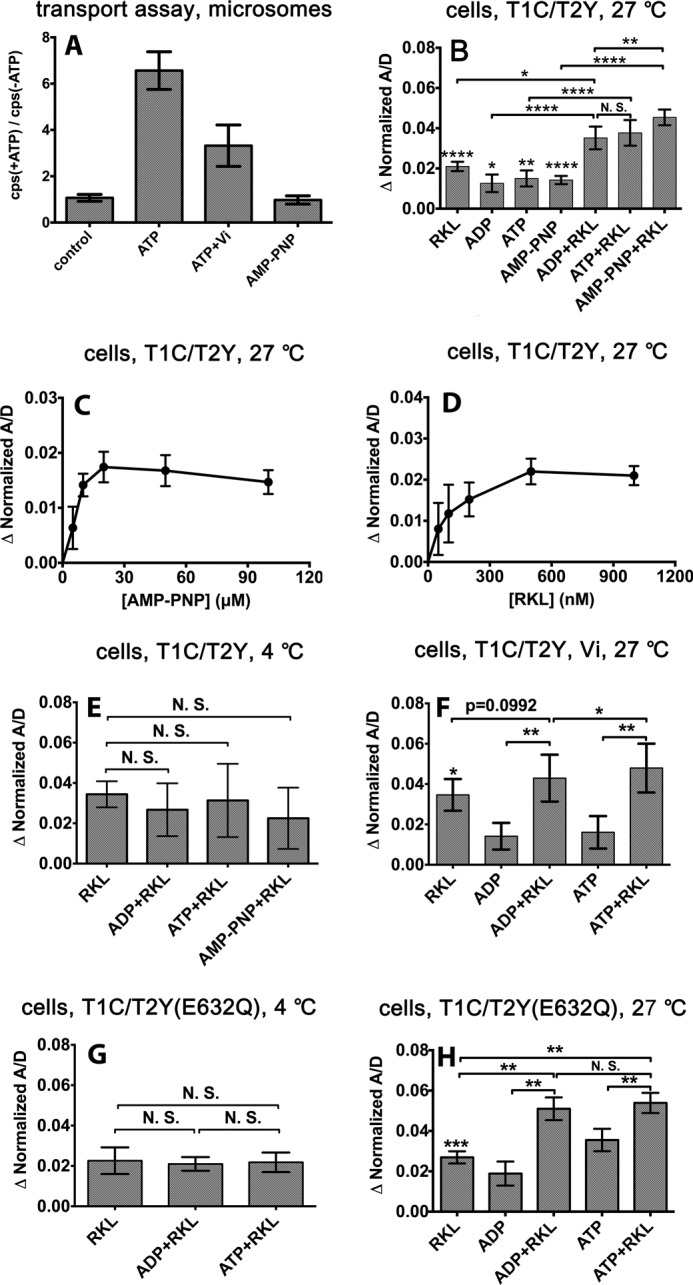
Effects of vanadate and AMP-PNP on peptide transport and combined effects of nucleotides and peptide on FRET efficiency changes in permeabilized cells expressing the indicated TAP complexes. A, peptide transport assays with microsomes expressing T1C-T2Y complexes in the absence or presence of ATP (5 mm), ATP and vanadate (5 mm each), or AMP-PNP (5 mm). Vanadate inhibits ATP-dependent peptide translocation, and AMP-PNP does not power peptide translocation. Representative data are shown from one of two experiments, each performed in triplicate. The fluorescence readings are indicated in counts/s (cps). B–H, FRET efficiency changes were measured in permeabilized cells expressing the T1C-T2Y or T1C-T2Y(E632Q) complexes in the presence of nucleotide alone, peptide RKL alone, nucleotide + the peptide RKL, or, additionally, in the presence of vanadate (Vi) at 4 °C or 27 °C as indicated. FRET efficiencies measured under the indicated conditions were normalized relative to the conditions in which cells were incubated in buffer alone (without nucleotide or peptide), except in F, where FRET efficiency was normalized relative to the buffer condition and 100 μm vanadate. Nucleotides and peptide were used at 100 and 1 μm, respectively, unless indicated otherwise. Error bars (B–F) represent the S.E. from at least three independent experiments, each analyzed in triplicate. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001. N.S., not significant.
