

Background: Phosphatidylserine floppase activity of ABCA1 is required for optimal cholesterol efflux, as demonstrated via a floppase-impaired ABCA1 mutation.
Results: Sphingomyelin depletion compensates for floppase-impaired ABCA1 and increases cell surface phosphatidylserine.
Conclusion: Sphingomyelin depletion inhibits flip of anionic phospholipids and thus promotes cholesterol efflux.
Significance: Flippase inhibition may serve as a novel drug target to increase cholesterol efflux.
Keywords: ABC Transporter, Apolipoproteins, Phosphatidylcholine, Phosphatidylethanolamine, Phosphatidylserine, Sphingolipid, Sphingomyelinase
Abstract
The phosphatidylserine (PS) floppase activity (outward translocation) of ABCA1 leads to plasma membrane remodeling that plays a role in lipid efflux to apolipoprotein A-I (apoAI) generating nascent high density lipoprotein. The Tangier disease W590S ABCA1 mutation has defective PS floppase activity and diminished cholesterol efflux activity. Here, we report that depletion of sphingomyelin by inhibitors or sphingomyelinase caused plasma membrane remodeling, leading to defective flip (inward translocation) of PS, higher PS exposure, and higher cholesterol efflux from cells by both ABCA1-dependent and ABCA1-independent mechanisms. Mechanistically, sphingomyelin was connected to PS translocation in cell-free liposome studies that showed that sphingomyelin increased the rate of spontaneous PS flipping. Depletion of sphingomyelin in stably transfected HEK293 cells expressing the Tangier disease W590S mutant ABCA1 isoform rescued the defect in PS exposure and restored cholesterol efflux to apoAI. Liposome studies showed that PS directly increased cholesterol accessibility to extraction by cyclodextrin, providing the mechanistic link between cell surface PS and cholesterol efflux. We conclude that altered plasma membrane environment conferred by depleting sphingomyelin impairs PS flip and promotes cholesterol efflux in ABCA1-dependent and -independent manners.
Introduction
The plasma membrane has an asymmetric leaflet-specific distribution of phospholipids with an exclusive enrichment of phosphatidylserine (PS)2 in the inner leaflet, whereas complex sphingolipids reside mainly on the outer leaflet (1–4). Several ABC (ATP-binding cassette) proteins mediate the translocation of lipids from the inner to outer leaflet (floppase activity), such as ABCA1, which transports PS to the outer leaflet (4), whereas P-type ATPases are responsible for translocating phospholipids from the outer to inner leaflet (flippase activity) of the plasma membrane (4–6). ABCA1 is a multiple transmembrane domain protein that assembles lipid-free apolipoprotein A-I (apoAI) with cellular lipids to generate nascent high density lipoprotein (HDL) that is released from the cell surface, and thus mediates the cellular efflux of cholesterol and phospholipids. ABCA1 has three distinct activities: PS floppase, apoAI binding, and partial unfolding of the apoAI N-terminal helix bundle, all of which are required for the efficient lipidation of apoAI and generation of nascent HDL (7–15). Maintenance of proper levels of HDL is important, as many studies have shown that low levels of HDL are associated with higher incidences of cardiovascular disease. Elevated levels of low density lipoprotein (LDL) in plasma lead to deposition of cholesterol-laden lipoproteins within the arterial wall, leading to the formation of macrophage foam cells, a hallmark of atherosclerosis. Macrophages accumulate large amounts of cholesterol due to their inability to down-regulate modified LDL uptake (16). Macrophage cholesterol balance is thus dependent on cholesterol efflux by the ABC transporters ABCA1, which generates nascent HDL, and ABCG1, which effluxes cholesterol to existing HDL (17–20). The removal of cholesterol and lipids from peripheral tissues, such as foam cells, initiated by ABCA1-apoAI-mediated cholesterol efflux, and the delivery to the liver for excretion in the bile is known as the reverse cholesterol transport pathway. The significance of this pathway was revealed upon discovery of mutations in the ABCA1 gene in Tangier disease patients (21–23). These patients have significantly lower levels of HDL and accumulate higher levels of cholesterol in their tissues. The W590S Tangier mutant allele of ABCA1 retains its ability to bind lipid-free lipoprotein apoAI, but has impaired PS floppase activity and efflux of phospholipids and cholesterol to apoAI (15, 24–26).
Cells detect changes in membrane composition and respond by modulating the net biosynthetic output and trans-bilayer translocation of various lipids. A complex network of signaling cascades are activated upon perturbing lipid homeostasis, leading to changes in levels of sphingolipids and phospholipids to maintain membrane structure and integrity (27, 28). A previous study using the potent sphingolipid biosynthesis inhibitor myriocin reported increased cholesterol efflux to apoAI, although the proposed mechanism for increased cholesterol efflux was increased ABCA1 trafficking to plasma membrane (29, 30). Myriocin inhibits serine-palmitoyltransferase subunit 1 (SPT1, encoded by the SPTLC1 gene), which catalyzes the rate-limiting first step in the biosynthetic pathway of sphingolipids (31).
In the present study, we report that reduction of cellular sphingomyelin (SM) levels by either decreased synthesis or increased catabolism led to increased cholesterol efflux by ABCA1-independent and -dependent pathways. We found that the inward translocation (flip) of phospholipids was diminished upon SM reduction leading to enhanced exposure of PS on the outer leaflet. We found that SM depletion by myriocin or sphingomyelinase (SMase) treatment could compensate for the defective PS floppase activity of the mutant W590S-ABCA1 isoform, restoring its cholesterol efflux activity to apoAI. Overall, our data indicates that SM depletion led to a redistribution of anionic phospholipids across the plasma membrane, decreased lipid rafts, increased cholesterol availability to cyclodextrin by a non-ABCA1 mediated pathway, and enhanced ABCA1-mediated cholesterol efflux to apoAI.
