

Background: MicroRNA-21 is important in hepatic fibrosis development, but the mechanism is unclear.
Results: MicroRNA-21 is predominantly up-regulated in activated hepatic stellate cells and could form a double negative feedback loop that links fibrogenic machinery.
Conclusion: The microRNA-21-mediated loop is a main driving force for hepatic fibrosis progression.
Significance: It suggests a mechanism for how microRNA-21 contributes to hepatic fibrosis.
Keywords: Fibrosis, Hepatic Stellate Cells, Liver Injury, MicroRNA, Transforming Growth Factor β (TGFβ)
Abstract
Sustained activation of hepatic stellate cells (HSCs) leads to hepatic fibrosis, which is characterized by excessive collagen production, and for which there is no available drug clinically. Despite tremendous progress, the cellular activities underlying HSC activation, especially the driving force in the perpetuation stage, are only partially understood. Recently, microRNA-21 (miR-21) has been found to be prevalently up-regulated during fibrogenesis in different tissues, although its detailed role needs to be further elucidated. In the present study, miR-21 expression was examined in human cirrhotic liver samples and in murine fibrotic livers induced by thioacetamide or carbon tetrachloride. A dramatic miR-21 increase was noted in activated HSCs. We further found that miR-21 maintained itself at constant high levels by using a microRNA-21/programmed cell death protein 4/activation protein-1 (miR-21/PDCD4/AP-1) feedback loop. Disrupting this loop with miR-21 antagomir or AP-1 inhibitors significantly suppressed fibrogenic activities in HSCs and ameliorated liver fibrosis. In contrast, reinforcing this loop with small interfering RNA (siRNA) against PDCD4 promoted fibrogenesis in HSCs. Further analysis indicated that the up-regulated miR-21 promoted the central transforming growth factor-β (TGF-β) signaling pathway underlying HSC activation. In summary, we suggest that the miR-21/PDCD4/AP-1 autoregulatory loop is one of the main driving forces for hepatic fibrosis progression. Targeting this aberrantly activated feedback loop may provide a new therapeutic strategy and facilitate drug discovery against hepatic fibrosis.
Introduction
Hepatic fibrosis is a wound healing process in response to chronic liver injuries that leads to unbalanced extracellular matrix (ECM)3 deposition and resolution. The persistent activation of wound healing responses causes quantitative and qualitative changes in the ECM components and could finally distort liver parenchyma and vascular architecture, which could impair liver function and potentially lead to liver failure and hepatocellular carcinoma. Following liver injury, quiescent hepatic stellate cells (HSCs) transdifferentiate into myofibroblast-like cells that are characterized by the expression of smooth muscle α-actin (α-SMA) and enhanced production of ECM (1). Despite the tremendous progress in understanding the mechanisms during fibrogenesis, the driving force underlying the persistent fibrogenic activities is still only partially understood.
Extensive studies have suggested the central role of feedback networks in dictating disease progression. Strikingly, these autofeedback loops could have the capacity to promote the irreversible activation of certain signaling pathways and stabilize the cellular phenotype switch under pathological settings, thus contributing to disease progression (2). One previous study sheds light on the miR-21/PDCD4/AP-1 autoregulatory loop, which is highly activated during tumorigenesis and could drive cell transformation and proliferation (3). In addition, elevated miR-21 expression has been noted in liver and other organs undergoing fibrosis (4–6), whereas its role in promoting hepatic fibrosis progression and moreover how miR-21 is dysregulated in the liver remain only slightly understood. MiR-21 harbors several transcription factors, and its expression could be modulated by different upstream effectors under different disease settings (7–9). In regard to liver fibrosis, a role for AP-1 is suggested because JNK is predominantly activated in myofibroblasts during hepatic fibrosis (10). Given that AP-1 could drive miR-21 expression and that miR-21-mediated PDCD4 down-regulation is necessary for maximal AP-1 activity (3), it is speculated that miR-21, PDCD4, and AP-1 may form a feedback loop during HSC activation and promote hepatic fibrosis progression.
Herein, the present study demonstrates that miR-21 expression is significantly increased in fibrotic livers, predominantly occurring in myofibroblasts. The up-regulated miR-21 maintains its own high level expression by utilizing a double negative feedback circuit in which miR-21 targets PDCD4, a well known AP-1 inhibitor, for degradation; the subsequent elevated AP-1 activity in turn promotes miR-21 expression. As a result, a high level of miR-21 efficiently depresses Smad7 expression and enhances p-Smad2/Smad2 activity. Altogether, once activated, this autofeedback paradigm could fuel HSC activation and promote the progression of liver fibrosis.
EXPERIMENTAL PROCEDURES
Reagents
Human recombinant TGF-β1 was purchased from PeproTech (Rocky Hill, NJ). The miR-21 precursor and scrambled precursor were purchased from Ambion (Foster City, CA). Antagomirs against miR-21 were designed, and the most efficient one (5′-CAACATCAGTCTGATAAGC-3′) was selected. Antagomir-21 and scrambled antagomirs with full phosphorothioate linkage were synthesized by SBS Genetech Co., Ltd. (Shanghai, China). The miRCURY LNA detection probe against miR-21 and the control scrambled probe were purchased from Exiqon (Woburn, MA). PDCD4 siRNA (sense, 5′-GCUGCUUUGGACAAGGCUATT-3′; antisense, 5′-UAGCCUUGUCCAAAGCAGCTT-3′), c-Jun siRNA (sense, 5′-GCCCUAGCUGAACUGCAUATT-3′; antisense, 5′-UAUGCAGUUCAGCUAGGGCTT-3′), and scrambled siRNA were designed and synthesized by Invitrogen and then verified. Dextran sulfate sodium (Mr 500,000) was purchased from Sigma-Aldrich. SR11302 was purchased from Tocris (Ellisville, MO). SP600125 was purchased from Beyotime (Nantong, China) for the cellular experiments and was further synthesized by Wuxi Apptec (Tianjing, China) for the animal experiments.
Animal Treatments
Male ICR mice (20 ± 2 g) of the same background were obtained from the Animal Centre of Yangzhou University (Yangzhou, China). The mice were randomly divided into different groups and had free access to water and laboratory chow. Mice received intraperitoneal injections of thioacetamide (TAA; 200 mg/kg of body weight) twice weekly for 8 weeks or carbon tetrachloride (CCl4; in oil, 1:3, 4 μl/g of body weight) twice weekly for 4 weeks, whereas the control group received saline or oil intraperitoneally. For the antagomir-21 or scrambled antagomir (antago-ctrl) groups, 2 days before the first saline/TAA or oil/CCl4 injection, mice received a single dose of antagomir-21 or antago-ctrl (10 mg/kg of body weight) dissolved in saline via the tail vein. During the following 8-week TAA or 4-week CCl4 treatments, the mice received an intravenous injection of antagomir-21 or antago-ctrl (10 mg/kg of body weight) every 10 days. For the SP600125 treatment group, 24 h after every TAA injection, a single dose of 100 μl of SP600125 (5 mg/ml) was administered intraperitoneally. Mice were sacrificed at the indicated time points. All of the experiments were conducted according to the use and care guidelines for experimental animals from Jiangsu Province, China.
Human Subjects
This study was approved by the local Institutional Review Board, and the patients provided written informed consent. Liver samples were obtained from 10 cirrhosis patients at Gulou Hospital (Nanjing, China). The patient information is included in Table 1.
TABLE 1.