Compared with ADP and peptide, the addition of ATP and peptide showed a small increase of NBD FRET efficiency at 27 °C, but this effect is not significant (Fig. 5B). Because the measured FRET efficiencies of bulk samples depend on the amplitudes of the various catalytic intermediates under conditions permissive for transport, the observed FRET efficiency in the presence of ATP and peptide (Fig. 5B) is likely the averaged signal resulting from the cycling of TAP between multiple conformational states. We expected that, in the presence of both the non-hydrolysable ATP analog AMP-PNP and peptide, TAP1/TAP2 might be arrested in a prehydrolysis reaction intermediate. We thus investigated whether binding of AMP-PNP and peptide led to a further increase in the A/D ratio. As seen in Fig. 5B, the addition of both AMP-PNP and peptide yielded changes in the A/D ratio that were increased significantly compared with the corresponding ADP and peptide condition. Overall, these findings indicate that, in the presence of substrate, binding of the non-hydrolysable ATP analog induces a population of TAP molecules with maximally closed NBDs. The FRET efficiency enhancements induced by AMP-PNP and peptide are saturating at 100 and 1 μm, respectively (Fig. 5, C and D), but both are significantly lower than that induced by the combination of AMP-PNP and peptide (B). Thus, it appears that there is a combined synergistic effect when both peptide and AMP-PNP are present.
Enhanced NBD closure induced by the combination of AMP-PNP and peptide relative to peptide alone or the combination of peptide and ADP is observed only at 27 °C (Fig. 5B) but not at 4 °C (E). Thus, conformational switches induced by the combination of peptide and nucleotide are strongly temperature-dependent. In the presence of peptide and vanadate, the addition of ATP also yielded significantly a higher FRET efficiency enhancement compared with that induced by the addition of ADP (Fig. 5F). The FRET assay was performed (Fig. 5F) at 27 °C, under which condition vanadate can be trapped by TAP as an ADP-sodium orthovanadate complex. Together, these findings indicate that both the non-hydrolysable ATP analog and the ATP-vanadate combination, when present together with peptide, induce stronger NBD closure compared with peptide or nucleotide alone.
We expected that the ATPase-deficient mutant TAP1-TAP2(E632Q) would also arrest the catalytic cycle of TAP in a prehydrolysis reaction intermediate in the presence of both ATP and peptide because TAP1-TAP2(E632Q) complexes are able to bind nucleotides and peptide with similar affinities as wild-type TAP (11). As shown in Fig. 2B, TAP1-TAP2(E632Q) complexes were impaired for peptide transport into sf9 cell microsomes because of a deduced deficiency in ATPase activity. We performed the FRET assay at both 4 °C and 27 °C using fluorescently tagged versions of the mutant complexes. The combined effects of nucleotides and peptide were again temperature-dependent. At 4 °C (Fig. 5G), similar FRET efficiencies were observed with T1C-T2Y(E632Q) complexes in the presence of both nucleotides and peptide, compared with that observed in the presence of peptide alone. At 27 °C, the addition of peptide together with ADP or ATP yielded a significantly stronger FRET efficiency compared with peptide alone (Fig. 5H). However, there was no significant difference between ADP and ATP.
DISCUSSION
In recent years, x-ray structures of several full-length ABC exporters have been published and have revealed large conformational changes during the substrate transport cycle (19, 22, 30–34). A number of biophysical and biochemical methods, including electron microscopy (35), pulsed EPR spectroscopy (36), fluorescent dye-based FRET spectroscopy (37), and cross-linking assays (18), were also adopted to investigate the conformations of ABC transporters under different conditions. All of these methods involve the use of purified/reconstituted protein, some of which may result in an altered or non-native conformational equilibrium of ABC transporters. For example, Sav1866 exhibits an unexpected outward-facing conformation in the presence of ADP (32). Therefore, a non-invasive approach is needed to study the conformational changes of the ABC transporter under physiological conditions. In this study, we have designed a system in which structural changes involving the NBDs are detected using full-length TAP proteins tagged with donor and acceptor fluorescent proteins for FRET spectroscopy, which allows us to monitor the relative distances between TAP1 and TAP2 NBDs under different conditions in a native cellular membrane environment.
