

Background: AT1R activation induces oxidative stress, promotes inflammation, and increases blood pressure.
Results: SNPs in AT1R promoter occur in linkage disequilibrium, forming two haplotypes. Transgenic mice with haplotype I have USF2-dependent AT1R overexpression, increased oxidative stress, and increased blood pressure.
Conclusion: Haplotype I leads to enhanced expression and pathophysiological effects of AT1R.
Significance: Polymorphisms in AT1R provide for genetic predisposition to hypertension.
Keywords: Angiotensin II, G Protein-coupled receptors (GPCR), Gene Regulation, Genetic Polymorphism, Hypertension, Molecular Genetics, Transcription, Transgenic Mice
Abstract
The renin-angiotensin system plays an important role in the regulation of blood pressure via angiotensin II and the angiotensin II receptor type 1 (AT1R). Human AT1R gene promoter has four SNPs: T/A at −777, T/G at −680, A/C at −214, and A/G at −119, that are in linkage disequilibrium. Variants −777T, −680T, −214A, and −119A almost always occur together (named haplotype I), and variants −777A, −680G, −214C, and −119G almost always occur together (named haplotype II) in Caucasian subjects. Genomic DNA analyses, from 388 normotensive and 374 hypertensive subjects, link haplotype I of the human AT1R (hAT1R) gene with hypertension in Caucasians (p = 0.004, χ2 = 8.46). Our results show increased basal promoter activity of the hAT1R gene in cells (H295R and A7r5) transfected with reporter construct containing haplotype I. We also show increased binding of the transcription factor, USF2, to oligonucleotide containing nucleoside −214A as opposed to −214C. Recombineering of a 166-kb bacterial artificial chromosome containing 68 kb of the 5′-flanking region, 45 kb of the coding sequence, and 53 kb of the 3′-flanking region of the hAT1R gene was employed to generate transgenic mice with either haplotype. We show that (a) hAT1R mRNA level is increased in the kidney and heart of transgenic mice containing haplotype I as compared with haplotype II; (b) USF2 binds more strongly to the chromatin obtained from the kidney of transgenic mice containing haplotype I as compared with haplotype II; and (c) blood pressure and oxidative stress are increased in transgenic mice containing haplotype I as compared with haplotype II.
Introduction
Hypertension is a risk factor for myocardial infarction, heart failure, vascular disease, stroke, and renal failure (1). The renin-angiotensin system is central to the regulation of blood pressure and plays an important role in the pathophysiology of hypertension (2). The octapeptide angiotensin II (ANGII)2 is synthesized from its precursor molecule, angiotensinogen by sequential proteolytic action of renin, and angiotensin-converting enzyme. ANGII is a potent vasoactive peptide that promotes adrenal aldosterone secretion and regulates the renal conservation of salt and water (3, 4). These biological responses of ANGII are mediated by its interaction with the high affinity G protein-coupled receptors localized on the surface of target cells (5). Two main ANGII receptor subtypes, the AT1 and AT2 receptors, have been identified (6). AT1 receptor (AT1R) is the principal mediator of the aforementioned physiological effects of ANGII in mammalian adults, and mounting evidence unequivocally shows the contributions of AT1R in cardiovascular pathologies (3). Ito et al. (7) have shown a 50% reduction in AT1a receptor expression and a significantly reduced systolic blood pressure in heterozygous AT1aR knock-out mice. On the other hand, Le et al. (8) have generated transgenic mice overexpressing the AT1R gene and have shown that increasing the gene copy number from 2 to 4 leads to doubling of AT1R mRNA expression in the vasculature and the heart of these mice. This resulted in hypertension, especially in female transgenic mice. Additionally, transgenic mice overexpressing AT1R in the cardiac myocytes demonstrate cardiac enlargement, heart failure, and early death (9, 10). Spontaneously hypertensive rats have also been shown to have dysfunctional AT1R activation in the brain (11), and increased ATRs are present in the brain of deoxycorticosterone acetate salt-sensitive rats (12).
Complex cardiovascular disorders, like hypertension, are polygenic in nature with strong familial inheritance. Polymorphisms in the regulatory regions of the genes regulating blood pressure contribute to this inheritance. Contextually, the AT1R gene is one of the prime candidates whose differential regulation may predispose to such disorders. An A/C polymorphism at position 1166 (rs5186) in the 3′-UTR of the AT1R gene has been associated with hypertension in the Finnish (13) (14) and the Caucasian populations (15). The role of this polymorphism, however, in human hypertension remains unconfirmed. On the other hand, in Caucasian hypertensive subjects we identified four novel SNPs, in linkage disequilibrium (LD), in the promoter region of the human AT1R (hAT1R) gene. The SNPs are: T/A at −777, T/G at −680, A/C at −214, and A/G at −119. Variants −777T, −680T, −214A, and −119A always occur together creating the haplotype I, and variants −777A, −680G, −214C, and −119G always occur together, creating the haplotype II. These SNPs may affect transcription factor binding, such as for upstream stimulatory factor-2 (USF2), and modulate gene expression. Thus, we generated transgenic (TG) mice with the either haplotype and tested the hypothesis that haplotype-dependent, differential regulation of the hAT1R gene will increase hAT1R expression and predispose to AT1R-induced hypertension.
We show here that the haplotype I is associated with hypertension in Caucasian subjects. We also show increased promoter activity, in both basal and USFco-transfected cells, and increased USF2 binding to the oligonucleotide corresponding to the haplotype I in adrenal (H295-R) and vascular smooth muscle (A7r5) cells. In vivo testing was done in TG mice containing either haplotype I or haplotype II of the hAT1R gene—using recombineering of a 166-kb BAC containing 5′-flanking region, coding sequence, and 3′-flanking region of the hAT1R gene. TG mice with haplotype I demonstrated enhanced USF2 chromatin binding with increased hAT1R expression in the cardiac and the renal tissues. Complementary experiments revealed significantly higher blood pressure; pro-inflammatory markers, including IL-6, TNFα, and CRP; and oxidative stress markers, such as NADPH oxidase, in TG mice containing haplotype I of the hAT1R gene as compared with TG mice with the haplotype II.
EXPERIMENTAL PROCEDURES
Patient Selection
We have analyzed the genomic DNA from 374 hypertensive (182 males and 192 females; mean age, 59 ± 10 years) and 388 normotensive subjects (182 males and 206 females; mean age, 58 ± 10 years). All of these subjects were recruited from the outpatient department of Westchester Medical Center (Valhalla, NY) or from the AmDec Foundation. All case and control subjects gave informed consent before participating in the research. The Institutional Review Board at the New York Medical College approved the research protocol for animal use. Individuals were excluded if they had a previous history of coronary artery disease, peripheral vascular disease, cerebrovascular disease, secondary hypertension, diabetes mellitus, or renal diseases. For a period of 30 min before blood pressure (BP) measurement, no exercise, alcohol, caffeine, or smoking were allowed. BP was measured by conventional mercury sphygmomanometer. Measurements by two different observers were taken at the left arm with individuals in the seated position after 15 min of resting. The criteria for hypertension was defined as a systolic blood pressure (SBP) > 140 mm Hg, a diastolic blood pressure (DBP) > 90 mm Hg, or under antihypertensive therapy. The normotensives (with SBP/DBP < 140/90 mm Hg) without a history of hypertension and without diabetes mellitus were recruited from the same population and matched for the sex and age. Clinical data of the patients were self-reported during a detailed prestudy examination and confirmed by medical charts provided by their physicians.