Human subjects
M, male; F, female.
| Number | Gender | Age | Description |
|---|---|---|---|
| 1 | M | 43 | Nodular cirrhosis; portal chronic inflammation |
| 2 | M | 47 | Nodular cirrhosis surrounding necrotic area in liver |
| 3 | F | 50 | Nodular cirrhosis |
| 4 | M | 71 | Nodular cirrhosis with necrosis |
| 5 | M | 58 | Nodular cirrhosis with inflammatory cell infiltration |
| 6 | M | 43 | Nodular cirrhosis; chronic hepatitis B |
| 7 | M | 40 | Nodular cirrhosis; cholestasis |
| 8 | F | 75 | Nodular cirrhosis; cholestasis |
| 9 | M | 45 | Nodular cirrhosis |
| 10 | F | 62 | Nodular cirrhosis; cholestasis |
Isolation of Primary Hepatic Stellate Cells
Firstly, 8–12-month-old Sprague-Dawley rats were anesthetized with an intraperitoneal injection of 15 mg/kg pentobarbital. The liver perfusion buffer and liver digest medium were prewarmed at 37 °C. A warm water circulation kept the buffer flow at a constant temperature of 37 °C. The liver was perfused in situ via the vena porta with a heparin sodium buffer while the inferior vena cava was used as the outflow until the liver became pale. The perfusion was continued with collagenase (Roche Applied Science; 3 mg/ml) and Pronase (Sigma; 3 mg/ml) buffer. The liver was carefully removed and dissociated in digestion medium II (0.5 mg/ml collagenase and Pronase, 0.2 mg/ml DNase I) for further digestion. Afterward, the liver cell suspension was filtered through gauze (100 μm) and centrifuged for 5 min at 50 × g at 4 °C to obtain the parenchyma cells. The non-parenchyma cells were collected from the 50 × g supernatant and washed twice before density gradient centrifugation. A discontinuous density gradient (15%, 10%) was made using OptiPrepTM following the manufacturer's instructions and centrifuged at 1400 × g at 4 °C for 15 min while allowing the rotor to decelerate without the brake. The HSCs at the interface of the mounting solution and low density barrier were collected. The viability of the isolated cells was determined using trypan blue staining. The purity of isolated quiescent HSCs was determined by vitamin A autofluorescence and routinely exceeded 90%.
For the isolation of primary activated HSCs from fibrotic livers, although HSCs lose vitamin A during activation, the activated HSCs could also be isolated by discontinuous density gradient centrifugation according to published protocols (11, 12). The mouse liver was perfused from the left ventricle of the heart, and the isolation procedure continued as described above. To obtain enough HSCs, the cells from two mice were mixed as one sample in the end. To isolate primary murine hepatocytes, the digest medium contained collagenase but not Pronase. Because the activated HSCs lost vitamin A during activation, the purity of isolated activated HSCs was determined by α-SMA/desmin double fluorescence staining; the purity exceeded 90%.
Cell Treatments
HSC lines (rat HSC-T6 cells) (13) or isolated primary rat HSCs were cultured in DMEM containing 10% FBS (Invitrogen) at 37 °C with 5% CO2 and humidity.
For HSC-T6 cells, 24 h after 50 nm antagomir-21/antago-ctrl or pre-miR-21/pre-ctrl transfection using Lipofectamine 2000, HSC-T6 cells were further stimulated with 10 ng/ml TGF-β1. For primary HSCs, during the 15-day self-activation process on plastic plates, transfections were conducted three times using 50 nm each. At the end of the process, the cells were analyzed.
Quantitative Real Time Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from cells or tissues, and the complementary DNA was synthesized. Transcript levels were detected via qRT-PCR following the manufacturer's instructions. Individual gene and microRNA expression was normalized to β-actin mRNA or small nuclear RNA (snRNA) U6 expression, respectively. The primers used are listed in Table 2.
TABLE 2.
PCR primers used in this study
R, reverse; F, forward.
| miR-21-R | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCAACATC |
| miR-21-F | ACACTCCAGCTGGGTAGCTTATCAGACTGA |
| URP | TGGTGTCGTGGAGTCG |
| U6-F | CTCGCTTCGGCAGCACA |
| U6-R | AACGCTTCACGAATTTGCGT |
| β-Actin-F | GACCTCTATGCCAACACAGTGC |
| β-Actin-R | GTACTCCTGCTTGCTGATCCAC |
| α-SMA(rat)-F | AACTGGTATTGTGCTGGACTCTG |
| α-SMA(rat)-R | GCTTTGGTGGATATTGTCGTAGT |
| Smad2(rat)-F | ACTCTCTCCCCTGTCAATCAC |
| Smad2(rat)-R | CGCACTCCCCTTCCTATA |
| COL1A1(rat)-F | TGGATGGCTGCACGAGTCA |
| COL1A1(rat)-R | AGATTGGGATGGAGGGAGTTTAC |
| Pdcd4(rat)-F | TGCTTTCTGACCTTTGCG |
| Pdcd4(rat)-R | GGTGATTGACAGGCTGCTG |
| α-SMA(mouse)-F | GAGCGTGAGATTGTCCGTGA |
| α-SMA(mouse)-R | GGTGCTGGGTGCGAGG |
| COL1A1(mouse)-F | TCCGGCTCCTGCTCCTCTTAG |
| COL1A1(mouse)-R | AGGCCATTGTGTATGCAGCTGAC |
| c-Jun(rat)-F | CCTTTTCTTTACGGTCTCGGT |
| c-Jun(rat)-R | GGGCTGTTCATCTGTTTGTCTT |
Western Blotting
Tissue homogenate or cell lysate was prepared. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes (Biotrace, Randburg, S.A.). Membranes were blocked using skim milk and then incubated with diluted primary antibody at 4 °C with gentle shaking overnight. After washing five times, secondary HRP-conjugated antibody was incubated for 1 h at room temperature. Positive signal was detected with fluorography using an enhanced chemiluminescence system (7003, Cell Signaling Technology Inc., Danvers, MA). The antibodies used are listed in Table 3.
TABLE 3.
Primary and secondary antibodies used in Western blotting
| Antigens | Poly/monoclonal | Manufacturer | Dilution |
|---|---|---|---|
| p-Smad2 | Polyclonal | Cell Signaling Technology Inc. (3101) | 1:1000 |
| Smad2/3 | Polyclonal | Cell Signaling Technology Inc. (3102) | 1:1000 |
| MADH7 | Polyclonal | Abcam Ltd., Cambridge, MA (ab90086) | 1:1000 |
| c-Jun | Monoclonal | Cell Signaling Technology Inc. (9165) | 1:1000 |
| Fra-1 | Polyclonal | Santa Cruz Biotechnology, Inc., Santa Cruz, CA (sc-183) | 1:200 |
| PTEN | Monoclonal | Cell Signaling Technology Inc. (9569) | 1:1000 |
| PDCD4 | Monoclonal | Cell Signaling Technology Inc. (9535) | 1:1000 |
| p-MEK1/2 | Monoclonal | Cell Signaling Technology Inc. (9154) | 1:1000 |
| COL1A1 | Polyclonal | Santa Cruz Biotechnology, Inc. (sc-25974) | 1:200 |
| α-SMA | Polyclonal | Abcam Ltd. (ab5694) | 1:200 |
| Desmin | Polyclonal | Santa Cruz Biotechnology, Inc. (sc-7559 | 1:200 |
| HRP-linked anti-rabbit IgG | Cell Signaling Technology Inc. (7104) | 1:1000 | |
| HRP-linked anti-goat IgG | Santa Cruz Biotechnology, Inc. (sc-2020) | 1:200 | |
| HRP-conjugated anti-GAPDH | Monoclonal | KangChen Bio-tech Inc., Shanghai, China (KG-5G5) | 1:1000 |
Sirius Red and H&E Staining
The paraffin-embedded liver tissues in each group were stained with H&E and Sirius Red, respectively. Briefly, the paraffin-embedded liver tissues were sectioned at 5 μm. After deparaffinization and hydration, sections were stained in 0.1% (w/v) Sirius Red (Direct Red 80, Sigma-Aldrich) in a saturated aqueous solution of picric acid for 1 h. Then the slides were rinsed twice for 15 min each in 0.01 n HCl to remove unbound dye. After dehydration, the slides were mounted.