In intact ABC transporters, the Walker A motif of one NBD is oriented toward the signature motif of the second NBD (reviewed in Ref. 5). Some nucleotide-bound, full-length ABC transporter structures reveal fully closed NBD conformations when ADP or ATP are sandwiched at the interface (32, 38), compared with open NBD conformations in nucleotide-free transporter structures (19). Other nucleotide-bound transporter structures reveal relative open NBD conformations similar to the apo state, even in the presence of non-hydrolysable ATP analogs (34). For TAP on native membranes, we found that the addition of nucleotides alone, including ADP, ATP, and AMP-PNP, enhances FRET efficiency at 27 °C, with the most significant enhancement induced by ATP, in the case of the ATPase-deficient mutant TAP complex (Fig. 3). Nonetheless, compared with other conditions, ATP alone was insufficient to induce maximal FRET efficiency (Fig. 5). A recent structure of TM287-TM288, an ABC transporter from Thermotoga maritima, also revealed an intermediate, partially closed, inward-facing conformation in the presence of AMP-PNP, which was bound only to the non-canonical, high-affinity ATP site rather than to the catalytic site (33).
The T1C-T2Y(E632Q) complex is more sensitive to nucleotides than the wild-type TAP complex, as suggested by the findings of ATP-induced enhancement in FRET efficiency, even at 4 °C (Fig. 3C), and a more significant enhancement in FRET at 27 °C compared with wild-type complexes (normalized FRET efficiency changes of ∼3.6 and ∼2%, respectively). These findings are consistent with our previous study, which showed that, compared with wild-type TAP1-TAP2, the TAP2(E632Q) mutation enhances labeling of both TAP1 and TAP2 with 8-azido-ATP (11). Additionally, in the context of the isolated ABC transporter NBD, the mutation is known to stabilize NBD dimers even in the absence of magnesium binding (16).
We found that nucleotide-induced TAP conformational change is temperature-dependent, indicated by higher FRET efficiencies of both the wild-type and T1C-T2Y(E632Q) mutant at 27 °C compared with 4 °C in the presence of nucleotide, both in the presence and in the absence of peptide. This finding is consistent with a previous study that showed that nucleotide occlusion by P-glycoprotein is also temperature-dependent (39). The temperature dependence of FRET efficiency changes caused by nucleotide binding might be due to increased membrane fluidity at 27 °C, which could enhance the interaction of NBDs. The influences of membrane fluidity on ABC transporter function have been examined previously for P-glycoprotein (40). In this case, drug transport is inhibited by compounds that increase membrane vesicle fluidity. These findings suggest that increased membrane fluidity might reduce, rather than enhance, conformational changes of TAP. It is possible that the temperature dependence of the nucleotide-induced TAP NBD FRET signal arises because of the temperature dependence of the elicited conformational changes or because of the temperature dependence of TAP-nucleotide binding, possibilities that need further study.
