

Background: Class III myosins contain both a motor and kinase domain.
Results: Phosphorylation of the kinase activation loop enhances MYO3A kinase activity, augmenting autophosphorylation-induced attenuation of motor and cellular activity.
Conclusion: MYO3A kinase activity mediates localization and function within actin protrusions.
Significance: Characterizing MYO3A kinase regulation enhances our understanding of the role of MYO3A in the maintenance of actin protrusions found in sensory epithelia.
Keywords: Actin, Hearing, Molecular Motors, Myosin, Phosphorylation
Abstract
Class III myosins are unique members of the myosin superfamily in that they contain both a motor and kinase domain. We have found that motor activity is decreased by autophosphorylation, although little is known about the regulation of the kinase domain. We demonstrate by mass spectrometry that Thr-178 and Thr-184 in the kinase domain activation loop and two threonines in the loop 2 region of the motor domain are autophosphorylated (Thr-908 and Thr-919). The kinase activity of MYO3A 2IQ with the phosphomimic (T184E) or phosphoblock (T184A) mutations demonstrates that kinase activity is reduced 30-fold as a result of the T184A mutation, although the Thr-178 site only had a minor impact on kinase activity. Interestingly, the actin-activated ATPase activity of MYO3A 2IQ is slightly reduced as a result of the T178A and T184A mutations suggesting coupling between motor and kinase domains. Full-length GFP-tagged T184A and T184E MYO3A constructs transfected into COS7 cells do not disrupt the ability of MYO3A to localize to filopodia structures. In addition, we demonstrate that T184E MYO3A reduces filopodia elongation in the presence of espin-1, whereas T184A enhances filopodia elongation in a similar fashion to kinase-dead MYO3A. Our results suggest that as MYO3A accumulates at the tips of actin protrusions, autophosphorylation of Thr-184 enhances kinase activity resulting in phosphorylation of the MYO3A motor and reducing motor activity. The differential regulation of the kinase and motor activities allows for MYO3A to precisely self-regulate its concentration in the actin bundle-based structures of cells.
Introduction
Class III myosins are unique in that they contain a conserved N-terminal kinase domain and central motor domain, whereas the C-terminal tail varies in sequence between isoforms (1, 2). The kinase domain is proposed to allow autoregulation of the myosin III motor (3), although regulation of the kinase domain is unknown. The first N-terminal kinase containing myosin was identified in Drosophila photoreceptors and is referred to as NINAC (neither inactivation nor after-potential C), because deletion of this gene disrupts the phototransduction electrophysiological response in a specific manner (4). The invertebrate kinase/motor myosins were recently classified as class XXI myosins due to their unique motor domain sequences (5). It is intriguing to suggest that the presence of an N-terminal kinase allows these kinase/motors to perform specific cellular functions, and further study is necessary to establish these mechanisms.
There are two isoforms of class III myosins expressed in vertebrates, MYO3A and MYO3B. Both isoforms are localized to actin bundle-based structures in sensory cells, including the calycal process of photoreceptors (2, 6, 7) and the stereocilia of inner ear hair cells (8). MYO3A and -3B are proposed to play a role in maintaining the length of the actin-bundled structures (9, 10). The ability of MYO3A and -3B to transport the actin regulatory protein, espin-1 (ESPN1), to the tips of actin bundle-based structures is thought to be critical for MYO3-based elongation of actin protrusions (9, 10). ESPN1 contains WH2 activity, which binds actin monomers and promotes filament elongation. Disruption of the MYO3A gene is associated with nonsyndromic deafness (DFNB30) (11), and a mouse model of DFNB30 results in age-dependent degeneration of the stereocilia of inner ear hair cells (12). Therefore, characterizing mechanisms of MYO3 motor and kinase regulation are critical for understanding its role in sensory cells.
Early studies found that class III myosins are not only expressed in the inner ear and retina but also in brain, intestine, and testes (2). Recently, modifications in the MYO3A gene were found to be associated with bladder and colon cancer and may be a risk factor for anxiety disorders. In a study that examined gene methylation markers, MYO3A was found along with five other genes to be a biomarker for bladder cancer (13). In addition, a genome-wide association study found that single nucleotide polymorphisms in the intronic region of the MYO3A gene were associated with an increased risk for colon cancer (14). Interestingly, the genome-wide association study found that MYO3A, along with several members of the MAPK pathway, were associated with colon cancer. Another study found single nucleotide polymorphisms in the MYO3A intronic region were associated with anxiety disorders (15). This study also found MYO3A mRNA expression in specific regions of the brain in humans, including cingulated cortex and amygdala. These studies highlight the importance of determining the kinase substrates for MYO3A and understanding its role in specific signaling pathways.
The kinase domain of vertebrate class III myosins is a member of the sterile-20 (STE20)3 family of kinases (16). Many members of the STE20 family of kinases as well as other kinase families are regulated by autophosphorylation (17). p21-activated protein kinases are well studied members of the STE20 family and contain a conserved kinase activation loop that requires phosphorylation to be fully activated (18–20). MYO3A and -3B contain several conserved threonines and serines in the putative activation loop sequence (Fig. 1). Therefore, we hypothesize that the kinase domain of class III myosins is regulated by phosphorylation of conserved residues in the activation loop.
FIGURE 1.
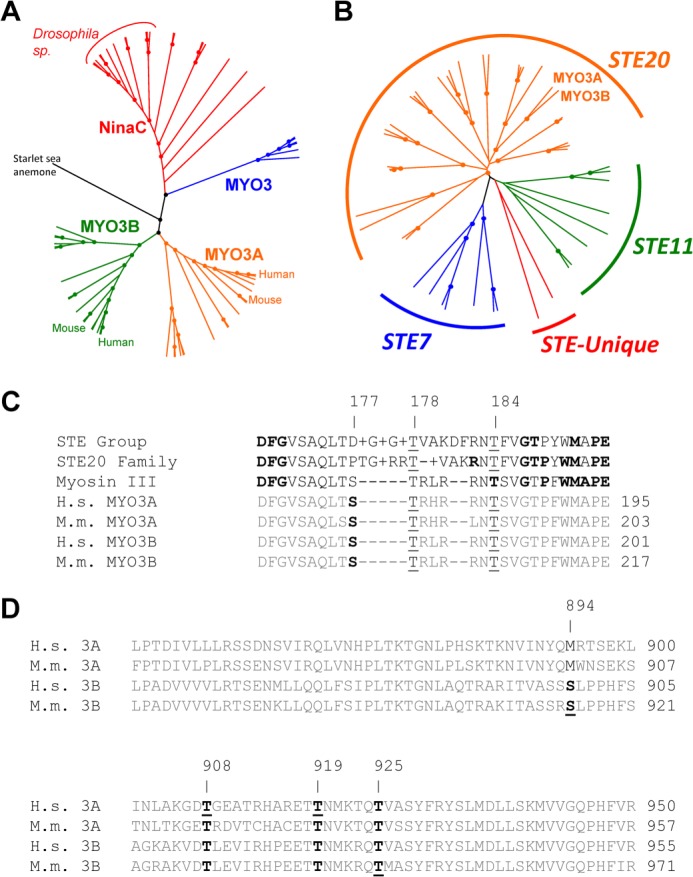
Sequence analysis of the MYO3A kinase domain. Unrooted phylogenetic trees of 61 class III myosins (A) and STE group kinases in the human genome (B) generated from ClustalW2 (default settings) alignments. Dots represent nodes validated by bootstrapping values of greater than 80%. C, alignments of the activation loop of MYO3A with consensus sequences from the STE group, STE20 family, and class III myosins. Boldface residues in the STE group, STE20 family, and class III consensus sequences represent 80% conservation at that position within that category. The underlines indicate well conserved phosphorylatable residues corresponding to positions 177, 178, and 184 of human MYO3A. Although threonine is the consensus residue at the 184 position, it is not completely conserved. However, all sequences except for MAP3K1 and COT contain a phosphorylatable residue within 1 amino acid of the 184 position. D, alignments of the human and mouse MYO3A and MYO3B loop 2 sequences from within the myosin motor domain. Boldface residues indicate well conserved phosphorylatable sequences in either MYO3A or MYO3B. Underlined residues indicate identified phosphorylation sites in human MYO3A from this study, and mouse MYO3A and MYO3B from a previous study (21).