EXPERIMENTAL PROCEDURES
Cell Culture
All cell culture incubations were performed at 37 °C, unless otherwise indicated, in a humidified 5% CO2 incubator. Cells were grown in DMEM with added antibiotics and 10% FBS. Drugs were added to growth media at the indicated concentrations and an equivalent amount of vehicle was added as control. Myriocin, sphingomyelin (catalog number S0756, from chicken egg yolk), methyl-β-cyclodextrin, and sphingomyelinase (from Staphylococcus aureus) were obtained from Sigma; fumonisin B1 was from Cayman Chemicals; and 18:1 NBD-PS, 18:1 NBD-phosphatidylethanolamine (PE), 25-NBD-cholesterol, NBD-sphingosine, and C2-dihydroceramide were obtained from Avanti Polar Lipids. Human apoAI was purified from human plasma as previously described (32).
Cholesterol Efflux Assay
On day 1, Abca1-GFP stably transfected HEK293 cells (15), non-transfected HEK cells, or RAW264.7 murine macrophages were plated in 24-well plates at a density of 400,000 cells per well. On day 2, the cells were labeled with [3H]cholesterol in 1% FBS in DMEM. To induce Abca1 expression in RAW264.7 cells, 0.3 mm 8-Br-cAMP (Sigma) was added on the evening of day 2. On day 3, the cells were washed and chased for 4 to 6 h in serum-free DMEM in the presence or absence of 5 μg/ml of apoAI. The radioactivity in the chase media was determined after a brief centrifugation to pellet any residual debris. Radioactivity in the cells was determined by extraction in hexane:isopropyl alcohol (3:2) with the solvent evaporated in a scintillation vial prior to counting. The percent of cholesterol efflux was calculated as 100 × (medium dpm)/(medium dpm + cell dpm).
DMPC-DPH Fluorescence Anisotropy Measurement
DMPC dissolved in chloroform:methanol (2:1, v/v) plus 0.2 mol % DPH was dried in a stream of nitrogen onto the sides of a glass culture tube and kept in vacuum overnight. The DMPC film was rehydrated at 5 mg/ml in PBS by extensive vortexing and alternating freeze/thaw in a dry ice/ethanol and 37 °C water bath. The resulting DMPC-DPH multilamellar vesicles (MLV) were warmed to 37 °C and extruded 11 times through a polycarbonate membrane using a mini-extruder (Avanti Polar Lipids) to derive large unilamellar vesicles (LUVs) of 100 nm diameter. DMPC:DPH (100:0.2 mol ratio) or DMPC:SM:DPH (75:25:0.2) LUVs were subjected to anisotropy measurements using a fluorimeter (PerkinElmer LS-50B) with excitation at 360 nm and emission at 430 nm. Measurements were performed over a temperature range (10–40 °C) in a water jacketed cuvette holder with a circulating chilling/heating water bath.
NBD-PS Translocation Assay in DMPC LUVs
The assay was adapted from an earlier study (33) describing rapid flip-flop of phospholipids in biological membranes. DMPC or DMPC:SM (75:25) LUVs were prepared as described above. Liposomes (120 μg of lipid/ml) were incubated with 25 μm NBD-PS for 5 min at 4 °C. To start the reaction, the labeled liposomes in 200-μl aliquots were transferred to a 96-well plate and incubated at 23 °C, the phase transition temperature of DMPC. At various time points NBD-PS fluorescence was measured after addition of either 20 μl of freshly prepared 1 m sodium dithionite (quench resistant signal) or 20 μl of 100 mm Tris, pH 10.0 (total signal). The fluorescent signal from samples was recorded using a fluorescent plate reader with excitation and emission wavelengths 470 and 530 nm, respectively. The quench-resistant signal resides on the inner leaflet of the LUV, and the percent of NBD-PS translocated was calculated as 100 × (quench resistant signal)/(total signal), and plotted against time to determine the rate of NBD-PS translocation.
Fluorescent Microscopy
To assess NBD-lipid trafficking, RAW264.7 cells were grown in 24-well plates for 24 h in growth media with 10 μm myriocin or DMSO vehicle. The cells were washed gently with PBS and incubated with fresh phenol red-free DMEM containing 25 μm NBD-labeled phospholipids at 37 °C for the indicated time period. The cells were washed and media was replaced with PBS followed immediately by epifluorescent microscopy. To assess lipid rafts, RAW cells grown in the presence of 10 μm myriocin (16 h pretreatment) or treated with SMase (treatment for 1 h with 3 units of SMase/ml media) were incubated with 1 μg/ml of cholera toxin B (CTB):Alexa 647 for 5 min at 37 °C prior to PBS washing and fluorescent microscopy at room temperature. All images were captured using an Olympus IX51 inverted epifluorescent microscope, Olympus LUCPlanFl ×40/0.6 lens, with a Q-Image EXi aqua camera and Olympus cellSens Dimension version 1.7 software. Post-imaging processing was performed in Adobe Photoshop CS2.
Flow Cytometry Analysis
To measure cell surface PS, cells were cultured in 6-well plates in growth media, washed twice with PBS, and resuspended in 500 μl of Annexin V binding buffer plus 1 μl of Annexin V-Cy5 (Biovision). The samples were incubated at room temperature for 5 min in the dark. Flow cytometry analysis was performed using a BD Biosciences LSRFortessa cytometer using a 639-nm excitation laser and emission at 650–670 nm; and, data were analyzed using FlowJo software.