Compared with all the tested nucleotides, a larger increase in FRET efficiency was seen upon addition of a specific peptide (normalized FRET efficiency change of maximally ∼3% for RKL compared with ∼2% for ATP at 27 °C). Peptides are known to bind TAP in an ATP-independent manner (12). TAPs displays a low basal ATPase activity in the absence of peptides, and peptide binding stimulates ATP hydrolysis (26, 29). The allosteric coupling between peptide binding and ATP hydrolysis suggested that peptide binding might induce a significant structural reorganization of the TAP complex, which triggers ATP hydrolysis. How peptide binding is coupled with ATP hydrolysis was unknown. Our results (Figs. 4 and 5) indicate that peptide alone, by interacting with the TAP transmembrane domains, induces a partial closure of the TAP NBD dimer (Fig. 6, A → B). We attribute the peptide binding-induced partial NBD closure to an intermediate pretranslocation state (Fig. 6B) (41). In contrast to the interaction of ATP analog with the resting state (Fig. 6B'), binding of an ATP analog to the pretranslocation state (Fig. 6C) drives progression to the maximally closed state (Fig. 6D), indicating that peptide binding lowers the energy barrier for closure of the NBD dimer, a necessary prerequisite for ATP hydrolysis. Peptide binds to the open form to induce the pretranslocation state, thus explaining the well known enhancement of ATP hydrolysis by peptide (26, 29). This property of TAP emphasizes the important role of the substrate in regulating the ATPase activity by stabilizing a specific conformation of the transporter through an induced-fit mechanism (42). During the preparation of this manuscript, two papers were published (43, 44), suggesting that substrates also facilitate NBD dimerization in MsbA and P-glycoprotein, findings that are highly consistent with the results from our study.
FIGURE 6.
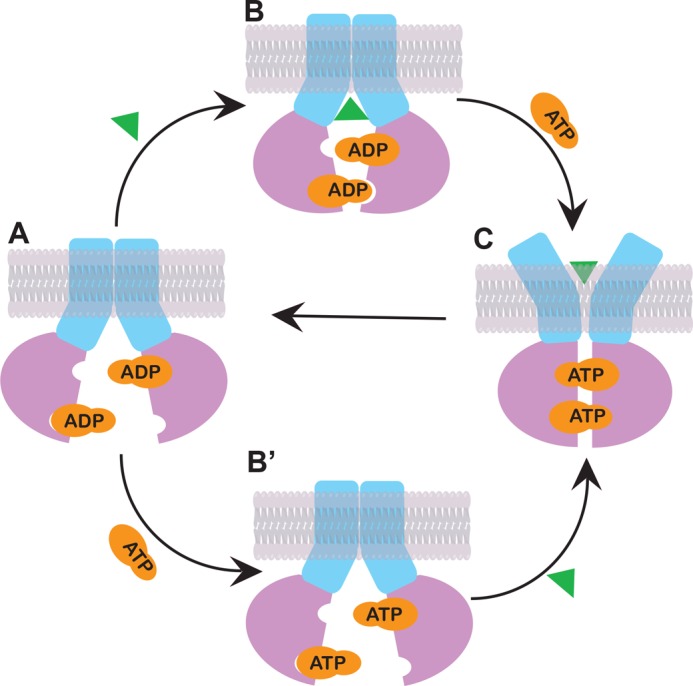
Model for the ATP-dependent peptide transport cycle of TAP. The cycle starts with the resting state, with ADP bound to the NBDs and the peptide-binding site exposed to the cytosol (A). Both peptide binding (B) and ADP-ATP exchange (B') could happen independently, inducing the NBDs to a partially closed conformation. ATP and peptide binding together result in maximal NBD closure and the peptide-binding site exposed to the ER lumen, ultimately causing release of peptide to the ER (C). ATP hydrolysis leads to phosphate release and resets TAP into the resting state (A).
Given that the putative peptide binding pockets are located in the transmembrane region (9), conformational changes of the TMD generated by peptide binding are most likely transmitted to the NBDs through non-covalent interactions between intracellular loops from TMDs and the Q-loop and/or X-loop from NBDs at the shared interface (45).
Maximal NBD closure occurs in both the prehydrolysis (AMP-PNP) and transition (ADP-sodium orthovanadate) states (Fig. 5, B and F). The structural details of the catalytic cycle are not yet fully understood, and it is under debate whether ATP binding or hydrolysis triggers the conformational changes needed to drive substrate translocation. The similarity of FRET efficiency changes induced by AMP-PNP or ATP-vanadate in the presence of peptide (Fig. 5, B and F, normalized FRET efficiency changes of 4.5 and 4.8%, respectively) indicates that the maximum observable NBD closure can be triggered by ATP binding and does not rely on hydrolysis. Interestingly, with T1C-T2Y(E632Q), ADP binding triggers NBD dimerization to the same extent as ATP in the presence of peptide. These findings are consistent with the possibility that the E632Q mutation of TAP2 lowers the energy barrier for the conformational change so that either ADP or ATP suffices to induce full closure.