Electrophoretic Mobility Shift Assays (EMSAs)
The nuclear proteins were extracted from the liver of mice receiving saline or TAA treatment using a nuclear protein extraction kit (P0028, Beyotime, Nantong, Jiangsu, China) according to the manufacturer's instructions. For EMSAs, the biotin-conjugated AP-1 consensus binding sequence (sense, 5′-CGC TTG ATG ACT CAG CCG GAA-3′; antisense, 3′-GCG AAC TAC TGA GTC GGC CTT-5′) (GS011B, Beyotime) was used. For DNA binding reactions, 2 μl (∼16 μg) of nuclear extract was added to a 10-μl reaction (final) containing 50 nm biotin-labeled oligonucleotide and 5 μg of poly(dI-dC) (GS005, Beyotime). At the same time, the unlabeled probe (GS011, Beyotime) or unlabeled mutated probe (GS011M, Beyotime) was used as a cold competitor. The binding reactions were incubated for 20 min at room temperature. Then the binding reactions were loaded on prerun 6% native PAGE gels in 0.5× Tris borate-EDTA at 6 mA and transferred onto a nylon membrane. The membrane was incubated with shaking in blocking buffer containing avidin-HRP enzyme and balanced with substrate balance buffer. The signal was detected on x-ray film using an enhanced chemiluminescence system (7003, Cell Signaling Technology Inc.)
Fluorescence in Situ Hybridization
Fluorescence in situ hybridization (FISH) was performed following published protocols (14).
Measurement of Hepatic Hydroxyproline Content
Hepatic hydroxyproline levels were biochemically measured and calculated according to the manufacturer's instructions (Jiancheng, Nanjing, China).
Determination of HSC Proliferation
Cell proliferation was measured with the CCK-8 kit (Dojindo). To observe cell proliferation, antagomiR-21 or pre-miR-21 and corresponding controls (50 nm) were transfected into target cells using Lipofectamine 2000. The transiently transfected cells were seeded in a 96-well plate at a cell density of 5 × 103 and then cultured at 24-h intervals for 3 days. Briefly, 10 μl of CCK-8 solution was added to each well of the plate, and the plate was incubated at 37 °C for 1 h. The absorbance was measured at 450 nm as an indicator of cell proliferation.
Determination of HSC Apoptosis
Apoptosis of HSC was induced by serum deprivation plus cycloheximide (CHX) treatment. HSC-T6 cells were pretransfected with antagomiR-21 or pre-miR-21 and corresponding controls (50 nm), and then 24 h after transfection, cells were exposed to CHX (Sigma). Following a 4-h incubation with CHX at 37 °C, apoptosis of cells was determined using an Annexin V/dead cell apoptosis kit for flow cytometry according to the manufacturer's instructions (V13241, Invitrogen).
Statistical Analysis
The results are given as the means ± S.D. Statistical analysis was performed using a one-way analysis of variance, and p < 0.05 was considered significant (denoted as * in each figure; ** and *** indicate p < 0.01 and p < 0.001, respectively).
RESULTS
MiR-21 Was Predominantly Up-regulated in HSCs in Human and Murine Fibrotic Liver
Elevated miR-21 expression has been reported in human fibrotic liver (4); however, the miR-21 expression profile in different liver cells remains elusive. To shed light on this issue, 10 human cirrhotic liver samples were collected (Table 1), and FISH was performed on liver sections to locate the up-regulated miR-21. The scrambled miR-21 probe showed no positive signal on the liver sections during the experiments. The results are presented in Fig. 1A, and a higher magnification image is shown in Fig. 1B. In all 10 collected human samples, miR-21 was predominantly up-regulated in the myofibroblasts as indicated by the co-localization of the miR-21 probe with α-SMA-positive cells.
FIGURE 1.

Up-regulated miR-21 expression in activated HSCs during liver fibrogenesis. A, representative FISH results on paraffin-embedded liver sections from human cirrhotic liver subjects. Scale bars, 100 μm. Panel i, miR-21; panel ii, α-SMA; panel iii, DAPI; panel iv, merge. B, representative FISH results from human subjects with higher magnification. Scale bars, 100 μm. Panel i, miR-21; panel ii, α-SMA; panel iii, DAPI; panel iv, merge. C, one of three independent FISH results on paraffin-embedded liver sections from 8-week saline/TAA-treated mice with similar results. Scale bars, 20 μm. Panel i, miR-21; panel ii, α-SMA; panel iii, DAPI; panel iv, merge. D, the miR-21 levels in the total liver of saline/TAA-treated mice were determined by qRT-PCR, and compared with the basal content before hepatic fibrosis was induced. E, miR-21 levels in the isolated activated HSCs from saline/TAA-treated mice were determined by qRT-PCR and compared with the basal content before fibrosis induction. F, c-Jun and Fra-1 protein levels were determined by Western blotting from murine fibrotic liver samples. EMSA was performed on liver nuclear extracts from the 8-week saline/TAA-treated mice. The data represent one of three cohorts with similar results. The data are expressed as the means ± S.D. of three independent experiments. Each group contained at least three samples, and the HSCs from two mice were mixed into one sample. *, p < 0.05; **, p < 0.01; ***, p < 0.001; N.S., not significant. Error bars represent S.D.
Then a murine liver fibrosis model was established, and FISH was performed on saline/TAA-treated liver sections. Consistent with the above findings, miR-21 exhibited a much higher accumulation in myofibroblasts (Fig. 1C). Furthermore, the miR-21 up-regulation in the total liver correlated with hepatic fibrosis progression (Fig. 1D). Activated HSCs are the major source of myofibroblasts in the liver during hepatic fibrosis (15–17). Thus, the primary activated HSCs were further isolated from saline/TAA-treated fibrotic livers according to published protocols (11, 12). The isolated activated HSCs were determined by α-SMA/desmin double fluorescence staining, and the purity routinely exceeded 90%. MiR-21 was continuously up-regulated in the HSCs during the development of hepatic fibrosis (Fig. 1E). The miR-21 change in HSCs was further validated in CCl4-induced hepatic fibrosis and showed similar continuous up-regulation in HSCs (data not shown). It is noted that enhanced AP-1 activity predominantly occurs in myofibroblasts during liver fibrosis (10), and AP-1 is one of the transcription factors of miR-21 among which heterodimers formed by c-Jun and Fra-1 have been reported to promote miR-21 expression (3, 18, 19). Consistently, c-Jun and Fra-1 expression was elevated during fibrosis progression, and the EMSA results confirmed the elevated AP-1 activity in the fibrotic liver (Fig. 1F).
Sequestering MiR-21 by Antagomir-21 Attenuated Collagen Deposition in the Livers of TAA- and CCl4-treated Mice
To further confirm that miR-21 participated in and promoted the progression of hepatic fibrosis, chemically synthesized antagomirs were administered to knock down miR-21 expression. The miR-21 levels were determined in isolated HSCs from mice after antagomir-21/antago-ctrl treatment. Antagomir-21 significantly reduced miR-21 content in HSCs in the TAA-induced model (Fig. 2A). Furthermore, α-SMA and COL1A1 mRNA expression was down-regulated in isolated primary HSCs after miR-21 knockdown (Fig. 2, B and C). Along with the reduced profibrogenic activities in HSCs, the total collagen deposition in the liver was much reduced (Fig. 2, D and E). Consistent with the histologic results, the hydroxyproline content was significantly down-regulated after antagomir-21 treatment (Fig. 2F). Similarly, in the CCl4-induced model, antagomir-21 greatly reduced miR-21 content in the HSCs and ameliorated hepatic fibrosis (data not shown). Accordingly, the results from animal models confirmed the involvement of miR-21 in HSC activation and in the pathogenesis of liver fibrosis.