Analysis of Genomic DNA
The genomic DNA was amplified using 5′-TGT AAA CTA CAG TCA CCC TCT CAC C-3′ as a forward primer and 5′-ATG AGG CAG TAT CAC CCT GA-3′ as a reverse primer to amplify the 238-bp 5′-flanking region of the hAT1R gene containing the T/G polymorphic site at the −680 position of the promoter. The amplified fragment was then analyzed by 3.5% agarose gel electrophoresis after HinfI restriction digestion. HinfI recognizes the nucleotide sequence GANTC and cleaves the amplified DNA when nucleoside A is present at −680 to produce 127- and 111-bp fragments. The results obtained by restriction analysis were confirmed by direct sequencing of representative amplified fragments.
Plasmid Construction
The reporter constructs pHAT1Rhap-1luc (haplotype I) and pHAT1Rhap-2luc (haplotype II) were synthesized by PCR amplification of the human AT1R gene using TGTAGGCTTTGTCCATTTTT as the forward primer and GTCCAGACGTCCTGTCACTCTCGAG as the reverse primer using DNA from human subjects containing either haplotype I or II of the hAT1R gene. The amplified fragment contained nucleotides −1174 to +55 and was subcloned in the pGL3-basic vector lacking eukaryotic promoter and enhancer sequences (Promega). Expression vector RSV-β-gal was obtained from Promega. Plasmid DNAs for transient transfection were prepared by Qiagen midi or maxi plasmid kits using conditions described by the manufacturer.
Cell Culture and Transient Transfection
Human adrenocortical carcinoma cells (H295-R) or vascular smooth muscle cells (A7r5) were grown as monolayers in DMEM/F-12 medium (Invitrogen) supplemented with 20% Nu-Serum IV, 1% ITS (BD Biosciences), 100 units/ml penicillin, and 100 μg/ml streptomycin in an atmosphere of 5% CO2. For transient transfections, reporter DNA (500 ng) and β-galactosidase DNA (10 ng) were mixed with pBluescript DNA to a final weight of 1 μg of DNA. Transient transfections were performed with FuGENE 6 transfection reagent (Roche Applied Science) following the manufacturer's protocol. For co-transfection experiments, expression vector pSV-USF2 (100 ng) was added to the reporter constructs. After 24 h of transfection, the cells were serum-starved for an additional 24 h. The cells were harvested 48 h post-transfection, and whole cell extracts were prepared by resuspension in 200 μl of lysis buffer (Promega). An aliquot of the cell extract was used to measure luciferase activity in a Turners Design 20/20 luminometer using a luciferase assay system (Promega) as described by the manufacturer. Luciferase activity was normalized to β-galactosidase activity that was determined using the β-glo assay system (Promega).
Gel Mobility Shift Assay
The probes for electrophoretic mobility shift assay were chemically synthesized by MWG Biotech (High Point, NC), annealed, and radiolabeled at the 5′-ends by polynucleotide kinase using [γ-32P]ATP. DNA fragments (100,000 cpm), poly(dI-dC) (2 μg), BSA (20 μg), and nuclear extract (5–10 μg) were incubated in a solution containing 25 mm HEPES (pH 7.5), 100 mm NaCl, 0.2 mm PMSF, 2 mm dithiothreitol, and 5% glycerol on ice for 30 min and separated on a 6% polyacrylamide gel in a cold room. After 2–3 h, the gel was dried under vacuum, and protein-nucleic acid complexes were identified by autoradiography. For supershift assay 5 μg of USF1 antibody (Santa Cruz Biotechnology) was added to the reaction mixture, which was incubated for 20 min and analyzed by electrophoretic mobility shift assay. Radioactive nucleotides were purified by chromaspin columns (BD Biosciences). Nuclear extracts for gel mobility shift assays were prepared by modification of a previously described method (16).
Oligonucleotides
Double-stranded oligonucleotides 214A and 214C were obtained by annealing CGGGACCACGTGAAC, and CGGGACCCCGTGAAC with their respective complementary oligonucleotides. Double-stranded oligonucleotide MLTF containing the consensus USF binding site was obtained by annealing CTAGTGTAGGCCACGTGACCGGG with its respective complementary oligonucleotide (USF binding site is underlined). Oligonucleotides containing the consensus HNF-3 binding site was obtained by annealing CTAGTATTATTGACTTAGGATC with its complementary oligonucleotide.
Animal Experiments
Procedures were approved by the University of Toledo Animal Care and Use Committee and performed according to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (publication no. 85-23, revised 1996).
Preparation and Modification of hAT1R BAC
BAC clone (CTD-3080C3) containing hAT1R gene was purchased from CalTech. The plasmid was grown in low salt LB medium containing 12.5 μg/ml chloramphenicol at 37 °C, and BAC DNA was purified using Nucleobond BAC maxiprep kit (BD Biosciences). The promoter sequence in the wild type hAT1R BAC was modified by recombineering using the galK two-step procedure in SW102 cells (17). SW102 cells were grown at 32 °C. First, the 1.7-kb promoter region of the hAT1R gene was deleted by inserting galK cassette. Next, the galK cassette was replaced with a 1.7-kb promoter region from the DNA of a subject containing mutant haplotype of the hAT1R gene. Wild type and modified BAC DNAs were analyzed by fingerprinting after treatment with SpeI (releasing 30 fragments varying in size from 500 bp to 16 kb). The modified region of the hAT1R BAC was finally confirmed by direct sequencing. The nucleotide sequence of the promoter, all the exons, and the 3′-flanking region were sequenced to confirm that the two BAC differ only in the promoter region and contained either haplotype I or II of the hAT1R gene.
Generation of Transgenic Mice, Containing Haplotype I and Haplotype II of the hAT1R Gene
To understand the regulation of human AT1R gene in an in vivo situation, we generated transgenic mice using 166-kb BAC plasmid DNA containing hAT1R gene. This BAC contains 68 kb of the 5′-flanking region, 45 kb of the coding region, and 53 kb of the 3′-flanking region of the hAT1R gene cloned in 7.4 kb of pBeloBAC11 vector (see Fig. 6A). We characterized the BAC by restriction digestion with SpeI enzyme followed by gel electrophoresis. This treatment produced 30 DNA fragments varying from 500 bp to 16 kb as predicted (see Fig. 6B). Nucleotide sequencing of the BAC revealed that it contains haplotype I of the hAT1R gene. The 1.2-kb promoter of the hAT1R gene in this BAC was modified to haplotype II by recombineering using the 1.2-kb promoter fragment from a human subject containing haplotype II of the hAT1R gene as described previously for hAGT gene (18). The nucleotide sequence of the resulting BAC was confirmed by sequence analysis. The linearized DNA fragments from the wild type and recombineered BACs were obtained after NotI treatment and were used for microinjection to produce transgenic mice. The presence of hAT1R gene was analyzed by PCR amplification of tail DNA of transgenic mice. The location of different regions of the BAC plasmid that were used for PCR amplification to confirm the integration of hAT1R gene in mouse genome is shown in Fig. 6C. Nucleotide sequences of primers used for the amplification of different regions of hAT1R gene in transgenic animals are presented in Table 1. Initially we obtained three transgenic lines from each construct, but after characterization we maintained only one TG line for each haplotype of hAT1R gene. These TG mice have single copy of the hAT1R gene as determined by quantitative PCR (19).