The synergy between nucleotide and peptide binding in inducing a conformational change parallels the well characterized acto-myosin ATPase cycle, which is an essential part of muscle contraction and cellular motility (reviewed in Ref. 46). The sliding of the acto-myosin cross-bridge accompanies a conformational change in the myosin NBD that is synergistic with actin binding and nucleotide exchange. Although the specific sequence of events and nucleotide states are distinct between acto-myosin and TAP-mediated peptide translocation, the conceptual elements are similar and provide for a broad framework in which to understand the coordination between nucleotide and peptide binding to drive the conformational changes that lead to peptide translocation in TAP complexes.
As a bridge between TAP and MHC class I molecules, tapasin has been found to be associated with the TAP subunits through interacting with N-terminal extensions of the TMD of both TAP subunits. Our previous work has shown that tapasin enhances the structural stability of the peptide binding site of TAP1-TAP2 complexes both in the presence and absence of nucleotides and the thermostability of both TAP subunits (47). However, tapasin is non-essential for peptide transport by TAP, and core TAP domains lacking a tapasin-binding site are competent for peptide transport (48). We expect that the effects described here of peptide and nucleotide-induced FRET signals will be largely conserved in the presence of tapasin, with more significant effects of peptide and nucleotide because of the increased stability of TAP.
With the foregoing evidence in mind, together with the structural analyses presented in this study, we propose a working model in Fig. 6. Overall, our results support the switch model rather than the constant contact model (6), given the measurable distance changes of the NBD when cycling between the inward-facing and outward-facing conformations. Our data suggest that, in the presence of ADP, TAP is in an inward-facing resting state conformation. The peptide-binding site is exposed to the cytoplasm, and the NBDs of the TAP subunits are separated (Fig. 6A). In the following step, peptide binding and ATP-ADP exchange could happen independently, and either one could take place first. Peptide binding would induce a pretranslocation complex, and the two NBDs of TAP rotate inward, forming a semiopen dimer (Fig. 6B). ATP binding to the pretranslocation state promotes a concerted conformational change involving full closure of the NBDs and rotation of the TMDs so that peptide can be released into the ER lumen (Fig. 6C). In another pathway, ADP can be exchanged to ATP (Fig. 6B'), and the following peptide-binding step results in a fully closed NBD dimer (Fig. 6C). The peptide is then transferred from TAP into the ER lumen, and two ATP molecules are positioned at the closed dimer interface for hydrolysis (Fig. 6C).
The ability of the FRET assays described here to measure conformational changes in a native cellular membrane environment offers unique insights into the functional mechanism of ABC transporters. Our experiments show that TAP NBDs are separated in the apo state and that the distance between the two NBDs is significantly reduced upon peptide binding and ATP binding. On the basis of these findings, we propose that there are at least three dynamically and structurally distinct intermediate states of TAP (Fig. 6B, B', and C). Three steps are involved in transferring peptide across the ER membrane. Peptide or ATP binding to TAP induces a partially closed pretranslocation NBD conformation. ATP and peptide binding together promote the formation of the fully closed conformation, likely the outward-facing state that transfers peptide from TAP to the ER. ATP hydrolysis resets the transporter to the inward-facing state to trap another peptide. The proposed steps may be general features of the functional mechanism of ABC transporters.
Acknowledgments
We thank Angela Harrivel, Christopher Perria, and Gayatri Raghuraman for early contributions to this project, Drs. Samuel Straight and Joel Swanson and members of the Sivaramakrishnan lab for many helpful discussions, and Dr. Rachelle Gaudet for the human TAP1-TAP2-encoding baculovirus.