FIGURE 2.

Attenuation of murine liver fibrosis by sequestering miR-21. Antagomir-21 or antago-ctrl was intravenously administered during TAA treatment in mice. A, the miR-21 content in the isolated HSCs was measured by qRT-PCR. The α-SMA mRNA (B) and COL1A1 mRNA (C) content in the isolated HSCs was determined by qRT-PCR. D, representative Sirius Red and H&E staining of mouse liver sections. Scale bars, 100 μm. Panel i, saline; panel ii, TAA; panel iii, TAA + antagomir-21; panel iv, TAA + antago-ctrl. E, semiquantitative analysis of the area of Sirius Red staining. F, the hydroxyproline content in mouse liver after antagomir-21/antago-ctrl treatment. The data are expressed as the means ± S.D. of three independent experiments. Each group contained at least three samples, and the isolated HSCs from two mice were mixed as one sample. ***, p < 0.001. Error bars represent S.D.
MiR-21 Was Up-regulated during HSC Activation
To confirm the miR-21 change during HSC activation, primary quiescent HSCs were harvested and cultured on plastic plates, which mimics the in vivo activation process (20). Quiescent HSCs begin to undergo activation on plastic plates and are positive for α-SMA until day 6, and the α-SMA levels peaked at ∼18 days, consistent with the reported results (21). Notably, the miR-21 level increased with the up-regulation of α-SMA expression (Fig. 3A), indicating that the miR-21 levels correlated to the activation status of HSCs.
FIGURE 3.
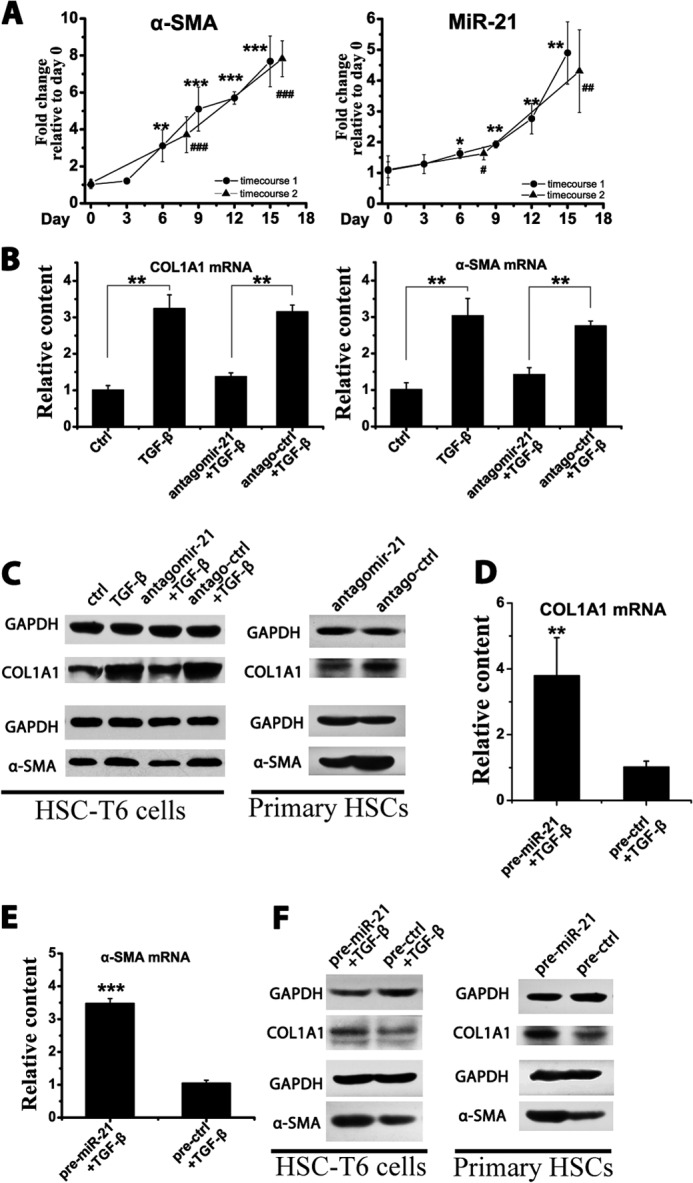
miR-21 could enhance HSC activation. A, isolated primary HSCs were cultured on plastic plates, and the α-SMA mRNA and miR-21 levels were measured by qRT-PCR after culture for the indicated number of days. B and C, HSC-T6 cells were transfected with 50 nm antagomirs and stimulated with 10 ng/ml TGF-β for 24 h, and isolated primary HSCs were cultured on plastic cells for 15 days during which 50 nm antagomirs were transfected three times. B, the COL1A1 and α-SMA mRNA levels in HSC-T6 cells were measured by qRT-PCR. C, the COL1A1 and α-SMA protein levels in HSC-T6 cells and primary HSCs were determined by Western blotting. D and E, HSC-T6 cells were transfected with 50 nm precursors and stimulated with 10 ng/ml TGF-β for 24 h. The COL1A1 (D) and α-SMA (E) mRNA levels were measured by qRT-PCR. F, the COL1A1 and α-SMA protein levels in HSC-T6 cells and primary HSCs were determined by Western blotting. Western blots were performed three times with similar results. The data are expressed as the means ± S.D. of three independent experiments, each containing at least three samples. * and #, p < 0.05; ** and ##, p < 0.01; *** and ###, p < 0.001. Error bars represent S.D.
To evaluate the involvement of miR-21 in HSC activation, expression of the HSC activation marker COL1A1 and α-SMA was analyzed by modulating miR-21 content. Antagomir-21 was first transfected to knock down miR-21 expression. Knocking down miR-21 expression down-regulated the COL1A1 and α-SMA mRNA levels in HSC-T6 cells (Fig. 3B). In line with the mRNA change, knocking down miR-21 reduced the COL1A1 and α-SMA protein levels in T6 cells, and this was further confirmed in primary HSCs (Fig. 3C).
Then the miR-21 content in HSCs was transiently increased by transfecting with precursor miR-21. Increasing the miR-21 content up-regulated the COL1A1 and α-SMA mRNA levels in HSC-T6 cells (Fig. 3, D and E) and the COL1A1 and α-SMA protein levels in HSC-T6 cells and primary HSCs (Fig. 3F).
MiR-21 Amplified the TGF-β1 Signaling Cascade by Down-regulating Inhibitory Smad7 and Up-regulating Smad2/3 Expression
Because the TGF-β signaling pathway is the major profibrogenic pathway during fibrosis, to determine the role of miR-21 in the TGF-β signaling pathway, antagomir-21/antago-ctrl or pre-miR-21/pre-ctrl was transfected into HSCs, and the expression profiles of Smad proteins were analyzed. Antagomir-21 attenuated Smad2 phosphorylation in HSC-T6 cells and primary HSCs (Fig. 4, A and B), whereas pre-miR-21 enhanced p-Smad2 levels (Fig. 4, C and D). Smad6/7 exhibits antagonistic effects on Smad2/3 phosphorylation (22) and is a direct target of miR-21 (4, 5). Antagomir-21 increased Smad7 levels, whereas pre-miR-21 decreased Smad7 expression in both HSC-T6 cells and primary HSCs (Fig. 4, E and F), suggesting a role for miR-21 in regulating Smad2 phosphorylation by modulating the Smad7 content.
FIGURE 4.