FIGURE 6.

Generation of transgenic mice containing haplotype I and II of the hAT1R gene: Transgenic mice were generated using 166-kb BAC plasmid DNA containing hAT1R gene by recombineering as described under “Experimental Procedures.” A, diagrammatic presentation of the 5′-UTR, coding region, and 3′-UTR of hAT1R gene of BAC construct in 7.4 kb of pBeloBAC11 vector. B, restriction digestion of BAC containing hAT1R gene with restriction enzyme SpeI followed by gel electrophoresis was performed to determine authenticity of the BAC. Lane 1 shows the molecular size marker of the 1-kb ladder DNA; lanes 2 and 3 show DNA fragments obtained from the restriction digestion of haplotypes I and II, respectively. C, location of primers designed from different regions of the BAC plasmid for the characterization of hAT1R gene.
TABLE 1.
Sequences of the primers used for the characterization of human AT1R gene in the hAT1R transgenic mice
TIS indicates transcription initiation site.
| Primer | Location | Sequence |
|---|---|---|
| 5′-Upstream-F | 43-kb 5′-TIS | GAGATGGAGTCTCACTCT |
| 5′-Upstream-R | CATGAGCAGACAATTCTC | |
| Exon 2-F | 10-kb 3′-TIS | CTCTTAAGATGCAGTGTGG |
| Exon 2-R | CTGCTTCCTCTACTTCT | |
| Exon 5-F | 43-kb 3′-TIS | TGGCATGGCAGTGGTGT |
| Exon 5-R | CAGATGCTCAAGAATGG | |
| 3′-Downstream-F | 70-kb 3′-TIS | GACACAGTGAGTGACAT |
| 3′-Downstream-R | CTATATGTTCCACAGGCA |
Quantitative RT-PCR for mRNA Analysis
Kidney and heart from 10–12-week-old hAT1R transgenic male mice were harvested and immediately frozen in liquid nitrogen for storage at −80 °C. RNA was isolated using RNeasy Plus mini kit (Qiagen). RNA (1 μg) was reverse-transcribed into cDNA using Revert Aid first strand cDNA synthesis kit (Fermentas). Quantitative RT-PCR was performed using primers for human AT1R, IL-6, TNFα, and CRP from SAB using standard protocol. Relative transcript abundance was calculated following normalization with mouse GAPDH.
In Vivo ChIP assay
The ChIP assay was performed using the EZ ChIP chromatin immunoprecipitation kit from Millipore. The mice were perfused with normal saline. Kidney was excised and washed in PBS; minced into smaller pieces; fixed with 1% formaldehyde for 20 min at room temperature; and washed with chilled PBS followed by their lysis. The DNA was fragmented by sonication, and 10 μl of the chromatin solution was saved as input. A 5-μg amount of USF2 or RNA pol II antibody or rabbit immunoglobulin G was added to the tubes containing 900 μl of sonicated chromatin solution, and the mixture was incubated overnight at 4 °C. The antibody complexes were captured with protein A-agarose beads and subjected to serial washes (as described in manufacturer's protocol). The chromatin fraction was extracted with SDS buffer and reverse cross-linked at 65 °C for 4–6 h. The DNA was then purified using Qiagen miniprep column. The immunoprecipitated DNA (1 μl) and the input DNA (1 μl) were subjected to 35 cycles of PCR amplification using −213for TCCGCAGGAAATGATACTCC as a forward and −213rev ACGAGGCTCTGTTTTGCATT as a reverse primer when USF2 antibody was used for immunoprecipitation. This amplified 217 bp fragment spanning the USF binding site located around −213 region of the human AT1R gene promoter −119for TGATAGTTGACACGGGACGA as a forward primer and −119rev TTTATAGTGAGGGGCGTTGC as a reverse primer when RNA pol II antibody was used. This amplified 178-bp fragment containing RNA pol II-binding region of the human AT1R gene promoter. The PCR-amplified products were analyzed on 2% agarose gel.
Blood Pressure Measurement in Transgenic Mice
All mice were fed standard mice chow and had access to water ad libitum. BP was measured in the conscious state by radio telemetry as described previously (20). After 4 days of basal blood pressure measurement, animals were treated with the AT1R blocker losartan (Sigma-Aldrich; 30 mg/kg/day) in drinking water as described previously (18). The blood pressure was then measured 24 h after losartan treatment. Mean BP values were calculated for every hour from the values taken over 24 h. The animals were housed in individual cages.
Statistical Analysis
The GraphPad statistical software package (GraphPad 3.00 for Windows; GraphPad Software, San Diego, CA) was used for calculating the differences in allele frequency between case and control subjects and comparison of promoter activity of different reporter constructs in transient transfection assays. Allele and genotype frequencies between study participants were estimated by gene counting and analyzed by means of the χ2 test and logistic regression using SPSS 10.0 (SPSS Inc., Chicago, IL). Data are expressed as the means ± S.E. Statistical significance was assessed using analysis of variance with a Tukey-Kramer post hoc test. Statistical significant results are marked by asterisks (p < 0.05).
RESULTS
−680T Allele of the hAT1R Gene Is Associated with Increased Risk of Hypertension in Caucasians
Because of LD of the SNPs in haplotypes, −680T or −680G was used as reporter polymorphism for haplotype I and II, respectively (Fig. 1). DNA from 374 hypertensive and 388 normotensive subjects was analyzed by restriction analysis followed by gel electrophoresis (Fig. 2). −680T allele of the hAT1R gene is associated with hypertension in Caucasian subjects (p = 0.004, χ2 = 8.46) (allele frequency of 680T was 0.80 in hypertensive subjects and 0.74 in normotensive subjects). All patients and control subjects were in Hardy-Weinberg equilibrium. The results of this analysis suggest that −680T allele of the AT1R gene to be a risk factor for hypertension in Caucasian subjects (Table 2).
FIGURE 1.

Nucleotide sequence of haplotypes I and II of the hAT1R gene. Human AT1R gene contains four polymorphic sites in its promoter that are in LD. Haplotype I contains nucleosides T, T, A, and A at −777, −680, −214, and −119, respectively, in its promoter. Haplotype II contains nucleosides A, G, C, and G at these positions.
FIGURE 2.