A previous study examined the phosphorylation sites in the kinase, motor, and tail domains of mouse MYO3A and MYO3B (21). In this study, they expressed the kinase domain only and determined its ability to autophosphorylate the kinase domain as well as phosphorylate polypeptides of the motor and tail domains. However, the regulatory role and potential biological function of the phosphorylation sites were not investigated. Based on studies of MYO3A, we have proposed a model of class III myosin regulation in the actin bundle-based structures (3). In this model, MYO3A translocates to the tips of actin bundle-based structures with an active dephosphorylated motor domain. Upon reaching the tip of the actin bundle, MYO3A will become more concentrated in the tip compartment and be more likely to undergo intermolecular autophosphorylation. The phosphorylated form of MYO3A contains reduced motor activity and is therefore more likely to be transported back to the cell body by diffusion or retrograde flow. In this study, we investigated how phosphorylation of specific sites in the kinase domain modulates kinase and motor activity. Our results suggest that a single phosphorylation site in the kinase activation loop is critical for regulating the kinase activity of MYO3A. We propose that phosphorylation-dependent regulation of the kinase and motor domains allows class III myosins to autoregulate their localization in actin bundle-based structures.
EXPERIMENTAL PROCEDURES
Reagents
ATP and ADP were prepared from powder (22). Radiolabeled [γ-32P]ATP was purchased from PerkinElmer Life Sciences and contained a specific activity of 10 Ci/mmol. All solution experiments were performed in KMg50 buffer (10 mm imidazole Cl, pH 7.0, 50 mm KCl, 1 mm EGTA, 1 mm MgCl2, 1 mm DTT).
Construction of Expression Plasmids
The previously generated construct of human MYO3A containing 2IQ domains (MYO3A 2IQ) (3, 23, 24) was modified with site-directed mutagenesis to introduce the phosphorylation site mutations (T178A and T178D; T184A and T184E) and a mutation to abolish motor activity (E722A). The T184E and T184A mutations were also introduced in the full-length GFP-tagged version of MYO3A for expression in COS7 cells (3). We generated a construct containing only the MYO3A kinase domain (MYO3 kinase, residues 1–339). The MYO3A 2IQ ΔK, K50R, and ESPN1 constructs were described previously (3). All baculovirus constructs contained a C-terminal FLAG tag for purification purposes.
Protein Expression and Purification
The MYO3A kinase and MYO3A 2IQ constructs were expressed with the baculovirus/SF9 cell system and purified as described (3, 23, 24). Protein purity was assessed by Coomassie-stained SDS-polyacrylamide gels, and protein concentration was determined by the Bradford assay using BSA as a standard or by absorbance using the predicted extinction coefficients (3, 23, 24). Actin was purified from rabbit skeletal muscle using an acetone powder method (25). The actin concentration was determined by absorbance at 290 nm (ϵ290 = 2.66 × 104 m−1·cm−1). A molar equivalent of phalloidin was added to stabilize F-actin.
Sequence Analysis
Sequences for 61 class III myosins were identified by Odronitz et al. (5). Sequences for the complement of human STE group and STE20 family proteins were identified from the supplemental information in Manning et al. (16). ClustalW2 (26) with the default settings was used to align the amino acid sequences, determine consensus sequences, and generate the unrooted phylogenetic tree. Bootstrapping was performed using SeaView 4.2.6 (27), and FigTree1.3.1 (28) was used to draw the tree.
Mass Spectrometry Analysis
Protein bands (n = 5 phosphorylated and n = 5 unphosphorylated) from one-dimensional SDS-polyacrylamide gels were digested in-gel with trypsin overnight. Digestions were terminated with formic acid, and the extracted tryptic peptides were centrifuged under vacuum until ∼1–2 μl of liquid remained. Fifteen microliters of 0.1% formic acid in water was then added. Reversed phase HPLC-tandem mass spectrometric analysis was performed on the MYO3A tryptic peptides using a hybrid linear ion trap-Orbitrap mass spectrometer (LTQ Orbitrap XL, Thermo, San Jose, CA) operated with the Xcalibur (version 2.0) data acquisition software. The MYO3A digests were loaded onto a 75-μm inner diameter × 2-cm capillary trap (ProteoPep II, New Objective), desalted with 3% acetonitrile, 0.1% formic acid for 5 min prior to injection onto a 75-μm inner diameter × 15-cm C18 (ProteoPep II, New Objective) analytical column. A 90-min linear gradient to 40% acetonitrile at 250 nl/min was provided by a two-dimensional Ultra nano-HPLC system (Eksigent, Dublin, CA). Full m/z survey scans were acquired in the Orbitrap mass analyzer at mass resolving power of 60,000 followed by parallel MS/MS linear ion trap analysis of the top five most intense precursor ions. Collision-induced dissociation in the linear ion trap was carried out using helium at a 3.0-μm isolation width and 35% normalized collision energy.
Database Searching
LC-MS/MS data were extracted by BioWorks version 3.3 and searched against the IPI human protein database containing both forward and reversed sequences (version 3.54, 150,852 entries) using the Mascot version 2.2 (Matrix Science, Boston) search algorithm. A fragment ion mass tolerance of 0.80 Da and a parent ion tolerance of 10.0 ppm were used. Additionally, trypsin was specified as the digestion enzyme with the possibility of one missed cleavage. Carbamidomethylation of cysteine was defined as a fixed modification, whereas oxidation of methionine and phosphorylation of serine, threonine, and tyrosine were defined as variable modifications. The software program Scaffold (version Scaffold-3_00_06, Proteome Software Inc., Portland, OR) was used to validate MS/MS-based peptide and protein identifications using the following criteria: peptide identifications were accepted at greater than 95.0% probability based on the Peptide Prophet algorithm (29); protein identification was accepted at greater than 99.0% probability based on the Protein Prophet algorithm (30) with at least two unique identified peptides. The Scaffold results were then exported as an mzIdentML file and loaded into Scaffold PTM (Proteome Software) to further characterize phosphorylation sites obtained from MS/MS data. The Ascore algorithm (31) incorporated within Scaffold PTM was used to re-analyze the Mascot search results by assigning an ambiguity score and a site location probability for each phosphorylation site.
Kinase Activity Assays
Autophosphorylation of MYO3A 2IQ was detected by kinase assay using [32P]ATP or anti-phosphothreonine Western blotting (3, 23, 24). Myosin at specified concentrations was allowed to react with 200 μm [32P]ATP at room temperature (∼22 °C) for specific time periods ranging from 0 to 60 min. The reaction was stopped at each time point by the addition of SDS loading buffer. Samples were run on an SDS-polyacrylamide gel, and the incorporation of [32P] into MYO3A was detected by phosphorimaging using the Typhoon 8600 Variable Mode Imager (GE Healthcare). Following phosphorimaging, the gel was Coomassie-stained to assess evenness of loading. Densitometry analysis using ImageJ software (National Institutes of Health) was used to determine band intensities that were compared relative to wild-type MYO3A 2IQ.
We also determined the number of moles of [32P] incorporation per mol of MYO3A by excising MYO3A 2IQ from the Coomassie-stained SDS-polyacrylamide gels, dissolving them into 0.5 ml of hydrogen peroxide for 24–48 h and examining them by scintillation counting. The samples were compared with a standard curve to determine moles of [32P] incorporation.
Steady-state ATPase Activity
Steady-state ATP hydrolysis by MYO3A 2IQ (50–100 nm) in the absence and presence of actin (0–60 μm) was examined by using the NADH-linked assay (3, 23, 24, 32) with a final MgATP concentration of 1 mm. The assay was performed in an Applied Photophysics stopped-flow spectrofluorometer (Surrey, UK) in which the NADH absorbance at 340 nm was monitored continuously for 200 s. The ATPase rate at each actin concentration was determined, and the Michaelis-Menten equation was used to calculate the kcat and KATPase (V0 + ((kcat [actin])/(KATPase + [actin]))), where V0 is the ATPase rate in the absence of actin; kcat is the maximal ATPase rate, and KATPase is the actin concentration at which the ATPase activity is one-half maximal. Uncertainties are reported as standard errors of the fits unless stated otherwise. The data at each actin concentration represent an average of 3–5 protein preparations.