To measure the inward translocation of various NBD-labeled phospholipids, RAW cells were incubated with phenol red-free DMEM containing 25 μm NBD-PS or NBD-PE for 5–15 min at 37 °C or RT. For neutral lipids, 2.5 μm NBD cholesterol and 50 μm NBD sphingosine was used. The media was then replaced with PBS. The cells were gently scraped and subjected to flow cytometry analysis first without sodium dithionite to calculate total NBD fluorescence, and then 5 min after the addition of 30 μl of a freshly prepared 1 m sodium dithionite solution to determine the fraction of the signal resistant to quenching (translocated signal). Flow cytometry analysis was performed as above using a 488-nm laser for excitation and emission at 505–525 nm.
To measure cell surface lipid rafts, CTB binding was assessed. RAW cells were treated with DMSO vehicle, 10 μm myriocin (16 h pretreatment), or 3 units of SMase/ml of media (1 h pretreatment) at 37 °C. These cells were then washed twice with PBS and gently scraped in 1.5 ml of PBS. Alexa 647:CTB was added at final concentration of 1 μg/ml and incubated for 1 min prior to flow cytometry as described above using a 639-nm excitation laser and emission at 650–670 nm.
SM Quantification
The levels of SM were determined by using the enzymatic assay described earlier (34, 35). SM was hydrolyzed by bacterial SMase and converted to N-acylsphingosine and phosphorylcholine. Alkaline phosphatase was used to convert phosphorylcholine to choline, which was then oxidized by choline oxidase and further acted upon peroxidase in the presence of DAOS (N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3,5-dimethoxyaniline, sodium salt), H2O2, and 4-aminoantipyrine to generate a blue colored dye. The SM standard solution was prepared by dissolving 5 mg of SM in 10 ml of 2% Triton X-100 in ethanol. Cells grown in 24-well plates were treated with 10 μm myriocin for 6 h or 3 units of SMase for 1 h at 37 °C. The cells were then washed twice with PBS and resuspended in 200 μl of Reagent 1 (2 units of SMase, 4.86 units of alkaline phosphatase, 2 mm DAOS, and 0.05% Triton X-100, 0.05 m Tris-HCl, 0.66 mm calcium chloride, pH 8). The cell suspension was then gently mixed by pipetting up and down several times and was transferred to a 96-well plate for a 20-min incubation at 37 °C. 100 μl of Reagent 2 (0.07 units choline oxidase, 3 units peroxidase, 0.72 mm 4-aminoantipyrine, 0.05 m Tris-HCl, 1 mm calcium chloride, pH 8) was added and incubated at 37 °C for 30 min. Absorption was measured at 620 nm using a plate spectrophotometer. The amount of SM was calculated by comparing it to a standard curve of SM and plotted per mg of cell protein determined using the BCA assay (Pierce).
ApoAI Lipid Solubilization Assay
MLVs containing DMPC or DMPC:SM (3:1 mol ratio) were prepared as described above. 20 μg of these MLVs in Tris-buffered saline:EDTA, pH 7.5, were incubated with 1 μg of human apoAI at 25 °C in a final volume of 200 μl. MLV solubilization by human apoAI was monitored by measuring sample turbidity (absorbance) at 325 nm using a plate spectrophotometer.
Total and Cell Surface ABCA1 Levels
RAW267.4 cells were cultured in DMEM with 10% FBS and treated with or without 300 μm 8-Br-cAMP to induce Abca1 for 16 h. Cells were incubated with apoAI (5 μg/ml) in the presence of either vehicle or 10 μm myriocin (4 h) or 3 units/ml of SMase (4 h, plus 1 h pretreatment). The cells were then incubated for 30 min on ice with phosphate-buffered saline (PBS) containing 1 mg/ml of sulfo-NHS-biotin (Pierce). The PBS-washed cell pellet was lysed in 200 μl of lysis buffer (150 mm sodium chloride, 5 mm EDTA, 50 mm Tris phosphate, pH 8.0, 1% Nonidet P-40, and 10% protease inhibitor). Cell surface ABCA1 was determined by purifying biotinylated proteins by overnight incubation of 250 μg of cell protein with 75 μl of UltraLink Plus Immobilized Streptavidin gel (Pierce) at 4 °C on a platform shaker in a final volume of 200 μl in PBS. The beads were spun down, washed, and resuspended by boiling in 100 μl of 1× NuPAGE LDS Sample buffer (Invitrogen). 45 μl of the bound protein samples along with 20 μg of total protein were run on NuPAGE 3–8% Tris acetate gels (Invitrogen) and transferred onto polyvinylidene fluoride membranes (Invitrogen). Blots were incubated sequentially with 1:500 mouse monoclonal antibody raised against ABCA1 (Santa Cruz Biotechnology, catalog number AC10) and 1:15,000 horseradish peroxidase-conjugated goat anti-mouse secondary antibody (Bio-Rad). The signal was detected with an enhanced chemiluminescent substrate (Pierce).
ATP Quantification
RAW267.4 cells were cultured in DMEM with 10% FBS and allowed to grow further in the presence of glucose or 2 mm deoxy-d-glucose for 4 h. Cell lysates were prepared using a buffer containing Nonidet P-40 and the postnuclear supernatant was assayed immediately using an ATP determination kit (Invitrogen, catalogue number A22066), per the manufacturer's protocol. Cellular ATP was calculated from an ATP standard curve and the levels of ATP were normalized by the supernatant protein content.