This work was supported, in whole or in part, by National Institutes of Health Grant AI044115 (to M. R.) and utilized the DNA sequencing cores of the Michigan Diabetes Research and Training Center funded by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institutes of Health Grant DK020572.
- TAP
- transporter associated with antigen processing
- ER
- endoplasmic reticulum
- ABC
- ATP-binding cassette
- NBD
- nucleotide-binding domain
- TMD
- transmembrane domain
- ECFP
- enhanced cyan fluorescent protein
- EYFP
- enhanced yellow fluorescent protein
- AMP-PNP
- adenosine 5′-(β,γ-imido)triphosphate
- A
- acceptor
- D
- donor.
REFERENCES
- 1. Raghavan M., Del Cid N., Rizvi S. M., Peters L. R. (2008) MHC class I assembly. Out and about. Trends Immunol. 29, 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schmitt L., Tampé R. (2000) Affinity, specificity, diversity. A challenge for the ABC transporter TAP in cellular immunity. ChemBioChem 1, 16–35 [DOI] [PubMed] [Google Scholar]
- 3. Lankat-Buttgereit B., Tampé R. (2002) The transporter associated with antigen processing. Function and implications in human diseases. Physiol. Rev. 82, 187–204 [DOI] [PubMed] [Google Scholar]
- 4. Higgins C. F. (2001) ABC transporters. Physiology, structure and mechanism. An overview. Res. Microbiol. 152, 205–210 [DOI] [PubMed] [Google Scholar]
- 5. Rees D. C., Johnson E., Lewinson O. (2009) ABC transporters. The power to change. Nat. Rev. Mol. Cell Biol. 10, 218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. George A. M., Jones P. M. (2012) Perspectives on the structure-function of ABC transporters. The Switch and Constant Contact models. Prog. Biophys. Mol. Biol. 109, 95–107 [DOI] [PubMed] [Google Scholar]
- 7. Gaudet R., Wiley D. C. (2001) Structure of the ABC ATPase domain of human TAP1, the transporter associated with antigen processing. EMBO J. 20, 4964–4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Procko E., Ferrin-O'Connell I., Ng S. L., Gaudet R. (2006) Distinct structural and functional properties of the ATPase sites in an asymmetric ABC transporter. Mol. Cell 24, 51–62 [DOI] [PubMed] [Google Scholar]
- 9. Corradi V., Singh G., Tieleman D. P. (2012) The human transporter associated with antigen processing. Molecular models to describe peptide binding competent states. J. Biol. Chem. 287, 28099–28111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lapinski P. E., Raghuraman G., Raghavan M. (2003) Nucleotide interactions with membrane-bound transporter associated with antigen processing proteins. J. Biol. Chem. 278, 8229–8237 [DOI] [PubMed] [Google Scholar]
- 11. Perria C. L., Rajamanickam V., Lapinski P. E., Raghavan M. (2006) Catalytic site modifications of TAP1 and TAP2 and their functional consequences. J. Biol. Chem. 281, 39839–39851 [DOI] [PubMed] [Google Scholar]
- 12. Lapinski P. E., Neubig R. R., Raghavan M. (2001) Walker A lysine mutations of TAP1 and TAP2 interfere with peptide translocation but not peptide binding. J. Biol. Chem. 276, 7526–7533 [DOI] [PubMed] [Google Scholar]
- 13. Neumann L., Tampé R. (1999) Kinetic analysis of peptide binding to the TAP transport complex. Evidence for structural rearrangements induced by substrate binding. J. Mol. Biol. 294, 1203–1213 [DOI] [PubMed] [Google Scholar]
- 14. Herget M., Oancea G., Schrodt S., Karas M., Tampé R., Abele R. (2007) Mechanism of substrate sensing and signal transmission within an ABC transporter. Use of a Trojan horse strategy. J. Biol. Chem. 282, 3871–3880 [DOI] [PubMed] [Google Scholar]