Cell Culture and Transient Transfection
COS7 cells were grown as described previously (3). Briefly, cell cultures were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum, 1 mm sodium pyruvate, 2 mm GlutaMAX (Invitrogen), and 100 units of penicillin/streptomycin. Cells were passaged using 0.25% trypsin/EDTA. Prior to transfection, the cells were plated onto acid-washed, 22-mm2, number 1.5 coverslips at ∼33,000 cells/coverslip (one coverslip/well of a 6-well dish) and allowed to adhere overnight. Cells were transiently transfected using FuGENE HD (Promega, Madison, WI). Briefly, 0.3 μg of plasmid DNA (0.6 μg total for co-transfection experiments) was diluted into 100 μl of Opti-MEM media (Invitrogen) without serum or antibiotics and mixed with 3 μl of FuGENE HD. After gentle mixing, the transfection complexes were allowed to form by incubation at room temperature for 15 min and then added dropwise to the well containing the cells to be transfected (2 ml of medium/well).
Imaging of Live Cells Expressing Fluorescent Proteins
Transfected cells were imaged for ∼18–30 h following transfection. The coverslips were assembled into Rose chambers containing imaging medium (Opti-MEM without phenol red supplemented with 5% fetal bovine serum and 100 units of penicillin/streptomycin). During imaging, a Nevtek ASI400 airstream incubator was used to maintain the cellular environment at ∼35 °C. Images were collected using a Nikon TE2000-PFS fluorescence microscope with a 60 × 1.4 N.A. phase objective. Images were obtained using a CoolSnap HQ2-cooled charge-coupled device digital camera (Photometrics, Tucson, AZ) and NIS Elements AR software (Nikon, Tokyo, Japan). For single images, exposure times were held constant at 400 ms for all fluorescence channels and 60 ms for the bright field channel (phase contrast). Images were collected for all three channels for all treatment groups.
Imaging of Fixed Cells Expressing Fluorescent Proteins
COS7 cells were plated and transfected using the protocol described above and then incubated for 16–24 h. Samples were fixed for 10 min in 3.7% paraformaldehyde in phosphate-buffered saline (PBS), permeabilized for 2 min in 0.5% Triton X-100 in PBS, counterstained with 6.6 nm Alexa Fluor-568 phalloidin (Invitrogen), and mounted in PBS with 50% glycerol and 3% n-propyl gallate. Images were obtained with the microscope and camera described above. The images for the GFP MYO3A and ESPN1 co-transfections were collected with Nikon Eclipse microscope and Ultraview confocal microscope with a 100 × 1.4 NA objective (PerkinElmer Life Sciences).
Data Analysis, Statistical Analysis, and Software Used
Both NIS Elements AR (Nikon) and Microsoft Excel 2007 were used for image analysis. The ratio of tip intensity to cell body intensity (rt/c) was calculated as described previously (3). Integrated intensity was measured for a 4 × 4-pixel region of the background (bb), and the filopodial tip (bt), the cell body (bc), and the rt/c values were calculated as shown in Equation 1,
 |
Filopodial density was calculated from phase contrast images by dividing the number of thin protrusions extending from a cell margin (not contacting another cell) by the length of that cell margin.
Data were compared by Tukey analysis using Minitab 12 software. Data are expressed as means ± S.E. Box-whisker plots in the figures represent the 90th and 10th percentiles with the whiskers, 75th and 25th percentiles with the box top and box bottom, median with the line across the box, and mean with a symbol (■) within the box. All images were prepared for publication using NIS-Elements AR, Metamorph Version 6.0, Photoshop CS, or some combination of these software packages.
RESULTS
Sequence Analysis
To identify well conserved regions within the kinase activation loop, we compared the entire core kinase domain of human MYO3A and MYO3B to 59 other class III myosin sequences available (Fig. 1A) (5) and the 47 other kinases within the STE group (Fig. 1B). We determined the consensus sequence for the activation loop of the STE kinase group, the STE20 kinase family, and class III myosins. By aligning the activation loops of human and mouse MYO3A and MYO3B with the consensus activation loops (Fig. 1C), we were able to identify putative well conserved phosphorylatable residues aligning with threonines 178 and 184 of the human MYO3A sequence.
Alignment also revealed putative phosphorylation sites in loop 2 of the myosin motor domain, which were often well conserved within subclasses of myosin III. The position corresponding to human MYO3A Thr-908 is phosphorylatable in all available MYO3B sequences as well as MYO3A sequences not from fish. The threonine at position 919 in human MYO3A was conserved across all MYO3A and -3B sequences. Interestingly, the corresponding residue in NINAC and invertebrate MYO3 sequences was found to be glutamine, with the exception of starlet sea anemone MYO3. The sequence corresponding to human MYO3A Thr-925 was conserved across all sequences available except for Limulus NinaC. Taken together, these data suggest that although phosphorylation may be a common mechanism of regulation for class III myosins, there may be differences in the specific sites that are regulatory.
Mass Spectrometry
To examine the sites in the kinase domain that are phosphorylated, we performed mass spectrometry analysis focusing on the phosphorylation sites in MYO3A 2IQ. The results from the mass spectrometry analysis are shown in Fig. 2 and Table 1. LC-MS/MS analysis was performed on five separate samples for each group, phosphorylated (incubated at room temperature for 60 min in the presence of 200 μm ATP) and unphosphorylated (incubated without ATP). To be stringent in terms of confidence of site identification and localization, the results were filtered based on identification of the phosphorylation site in at least two samples out of five; the phosphopeptide identification probability was 95%, and the site location probability was at least 75%. With these criteria, five unique phosphorylation sites were identified, including Ser-177, Thr-178, and Thr-184 for unphosphorylated and Thr-184, Thr-908, and Thr-919 for phosphorylated samples. It is important to note that it was difficult to unambiguously resolve the phosphorylation site location in the phosphopeptide containing Ser-177 and Thr-178 given the limited amount of fragment ion information representing the C-terminal region in the unphosphorylated samples for certain MS/MS spectra. However, the total spectral counts (SC) for MS/MS spectra associated with Thr-178 assignment was greater (SC = 7) than for S177 (SC = 2), suggesting the phosphorylation site most likely resides on Thr-178 based on the consistency of this site location assignment across multiple replicates. Additionally, given the limited amount of fragment ion representation of the N-terminal region in the tryptic peptide that contained Thr-184, it was sometimes difficult to unambiguously assign the phosphorylation site to Thr-184 given that the proximal residue, Ser-185, could also be phosphorylated based on certain MS/MS site assignments by Scaffold. However, using our stringent filtering criteria as well as spectral count information, we determined the phosphorylation site to be localized to Thr-184. Therefore, we focused on examining the two most likely phosphorylation sites in the kinase activation loop (Thr-178 and Thr-184).
FIGURE 2.
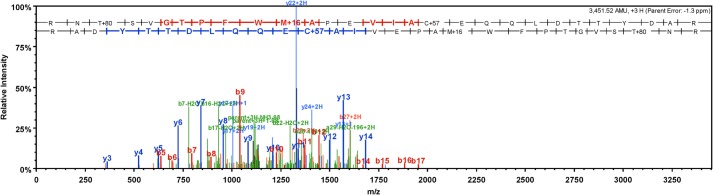
Identification of Thr-184 phosphorylation by mass spectrometry. MS/MS spectrum showing the annotated fragment ions of RNpTSVGTPFWoxMAPEVIAC*EQQLDTTYDAR, where C* indicates carbamidomethylation of cysteine. Site localization (to Thr-184) was assigned using the Ascore algorithm.
TABLE 1.