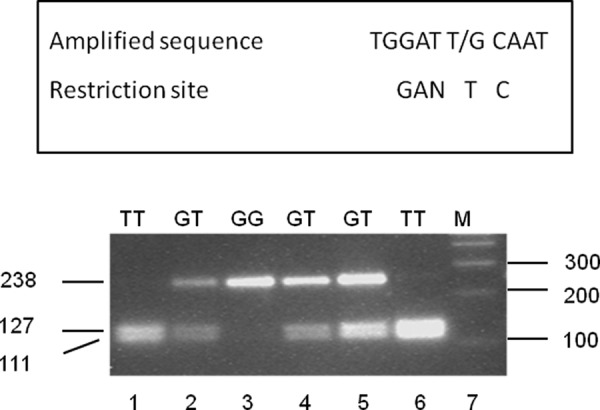
Restriction analysis to determine the frequency of haplotypes I and II of the hAT1R gene. Because four polymorphisms in the hAT1R promoter are in LD, the frequency of each haplotype may be calculated by measuring frequency of any polymorphism. Restriction enzyme HinfI digests the PCR-amplified fragment only when −680T is present. Therefore, genomic DNA from hypertensive patients and normotensive controls was amplified to produce 238-bp fragments as described under “Experimental Procedures.” The partial nucleotide sequence of the amplified fragment around the T/G polymorphic site at −680 of the AT1R gene is shown in the first line. The HinfI restriction site (which will cleave the amplified DNA if nucleoside T is present at −680) is shown in the second line. The amplified DNA fragments were treated with HinfI and separated on a 3.5% agarose gel. Lanes 1 and 6, samples from TT homozygotes; lanes 2, 4, and 5, samples from GT heterozygotes; lane 3, sample from a GG homozygote; lane 7, positions of DNA markers.
TABLE 2.
Analysis of human AT1R gene polymorphism at −680 in Caucasian subjects
| Total subjects | TT | GT | GG | p | |
|---|---|---|---|---|---|
| Hypertensive | 374 | 244 | 114 | 16 | 0.004 |
| Normotensive | 388 | 208 | 159 | 21 |
Reporter Construct Containing Haplotype I of the hAT1R Gene Has Increased Promoter Activity as Compared with the Reporter Construct Containing Haplotype II on Transient Transfection
Reporter constructs with a 1.2-kb region of the hAT1R gene promoter of haplotypes I and II were attached in front of the luciferase gene in the basic vector pGL3-luc. We next performed transient transfections of the reporter constructs pHAT1Rhap-1luc (haplotype I) and hAT1Rhap-2luc (haplotype II) in H295R cells. These cells have been shown to contain functional angiotensin II receptors (21). Our results show ∼4-fold increased (p < 0.05) promoter activity of the haplotype I reporter construct as compared with haplotype II (Fig. 3A). Similar results were seen in A7r5 cells. AT1R gene is expressed in these cells, which were transfected with either pHAT1Rhap-1luc or pHAT1Rhap-2luc. Increased promoter activity of the reporter construct with haplotype I, ∼1.9-fold (p < 0.05), was observed as opposed to the promoter activity of haplotype II (Fig. 3B).
FIGURE 3.
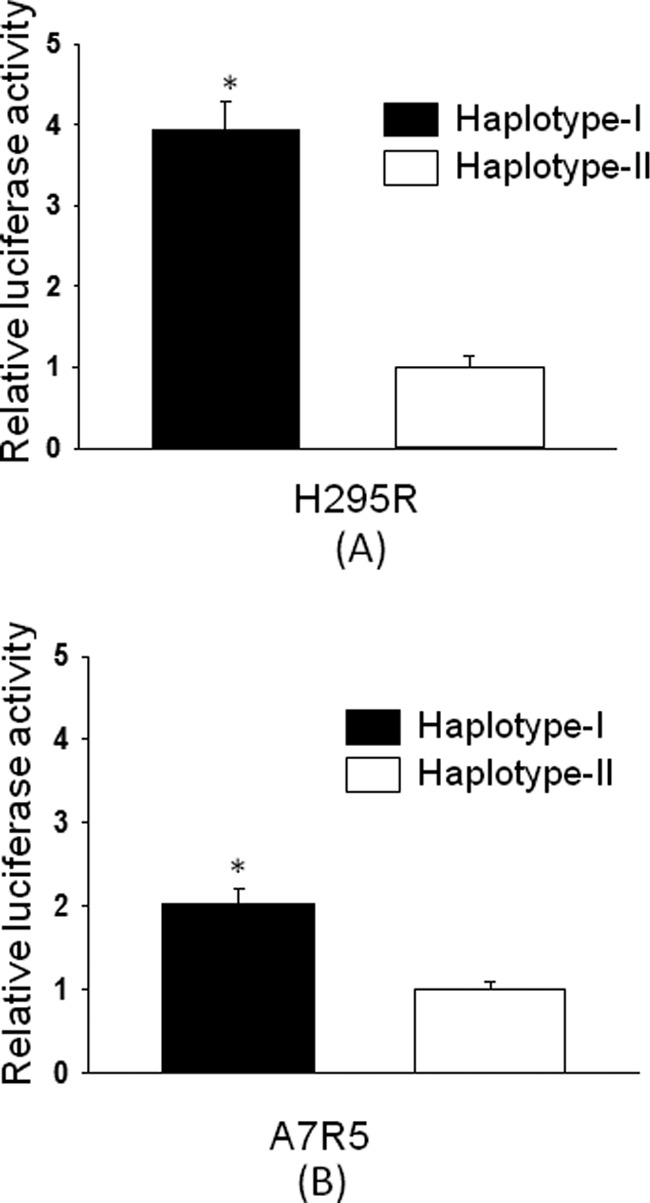
Basal promoter activity of reporter constructs containing haplotype I of the human AT1R gene promoter is increased in H295R (A) and A7r5 (B) cells as compared with haplotype II. Basal promoter activity of reporter constructs containing either haplotype I or II was determined by transient transfection in H295R or A7r5 cells. Relative luciferase activity of the reporter construct containing haplotype I (solid bars) was calculated by assuming the luciferase activity of the reporter construct containing haplotype II (empty bars) as 1. The luciferase activity of each construct is the average of three sets of triplicate experiments. Error bars show S.E. *, p < 0.05 versus haplotype II.
Nucleotide Sequence of hAT1R Gene Promoter Containing −214A Binds Strongly to Transcription Factor USF as Compared with −214C
The USF consensus sequence, CANNTG, exhibits greater sequence homology to the hAT1R promoter of haplotype I when compared with that of haplotype II. Site −214 is the principal determinant of this preference, where −214A of haplotype I could provide for stronger USF binding than −214C of haplotype II (Fig. 4A). The hypothesis was tested with gel shift assay performed using radioactive oligonucleotides −214A and −214C as probes and H295R extract as a source of transcription factors. Competition was then performed in the presence of cold oligonucleotides 214A, 214C, consensus USF site, nonspecific oligonucleotide containing HNF-3 binding site, and in the presence or absence of USF1 antibody (Fig. 4B). Radioactice oligonucleotide 214A formed a complex (shown by an arrow) (lane 1) that was competed out with self-oligonucleotide (lanes 2 and 3) or an oligonucleotide-containing consensus USF binding site (lane 6) but not with 214C or nonspecific oligonucleotides (lanes 4, 5, and 8). This complex was also removed in the presence of USF1 antibody (lane 7). On the other hand, radioactive oligonucleotide 214C did not form this complex but formed a faster moving complex (lane 9). These results allude to a stronger USF binding to haplotype I of the hAT1R gene.