RESULTS
Myriocin Enhances Cholesterol Efflux and Restores Efflux Activity of the W590S Abca1 Mutation
We assessed the effects of myriocin, a potent inhibitor of the first step in sphingolipid synthesis on cholesterol efflux to apoAI, a process that is mediated by ABCA1, in stably transfected HEK293 cells expressing equivalent levels of wild type (WT) murine and two mutant ABCA1 isoforms (15). Baseline efflux to apoAI in the absence of myriocin was low in non-transfected HEK cells and significantly induced by 7.6-fold in cells expressing WT Abca1 (p < 0.0001 by ANOVA post test, Fig. 1A). As previously observed (15, 24–26), cholesterol efflux to apoAI from cells expressing either the W590S (PS translocation deficient) or C1477R (apoAI binding deficient) ABCA1 mutant isoforms was partially, but not completely impaired compared with the WT isoform (p < 0.001 compared with control HEK and WT ABCA1 by ANOVA post test). Upon treatment with 10 μm myriocin, cholesterol efflux was significantly increased in all four cell types, however, the net increase was greatest for the W590S mutant isoform (2.34% increase, 1.72-fold), with the wild type ABCA1 isoform having a 1.44% net increase, and the other two cell lines having <1% net increases. Thus, myriocin treatment could overcome much of the impaired cholesterol efflux activity of the W590S Abca1 mutation, although not greatly improving efflux from the C1477R mutation. To check if the myriocin effect was reproducible in cells expressing ABCA1 endogenously, we performed similar experiments using the murine macrophage cell line RAW264.7, in which the expression of ABCA1 is inducible by cAMP analogues (36, 37). Treatment of RAW cells with 10 μm myriocin during the apoAI chase led to a small but significant increase in efflux in the absence of Abca1 induction, but had a larger (4% net increase) effect on cholesterol efflux to apoAI when Abca1 was induced (Fig. 1B).
FIGURE 1.
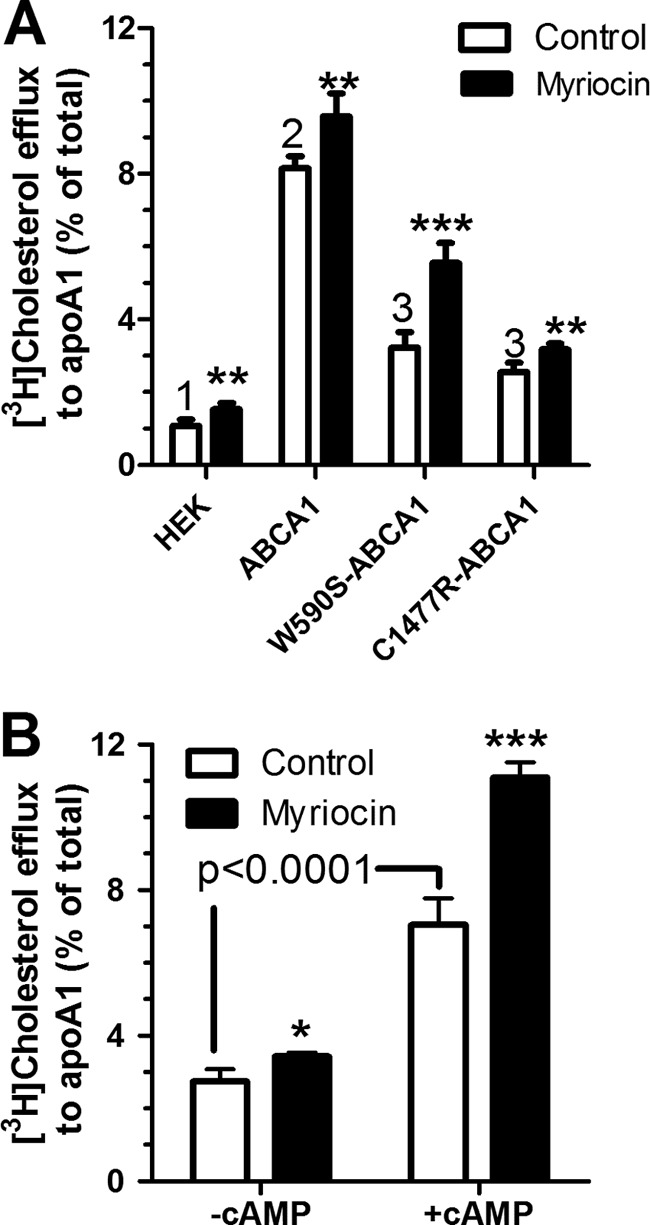
Myrocin induces cholesterol efflux. A, myriocin compensates for the W590S-Abca1 mutation. Non-transfected HEK293 cells along with HEK cells transfected with WT-Abca1-GFP, W590S-Abca1-GFP, or C1477R-Abca1-GFP isoforms were incubated with radiolabeled cholesterol and subsequently chased with apoAI (5 μg/ml) for 6 h. The chase media contained either 10 μm myriocin (black bars) or DMSO vehicle (open bars). B, RAW264.7 murine macrophages pretreated with or without 8-Br-cAMP to induce Abca1 were loaded with cholesterol and chased as described above. Values are % cholesterol efflux mean ± S.D., n = 4. Different numbers above the bars show p < 0.001 for the samples treated in the absence of myriocin by ANOVA with a Bonferroni post test. Asterisks represent the effect of myriocin in two-tailed t tests; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Sphingomyelin Depletion Enhances Cholesterol Efflux Similar to Myriocin
Myriocin inhibits the first step in sphingolipid synthesis and thus blocks the formation of all sphingolipids including ceramide and SM. Additionally, a prior study implicated that SPT1, the target of myriocin, interacts directly with ABCA1 retaining it in the ER, and that myriocin treatment disrupts the SPT1-ABCA1 interaction allowing ABCA1 to traffic to the plasma membrane (29). Thus, we examined whether the myriocin effect on cholesterol efflux was specific to this SPT1 interacting drug, and if not, which sphingolipid species was responsible for this effect. The pathway of sphingolipid synthesis and the specific inhibitors used are shown in Fig. 2. First we tested fumonisin B1, an inhibitor of ceramide synthase (38, 39), and similar to myriocin, it induced efflux to Abca1-induced RAW264.7 cells (Fig. 3A), demonstrating that the myriocin effect was due to sphingolipid depletion rather than the direct effect of myriocin on ABCA1 trafficking. To determine whether SM was responsible for the myriocin and fumonisin B1 effects on cholesterol efflux, we used SMase treatment to directly cleave the phosphocholine headgroup from SM and create ceramide. In 8-Br-cAMP-induced RAW264.7 cells, SMase, as well as myriocin significantly increased cholesterol efflux to apoAI to the same extent (Fig. 3A), thus demonstrating that SM depletion is responsible for the observed effects as it is the only sphingolipid change predicted to be shared by myriocin, fumonisin B1, and SMase treatments (Fig. 2).