Phosphorylation sites identified by mass spectrometry
Phos, phosphorylated; UnPhos, unphosphorylated. C* indicates carbamidomethylation of cysteine.
| Condition | Peptide sequence | Modified site | Best A score | Localization probability | Best Mas-cot score | Mascot identity thres-hold | Total spectral counts |
|---|---|---|---|---|---|---|---|
| % | |||||||
| UnPhos | LVDFGVSAQLTpSTR | Ser-177 | 9.07 | 79 | 57.6 | 35.1 | 2 |
| UnPhos | LVDFGVSAQLTSpTR | Thr-178 | 9.07 | 79 | 73.7 | 35.2 | 7 |
| UnPhos | RNpTSVGTPFWoxMAPEVIAC*EQQLDTTYDAR | Thr-184 | 7.38 | 100 | 50.6 | 34.6 | 3 |
| Phos | RNpTSVGTPFWoxMAPEVIAC*EQQLDTTYDAR | Thr-184 | 7.38 | 100 | 55.0 | 34.6 | 4 |
| Phos | LINLAKGDpTGEATR | Thr-908 | 42.92 | 100 | 53.7 | 35.2 | 18 |
| Phos | ETpTNoxMKTQTVASYFR | Thr-919 | 6.4 | 100 | 34.7 | 35.1 | 3 |
Kinase Activity
We examined the kinase activity of MYO3A using [32P]ATP assays with the MYO3A 2IQ and MYO3A kinase constructs. Assays were performed with 2 μm MYO3A and 200 μm [32P]ATP over a 60-min time period. All of the phosphorylation time courses examined by scanned SDS-polyacrylamide gels and scintillation counting demonstrated that phosphorylation reached saturation during the 60-min period. We found that there was ∼1.3 phosphates incorporated into WT MYO3A 2IQ during the time course (Table 2), which is less than the four sites identified by mass spectrometry (Table 1). The results are in agreement with the kinase activation loop sites being at least partially phosphorylated prior to the time course (Table 1). The number of phosphates incorporated into T184E MYO3A 2IQ was similar to WT, suggesting that this site is mostly phosphorylated during expression/purification. The number of phosphates incorporated into T178A or T178E MYO3A 2IQ was reduced (0.6 and 0.8, respectively) compared with WT, suggesting that although this site may be partially phosphorylated prior to the time course there is an increase in phosphorylation at this site during the time course. Taken together, the number of phosphates incorporated into the Thr-178 mutants may be due to phosphorylation of the motor sites (Thr-908 and Thr-919), because Thr-184 appears to be completely phosphorylated prior to the time course. Thus, our results suggest phosphorylation of the motor sites may be less efficient (i.e. 100% phosphorylation may not be achieved at these sites).
TABLE 2.
Kinase activity of MYO3A constructs
| Construct | Kinase activitya | Moles of 32P/mol of MYO3Ab | One-half timec |
|---|---|---|---|
| min−1 | min | ||
| WT MYO3A 2IQ | 0.173 ± 0.004 | 1.28 ± 0.35 | 2.93 ± 0.17 |
| MtDd MYO3A 2IQ | 0.113 ± 0.001 | 1.50 ± 0.22 | 3.44 ± 0.48 |
| T184E MYO3A 2IQ | 0.173 ± 0.030 | 1.28 ± 0.47 | 2.85 ± 0.16 |
| T184A MYO3A 2IQ | 0.006 ± 0.001 | 0.33 ± 0.22 | 12.62 ± 1.15 |
| T178D MYO3A 2IQ | 0.094 ± 0.007 | 0.82 ± 0.07 | 3.32 ± 0.68 |
| T178A MYO3A 2IQ | 0.034 ± 0.001 | 0.58 ± 0.22 | 4.79 ± 0.23 |
| MYO3A kinase | 0.026 ± 0.002 | 0.54 ± 0.13 | 5.22 ± 0.75 |
We demonstrate that WT and T184E MYO3A 2IQ have similar rates of kinase activity, although T184A MYO3A 2IQ is significantly reduced (Fig. 3 and Table 2). We found that the kinase activity of T184A was about 30-fold slower when compared with WT MYO3A 2IQ, and T184A MYO3A 2IQ only achieved a fraction of phosphate incorporation (0.33 mol of 32P/mol of MYO3A 2IQ) after 60 min. We performed a similar analysis of the Thr-178 site and found that preventing phosphorylation at this site resulted in a more modest 3–5-fold reduction in kinase activity (Fig. 4 and Table 2).
FIGURE 3.

Phosphorylation of Thr-184 regulates MYO3A kinase activity. A, phosphorylation was measured by examining [32P] incorporation with phosphorimaging following SDS-PAGE. Gels were also Coomassie-stained to determine evenness of loading, and bands were excised and examined by scintillation counting (Table 2). B, phosphorylation time course demonstrates the relative intensity of the bands from the phosphorimage during the 60-min time course. The phosphorylation time courses of T184E, T184A, and motor-dead (MtDd) are compared with WT MYO3A. The intensities are plotted relative to the WT MYO3A 60-min time point for comparison. Error bars represent the standard error from four experiments.
FIGURE 4.

Phosphorylation of Thr-178 does not play a major role in MYO3A kinase regulation. A, phosphorylation was measured by examining [32P] incorporation with phosphorimaging following SDS-PAGE. Gels were also Coomassie-stained to determine evenness of loading, and bands were excised and examined by scintillation counting (Table 2). B, phosphorylation time course demonstrates the relative intensity of the bands from the phosphorimage during the 60-min time course. The intensity is plotted relative to the WT MYO3A 60-min time point for comparison. Error bars represent the standard error from three experiments.
To determine how inactivation of the motor domain impacts kinase activity, we generated a motor-dead MYO3A 2IQ construct. The point mutation E722A, located in the conserved switch II region, was utilized to prevent ATP hydrolysis as demonstrated in other myosins (33–35). The E722A MYO3A 2IQ (motor-dead) had no actin-activated ATPase activity as examined in the NADH assay in the presence of 20 μm actin (0.06 s−1) and absence of actin (0.07 s−1). The kinase activity of motor-dead MYO3A 2IQ was slightly reduced compared with WT MYO3A 2IQ, although the amount of [32P] incorporation after 60 min was similar (Fig. 3 and Table 2).
We examined the steady-state kinetic properties of the MYO3A 2IQ kinase activity as a function of MYO3A 2IQ concentration. The initial velocity of the kinase reaction at each MYO3A 2IQ concentration was examined by focusing on the first 2–2.5 min of the reaction (Fig. 5, inset). The plot of the initial velocity as a function of concentration was hyperbolic, which allowed us to fit the data to Michaelis-Menten kinetics (Fig. 5). This analysis allowed us to determine the Km value for MYO3A 2IQ (1.6 ± 0.4 μm) and maximum kinase activity (kcat = 0.30 ± 0.03 min−1).
FIGURE 5.

Steady-state kinase activity of WT MYO3A 2IQ. The initial velocity of the kinase reaction ([32P] incorporation) was monitored as a function of WT MYO3A 2IQ concentration and fit to the Michealis-Menten relationship to determine kcat and Km values. The inset demonstrates the linear fits of the kinase reaction time courses that determined the initial velocity of the kinase reaction for four different concentrations of WT MYO3A 2IQ. The error bars represent the standard errors from the linear fits of the initial velocity measurements.
ATPase Activity
To examine the impact of the phosphorylation site mutations (T178D, T178E, T184E, and T184A) on MYO3A motor activity, we performed actin-activated motor ATPase activity assays. We determined the ATPase activity in the presence of varying concentrations of actin (0–60 μm) using the NADH-coupled assay (3, 23, 24, 32) at 25 °C (Fig. 6). Our results demonstrate that the ATPase activity in the absence of actin was fairly similar between all mutations and wild-type MYO3A 2IQ. The actin concentration dependence of the ATPase activity allowed us to fit the data to a hyperbolic relationship to determine the maximum ATPase activity (kcat) and actin concentration at which the ATPase activity is one-half maximal (KATPase). There was a 2-fold reduction in kcat with both phosphoblock mutations (T178A and T184A), although the phosphomimic mutations resulted in a slight increase in kcat. Interestingly, the KATPase was increased 2-fold in both mutations at the Thr-184 site. The catalytic efficiency as examined by kcat/KATPase was altered by no more than 2-fold as a result of the phosphorylation site mutations. Our results suggest that there is coupling between the motor and kinase activities of MYO3A, because mutations in the kinase domain impact the motor ATPase activity and actin affinity.
FIGURE 6.

Actin-activated ATPase activity of MYO3A 2IQ phosphorylation site mutations. Both T184A MYO3A2IQ and T184E MYO3A2IQ displayed an increased KATPase when compared with WT MYO3A2IQ. The T184A and T178A MYO3A2IQ mutants displayed a decrease in kcat compared with WT MYO3A 2IQ. Data represent averages from three to five separate experiments (error bars represent mean ± S.E.) and are summarized in Table 3.
To further demonstrate the reduction in motor activity observed upon phosphorylation of the motor domain of MYO3A 2IQ, we examined the actin-activated ATPase of unphosphorylated MYO3A 2IQ ΔK compared with phosphorylated MYO3A 2IQΔK (Fig. 7). The phosphorylated MYO3A 2IQΔK (2 μm) was incubated with MYO3A kinase (4 μm) for 60 min, and the unphosphorylated MYO3A 2IQ ΔK was incubated in parallel without MYO3A kinase. The degree of phosphorylation was examined by antiphosphothreonine Western blotting (Fig. 7B). The impact of phosphorylation resulted in a 2-fold reduction in kcat and 5-fold increase in KATPase, which resulted in a 10-fold reduced catalytic efficiency (Table 3).