FIGURE 4.
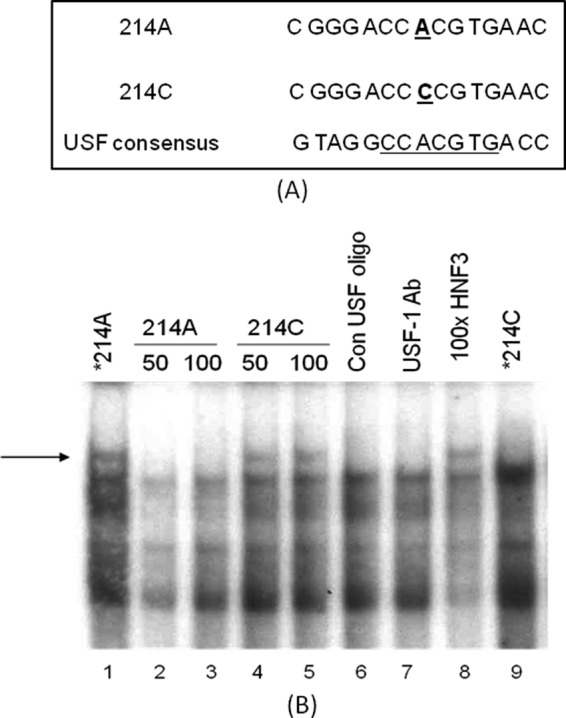
A, nucleotide sequence of hAT1R promoter containing −214A has stronger homology with USF binding site. B, oligonucleotide containing −214A binds strongly to USF as compared with the oligonucleotide containing −214C. Oligonucleotide 214A formed a complex (shown by an arrow) (lane 1), which was competed out with self-oligonucleotide (lanes 2 and 3), USF consensus oligonucleotide (Con USF oligo, lane 6), but not with an oligonucleotide containing −214C (lanes 4 and 5) or a nonspecific oligonucleotide (lane 8). This complex was also removed in the presence of USF1 antibody (lane 7). On the other hand, radioactive oligonucleotide 214C did not form this complex but formed a faster moving complex (lane 9).
USF2 Increases the Promoter Activity of Reporter Construct Containing Haplotype I of the hAT1R Gene, as Compared with Haplotype II in H295R Cells
In line with our earlier results, we examined effects of USF on promoter activity of constructs with either haplotype. The expression vector containing a coding region of the USF2 was co-transfected with reporter constructs hAT1Rluchap-1 or hAT1Rluchap-2 in H295R cells. USF2 increased (p < 0.05) the promoter activity of reporter construct containing haplotype I as compared with haplotype II of the hAT1R gene (results are expressed as fold increase relative to basal promoter activity) (Fig. 5).
FIGURE 5.
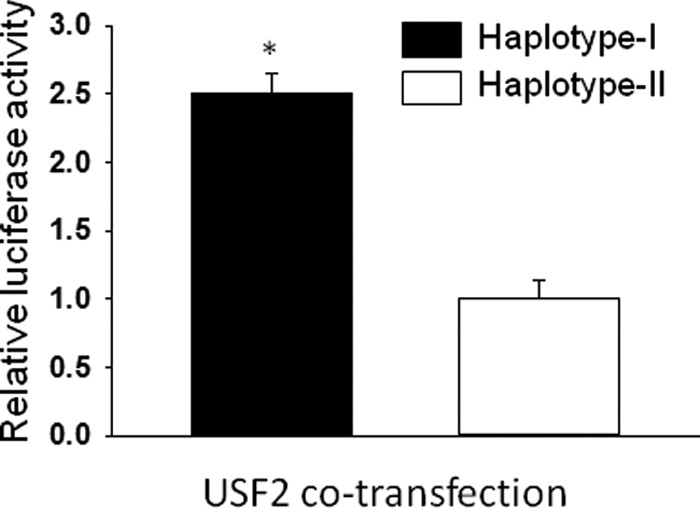
Co-transfection of USF2 increases the promoter activity of reporter construct containing haplotype I of the hAT1R gene as compared with haplotype II on transient transfection in H295R cells. Reporter constructs containing either haplotype I or II of the hAT1R gene was co-transfected with an expression vector containing USF2 coding region, and luciferase activity was determined after transient transfection in H295R cells. Luciferase activity of reporter construct containing haplotype I is shown by a solid bar, and that of haplotype II is shown by an empty bar. The promoter activity after co-transfection of expression vectors was calculated by assuming the basal promoter activity (control) as one. The luciferase activity of each construct is the average of three sets of triplicate experiments. Error bars are S.E. *, p < 0.05 versus haplotype II
TG Mice with Haplotype I Show Increased hAT1R Expression, in Renal and Cardiac Tissues, as Compared with TG Mice with Haplotype II
Haplotype-dependent, physiological regulation and the role of the hAT1R gene were assessed in TG mice generated with each haplotype. The AT1R gene is not uniformly expressed in all mammalian tissues, with high base-line expression in the renal and cardiac tissues. To preserve this tissue-specific regulation of the transfected gene, BAC containing 68 kb of the 5′-flanking region, 45 kb of the coding region, and 53 kb of the 3′-flanking region of the hAT1R gene was used to generate TG mice (Fig. 6). Quantitative RT-PCR analysis was performed to quantify hAT1R expression in renal and cardiac tissues of the two haplotypes. Our results show increased renal (∼6-fold, p < 0.05) and cardiac (∼3-fold, p < 0.05) hAT1R gene expression in TG mice with haplotype I as compared with ones with haplotype II (Fig. 7, A and B). Complementary experiments show no significant difference in the mRNA levels of the mAT1R in these tissues (data not shown).
FIGURE 7.
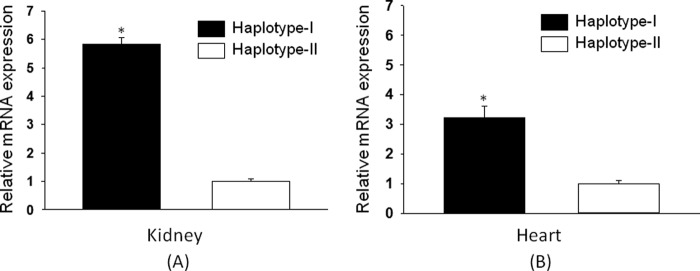
hAT1R mRNA shows increased expression in transgenic mice containing haplotype I. Relative mRNA levels of hAT1R were analyzed from the kidney (A) and heart (B) of transgenic mice containing haplotype I and haplotype II of the hAT1R gene using quantitative RT-PCR. Results were normalized to mouse GAPDH. Error bars are S.E. (n = 6). *, p < 0.05 versus haplotype II.