FIGURE 2.
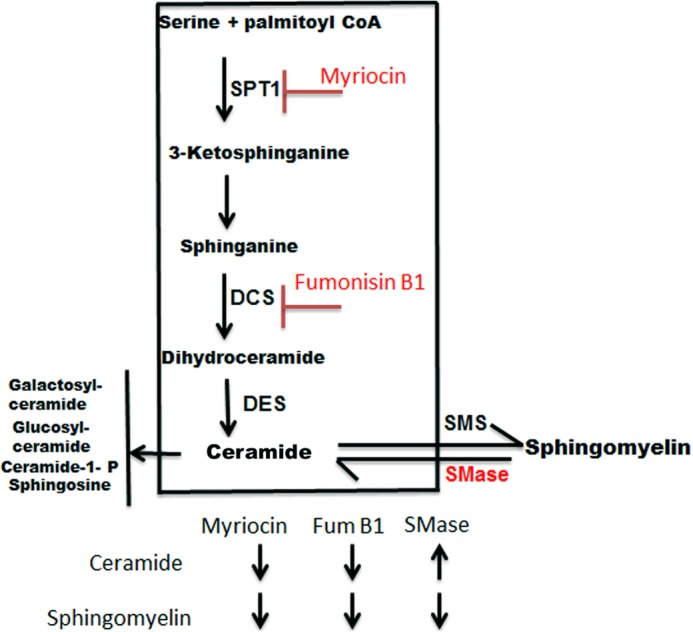
Sphingolipid biosynthetic pathway and SM inhibitors. Schematic depicting SM depletion by different treatments. Myriocin blocks SPT1 that catalyzes the first and rate-limiting step in SM biosynthesis. Fumonisin B1 inhibits dihydroceramide synthase (DCS). SMase catalyzes hydrolytic cleavage of SM, generating ceramide and phosphocholine. Depletion of SM is common to all three treatments, whereas ceramide is decreased by the first two, and increased by the third treatment.
FIGURE 3.

A sphingomyelinase treatment increases cholesterol efflux and decreases a marker of lipid rafts. A, sphingomyelin depletion leads to increased apoAI-mediated cholesterol efflux in RAW cells. 8-Br-cAMP pretreated RAW264.7 macrophages were cholesterol labeled and then chased with apoAI (5 μg/ml) in the presence of 3 units/ml of SMase, 10 μm myriocin (Myr), or 10 μm fumonisin B1. Values plotted are % cholesterol efflux, n = 3, mean ± S.D., different numbers above the bars show p < 0.01, by ANOVA with a Bonferroni post test. B, myriocin or SMase treatment do not increase total or cell surface ABCA1 levels. RAW267.4 cells were incubated with or without 8-Br-cAMP to induce endogenous Abca1. These cells were either treated with vehicle or 10 μm myriocin for 16 h or pretreated with 3 units/ml of SMase for 1 h at 37 °C. Cell surface proteins were labeled with biotin as described under “Experimental Procedures.” The cells extracts were prepared and biotinylated proteins were isolated on streptavidin-agarose beads. Total and cell surface protein fractions were resolved on SDS-PAGE gel, transferred to a membrane, and probed for ABCA1. Position of the molecular mass markers in kDa are shown to the left of each blot. C, cell surface GM1 levels, a marker for lipid rafts, were assessed by binding of cholera toxin B (CTB) in SM-depleted cells. RAW cells were treated with 10 μm myriocin (24 h) or 3 units/ml of SMase (1 h) at 37 °C and washed twice with PBS. 1 μg/ml of Alexa 647-labeled CTB was added and cells were incubated for 1 min at RT. The cells were washed again with PBS and visualized by epifluorescent microscopy. D, for quantification of CTB binding, RAW cells treated with myriocin or SMase were washed with PBS and gently scraped from the wells in PBS. 1 μg/ml of Alexa 647-labeled CTB was added to cells and incubated for 1 min before subjecting to flow cytometry analysis. Values are the mean ± S.D. of the median fluorescence from 3 independent wells, different numbers above the bars show p < 0.01, by ANOVA with a Bonferroni post test. E, primary flow cytometry data showing decreased binding of CTB to SM-depleted RAW264.7 cells. Red indicates untreated cells, green and blue indicate SMase- and myriocin-treated cells, respectively. F, myriocin and SMase effects on cellular SM levels. RAW267.4 cells or HEK cells transfected with either WT-Abca1 or W590S-Abca1 isoforms were treated with either 10 μm myriocin for 6 h or 3 units/ml of SMase for 1 h at 37 °C. SM levels were determined by an enzymatic assay as described under “Experimental Procedures” (n = 3, mean ± S.D., different numbers above the bars show p < 0.0001, by ANOVA with a Bonferroni post test). G, SMase dose-response on cholesterol efflux to apoAI. HEK cells stably transfected with either WT-Abca1-GFP or W590S-Abca1-GFP isoforms were loaded with radiolabeled cholesterol and chased with apoAI (5 μg/ml) for 6 h. Different units/ml of SMase were added during the chase. Values are % cholesterol efflux mean ± S.D. of duplicate wells.