Regulation of the TGF-β/Smad signaling pathway by miR-21. HSC-T6 cells were transfected with 50 nm antagomirs or precursors and stimulated with 10 ng/ml TGF-β for 24 h. Isolated primary HSCs were cultured on plastic cells for 15 days, during which 50 nm antagomirs were transfected three times. A, the p-Smad2 and Smad2 protein levels in HSC-T6 cells of the antagomir group were measured by Western blotting, and the Smad2 mRNA levels in HSC-T6 cells were determined by qRT-PCR. B, the p-Smad2 and Smad2 protein levels in the primary HSCs of the antagomir group were measured by Western blotting. C, the p-Smad2 and Smad2 protein levels in HSC-T6 cells of the precursor group were measured by Western blotting, and the Smad2 mRNA levels in HSC-T6 cells were determined by qRT-PCR. D, the p-Smad2 and Smad2 protein levels in the primary HSCs of the precursor group were measured by Western blotting. E and F, the Smad7 protein levels in HSC-T6 cells (E) and primary HSCs (F) were measured by Western blotting. All of the Western blot experiments were repeated twice with a similar trend. The data are expressed as the means ± S.D. of three independent experiments; n = 3 per group. *, p < 0.05; **, p < 0.01; N.S., not significant. Error bars represent S.D.
Regulation of PTEN and p-MEK by MiR-21
Interestingly, reduced Smad2/3 protein levels were observed when knocking down miR-21 in HSC-T6 cells (Fig. 4A). The phosphorylation of MEK has been reported to enhance Smad2/3 expression (23). Furthermore, the phosphorylation of MEK could be depressed by PTEN, a direct target of miR-21 (24, 25). Therefore, miR-21 was hypothesized to regulate Smad2 levels by modulating the activities of PTEN and p-MEK. TGF-β1 significantly promoted the phosphorylation of MEK1/2. Antagomir-21 negated this effect, whereas PTEN was still maintained at a high level (Fig. 5, A and B). In contrast, pre-miR-21 reduced PTEN protein levels and potentiated MEK1/2 phosphorylation (Fig. 5, C and D).
FIGURE 5.
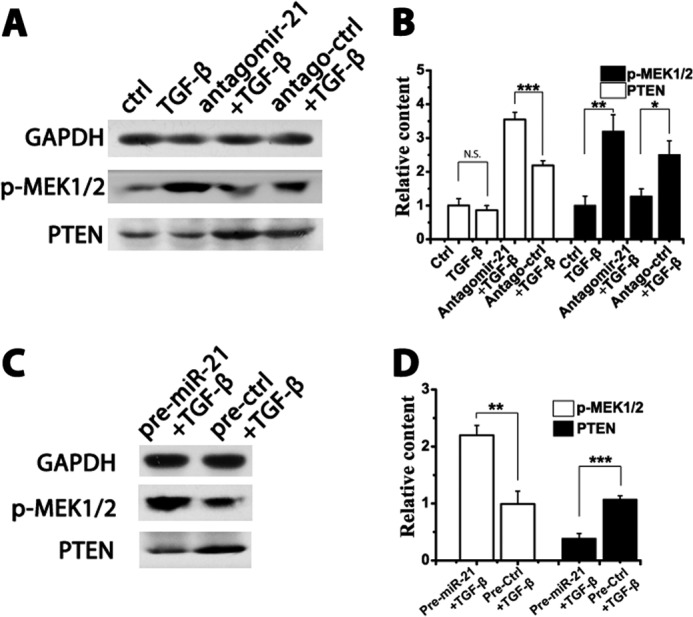
Regulation of PTEN and p-MEK by miR-21. HSC-T6 cells were transfected with 50 nm antagomirs or precursors and stimulated with 10 ng/ml TGF-β for 24 h. A, the protein levels of PTEN and p-MEK in the antagomir group were measured by Western blotting. B, densitometric quantification of the Western results in the antagomir group. C, Western blot results of the PTEN and p-MEK levels in the precursor group. D, densitometric quantification in the precursor group. All Western blot data represent one of three independent experiments. The data are expressed as the means ± S.D. of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; N.S., not significant. Error bars represent S.D.
MiR-21, PDCD4, and AP-1 Formed an Autoregulatory Feedback Loop in HSCs
Primary HSCs were harvested and cultured on uncoated plastic plates. Primary HSCs were cultured with two different AP-1 inhibitors: SR11302, an intrinsic AP-1 inhibitor, and SP600125, which interferes with c-Jun phosphorylation (26, 27). The miR-21 content was significantly down-regulated in SR11302- and SP600125-treated primary HSCs compared with the DMSO solvent-treated group (Fig. 6A). Moreover, to exclude the off-target effects of chemical inhibitors, an siRNA against c-Jun, the key member in the AP-1 family, was transfected into primary HSCs; similarly, the miR-21 levels were down-regulated (Fig. 6A). The co-culture of AP-1 inhibitors with HSC-T6 cells achieved a similar inhibition of miR-21 expression (data not shown). Therefore, inhibiting AP-1 activity suppressed miR-21 expression in HSCs.
FIGURE 6.

A double negative feedback loop mediated by miR-21, PDCD4, and AP-1 in HSCs. Isolated primary HSCs were cultured on plastic plates for 15 days during which cells were treated as indicated. HSC-T6 cells were transfected with 50 nm antagomirs or precursors and stimulated with 10 ng/ml TGF-β for 24 h. A, the miR-21 levels in primary HSCs were measured by qRT-PCR after co-culture with AP-1 inhibitors (10 μm SR11302, 15 μm SP600125, or DMSO vehicle) or after transfection with 50 nm c-Jun/ctrl siRNA. B, the miR-21 levels in primary HSCs were measured by qRT-PCR after transfection with 50 nm PDCD4/ctrl siRNA. The PDCD4 levels in HSC-T6 cells (C) and primary HSCs (D) were determined by Western blotting. The c-Jun and Fra-1 levels in HSC-T6 cells (E) and primary HSCs (F) were measured by Western blotting. G, HSC-T6 cells were transfected with antagomir-21 or pre-miR-21, and cell growth was determined at 12-h intervals after transfection. H, HSC-T6 cells were transfected with antagomir-21 or pre-miR-21, and 24 h after transfection, cells were exposed to serum-free medium (SF) plus 50 μm CHX for 4 h. Apoptosis in HSCs was determined by flow cytometry in three independent experiments. The Western blot data represent one of three independent experiments. The data are expressed as the means ± S.D. of three independent experiments, each containing at least three samples. *, p < 0.05; **, p < 0.01; ***, p < 0.001. *Control indicates the isolated primary HSCs cultured on plastic for 15 days without treatment. Error bars represent S.D.
It has been shown that PDCD4 has the ability to inhibit AP-1 activity (28, 29). A PDCD4 siRNA was transfected into primary HSCs and HSC-T6 cells. Along with the down-regulation of PDCD4 mRNA, the expression of miR-21 was up-regulated in the primary HSCs (Fig. 6B) and in HSC-T6 cells (data not shown).
MiR-21 can target PDCD4 for degradation (30). Antagomir-21 reversed the down-regulation of PDCD4 mediated by miR-21, whereas pre-miR-21 promoted the degradation of PDCD4 in HSC-T6 cells (Fig. 6C) and in primary HSCs when cultured on plastic plates (Fig. 6D).
Furthermore, the effects of modulating miR-21 levels on the expression of c-Jun and Fra-1 were determined in both HSC-T6 cells (Fig. 6E) and primary HSCs (Fig. 6F). Antagomir-21 decreased c-Jun and Fra-1 expression, whereas pre-miR-21 further enhanced c-Jun and Fra-1 expression.