USF2 and RNA Polymerase Have Higher Affinity to the Chromatin of Kidney from TG Mice Containing Haplotype I of the hAT1R Gene as Compared with Haplotype II
In vitro results, where USF2 increased promoter activity of the reporter construct containing a 1.2-kb promoter with haplotype I of the hAT1R gene, were confirmed in vivo by ChIP analysis. As shown in Fig. 8 (A and C), a 2.2-fold increase (p < 0.05) in USF2 binding is seen to the nucleotide sequence around −214-region of the chromatin from TG mice containing haplotype I. Because the hAT1R mRNA level is increased in the kidneys of TG animals containing haplotype I of the hAT1R gene, we also performed a ChIP assay using antibody against RNA polymerase II. The results of this experiment showed that RNA polymerase II has stronger affinity to the nucleotide sequence present in the chromatin of transgenic animals containing haplotype I of the hAT1R gene as compared with haplotype II by 1.6-fold (Fig. 8, B and D).
FIGURE 8.

Chromatin immunoprecipitation assay shows stronger binding of USF2 and pol II to the promoter of hAT1R gene from the kidney chromatin of TG mice containing haplotype I as compared with haplotype II. A and B show the PCR amplification of immunoprecipitated DNA from the two haplotypes in the presence of antibodies against USF2, pol II, and IgG (mock control). Non specific primer shows amplification using nonspecific primers from a region not containing the respective binding site. C and D show the relative band intensities for both the haplotypes for USF2 and pol II ChIP assays that were normalized to the band intensities from the input DNA. Error bars are S.E. (n = 4). *, p < 0.05 versus haplotype II.
Expression of Pro-inflammatory and Oxidative Markers Is Increased in the Kidney of Transgenic Animals Containing Haplotype I as Compared with Haplotype II of the hAT1R Gene
Functional relevance of the up-regulated hAT1R was assessed by expression analysis of the pro-inflammatory/oxidative markers, including IL-6, TNFα, CRP, and NADPH-oxidase. ANGII, via AT1R activation, induces synthesis and secretion of these inflammatory cytokines. Renal homogenates show an increase in the mRNA levels of IL-6 (2.05-fold), TNFα (2.8-fold), and CRP (2.2-fold) in the kidney of male hAT1R-TG mice containing haplotype I as compared with haplotype II (Fig. 9A; p < 0.05). Complementary experiments show up-regulation of NADPH-oxidase mRNA (Fig. 9B) in the kidneys of male hAT1R TG mice containing haplotype I as compared with haplotype II (p < 0.05).
FIGURE 9.
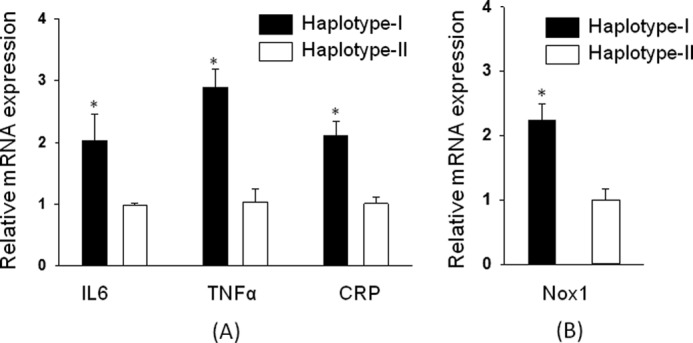
Expression of pro-inflammatory markers and oxidative markers is increased in transgenic mice containing haplotype I of hAT1R gene. Functional relevance of the up-regulated hAT1R was assessed by relative mRNA expression analysis of the pro-inflammatory genes including IL-6, TNFα, CRP (A), and oxidative marker Nox-1 (B) in the kidney of transgenic mice containing haplotype I (solid bars) and haplotype II (empty bars) of hAT1R gene using quantitative RT-PCR. Results were normalized to mouse GAPDH. Error bars are S.E. (n = 6). *, p < 0.05 versus haplotype II.
Blood Pressure Is Increased in Transgenic Mice Containing Haplotype I of the hAT1R Gene as Compared with Mice Containing Haplotype II
Increased AT1R density is expected to modulate blood pressure by sensitizing effector organs to the effects of circulating ANGII. Blood pressure recordings using telemetry probes were obtained in both haplotypes of TG mice. SBP was measured over a period of 4 days in 24-h durations. As shown in Fig. 10A, mean SBP of TG mice containing haplotype I was significantly higher (p < 0.05) from that in TG mice containing haplotype II. To confirm the role of increased hAT1R in bringing about this difference in SBP, experiments were conducted in the absence and in the presence of an AT1R-selective blocker (losartan). In animals treated with losartan, the haplotypic difference in SBP was not observed (Fig. 10B).
FIGURE 10.
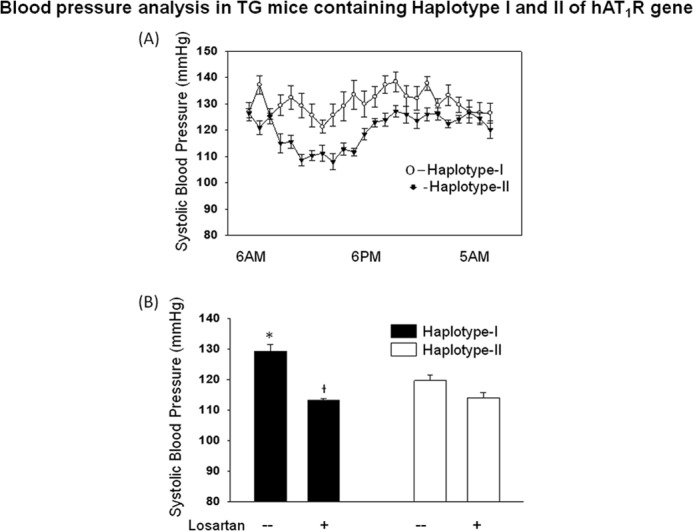
Blood pressure is increased in male transgenic mice containing haplotype I of hAT1R gene. A, systolic blood pressure of hAT1R-TG (TG) mice taken over 24 h by telemetry. Each point represent hourly mean from six male animals taken over 4 days. B, comparison of 24 h of average systolic blood pressure of transgenic mice containing either haplotype I (solid bars) and haplotype II (empty bars). To confirm the role of increased hAT1R in bringing about this difference in SBP, animals were treated with losartan for 24 h, and BP was measured by telemetry. Error bars are S.E. (n = 6). *, p < 0.05 versus haplotype II. † indicates p < 0.05 versus haplotype I without losartan.
DISCUSSION
ANGII activates AT1R and regulates the mean arterial pressure via vascular and renal effects. This study demonstrates for the first time an increased prevalence of a haplotype characterized by AT1R overexpression in hypertensive Caucasians. The first key finding of the study identifies two distinct haplotypes of the human AT1R gene with four SNPs (−777T/A, −680T/G, −214A/C, and −119A/G) always occurring in LD. Haplotype I, with TTAA at these positions, occurs with a significantly higher frequency in hypertensive subjects than haplotype II. This is the first report characterizing these haplotypes and demonstrating haplotype-dependent association with human hypertension. This association is accompanied by increased AT1R expression in adrenal and vascular smooth muscle cells transfected with haplotype I. Increased promoter activity of haplotype I, when compared with haplotype II, is the causative factor for AT1R overexpression in this haplotype.