FIGURE 7.

Actin-activated ATPase activity of phosphorylated MYO3A 2IQ ΔK. A, comparison of phosphorylated MYO3A 2IQ ΔK (treated with MYO3A kinase for 60 min) and control-treated MYO3A 2IQ ΔK demonstrates phosphorylation reduces motor ATPase activity. B, antiphosphothreonine Western blot demonstrates MYO3A 2IQ ΔK phosphorylation compared with control treated. Data represent averages from two separate experiments (error bars represent mean ± S.E.).
TABLE 3.
Actin-activated ATPase activity of MYO3A 2IQ constructs
Phos, phosphorylated; UnPhos, unphosphorylated.
| Construct | V0 | kcat | KATPase | kcat/KATPase |
|---|---|---|---|---|
| s−1 | s−1 | μm | μm−1·s−1 | |
| WT MYO3A 2IQ | 0.15 ± 0.02 | 1.1 ± 0.1 | 20.5 ± 5.1 | 0.054 ± 0.014 |
| T184E MYO3A 2IQ | 0.11 ± 0.02 | 1.6 ± 0.1 | 46.7 ± 5.5 | 0.035 ± 0.005 |
| T184A MYO3A 2IQ | 0.11 ± 0.04 | 1.0 ± 0.1 | 55.3 ± 18.2 | 0.018 ± 0.006 |
| T178D MYO3A 2IQ | 0.13 ± 0.03 | 1.3 ± 0.1 | 28.1 ± 6.8 | 0.046 ± 0.012 |
| T178A MYO3A 2IQ | 0.10 ± 0.03 | 0.7 ± 0.1 | 22.6 ± 7.4 | 0.031 ± 0.011 |
| MYO3A 2IQ ΔK UnPhos | 0.08 ± 0.01 | 1.8 ± 0.1 | 4.0 ± 0.9 | 0.450 ± 0.110 |
| MYO3A 2IQ ΔK Phos | 0.11 ± 0.01 | 1.0 ± 0.1 | 20.4 ± 3.9 | 0.049 ± 0.011 |
COS7 Cell Localization
COS7 cells transfected with full-length GFP-tagged MYO3A containing the T184A or T184E mutation were examined for their ability to localize to filopodia tips. Fig. 8A demonstrates the filopodia tip localization of the MYO3A constructs. Similar to our previous work, we find that wild-type MYO3A localizes along the length of the filopodia, and the kinase-deleted construct (MYO3A ΔK) demonstrates increased tip localization. The activation loop mutations did not have an impact on the ability of MYO3A to localize to filopodia tips as examined by the tip to cell body ratio measurements (Fig. 8B). We previously demonstrated that MYO3A ΔK can enhance filopodia density in COS7 cells (3), although the activation loop mutations did not impact the density of filopodia along the cell periphery (Fig. 8C).
FIGURE 8.

Impact of the Thr-184 site on localization to filopodia structures in COS7 cells. A, GFP-tagged full-length Myo3A localizes along the length of filopodia in COS7 cells, unlike GFP-MYO3A ΔK (lacking the kinase domain) that shows concentration at filopodial tips. Neither the T184A nor the T184E mutations in GFP-MYO3A result in increased tip localization. Representative images are epifluorescence of paraformaldehyde-fixed and phalloidin-stained COS7 cells. GFP constructs are shown in cyan, and Alexa 568-phalloidin is shown in yellow. All images were captured under identical imaging conditions and scaled identically for brightness. Scale bar, 10 μm. B, T184A and T184E mutations do not alter the tip localization of MYO3A as indicated by the ratio of tip to cell body fluorescence. C, similarly, expression of GFP-MYO3A-T184A and GFP-MYO3A-T184E did not increase the density of filopodia along the cell periphery. B, n >46 filopodial tips from 10 or more cells per condition; C, n >24 cells per condition. * indicates significant difference (p < 0.003, Tukey test) from all other constructs.
We also examined the elongation of filopodia actin in the presence of ESPN1 (Fig. 9). The Thr-184 mutation constructs were co-transfected with ESPN1 and compared with kinase-dead (K50R), ΔK, and wild-type MYO3A. Our results mirror our previous work that demonstrated collaboration between ESPN1 and MYO3A ΔK more efficiently elongates filopodia compared with K50R and wild-type MYO3A, although K50R was more efficient than wild-type MYO3A. Interestingly, the T184A mutation that dramatically reduces kinase activity was similar to K50R in elongation ability. The T184E mutant, which contains a constitutively active kinase domain, was the least efficient at elongating filopodia. The difference between the K50R MYO3A and MYO3A ΔK reflects the 2-fold enhanced motor activity of MYO3A ΔK that we previously reported, which is proposed to increase the ESPN1 transport to the filopodia tips (9). Our results are in agreement with our working model in which MYO3A undergoes autophosphorylation at the tips of actin protrusions, which reduces its ability to remain tip-localized because of a reduced affinity for actin and reduced motor ATPase activity.
FIGURE 9.
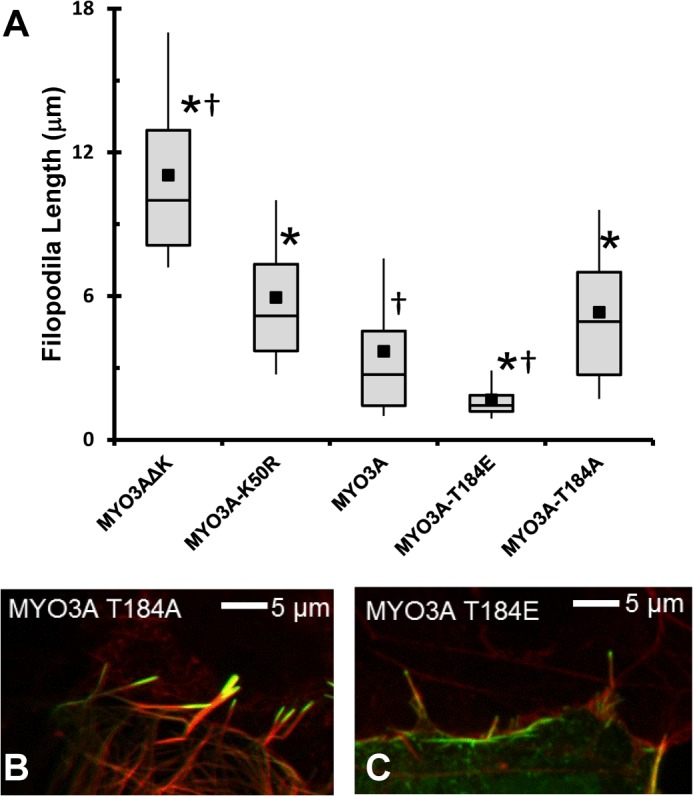
Impact of the Thr-184 site on MYO3A filopodia elongation activity in the presence of ESPN1. Expressing the GFP-tagged versions of the MYO3A mutations in combination with ESPN1 revealed that kinase activity was required for regulation of filopodial length. The reduced kinase activity of T184A failed to regulate filopodial length (similar to K50R), although the enhanced kinase activity of T184E resulted in shorter filopodia than wild-type MYO3A. * indicates significantly different from MYO3A (p < 0.008, Tukey test). † indicates significantly different from GFP-MYO3A-K50R (p < 0.0001, Tukey test).
DISCUSSION
Kinases are known to have different mechanisms of regulation, including dimerization, proteolytic cleavage, association with other proteins, as well as conformational changes in the activation segment (17). Elucidating the regulatory mechanism of a kinase has proven to be extremely valuable in understanding its role in the cell and designing strategies to inhibit or constitutively activate specific kinases. We have determined that phosphorylation of a conserved threonine (Thr-184) in the activation loop of MYO3A is critical for activating the kinase domain. We also demonstrate the functional consequences of phosphorylation of Thr-184 in the context of the MYO3A localization and in the elongation of actin bundle-based protrusions.
MYO3 Kinase Structure-Function
Our sequence analysis results suggest that class III myosins have features classically found in active kinases (17). The MYO3A kinase domain contains several motifs that are classically associated with an active kinase, including the glycine-rich loop (GXGXXG, residues 28–33), β3 lysine (XXXK, residues 47–50), catalytic and magnesium-binding motif (XXDXK, residues 150–154), and magnesium-binding motif activation segment (DFG, residues 168–170). Our previous work in which we mutated the β3 lysine to arginine demonstrates that this dramatically reduces kinase activity (3, 9), which is consistent with MYO3A containing an established kinase catalytic mechanism.