- 15. Malik R. U., Ritt M., DeVree B. T., Neubig R. R., Sunahara R. K., Sivaramakrishnan S. (2013) Detection of G-protein selective G-protein coupled receptor (GPCR) conformations in live cells. J. Biol. Chem. 288, 17167–17178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith P. C., Karpowich N., Millen L., Moody J. E., Rosen J., Thomas P. J., Hunt J. F. (2002) ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell 10, 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tombline G., Bartholomew L. A., Urbatsch I. L., Senior A. E. (2004) Combined mutation of catalytic glutamate residues in the two nucleotide binding domains of P-glycoprotein generates a conformation that binds ATP and ADP tightly. J. Biol. Chem. 279, 31212–31220 [DOI] [PubMed] [Google Scholar]
- 18. Loo T. W., Bartlett M. C., Detty M. R., Clarke D. M. (2012) The ATPase activity of the P-glycoprotein drug pump is highly activated when the N-terminal and central regions of the nucleotide-binding domains are linked closely together. J. Biol. Chem. 287, 26806–26816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ward A., Reyes C. L., Yu J., Roth C. B., Chang G. (2007) Flexibility in the ABC transporter MsbA. Alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miyawaki A., Tsien R. Y. (2000) Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 327, 472–500 [DOI] [PubMed] [Google Scholar]
- 21. Willemse M., Janssen E., de Lange F., Wieringa B., Fransen J. (2007) ATP and FRET. A cautionary note. Nat. Biotechnol. 25, 170–172 [DOI] [PubMed] [Google Scholar]
- 22. Dawson R. J., Locher K. P. (2007) Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581, 935–938 [DOI] [PubMed] [Google Scholar]
- 23. Oldham M. L., Chen J. (2011) Snapshots of the maltose transporter during ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 108, 15152–15156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu G., Westbrooks J. M., Davidson A. L., Chen J. (2005) ATP hydrolysis is required to reset the ATP-binding cassette dimer into the resting-state conformation. Proc. Natl. Acad. Sci. U.S.A. 102, 17969–17974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zaitseva J., Oswald C., Jumpertz T., Jenewein S., Wiedenmann A., Holland I. B., Schmitt L. (2006) A structural analysis of asymmetry required for catalytic activity of an ABC-ATPase domain dimer. EMBO J. 25, 3432–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gorbulev S., Abele R., Tampé R. (2001) Allosteric crosstalk between peptide-binding, transport, and ATP hydrolysis of the ABC transporter TAP. Proc. Natl. Acad. Sci. U.S.A. 98, 3732–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Momburg F., Roelse J., Howard J. C., Butcher G. W., Hämmerling G. J., Neefjes J. J. (1994) Selectivity of MHC-encoded peptide transporters from human, mouse and rat. Nature 367, 648–651 [DOI] [PubMed] [Google Scholar]
- 28. Schumacher T. N., Kantesaria D. V., Heemels M. T., Ashton-Rickardt P. G., Shepherd J. C., Fruh K., Yang Y., Peterson P. A., Tonegawa S., Ploegh H. L. (1994) Peptide length and sequence specificity of the mouse TAP1/TAP2 translocator. J. Exp. Med. 179, 533–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen M., Abele R., Tampé R. (2003) Peptides induce ATP hydrolysis at both subunits of the transporter associated with antigen processing. J. Biol. Chem. 278, 29686–29692 [DOI] [PubMed] [Google Scholar]
- 30. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., Chang G. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ward A. B., Szewczyk P., Grimard V., Lee C. W., Martinez L., Doshi R., Caya A., Villaluz M., Pardon E., Cregger C., Swartz D. J., Falson P. G., Urbatsch I. L., Govaerts C., Steyaert J., Chang G. (2013) Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. U.S.A. 110, 13386–13391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dawson R. J., Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 33. Hohl M., Briand C., Grütter M. G., Seeger M. A. (2012) Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat. Struct. Mol. Biol. 19, 395–402 [DOI] [PubMed] [Google Scholar]