The nucleotide sequence of the hAT1R promoter containing nucleoside A at −214 has greater homology with E-box (CANNTG) that is recognized by helix-loop-helix family of transcription factors (22). USF1 is a member of the basic helix-loop-helix leucine zipper family of transcription factors (23). USF1 is a ubiquitous transcription factor that was originally discovered by its ability to bind to the adenovirus major late promoter and regulate its expression. USF1 binds as a homodimer, or as a heterodimer with the related transcription factor USF2 to E boxes and controls the expression of a number of genes (24, 25). USF has been shown to increase the expression of genes involved in glucose and lipid metabolism (26). Overexpression of USF2 in transgenic mice influences metabolic traits such as obesity, lipid profiles, and glucose/insulin ratio (27). Polymorphisms in the USF1 gene have been identified in human linkage studies with diabetic nephropathy (28). Additionally, earlier studies have established SNP-dependent differential gene regulation by USF. In this regard, we have reported SNP in USF binding site at −20 position in the promoter of human angiotensinogen gene that modulates the activity of this gene (29). Other investigators have confirmed the role of SNPs in USF1-binding sites in the human angiotensinogen gene expression in adipose tissue (30). Genes, including the C-reactive protein, also encode two SNPs that modulate USF binding and account for differences in base-line CRP levels in apparently healthy people (31). In line with these studies, we show here increased USF binding to the −214A site corresponding to haplotype I, as compared with −214C site analogous to haplotype II. Complementary studies confirm USF-dependent up-regulation of hAT1R expression in cells co-transfected with USF and hAT1R. Because USF is the only transcription factor that differentially binds to −214A and −214C alleles of the hAT1R gene, we have analyzed its role in transcriptional regulation in the present communication. However, the role of other transcription factors that may interact with USF or bind to other polymorphic sites remains to be examined.
A homologous set of transcription factors in mice and humans provides for a unique opportunity to study individual human gene regulation in an in vivo setting. The second key finding of the study demonstrates haplotype-specific regulation of the hAT1R gene in TG mice, where haplotype I favors increased expression of hAT1R in renal and cardiac tissues of these mice. The absence of such a difference in the expression of mAT1R points to the haplotype-dependent differential regulation of the hAT1R. Complementary experiments show increased USF and RNA polymerase II binding to chromatin extracts from renal tissues of TG mice with haplotype I. These results are in agreement with our in vitro findings and implicate USF-directed enhanced transcription of the hAT1R gene in bringing about higher hAT1R expression in haplotype I versus haplotype II. This is the first report citing haplotype-dependent differences in expression of the hAT1R gene in TG mice, where genetic constitution of the mice with two haplotypes is the same except for the 1.2-kb promoter region containing the aforementioned SNPs. This provides for a strong argument for USF-mediated up-regulation of the hAT1R gene whose SNP-defined haplotypes favor differential binding of the transcription factors and promotes gene expression in one haplotype over the other.
The physiological significance of these haplotypes and their derivative differential regulation of hAT1R expression is provided by the examination of the end effects of hAT1R activation. In this regard, the third key finding of the study shows elevation of the systolic blood pressure and development of a pro-inflammatory/pro-oxidative state in TG mice with increased expression of hAT1R. AT1R is a G protein-coupled receptor and is ubiquitously expressed in mammalian tissues. ANGII-mediated AT1R-dependent physiological effects of the RAS include regulation of renal salt and water balance, vascular tone, and cardiac structure and contractility. Together, these variables govern long term set point for the mean arterial pressure. Additionally, chronic overactivation of AT1R promotes cellular oxidative stress, primarily via activation of NADPH oxidase, and secretion of pro-inflammatory/pro-fibrotic cytokines (for a review see Ref 32). Increased expression of cytokines, including IL-6, TNFα, and CRP, and increased levels of the NADPH oxidase component NOX1 confirm the functional role of increased hAT1R activation in TG mice with haplotype I. Both mAT1R and hAT1R are amenable to activation by mouse ANGII, and the haplotype-dependent differential effects in measured physiological parameters corroborate the contributions of increased hAT1R density in TG mice with haplotype I. Higher AT1R expression will allow for more spare receptors, thus sensitizing the tissues to circulating levels of ANGII. Elevation of the SBP in TG mice with haplotype I supports this notion. Reversal of this elevated SBP by an AT1R blocker is supportive of earlier evidence linking increased AT1R expression to heightened pathophysiological response to endogenous RAS. Because our main objective was to examine the effect of these haplotypes of the human AT1R gene on its expression and to understand their role in blood pressure regulation, we did not use wild type C57 animals as control.
In conclusion, we provide the first evidence linking human hypertension with haplotype-dependent variability in AT1R expression. We provide in vitro and in vivo evidence of USF-mediated transcriptional up-regulation of the hAT1R gene in haplotype I, as opposed to haplotype II. Finally, our results showing increased tissue expression of inflammatory/oxidative markers and an elevated SBP in haplotype I provide pathophysiological perspectives to the study. Where chronic activation of RAS is frequently associated with cardiovascular and renal abnormalities, as seen in animals with Goldblatt's hypertension, increased AT1R expression has the potential to bring about similar pathophysiological alterations. This study provides evidence for the notion that genetic susceptibility toward increased AT1R-density will alter RAS-regulated physiological systems by sensitizing them to endogenous levels of ANGII, which may or may not be elevated. We are currently pursuing detailed physiological studies to identify the impact of these haplotypes on cardiac, vascular, and renal function.
Acknowledgment
We are thankful to Dr. M. Sawadogo for USF2 expression vector.
This work was supported, in whole or in part, by National Institutes of Health Grants HL81752, HL105113, and HL092558.
- ANGII
- angiotensin II
- AT1R
- angiotensin II receptor type 1
- BAC
- bacterial artificial chromosome
- hAT1R
- human AT1R
- LD
- linkage disequilibrium
- USF
- upstream stimulatory factor
- TG
- transgenic
- BP
- blood pressure
- SBP
- systolic blood pressure
- DBP
- diastolic blood pressure
- pol
- polymerase.