Phosphorylation Sites in the Kinase Domain and Kinase Regulation
Our work and the work of others have demonstrated that class III myosins are capable of autophosphorylation (3, 21, 23, 24, 36). In this work, we determined the phosphorylation sites in the motor and kinase domain by mass spectrometry with and without undergoing in vitro autophosphorylation (Table 1). We found two sites in the kinase domain located in the kinase activation loop. In the previous mouse MYO3A and -3B studies, phosphorylation sites in the kinase domain were examined in vitro using the purified MYO3A or -3B kinase domain and allowing autophosphorylation to take place (21). They also identified sites in the kinase activation loop located at the corresponding site to Thr-184, although the second phosphorylation site was at the site corresponding to serine 177, adjacent to Thr-178. In our work, we demonstrate that the Thr-184 site was phosphorylated in our samples that did not undergo autophosphorylation (“unphosphorylated”), which suggests that this site can be phosphorylated either by MYO3A or another cellular kinase during the expression/purification process. The Thr-184 site is highly conserved in kinases that are activated by phosphorylation of the activation loop, including the well characterized STE20 member, PAK1. The Thr-184 site contains P(−2), P(−3), and P(−5) arginines (where P is associated with the residue position relative to the phosphorylation site at P0) that are associated with the known substrate specificity for other basophilic kinases such as PKA (37). Therefore, the Thr-184 site can likely be phosphorylated by other kinases as well as through autophosphorylation. A critical point is that we directly demonstrate that preventing phosphorylation at Thr-184 dramatically reduces the kinase activity 30-fold (Fig. 3). The phosphorylation at Thr-178 was also found in the unphosphorylated samples we examined by mass spectrometry, although this site does not contain the obvious features associated with substrate specificity for other kinases. Preventing phosphorylation at the Thr-178 site only altered the kinase 3–5-fold (Fig. 4) and therefore may play a more minor role in kinase regulation.
In our previous work, we demonstrated that autophosphorylation of MYO3A is concentration-dependent and therefore occurs by a trans-autophosphorylation mechanism (3). Phosphorylation of the kinase activation loop is a common mechanism for regulating kinase catalytic activity (38, 39). As described above, the regulatory mechanism for MYO3A could occur through another kinase that is capable of phosphorylating Thr-184. Another possible mechanism is that the MYO3A kinase is able to adopt an active conformation even in the dephosphorylated state. The structural and biochemical analysis of the β3 lysine mutant (K299R) in PAK1 kinase provides support for this mechanism (40). Indeed, our results demonstrate that in the unphosphorylated state (T184A), there is some, albeit very low, kinase activity (Fig. 3 and Table 2). Alternatively, phosphorylation could occur through an activation loop exchange mechanism demonstrated in checkpoint kinase 2 and STE-like kinase (41, 42). High resolution structural studies of the MYO3 kinase domain will help define the structural basis of MYO3 kinase regulation.
Substrate Specificity of MYO3A Kinase
The external cellular substrates of class III myosins are currently unknown, and determining the substrate specificity of a kinase can provide insight into its ability to participate in specific signaling cascades. The previous work with mouse MYO3A and -3B found that there may be substrate specificity in which there is preference for a basic residue at P(−3) or P(+2), or a hydrophobic residue at P(+1). Although these results do not necessarily fit into the proposed substrate specificity, we did find that two phosphorylation sites in loop 2 (Thr-908 and Thr-919) contain a basic residue at P(−3). However, evidence suggests that preference for a basic residue at position P(−3) requires a single conserved acidic residue in the catalytic region (37), although MYO3A contains a serine (Ser-107) at this position, and indeed most STE kinases contain an acidic residue at this position. Substrate preference for a basic residue at P(−2) is thought to require two conserved acidic residues in the catalytic site, and MYO3A only contains one (Glu-223) but lacks a second acidic residue (Asn-154) at these positions, which is consistent with MYO3A lacking P(−2) specificity.
Kinase Catalytic Activity
Our results demonstrate along with our previous work that the kinase autophosphorylation activity is relatively slow (t½ = 2.9 ± 0.2 min). We also determined the kcat value with respect to autophosphorylation in this study and found that it is 5000-fold slower than the structurally related PAK1 kinase examined by autophosphorylation (43) or with a peptide substrate (40). The Km values for MYO3A were about 100-fold lower than the reported values for PAK1 determined with a peptide substrate (40). The steady state kinetic parameters (kcat and Km) determined for other kinases vary considerably, although the values for MYO3A are similar to those reported for mammalian target of rapamycin (44) and MST1 (45). Many kinases are auto-inhibited by the C- or N-terminal domains and utilize protein-protein interactions to relieve autoinhibition (17). Therefore, we investigated the possibility that the presence of the motor domain influences the catalytic activity of the kinase domain. Interestingly, the kinase domain alone had reduced kinase activity compared with MYO3A 2IQ (Table 2), demonstrating that the motor domain does not serve an inhibitory role. Future studies will define the structural basis of MYO3A kinase activity and investigate other possible regulatory mechanisms such as interactions with cellular binding partners.
Phosphorylation Sites in the Motor Domain and Motor Regulation
The two phosphorylation sites in the motor domain mapped to the loop 2 region of the MYO3A motor. Previous work with mouse MYO3A used peptides that corresponded to the loop 2 region to demonstrate that residues in this region can be phosphorylated in vitro by the mouse kinase domain (21). Our work utilized purified MYO3A containing both the kinase and motor domains to identify the loop 2 phosphorylation sites in the motor domain. Interestingly, the phosphorylation sites identified are not the same as those found using the peptides and mouse MYO3A. Kinases are known to prefer phosphorylation of less well ordered and surface-exposed regions of proteins (17), which is consistent with studies that demonstrate loop 2 is highly disordered in myosins (46). Studies have demonstrated that the degree of positive charge in loop 2 can impact actin-activated ATPase activity and actin affinity of myosins (47–49). The additional negative charges associated with phosphorylation are well positioned to reduce actin affinity and ATPase activity. Indeed, we previously reported a 30–40% reduction in maximum actin-activated ATPase activity (kcat) and a 2-fold reduced actin affinity in the phosphorylated compared with the unphosphorylated MYO3A 2IQ (3). We add to these studies by demonstrating a more dramatic 2-fold reduction in the kcat of MYO3A 2IQ ΔK in the phosphorylated state (Fig. 7). In addition, the actin concentration dependence of ATPase activity (KATPase) is increased 5-fold in phosphorylated MYO3A ΔK 2IQ, consistent with a weaker actin affinity. The biochemical analysis is in agreement with cell biological localization studies that found filopodia tip localization of GFP-tagged MYO3A ΔK is reduced 2–3-fold when co-transfected with kinase-active MYO3A, which is capable of phosphorylating and down-regulating MYO3A ΔK (3). Thus, the phosphorylation sites in loop 2 (Thr-908 and Thr-919) are likely responsible for the down-regulation of MYO3A motor activity observed in our current and previous work (3).
The actin-activated ATPase results suggest that activation loop phosphorylation may alter allosteric motor-kinase interactions that have a minor impact on MYO3A motor activity. Interestingly, both mutations of Thr-184 increase KATPase 2-fold, although the T184A mutation reduces kcat 30–40% compared with T184E. The overall catalytic efficiency difference between T184E and T184A MYO3A (2-fold) is not as dramatic as the difference associated with phosphorylation of the MYO3A ΔK 2IQ (10-fold). We expected that the T184E mutant would display identical motor ATPase properties to WT MYO3A 2IQ, because Thr-184 appears to be mostly phosphorylated in the WT MYO3A 2IQ. The difference may reflect the impact of having a phosphate group at Thr-184 versus a mutation and suggests that the phosphate group more efficiently blocks the interactions between the motor and kinase that enhance actin affinity. The Thr-178 site mutations also appear to have similar overall impact on catalytic efficiency, although the changes in actin dependence were not observed. Overall, the introduction of acidic amino acids in the kinase activation loop modestly enhances catalytic efficiency suggesting phosphorylation prevents inhibitory kinase-motor interactions.