Altogether, miR-21-mediated PDCD4 degradation maintained the maximal AP-1 activity in HSCs that could further promote miR-21 expression. Furthermore, miR-21-mediated PDCD4 down-regulation may affect HSC proliferation and protect activated HSCs from apoptosis as PDCD4 is a well known mediator in these processes (3, 31). HSC-T6 cells were transfected with antagomir-21 or pre-miR-21, and the cell growth index was analyzed accordingly. Increasing the miR-21 content enhanced HSC proliferation, whereas antagomir-21 slowed down cell growth (Fig. 6G). Furthermore, apoptosis in HSC-T6 cells was induced by serum deprivation plus CHX treatment (32). A significant increase in apoptosis was achieved in HSCs after 4-h incubation with CHX. Pre-miR-21 reduced apoptosis, whereas antagomir-21 potentiated apoptosis in CHX-treated HSCs (Fig. 6H).
Inhibiting AP-1 Activity Ameliorated HSC Activation and Hepatic Fibrosis
As noted, COL1A1 mRNA expression showed the same trend as miR-21 change when primary HSCs were co-cultured with AP-1 inhibitors (Fig. 7A). Furthermore, COL1A1 mRNA was down-regulated in primary HSCs after transfection with c-Jun siRNA (Fig. 7B). In addition, COL1A1 mRNA in primary HSCs increased after transfection with PDCD4 siRNA during culture on plastic (Fig. 7C). All these experiments were conducted in the HSC-T6 cell line as well and achieved similar results (data not shown).
FIGURE 7.

Attenuation of fibrogenic events by inhibiting AP-1 activity. Isolated primary HSCs were cultured on plastic plates for 15 days during which cells were treated as indicated. A, the COL1A1 mRNA levels in primary HSCs were measured by qRT-PCR after co-culture with 10 μm SR11302, 15 μm SP600125, or DMSO vehicle. B, the COL1A1 mRNA levels in primary HSCs were measured by qRT-PCR after transfection with c-Jun/ctrl siRNA. C, the COL1A1 mRNA levels in primary HSCs were measured by qRT-PCR after transfection with PDCD4/ctrl siRNA. D–F, during 8-week TAA-induced hepatic fibrosis, the mice were treated with SP600125. D, the miR-21 levels in the liver were measured by qRT-PCR after SP600125 treatment. E, representative image of Sirius Red staining of mouse liver sections after SP600125 treatment. Scale bars, 100 μm. Panel i, saline; panel ii, TAA; panel iii, TAA + SP600125; panel iv, TAA + DMSO. F, semiquantitative analysis of the area of Sirius Red staining and the hydroxyproline content in mouse liver after SP600125 treatment. One of three independent results is presented. The data are expressed as the means ± S.D. of three independent experiments, each containing at least three samples. *, p < 0.05; **, p < 0.01; ***, p < 0.001. *Control indicates the isolated primary HSCs cultured on plastic for 15 days without treatment. Error bars represent S.D.
To analyze whether modulating components in this feedback loop could affect hepatic fibrosis progression, mice were further treated with the AP-1 inhibitor SP600125 during TAA induction. In the SP600125-treated mouse liver, miR-21 was significantly down-regulated (Fig. 7D). Additionally, the mice treated with SP600125 exhibited much lower collagen deposition (Fig. 7E) and much lower hydroxyproline content (Fig. 7F). This autoregulatory feedback loop is illustrated in Fig. 8.
FIGURE 8.
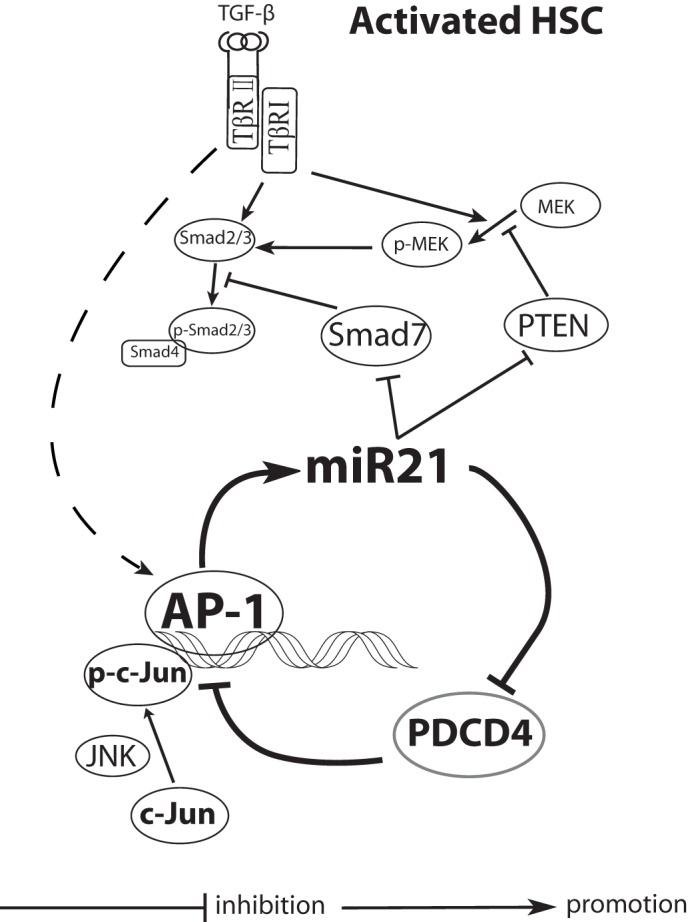
Schematic of the miR-21/PDCD4/AP-1 autoregulatory loop in perpetuating HSC activation. MiR-21-mediated PDCD4 degradation promoted AP-1 activity, which in turn maintained a constant high level of miR-21 in HSCs, finally leading to sustained HSC activation by modulating several targets. TβRI and TβRII, type I and type II TGF-β receptor, respectively.
DISCUSSION
During activation, HSCs undergo two stages: initiation and perpetuation. Maintaining the initial activation state can lead to the perpetuation state. TGF-β plays significant roles in both stages. However, recent evidence has suggested that TGF-β may not be the only driving force in the perpetuation stage. First, anti-TGF-β therapy failed to achieve positive outcomes in the clinic (33, 34). Second, activated HSCs display decreased surface receptors for TGF-β (35). Lastly, quiescent HSCs respond to TGF-β treatment by activation of R-Smads and display a functional negative feedback regulation via Smad7 induction, whereas after activation, in the perpetuation state, the activated HSCs are fully stimulated and display strong intrinsic R-Smad activation and, more importantly, lack Smad7 up-regulation in response to TGF-β stimulation (35). This implies that other mechanisms that suppress the anti-fibrotic Smad7 expression in the perpetuation stage exist. Taken altogether, in addition to TGF-β, these findings point to other driving forces underlying the perpetuation state in HSCs.
High levels of miR-21 represent a common feature of pathological cell proliferation and cellular stress (7). MiR-21 was found to be significantly up-regulated in liver fibrosis of different etiologies (4, 36, 37). However, in vivo, it remains elusive which liver cell type exhibits up-regulation of miR-21 expression, especially given that existing reports about the expression of miR-21 in in vitro activated HSCs are not consistent (4, 38). For the first time, we report that the up-regulated miR-21 predominantly occurs in activated HSCs in both human subjects and animal models. In addition, to date, there is no study addressing how miR-21 expression is elevated during hepatic fibrosis and, moreover, how miR-21 overexpression is linked to disease progression. JNK activation, whose effects are mainly mediated through AP-1 transcription factor, predominantly occurs in myofibroblasts during liver fibrogenesis (10), and AP-1 is one of the key transcription factors of miR-21. Moreover, PDCD4, as a direct target of miR-21, can inhibit AP-1 activity endogenously (3, 39). Thus, miR-21 could enhance the activity of AP-1 by down-regulating PDCD4 expression, which in turn results in the self-promoted miR-21 expression. In the present study, we proved that miR-21 was significantly up-regulated in activated HSCs during liver fibrogenesis in animal models, and this was further confirmed in human cirrhotic liver samples. Additionally, during activation in cultured primary HSCs, the miR-21 level correlated with the activation status of HSCs. In vivo data also demonstrated a close positive correlation between miR-21 levels in HSCs and hepatic fibrosis progression. Furthermore, titrating components in this loop with PDCD4 siRNA or by inhibiting AP-1 activity successfully modulated miR-21 levels and fibrogenic activities. Thus, by manipulating this feedback loop, once the expression has been elevated, miR-21 will maintain its expression at relatively high levels as well as the maximal AP-1 activity in HSCs.