- 34. Shintre C. A, Pike A.C., Li Q., Kim J. I., Barr A. J., Goubin S., Shrestha L., Yang J., Berridge G., Ross J., Stansfeld P. J., Sansom M. S., Edwards A. M., Bountra C., Marsden B. D., von Delft F., Bullock A. N., Gileadi O., Burgess-Brown N. A., Carpenter E. P. (2013) Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc. Natl. Acad. Sci. U.S.A. 110, 9710–9715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee J. Y., Urbatsch I. L., Senior A. E., Wilkens S. (2008) Nucleotide-induced structural changes in P-glycoprotein observed by electron microscopy. J. Biol. Chem. 283, 5769–5779 [DOI] [PubMed] [Google Scholar]
- 36. Hellmich U. A., Lyubenova S., Kaltenborn E., Doshi R., van Veen H. W., Prisner T. F., Glaubitz C. (2012) Probing the ATP hydrolysis cycle of the ABC multidrug transporter LmrA by pulsed EPR spectroscopy. J. Am. Chem. Soc. 134, 5857–5862 [DOI] [PubMed] [Google Scholar]
- 37. Verhalen B., Ernst S., Börsch M., Wilkens S. (2012) Dynamic ligand-induced conformational rearrangements in P-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy. J. Biol. Chem. 287, 1112–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oldham M. L., Khare D., Quiocho F. A., Davidson A. L., Chen J. (2007) Crystal structure of a catalytic intermediate of the maltose transporter. Nature 450, 515–521 [DOI] [PubMed] [Google Scholar]
- 39. Sauna Z. E., Nandigama K., Ambudkar S. V. (2006) Exploiting reaction intermediates of the ATPase reaction to elucidate the mechanism of transport by P-glycoprotein (ABCB1). J. Biol. Chem. 281, 26501–26511 [DOI] [PubMed] [Google Scholar]
- 40. Sinicrope F. A., Dudeja P. K., Bissonnette B. M., Safa A. R., Brasitus T. A. (1992) Modulation of P-glycoprotein-mediated drug transport by alterations in lipid fluidity of rat liver canalicular membrane vesicles. J. Biol. Chem. 267, 24995–25002 [PubMed] [Google Scholar]
- 41. Oldham M. L., Chen J. (2011) Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science 332, 1202–1205 [DOI] [PubMed] [Google Scholar]
- 42. Loo T. W., Bartlett M. C., Clarke D. M. (2003) Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J. Biol. Chem. 278, 13603–13606 [DOI] [PubMed] [Google Scholar]
- 43. Marcoux J., Wang S. C., Politis A., Reading E., Ma J., Biggin P. C., Zhou M., Tao H., Zhang Q., Chang G., Morgner N., Robinson C. V. (2013) Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc. Natl. Acad. Sci. U.S.A. 110, 9704–9709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Doshi R., van Veen H. W. (2013) Substrate binding stabilizes a pre-translocation intermediate in the ATP-binding cassette transport protein MsbA. J. Biol. Chem. 288, 21638–21647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oancea G., O'Mara M. L., Bennett W. F., Tieleman D. P., Abele R., Tampé R. (2009) Structural arrangement of the transmission interface in the antigen ABC transport complex TAP. Proc. Natl. Acad. Sci. U.S.A. 106, 5551–5556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Spudich J. A., Sivaramakrishnan S. (2010) Myosin VI. An innovative motor that challenged the swinging lever arm hypothesis. Nat. Rev. Mol. Cell Biol. 11, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Raghuraman G., Lapinski P. E., Raghavan M. (2002) Tapasin interacts with the membrane-spanning domains of both TAP subunits and enhances the structural stability of TAP1 × TAP2 Complexes. J. Biol. Chem. 277, 41786–41794 [DOI] [PubMed] [Google Scholar]
- 48. Procko E., Raghuraman G., Wiley D. C., Raghavan M., Gaudet R. (2005) Identification of domain boundaries within the N-termini of TAP1 and TAP2 and their importance in tapasin binding and tapasin-mediated increase in peptide loading of MHC class I. Immunol. Cell Biol. 83, 475–482 [DOI] [PubMed] [Google Scholar]