Regulation of Actin Protrusion Elongation
The stability and maintenance of actin bundle protrusions in photoreceptors and inner ear hair cells are critical for maintaining normal sensory transduction, ultrastructure, and preventing degeneration (50–54). MYO3A was found to play a role in maintaining stereocilia ultrastructure as the DFNB30 mouse displays age-dependent stereocilia degeneration (12). Our working hypothesis is that the localization and activity of class III myosins play a role in transport within actin bundle protrusions. This is highlighted by our previous results that demonstrate MYO3A and MYO3B can transport ESPN1 to the stereocilia tips (9, 10). We proposed the concentration-dependent regulation by autophosphorylation mechanism for how MYO3 transport and tip localization are regulated (3), and our current results add more detail to this model (Fig. 10). In this model, the kinase- and motor-dephosphorylated MYO3A moves to the tips of actin protrusions. When MYO3A accumulates at the actin bundle tips, the kinase activation loop (Thr-184) gets autophosphorylated, which activates the kinase domain. The slow kinase activity of the dephosphorylated kinase may still be able to trans-phosphorylate the neighboring MYO3A kinase domains crowded at the tip. The fully activated MYO3A kinase will be more likely to trans-phosphorylate loop 2 sites (Thr-909 and Thr-919) in the motor and down-regulate motor activity. The phosphorylated MYO3A motor, with reduced motor activity, has an increased chance of being returned to the cell body by retrograde flow or diffusion. Our actin protrusion elongation results support our proposed model because the T184A and K50R mutations enhance elongation and both have reduced kinase activity, which enhances the ability of MYOA to transport ESPN1 to the tips and induce elongation. In contrast, the T184E mutation reduces elongation because the kinase is constitutively active and has a higher probability of trans-phosphorylating the loop 2 sites in the motor, which reduces the efficiency of transporting ESPN1 to the actin protrusion tips.
FIGURE 10.
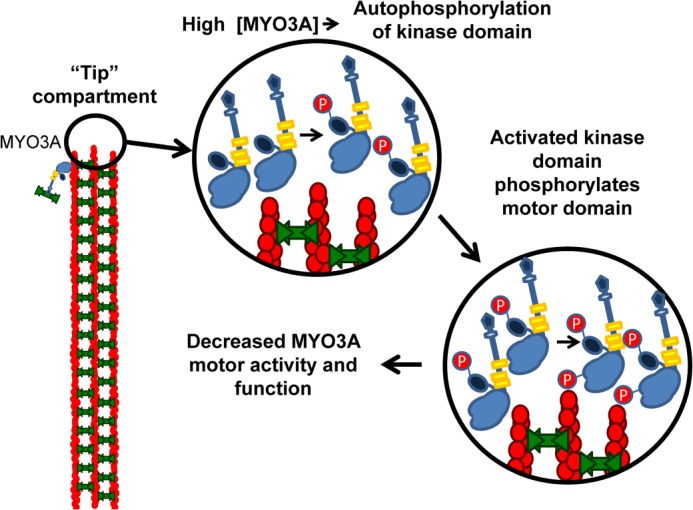
Model of MYO3A regulation in actin protrusions. We suggest a model of concentration-dependent regulation by autophosphorylation at the tips of actin protrusions that consists of a multistep process where increased concentration of MYO3A in the tip compartment results in activation of the MYO3A kinase domain by autophosphorylation. Active kinase is then capable of trans-phosphorylation of the motor domain of myosin. Reducing myosin motor activity leads to a loss of MYO3A protein from the tip compartment, thereby regulating the stability of the actin-based structure.
In summary, we demonstrate the phosphorylation-dependent regulatory mechanism of the MYO3A kinase domain. We find that altering kinase regulation can impact the MYO3A cellular function of mediating elongation of actin protrusions. Future studies will investigate if another cellular kinase is responsible for phosphorylating Thr-184 and activating the kinase domain as well as determining the phosphatase responsible for dephosphorylating class III myosins. Determining how MYO3A kinase activity correlates with the structure and function of stereocilia and calycal processes will provide an improved understanding of how these critically important actin-based structures are maintained throughout a lifetime.
Acknowledgments
We thank Manmeet Raval, Darshan Trivedi, and Anja Swenson for helpful discussions and Audrey Stokes for performing preliminary ATPase experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants EY0181416 (to C. M. Y.) and K01CA160667 (to O. A. Q.). This work was also supported by start-up funds from Penn State College of Medicine.
- STE20
- sterile-20 protein kinase.
REFERENCES
- 1. Dosé A. C., Burnside B. (2000) Cloning and chromosomal localization of a human class III myosin. Genomics 67, 333–342 [DOI] [PubMed] [Google Scholar]
- 2. Dosé A. C., Hillman D. W., Wong C., Sohlberg L., Lin-Jones J., Burnside B. (2003) Myo3A, one of two class III myosin genes expressed in vertebrate retina, is localized to the calycal processes of rod and cone photoreceptors and is expressed in the sacculus. Mol. Biol. Cell 14, 1058–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quintero O. A., Moore J. E., Unrath W. C., Manor U., Salles F. T., Grati M., Kachar B., Yengo C. M. (2010) Intermolecular autophosphorylation regulates myosin IIIa activity and localization in parallel actin bundles. J. Biol. Chem. 285, 35770–35782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Montell C., Rubin G. M. (1988) The Drosophila ninaC locus encodes two photoreceptor cell specific proteins with domains homologous to protein kinases and the myosin heavy chain head. Cell 52, 757–772 [DOI] [PubMed] [Google Scholar]
- 5. Odronitz F., Becker S., Kollmar M. (2009) Reconstructing the phylogeny of 21 completely sequenced arthropod species based on their motor proteins. BMC Genomics 10, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin-Jones J., Parker E., Wu M., Dosé A., Burnside B. (2004) Myosin 3A transgene expression produces abnormal actin filament bundles in transgenic Xenopus laevis rod photoreceptors. J. Cell Sci. 117, 5825–5834 [DOI] [PubMed] [Google Scholar]
- 7. Sahly I., Dufour E., Schietroma C., Michel V., Bahloul A., Perfettini I., Pepermans E., Estivalet A., Carette D., Aghaie A., Ebermann I., Lelli A., Iribarne M., Hardelin J. P., Weil D., Sahel J. A., El-Amraoui A., Petit C. (2012) Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J. Cell Biol. 199, 381–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schneider M. E., Dosé A. C., Salles F. T., Chang W., Erickson F. L., Burnside B., Kachar B. (2006) A new compartment at stereocilia tips defined by spatial and temporal patterns of myosin IIIa expression. J. Neurosci. 26, 10243–10252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Salles F. T., Merritt R. C., Jr., Manor U., Dougherty G. W., Sousa A. D., Moore J. E., Yengo C. M., Dosé A. C., Kachar B. (2009) Myosin IIIa boosts elongation of stereocilia by transporting espin 1 to the plus ends of actin filaments. Nat. Cell Biol. 11, 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Merritt R. C., Manor U., Salles F. T., Grati M., Dose A. C., Unrath W. C., Quintero O. A., Yengo C. M., Kachar B. (2012) Myosin IIIB uses an actin-binding motif in its espin-1 cargo to reach the tips of actin protrusions. Curr. Biol. 22, 320–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Walsh T., Walsh V., Vreugde S., Hertzano R., Shahin H., Haika S., Lee M. K., Kanaan M., King M. C., Avraham K. B. (2002) From flies' eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc. Natl. Acad. Sci. U.S.A. 99, 7518–7523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walsh V. L., Raviv D., Dror A. A., Shahin H., Walsh T., Kanaan M. N., Avraham K. B., King M. C. (2011) A mouse model for human hearing loss DFNB30 due to loss of function of myosin IIIA. Mamm. Genome 22, 170–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chung W., Bondaruk J., Jelinek J., Lotan Y., Liang S., Czerniak B., Issa J. P. (2011) Detection of bladder cancer using novel DNA methylation biomarkers in urine sediments. Cancer Epidemiol. Biomarkers Prev. 20, 1483–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lascorz J., Försti A., Chen B., Buch S., Steinke V., Rahner N., Holinski-Feder E., Morak M., Schackert H. K., Görgens H., Schulmann K., Goecke T., Kloor M., Engel C., Büttner R., Kunkel N., Weires M., Hoffmeister M., Pardini B., Naccarati A., Vodickova L., Novotny J., Schreiber S., Krawczak M., Bröring C. D., Völzke H., Schafmayer C., Vodicka P., Chang-Claude J., Brenner H., Burwinkel B., Propping P., Hampe J., Hemminki K. (2010) Genome-wide association study for colorectal cancer identifies risk polymorphisms in German familial cases and implicates MAPK signalling pathways in disease susceptibility. Carcinogenesis 31, 1612–1619 [DOI] [PubMed] [Google Scholar]