We performed cell surface biotinylation and examined total and cell surface ABCA1 levels by Western blot. Treatment with myriocin or SMase did not appreciably increase total or cell surface ABCA1 levels in 8-Br-cAMP-induced RAW264.7 cells, thus ruling out increased cell surface ABCA1 as the mechanism for increased cholesterol efflux to apoAI in these studies (Fig. 3B). We also determined the effects of myriocin and SMase on cellular lipid rafts by staining for the ganglioside GM1 with Alexa 647-labeled cholera toxin B as assessed by fluorescence microscopy and flow cytometry; and, as expected, both treatments significantly reduced the cellular lipid rafts (Fig. 3, C–E). We directly measured SM levels in RAW264.7 and HEK-Abca1 cells, and found that 10 μm myriocin or 3 units/ml of SMase both led to significant and similar ∼70% reductions in cellular SM levels (Fig. 3F).
We then compared the effects of depleting sphingomyelin on the cholesterol efflux activity of the WT and W590S isoforms of ABCA1 in stably transfected HEK cells. Increasing levels of SMase led to increased cholesterol efflux to apoAI for both isoforms, such that at the highest dose of SMase, the W590S isoform had as much efflux activity as the WT isoform in the absence of SMase (Fig. 3G). Thus, we could obtain complete restoration of the efflux activity of the ABCA1 W590S isoform with sufficient SM reduction, compared with the partial restoration observed in Fig. 1A.
Although it has been previously reported that treatment of cells with exogenous C2-dihydroceramide could induce ABCA1-mediated cholesterol efflux (40), we did not observe any effect of C2-dihydroceramide treatment on the ABCA1-mediated cholesterol efflux in RAW264.7 cells (data not shown). Taken together, our data strongly indicate that decreased SM, rather than increased ceramide, is responsible for the induction of cholesterol efflux, which can compensate for the impaired PS translocation in the W590S ABCA1 mutation.
SM Depletion Increases Cholesterol Efflux to Nonspecific Acceptors
To determine whether SM depletion altered non-ABCA1-mediated cholesterol efflux, we incubated uninduced RAW264.7 cells with the nonspecific cholesterol acceptor β-methyl cyclodextrin for 5 min at 23 °C. These short incubations at room temperature are used to assess the level of available cholesterol on the outer leaflet of the plasma membrane (41). Pretreatment with myriocin or SMase significantly up-regulated cholesterol efflux to cyclodextrin by >3-fold (Fig. 4A). To determine whether SM depletion altered cholesterol efflux in non-transfected HEK cells that do not express ABCA1, we incubated these cells with the weak detergent sodium taurocholate (NaTC), a nonspecific cholesterol acceptor. NaTC increased cholesterol efflux by >3-fold, which was augmented by another 1.6-fold upon myriocin pretreatment (Fig. 4B). Although WT ABCA1 expression increased cholesterol efflux to NaTC to almost 6-fold, expression of the W590S ABCA1 isoform increased efflux to NaTC by only 3.1-fold, as previously observed (15, 25). Myriocin increased cholesterol efflux from cells expressing either ABCA1 isoforms (1.6-fold for the WT and 2.1-fold for the W590S isoform), such that the myriocin-treated W590S-expressing cells exhibited comparable efflux to NaTC as seen in the myriocin-untreated WT-ABCA1 expressing cells (see gray bars in Fig. 4B). Thus, the SM depletion effect on cholesterol efflux is observed both in the absence and presence of ABCA1 expression, and can compensate for the W590S Abca1 mutation even with a non-selective acceptor.
FIGURE 4.