Smad7, as an intrinsic inhibitor of the TGF-β/Smad2/3 pathway, competes with R-Smads for type I TGF receptor and intervenes in TGF-β/Smad2/3 signal transduction. Overexpressing Smad7 in rat livers inhibited collagen deposition and α-SMA expression in hepatic stellate cells (22). In recent studies, the down-regulation of Smad7 in tissue fibrosis was attributed to the high level miR-21 in fibrogenic cells. Thus, miR-21 generated from the miR-21/PDCD4/AP-1 feedback loop can directly target the antifibrotic Smad7 during fibrogenesis, connecting this self-promoting feed-forward loop with the fibrogenic machinery. Furthermore, a single microRNA may have multiple targets. Besides Smad7, in the present study, we demonstrated that PTEN was also significantly down-regulated by miR-21 in activated HSCs. PTEN has been implicated in inhibiting the formation of functional R-Smad complexes (40). In addition, based on the present study, PTEN may also regulate Smad2/3 expression via the PTEN/p-MEK pathway. Smad2/3 is the main signal mediator of the TGF-β signaling pathway and plays a pivotal role in profibrogenic gene expression. Targeted deletion of Smad2/3 could prevent or halt the progression of hepatic fibrosis in animals (41). This implies that high miR-21 content could potentially promote fibrogenic activities by directly up-regulating R-Smad expression. Together with the reported findings that AP-1 can induce autocrine expression of TGF-β and synergize with Smad2/3 in transcription of profibrogenic genes (42, 43), the miR-21/PDCD4/AP-1 feedback loop could greatly promote fibrogenic activities in HSCs during the perpetuation stage. In addition, studies have reported that miR-21 affects PDCD4 expression and thereby affects both proliferation and apoptosis (3, 31). The effect of miR-21 on HSC activation could further be achieved by protecting the fully activated HSCs from apoptosis, thus resulting in an increased presence of these cells. Taken altogether, the miR-21/PDCD4/AP-1 feedback loop could promote HSC activation and disease progression by affecting several targets, and this may also provide a possible explanation for how HSCs could undergo self-activation without cytokine stimulation. Therefore, the miR-21/PDCD4/AP-1 feedback loop could work as a major impetus that maintains the phenotype switch of HSCs, resulting in the continuous production of fibrogenic ECMs.
Our data herein shed further light on a possible common paradigm in shaping phenotypic outputs under different disease settings. MiR-21, which is overexpressed in almost all tumors analyzed so far (7), is also up-regulated in osteoclastogenesis (39) and fibrotic diseases. In addition, AP-1 activation is closely associated with cancer development, and inhibition of JNK activity, which is effective in cancer therapy, could also attenuate liver fibrosis (44). Therefore, the experimental outcomes from studies on different pathological processes (cancer, osteoclastogenesis, and fibrosis) suggest that the self-promoted miR-21/PDCD4/AP-1 feedback loop may be a common driving force behind different progressive diseases. Through coordinately regulating a suite of targets, this feedback loop thereby could drive the pathological machinery in response to continued pressure overload.
In summary, in activated HSCs, miR-21, PDCD4, and AP-1 form a double negative feedback circuit during liver fibrogenesis. Once activated, the circuit maintains the stable conversion of HSCs from the quiescent state into the large ECM-producing state. This finding provides additional insight into the phenotypes in fully activated HSCs. In this regard, therapeutics modulating this feedback could be further developed to treat hepatic fibrosis.
This work was supported by National Science Fund for Distinguished Young Scholars Grant 81025019; National Basic Research Program of China Grant 2012CB517603; National Natural Science Foundation of China Grants J1103512, J1210026, 31070722, 31271013, 31071232, 31170751, 31200695, 51173076, 91129712, and 81102489; Key Project of the Chinese Ministry of Education Grant 108059; Ph.D. Programs Foundation of the Ministry of Education of China Grant 20100091120020; and the Fundamental Research Funds for the Central Universities.
- ECM
- extracellular matrix
- α-SMA
- smooth muscle α-actin
- AP-1
- activation protein-1
- COL1A1
- collagen 1a1
- HSC
- hepatic stellate cell
- miRNA or miR
- microRNA
- PDCD4
- programmed cell death protein 4
- PTEN
- phosphatase and tensin homolog
- p-MEK
- phosphorylated MEK
- Smad
- small mothers against decapentaplegic homolog
- TAA
- thioacetamide
- antago-ctrl
- scrambled antagomir
- ctrl
- control
- qRT-PCR
- quantitative real time reverse transcription-polymerase chain reaction
- CHX
- cycloheximide
- R-Smad
- receptor-regulated Smad.
REFERENCES
- 1. Friedman S. L. (2008) Mechanisms of hepatic fibrogenesis. Gastroenterology 134, 1655–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mendell J. T., Olson E. N. (2012) MicroRNAs in stress signaling and human disease. Cell 148, 1172–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Talotta F., Cimmino A., Matarazzo M. R., Casalino L., De Vita G., D'Esposito M., Di Lauro R., Verde P. (2009) An autoregulatory loop mediated by miR-21 and PDCD4 controls the AP-1 activity in RAS transformation. Oncogene 28, 73–84 [DOI] [PubMed] [Google Scholar]
- 4. Marquez R. T., Bandyopadhyay S., Wendlandt E. B., Keck K., Hoffer B. A., Icardi M. S., Christensen R. N., Schmidt W. N., McCaffrey A. P. (2010) Correlation between microRNA expression levels and clinical parameters associated with chronic hepatitis C viral infection in humans. Lab. Invest. 90, 1727–1736 [DOI] [PubMed] [Google Scholar]
- 5. Liu G., Friggeri A., Yang Y., Milosevic J., Ding Q., Thannickal V. J., Kaminski N., Abraham E. (2010) MiR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 207, 1589–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chung A. C., Dong Y., Yang W., Zhong X., Li R., Lan H. Y. (2013) Smad7 suppresses renal fibrosis via altering expression of TGF-β/Smad3-regulated microRNAs. Mol. Ther. 21, 388–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krichevsky A. M., Gabriely G. (2009) MiR-21: a small multi-faceted RNA. J. Cell. Mol. Med. 13, 39–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thum T., Gross C., Fiedler J., Fischer T., Kissler S., Bussen M., Galuppo P., Just S., Rottbauer W., Frantz S., Castoldi M., Soutschek J., Koteliansky V., Rosenwald A., Basson M. A., Licht J. D., Pena J. T., Rouhanifard S. H., Muckenthaler M. U., Tuschl T., Martin G. R., Bauersachs J., Engelhardt S. (2008) MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456, 980–984 [DOI] [PubMed] [Google Scholar]
- 9. Noetel A., Kwiecinski M., Elfimova N., Huang J., Odenthal M. (2012) microRNA are central players in anti- and profibrotic gene regulation during liver fibrosis. Front. Physiol. 3, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kluwe J., Pradere J. P., Gwak G. Y., Mencin A., De Minicis S., Osterreicher C. H., Colmenero J., Bataller R., Schwabe R. F. (2010) Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology 138, 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bataller R., Sancho-Bru P., Ginès P., Lora J. M., Al-Garawi A., Solé M., Colmenero J., Nicolás J. M., Jiménez W., Weich N., Gutiérrez-Ramos J. C., Arroyo V., Rodés J. (2003) Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 125, 117–125 [DOI] [PubMed] [Google Scholar]