REFERENCES
- 1. Galis Z. S., Thrasher T., Reid D. M., Stanley D. V., Oh Y. S. (2013) Investing in high blood pressure research. A National Institutes of Health perspective. Hypertension 61, 757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Griendling K. K., Murphy T. J., Alexander R. W. (1993) Molecular biology of the renin-angiotensin system. Circulation 87, 1816–1828 [DOI] [PubMed] [Google Scholar]
- 3. Griendling K. K., Ushio-Fukai M., Lassègue B., Alexander R. W. (1997) Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension 29, 366–373 [DOI] [PubMed] [Google Scholar]
- 4. Peach M. J., Dostal D. E. (1990) The angiotensin II receptor and the actions of angiotensin II. J. Cardiovasc. Pharmacol. 16, (Suppl. 4) S25–S30 [DOI] [PubMed] [Google Scholar]
- 5. Mendelsohn F. A. (1985) Localization and properties of angiotensin receptors. J. Hypertens. 3, 307–316 [DOI] [PubMed] [Google Scholar]
- 6. Timmermans P. B., Wong P. C., Chiu A. T., Herblin W. F., Benfield P., Carini D. J., Lee R. J., Wexler R. R., Saye J. A., Smith R. D. (1993) Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol. Rev. 45, 205–251 [PubMed] [Google Scholar]
- 7. Ito M., Oliverio M. I., Mannon P. J., Best C. F., Maeda N., Smithies O., Coffman T. M. (1995) Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc. Natl. Acad. Sci. U.S.A. 92, 3521–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Le T. H., Kim H. S., Allen A. M., Spurney R. F., Smithies O., Coffman T. M. (2003) Physiological impact of increased expression of the AT1 angiotensin receptor. Hypertension 42, 507–514 [DOI] [PubMed] [Google Scholar]
- 9. Hein L., Stevens M. E., Barsh G. S., Pratt R. E., Kobilka B. K., Dzau V. J. (1997) Overexpression of angiotensin AT1 receptor transgene in the mouse myocardium produces a lethal phenotype associated with myocyte hyperplasia and heart block. Proc. Natl. Acad. Sci. U.S.A. 94, 6391–6396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paradis P., Dali-Youcef N., Paradis F. W., Thibault G., Nemer M. (2000) Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc. Natl. Acad. Sci. U.S.A. 97, 931–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Veerasingham S. J., Raizada M. K. (2003) Brain renin-angiotensin system dysfunction in hypertension. Recent advances and perspectives. Br. J. Pharmacol. 139, 191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gutkind J. S., Kurihara M., Saavedra J. M. (1988) Increased angiotensin II receptors in brain nuclei of DOCA-salt hypertensive rats. Am. J. Physiol. 255, H646–H650 [DOI] [PubMed] [Google Scholar]
- 13. Perola M., Kainulainen K., Pajukanta P., Terwilliger J. D., Hiekkalinna T., Ellonen P., Kaprio J., Koskenvuo M., Kontula K., Peltonen L. (2000) Genome-wide scan of predisposing loci for increased diastolic blood pressure in Finnish siblings. J. Hypertens. 18, 1579–1585 [DOI] [PubMed] [Google Scholar]
- 14. Kainulainen K., Perola M., Terwilliger J., Kaprio J., Koskenvuo M., Syvänen A. C., Vartiainen E., Peltonen L., Kontula K. (1999) Evidence for involvement of the type 1 angiotensin II receptor locus in essential hypertension. Hypertension 33, 844–849 [DOI] [PubMed] [Google Scholar]
- 15. Bonnardeaux A., Davies E., Jeunemaitre X., Féry I., Charru A., Clauser E., Tiret L., Cambien F., Corvol P., Soubrier F. (1994) Angiotensin II type 1 receptor gene polymorphisms in human essential hypertension. Hypertension 24, 63–69 [DOI] [PubMed] [Google Scholar]
- 16. Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Warming S., Costantino N., Court DL, Jenkins N. A., Copeland N. G. (2005) Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jain S., Tillinger A., Mopidevi B., Pandey V. G., Chauhan C. K., Fiering S. N., Warming S., Kumar A. (2010) Transgenic mice with −6A haplotype of the human angiotensinogen gene have increased blood pressure compared with −6G haplotype. J. Biol. Chem. 285, 41172–41186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. D'haene B., Vandesompele J., Hellemans J. (2010) Accurate and objective copy number profiling using real-time quantitative PCR. Methods 50, 262–270 [DOI] [PubMed] [Google Scholar]
- 20. Butz G. M., Davisson R. L. (2001) Long-term telemetric measurement of cardiovascular parameters in awake mice. A physiological genomics tool. Physiol. Genomics 5, 89–97 [DOI] [PubMed] [Google Scholar]
- 21. Bird I. M., Mason J. I., Rainey W. E. (1994) Regulation of type 1 angiotensin II receptor messenger ribonucleic acid expression in human adrenocortical carcinoma H295 cells. Endocrinology 134, 2468–2474 [DOI] [PubMed] [Google Scholar]
- 22. Massari M. E., Murre C. (2000) Helix-loop-helix proteins. Regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 20, 429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sawadogo M., Roeder R. G. (1985) Interaction of a gene-specific transcription factor with the adenovirus major late promoter upstream of the TATA box region. Cell 43, 165–175 [DOI] [PubMed] [Google Scholar]
- 24. Sawadogo M. (1988) Multiple forms of the human gene-specific transcription factor USF. II. DNA binding properties and transcriptional activity of the purified HeLa USF. J. Biol. Chem. 263, 11994–12001 [PubMed] [Google Scholar]
- 25. Sirito M., Lin Q., Maity T., Sawadogo M. (1994) Ubiquitous expression of the 43- and 44-kDa forms of transcription factor USF in mammalian cells. Nucleic Acids Res. 22, 427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vallet V. S., Casado M., Henrion A. A., Bucchini D., Raymondjean M., Kahn A., Vaulont S. (1998) Differential roles of upstream stimulatory factors 1 and 2 in the transcriptional response of liver genes to glucose. J. Biol. Chem. 273, 20175–20179 [DOI] [PubMed] [Google Scholar]
- 27. Wu S., Mar-Heyming R., Dugum E. Z., Kolaitis N. A., Qi H., Pajukanta P., Castellani L. W., Lusis A. J., Drake T. A. (2010) Upstream transcription factor 1 influences plasma lipid and metabolic traits in mice. Hum. Mol. Genet. 19, 597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ewens K. G., George R. A., Sharma K., Ziyadeh F. N., Spielman R. S. (2005) Assessment of 115 candidate genes for diabetic nephropathy by transmission/disequilibrium test. Diabetes 54, 3305–3318 [DOI] [PubMed] [Google Scholar]
- 29. Zhao Y. Y., Zhou J., Narayanan C. S., Cui Y., Kumar A. (1999) Role of C/A polymorphism at −20 on the expression of human angiotensinogen gene. Hypertension 33,108–115 [DOI] [PubMed] [Google Scholar]
- 30. Dickson M. E., Tian X., Liu X., Davis D. R., Sigmund C. D. (2008) Upstream stimulatory factor is required for human angiotensinogen expression and differential regulation by the A-20C polymorphism. Circ. Res. 103, 940–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Szalai A. J., Wu J., Lange E. M., McCrory M. A., Langefeld C. D., Williams A., Zakharkin S. O., George V., Allison D. B., Cooper G. S., Xie F., Fan Z., Edberg J. C., Kimberly R. P. (2005) Single-nucleotide polymorphisms in the C-reactive protein (CRP) gene promoter that affect transcription factor binding, alter transcriptional activity, and associate with differences in baseline serum CRP level. J. Mol. Med. 83, 440–447 [DOI] [PubMed] [Google Scholar]
- 32. Mehta P. K., Griendling K. K. (2007) Angiotensin II cell signaling. Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 292, C82–C97 [DOI] [PubMed] [Google Scholar]