- 15. Unschuld P. G., Ising M., Specht M., Erhardt A., Ripke S., Heck A., Kloiber S., Straub V., Brueckl T., Muller-Myhsok B., Holsboer F., Binder E. B. (2009) Polymorphisms in the GAD2 gene-region are associated with susceptibility for unipolar depression and with a risk factor for anxiety disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 1100–1109 [DOI] [PubMed] [Google Scholar]
- 16. Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 17. Endicott J. A., Noble M. E., Johnson L. N. (2012) The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 81, 587–613 [DOI] [PubMed] [Google Scholar]
- 18. Zenke F. T., King C. C., Bohl B. P., Bokoch G. M. (1999) Identification of a central phosphorylation site in p21-activated kinase regulating autoinhibition and kinase activity. J. Biol. Chem. 274, 32565–32573 [DOI] [PubMed] [Google Scholar]
- 19. Wu H., Wang Z. X. (2003) The mechanism of p21-activated kinase 2 autoactivation. J. Biol. Chem. 278, 41768–41778 [DOI] [PubMed] [Google Scholar]
- 20. Chong C., Tan L., Lim L., Manser E. (2001) The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. J. Biol. Chem. 276, 17347–17353 [DOI] [PubMed] [Google Scholar]
- 21. Dalal J. S., Stevens S. M., Jr., Alvarez S., Munoz N., Kempler K. E., Dosé A. C., Burnside B., Battelle B. A. (2011) Mouse class III myosins: kinase activity and phosphorylation sites. J. Neurochem. 119, 772–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De La Cruz E. M., Ostap E. M. (2009) Kinetic and equilibrium analysis of the myosin ATPase. Methods Enzymol. 455, 157–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dosé A. C., Ananthanarayanan S., Moore J. E., Corsa A. C., Burnside B., Yengo C. M. (2008) The kinase domain alters the kinetic properties of the myosin IIIA motor. Biochemistry 47, 2485–2496 [DOI] [PubMed] [Google Scholar]
- 24. Dosé A. C., Ananthanarayanan S., Moore J. E., Burnside B., Yengo C. M. (2007) Kinetic mechanism of human myosin IIIA. J. Biol. Chem. 282, 216–231 [DOI] [PubMed] [Google Scholar]
- 25. Pardee J. D., Spudich J. A. (1982) Purification of muscle actin. Methods Enzymol. 85, 164–181 [DOI] [PubMed] [Google Scholar]
- 26. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X Version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 27. Galtier N., Gouy M., Gautier C. (1996) SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput. Appl. Biosci. 12, 543–548 [DOI] [PubMed] [Google Scholar]
- 28. Drummond A. J., Suchard M. A., Xie D., Rambaut A. (2012) Bayesian phylogenetics with BEAUti and BEAST 1.7. Mol. Biol. Evol. 8, 1969–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 30. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 31. Beausoleil S. A., Villén J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 32. De La Cruz E. M., Sweeney H. L., Ostap E. M. (2000) ADP inhibition of myosin V ATPase activity. Biophys. J. 79, 1524–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kad N. M., Rovner A. S., Fagnant P. M., Joel P. B., Kennedy G. G., Patlak J. B., Warshaw D. M., Trybus K. M. (2003) A mutant heterodimeric myosin with one inactive head generates maximal displacement. J. Cell Biol. 162, 481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sasaki N., Ohkura R., Sutoh K. (2002) Dictyostelium myosin II as a model to study the actin-myosin interactions during force generation. J. Muscle Res. Cell Motil. 23, 697–702 [DOI] [PubMed] [Google Scholar]
- 35. Trivedi D. V., David C., Jacobs D. J., Yengo C. M. (2012) Switch II mutants reveal coupling between the nucleotide- and actin-binding regions in myosin V. Biophys. J. 102, 2545–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Komaba S., Inoue A., Maruta S., Hosoya H., Ikebe M. (2003) Determination of human myosin III as a motor protein having a protein kinase activity. J. Biol. Chem. 278, 21352–21360 [DOI] [PubMed] [Google Scholar]
- 37. Zhu G., Fujii K., Liu Y., Codrea V., Herrero J., Shaw S. (2005) A single pair of acidic residues in the kinase major groove mediates strong substrate preference for P-2 or P-5 arginine in the AGC, CAMK, and STE kinase families. J. Biol. Chem. 280, 36372–36379 [DOI] [PubMed] [Google Scholar]
- 38. Delpire E. (2009) The mammalian family of sterile 20p-like protein kinases. Pflugers Arch. 458, 953–967 [DOI] [PubMed] [Google Scholar]
- 39. Glantschnig H., Rodan G. A., Reszka A. A. (2002) Mapping of MST1 kinase sites of phosphorylation. Activation and autophosphorylation. J. Biol. Chem. 277, 42987–42996 [DOI] [PubMed] [Google Scholar]
- 40. Wang J., Wu J. W., Wang Z. X. (2011) Structural insights into the autoactivation mechanism of p21-activated protein kinase. Structure 19, 1752–1761 [DOI] [PubMed] [Google Scholar]
- 41. Pike A. C., Rellos P., Niesen F. H., Turnbull A., Oliver A. W., Parker S. A., Turk B. E., Pearl L. H., Knapp S. (2008) Activation segment dimerization: a mechanism for kinase autophosphorylation of non-consensus sites. EMBO J. 27, 704–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oliver A. W., Paul A., Boxall K. J., Barrie S. E., Aherne G. W., Garrett M. D., Mittnacht S., Pearl L. H. (2006) Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. EMBO J. 25, 3179–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang J., Wu J. W., Wang Z. X. (2011) Mechanistic studies of the autoactivation of PAK2: a two-step model of cis initiation followed by trans-amplification. J. Biol. Chem. 286, 2689–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tao Z., Barker J., Shi S. D., Gehring M., Sun S. (2010) Steady-state kinetic and inhibition studies of the mammalian target of rapamycin (mTOR) kinase domain and mTOR complexes. Biochemistry 49, 8488–8498 [DOI] [PubMed] [Google Scholar]
- 45. Anand R., Kim A. Y., Brent M., Marmorstein R. (2008) Biochemical analysis of MST1 kinase: elucidation of a C-terminal regulatory region. Biochemistry 47, 6719–6726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sweeney H. L., Houdusse A. (2010) Structural and functional insights into the myosin motor mechanism. Annu. Rev. Biophys. 39, 539–557 [DOI] [PubMed] [Google Scholar]
- 47. Joel P. B., Sweeney H. L., Trybus K. M. (2003) Addition of lysines to the 50/20-kDa junction of myosin strengthens weak binding to actin without affecting the maximum ATPase activity. Biochemistry 42, 9160–9166 [DOI] [PubMed] [Google Scholar]
- 48. Furch M., Remmel B., Geeves M. A., Manstein D. J. (2000) Stabilization of the actomyosin complex by negative charges on myosin. Biochemistry 39, 11602–11608 [DOI] [PubMed] [Google Scholar]
- 49. Yengo C. M., Sweeney H. L. (2004) Functional role of loop 2 in myosin V. Biochemistry 43, 2605–2612 [DOI] [PubMed] [Google Scholar]
- 50. Kennedy B., Malicki J. (2009) What drives cell morphogenesis: a look inside the vertebrate photoreceptor. Dev. Dyn. 238, 2115–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kremer H., van Wijk E., Märker T., Wolfrum U., Roepman R. (2006) Usher syndrome: molecular links of pathogenesis, proteins, and pathways. Hum. Mol. Genet. 15, R262–R270 [DOI] [PubMed] [Google Scholar]
- 52. Sekerková G., Richter C. P., Bartles J. R. (2011) Roles of the espin actin-bundling proteins in the morphogenesis and stabilization of hair cell stereocilia revealed in CBA/CaJ congenic jerker mice. PLoS Genet. 7, e1002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ambati J., Fowler B. J. (2012) Mechanisms of age-related macular degeneration. Neuron 75, 26–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Manor U., Kachar B. (2008) Dynamic length regulation of sensory stereocilia. Semin. Cell Dev. Biol. 19, 502–510 [DOI] [PMC free article] [PubMed] [Google Scholar]