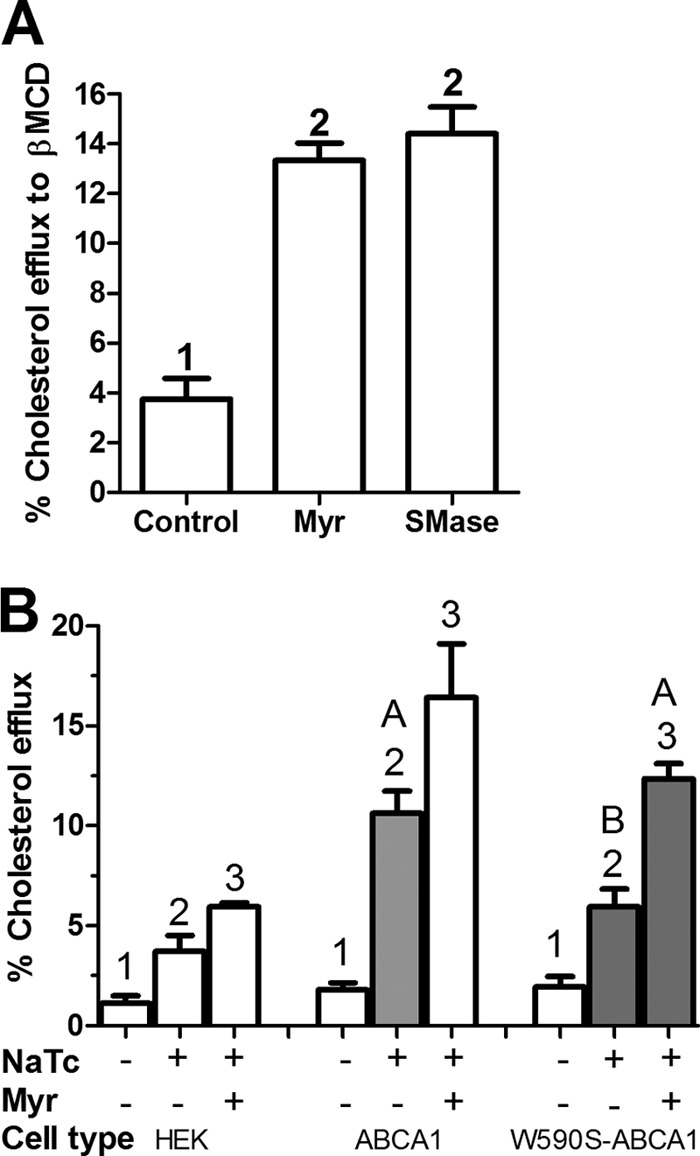
Sphingomyelin depletion induces ABCA1-independent cholesterol efflux. A, RAW macrophages were incubated with radiolabeled cholesterol. Control samples were treated with DMSO vehicle, whereas other samples were treated with either 10 μm myriocin for 16 h or 3 units/ml of SMase for 1 h and then subjected to cholesterol efflux to 2 mm β-methyl cyclodextrin for 5 min at 23 °C. Values are % cholesterol efflux mean ± S.D., n = 3, different numbers above the bars show p < 0.0001 by ANOVA with a Bonferroni post test. B, SM depletion by myriocin increases ABCA1-independent and -dependent cholesterol efflux to NaTC. HEK293 cells or stably transfected lines with either WT or W590S isoform of Abca1 were incubated with radiolabeled cholesterol and subsequently treated with or without 10 μm myriocin for 12 h. Cholesterol efflux to 1 mm NaTC for 4 h (continued presence of myriocin when indicated) was determined. Values are % cholesterol efflux mean ± S.D., n = 3, different numbers above the bars show p < 0.05 within each cell type by ANOVA with a Bonferroni post test. We also compared efflux to NaTC in the three conditions shown in the gray bars. Here, different letters above the bars show p < 0.01 by ANOVA with a Bonferroni post test.
SM Depletion Increases Cell Surface PS
The above data indicate that depletion of SM increased cholesterol efflux to apoAI as well as non-apoAI acceptors, but the mechanism was unclear. Because the ABCA1 W590S mutation has defective translocation of PS to the outer leaflet of the plasma membrane (11, 24), we assessed whether SM depletion altered the translocation of phospholipids across the plasma membrane. Outer leaflet PS was quantified by Annexin-V staining via flow cytometry. The WT-Abca1 expressing HEK cells showed significantly higher (1.7-fold) cell surface PS as compared with control HEK cells, indicating that ABCA1 expression led to increased PS translocation as expected (Fig. 5A). As reported earlier, the W590S-Abca1 expressing HEK cells showed decreased cell surface PS as compared with the WT isoform (15, 24, 25), only 1.3-fold higher than control HEK cells. However, upon treatment of the W590S-ABAC1 cells with myriocin, there was an almost complete restoration of cell surface PS (1.55-fold versus control HEK cells) (Fig. 5, A and B). The myriocin-treated control HEK cells also exhibited a small (1.2-fold) but significant increase in cell surface PS, again demonstrating an ABCA1 independent activity due to SM depletion (Fig. 5, A and B). To examine if myriocin treatment led to similar effects in cells with physiological expression of ABAC1, we used RAW264.7 macrophages. Uninduced RAW cells showed basal levels of PS exposure, which, as expected, were increased by 2-fold upon induction of Abca1 expression by 8-Br-cAMP treatment (Fig. 5, C and D). Myriocin led to significant increases in cell surface PS in RAW264.7 cells with or without expression of endogenous ABCA1, again demonstrating an ABCA1-independent activity. We measured dead cell levels in RAW 264.7 cells treated with myriocin or SMase by flow cytometry. We observed practically no dead cells upon SM depletion by myriocin or SMase treatment (Fig. 5E).
FIGURE 5.