- 12. Lee J. I., Lee K. S., Paik Y. H., Nyun Park Y., Han K. H., Chon C. Y., Moon Y. M. (2003) Apoptosis of hepatic stellate cells in carbon tetrachloride induced acute liver injury of the rat: analysis of isolated hepatic stellate cells. J. Hepatol. 39, 960–966 [DOI] [PubMed] [Google Scholar]
- 13. Vogel S., Piantedosi R., Frank J., Lalazar A., Rockey D. C., Friedman S. L., Blaner W. S. (2000) An immortalized rat liver stellate cell line (HSC-T6): a new cell model for the study of retinoid metabolism in vitro. J. Lipid Res. 41, 882–893 [PubMed] [Google Scholar]
- 14. de Planell-Saguer M., Rodicio M. C., Mourelatos Z. (2010) Rapid in situ codetection of noncoding RNAs and proteins in cells and formalin-fixed paraffin-embedded tissue sections without protease treatment. Nat. Protoc. 5, 1061–1073 [DOI] [PubMed] [Google Scholar]
- 15. Taura K., Miura K., Iwaisako K., Osterreicher C. H., Kodama Y., Penz-Osterreicher M., Brenner D. A. (2010) Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 51, 1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chu A. S., Diaz R., Hui J. J., Yanger K., Zong Y., Alpini G., Stanger B. Z., Wells R. G. (2011) Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology 53, 1685–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Higashiyama R. (2009) Negligible contribution of bone marrow-derived cells to collagen production during hepatic fibrogenesis in mice. Gastroenterology 137, 1459–1466.e1 [DOI] [PubMed] [Google Scholar]
- 18. Frezzetti D., De Menna M., Zoppoli P., Guerra C., Ferraro A., Bello A. M., De Luca P., Calabrese C., Fusco A., Ceccarelli M., Zollo M., Barbacid M., Di Lauro R., De Vita G. (2011) Upregulation of miR-21 by Ras in vivo and its role in tumor growth. Oncogene 30, 275–286 [DOI] [PubMed] [Google Scholar]
- 19. Fujita S., Ito T., Mizutani T., Minoguchi S., Yamamichi N., Sakurai K., Iba H. (2008) MiR-21 gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J. Mol. Biol. 378, 492–504 [DOI] [PubMed] [Google Scholar]
- 20. Friedman S. L. (1993) Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. N. Engl. J. Med. 328, 1828–1835 [DOI] [PubMed] [Google Scholar]
- 21. Thompson K. C., Trowern A., Fowell A., Marathe M., Haycock C., Arthur M. J., Sheron N. (1998) Primary rat and mouse hepatic stellate cells express the macrophage inhibitor cytokine interleukin-10 during the course of activation in vitro. Hepatology 28, 1518–1524 [DOI] [PubMed] [Google Scholar]
- 22. Dooley S., Hamzavi J., Breitkopf K., Wiercinska E., Said H. M., Lorenzen J., Ten Dijke P., Gressner A. M. (2003) Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 125, 178–191 [DOI] [PubMed] [Google Scholar]
- 23. Ross K. R., Corey D. A., Dunn J. M., Kelley T. J. (2007) SMAD3 expression is regulated by mitogen-activated protein kinase kinase-1 in epithelial and smooth muscle cells. Cell. Signal. 19, 923–931 [DOI] [PubMed] [Google Scholar]
- 24. Vinciguerra M., Sgroi A., Veyrat-Durebex C., Rubbia-Brandt L., Buhler L. H., Foti M. (2009) Unsaturated fatty acids inhibit the expression of tumor suppressor phosphatase and tensin homolog (PTEN) via microRNA-21 up-regulation in hepatocytes. Hepatology 49, 1176–1184 [DOI] [PubMed] [Google Scholar]
- 25. Meng F., Henson R., Wehbe-Janek H., Ghoshal K., Jacob S. T., Patel T. (2007) MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133, 647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang C., Ma W. Y., Dawson M. I., Rincon M., Flavell R. A., Dong Z. (1997) Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc. Natl. Acad. Sci. U.S.A. 94, 5826–5830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bennett B. L., Sasaki D. T., Murray B. W., O'Leary E. C., Sakata S. T., Xu W., Leisten J. C., Motiwala A., Pierce S., Satoh Y., Bhagwat S. S., Manning A. M., Anderson D. W. (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U.S.A. 98, 13681–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang H. S., Knies J. L., Stark C., Colburn N. H. (2003) Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene 22, 3712–3720 [DOI] [PubMed] [Google Scholar]
- 29. Wang Q., Sun Z., Yang H. S. (2008) Downregulation of tumor suppressor Pdcd4 promotes invasion and activates both β-catenin/Tcf and AP-1-dependent transcription in colon carcinoma cells. Oncogene 27, 1527–1535 [DOI] [PubMed] [Google Scholar]
- 30. Frankel L. B., Christoffersen N. R., Jacobsen A., Lindow M., Krogh A., Lund A. H. (2008) Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 283, 1026–1033 [DOI] [PubMed] [Google Scholar]
- 31. Lankat-Buttgereit B., Göke R. (2009) The tumour suppressor Pdcd4: recent advances in the elucidation of function and regulation. Biol. Cell 101, 309–317 [DOI] [PubMed] [Google Scholar]
- 32. Issa R., Williams E., Trim N., Kendall T., Arthur M. J., Reichen J., Benyon R. C., Iredale J. P. (2001) Apoptosis of hepatic stellate cells: involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut 48, 548–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hernandez-Gea V., Friedman S. L. (2011) Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 6, 425–456 [DOI] [PubMed] [Google Scholar]
- 34. Friedman S. L. (2010) Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 7, 425–436 [DOI] [PubMed] [Google Scholar]
- 35. Dooley S., Delvoux B., Lahme B., Mangasser-Stephan K., Gressner A. M. (2000) Modulation of transforming growth factor β response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology 31, 1094–1106 [DOI] [PubMed] [Google Scholar]
- 36. Jiang J., Gusev Y., Aderca I., Mettler T. A., Nagorney D. M., Brackett D. J., Roberts L. R., Schmittgen T. D. (2008) Association of microRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clin. Cancer Res. 14, 419–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X. W., Heegaard N. H., Orum H. (2012) MicroRNAs in liver disease. Gastroenterology 142, 1431–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maubach G., Lim M. C., Chen J., Yang H., Zhuo L. (2011) MiRNA studies in in vitro and in vivo activated hepatic stellate cells. World J. Gastroenterol. 17, 2748–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sugatani T., Vacher J., Hruska K. A. (2011) A microRNA expression signature of osteoclastogenesis. Blood 117, 3648–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hjelmeland A. B., Hjelmeland M. D., Shi Q., Hart J. L., Bigner D. D., Wang X. F., Kontos C. D., Rich J. N. (2005) Loss of phosphatase and tensin homologue increases transforming growth factor β-mediated invasion with enhanced SMAD3 transcriptional activity. Cancer Res. 65, 11276–11281 [DOI] [PubMed] [Google Scholar]
- 41. Schnabl B., Kweon Y. O., Frederick J. P., Wang X. F., Rippe R. A., Brenner D. A. (2001) The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology 34, 89–100 [DOI] [PubMed] [Google Scholar]
- 42. Kim S. J., Angel P., Lafyatis R., Hattori K., Kim K. Y., Sporn M. B., Karin M., Roberts A. B. (1990) Autoinduction of transforming growth factor β1 is mediated by the AP-1 complex. Mol. Cell. Biol. 10, 1492–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chinenov Y., Kerppola T. K. (2001) Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 20, 2438–2452 [DOI] [PubMed] [Google Scholar]
- 44. Wagner E. F., Nebreda A. R. (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev.. Cancer 9, 537–549 [DOI] [PubMed] [Google Scholar]