Myriocin treatment enhances ABCA1 independent exposure of PS at the plasma membrane outer leaflet. A, HEK293 cells and lines stably transfected with different isoforms of Abca1 were treated with either 10 μm myriocin or vehicle for 24 h. PS exposure was determined by flow cytometry via annexin V binding. Values are the mean ± S.D. of the median fluorescence from 3 independent wells, different numbers above the bars show p < 0.05 by ANOVA with a Bonferroni post test. B, primary flow cytometry data showing shift in annexin V signal in HEK293, HEK-Abca1, and HEK-W590S-Abca1 cells with or without treatment with myriocin. C, RAW macrophages were incubated with or without 8-Br-cAMP to induce Abca1 and with or without 10 μm myriocin for 24 h. PS exposure was determined by flow cytometry via annexin V binding. Values are the mean ± S.D. of the median fluorescence from 3 independent wells, different numbers above the bars show p < 0.01, by ANOVA with a Bonferroni post test. D, primary flow cytometry data showing shift in annexin V signal in RAW264.7 cells after treatment with cAMP or myriocin or both. E, flow cytometry analysis of dead cells using the Molecular Probes Dead Cell UV dye. The dark blue line shows cells with no dye. The red line indicates control cells with no treatment, whereas the brown and green lines indicate SMase (1 h) and myriocin treatments (4 h), respectively. The light blue line indicates the positive control using heat-treated dead cells.
SM Depletion Impairs Inward Translocation of Anionic Phospholipids
The increased levels of cell surface PS upon myriocin treatment could be due to an increased rate of PS flopping (outward translocation) or a decreased rate of PS flipping (inward translocation). We qualitatively and directly visualized the flip of exogenously added NBD headgroup-labeled PS in RAW264.7 cells by fluorescent microscopy. In the absence of myriocin, much of the exogenous NBD-PS moved to intracellular compartments over a 10-min 37 °C incubation period (Fig. 6A). However, in myriocin-treated cells, more of the label was observed in the plasma membrane. The flipping defect in myriocin-treated cells was still apparent at the 20-min incubation period, although some myriocin-treated cells showed intracellular accumulation of NBD-PS (Fig. 6A). To quantitatively measure flipping, we took advantage of the membrane-impermeable NBD quenching reagent sodium dithionite. Control RAW cells, or those depleted of SM by treatment with myriocin or SMase, were incubated with different NBD-labeled phospholipids and the amount flipped into the cells was calculated by measuring the % of NBD fluorescence resistant to quenching by sodium dithionite using flow cytometry. We performed a time course for NBD-PS flipping into the cells. Over a 15-min 37 °C incubation period almost 80% of exogenously added NBD-PS was flipped into the cells, but in SM-depleted cells, only ∼44% (myriocin) and 41% (SMase) of NBD-PS was flipped (p < 0.01 by ANOVA, Fig. 6B). A similar result was obtained using NBD-PE, in which untreated cells flipped 75%, whereas SM-depleted cells flipped 53 (myriocin) and 50% (SMase) (p < 0.01 by ANOVA, Fig. 6C). We observed a similar inhibitory effect of SM depletion on NBD-PS flipping at 23 °C, a temperature at which most endocytosis is halted (42) (Fig. 6D). A previous study also showed that uptake of NBD-PS is via a non-endocytic pathway (43). We also examined the flipping of two neutral NBD-labeled lipids. Both NBD-sphingosine and NBD-cholesterol were flipped efficiently into cells during a 20-min incubation at 37 °C; however, unlike the anionic phospholipids, there was no inhibitory effect of myriocin on the flip of these neutral lipids (Fig. 6, E and F). Taken together, these results show that SM depletion specifically inhibited anionic phospholipid flipping, which cannot be accounted for by an effect on endocytosis.
FIGURE 6.

SM depletion impairs anionic PL flip in RAW264.7 cells. A, myriocin impairs NBD-PS flip across the plasma membrane. RAW cells were treated with 10 μm myriocin or vehicle for 24 h. These cells were then incubated with 25 μm NBD-PS at RT for the indicated time, washed with PBS, and subjected to fluorescent microscopy. B, quantification of NBD-PS translocated inside the cells upon SM depletion. RAW macrophages were treated with either 10 μm myriocin or vehicle for 24 h, or incubated with 3 units/ml of SMase for 1 h. These cells were then incubated with 25 μm NBD-PS at 37 °C for different time periods (minutes) in phenol red-free DMEM. The cells were gently scraped and then subjected to flow cytometry analysis in the presence or absence of sodium dithionite as described under “Experimental Procedures” to calculate the % flipped. Values are the % NBD-PS translocated into the cells, mean ± S.D. of the median fluorescence from 3 independent wells, *, p < 0.0001 versus treated cells by ANOVA with a Bonferroni post test for each time point. C, quantification of NBD-PE translocated inside the cells upon SM depletion. RAW macrophages were treated as above and then incubated with 25 μm NBD-PE at 37 °C for 10 min in phenol red-free DMEM. Values are the % NBD-PE translocated into the cells, mean ± S.D. of the median fluorescence from 3 independent wells, different numbers above the bars show p < 0.01, by ANOVA with a Bonferroni post test. D, same as in B, but the experiment was performed at 23 °C. E and F, SM depletion did not alter inward translocation of NBD-cholesterol or NBD-sphingosine. RAW macrophages were treated with either 10 μm myriocin or vehicle for 16 h in DMEM with 10% FBS. The cells were then incubated with 2.5 μm 25-NBD-cholesterol (NBD-Cho) or 50 μm NBD-sphingosine (NBD-Sph) at 37 °C for 20 min in FBS-free DMEM. The cells were first washed with PBS and then gently scrapped from the plates in phenol red-free DMEM. These cells were then subjected to flow cytometry analysis as described for the NBD-PS translocation assay. The graphs shows % NBD-cholesterol (E) or % NBD-sphingosine (F) flipped into the cell (n = 3, mean ± S.D. of the median fluorescence from 3 independent wells; NS, not significant by two-tailed t test).
SM Increases the Inward Translocation of PS in Liposomes and Impairs ApoAI Solubilization
To determine whether there were inherent membrane effects of SM on PS flipping, we measured NBD-PS translocation in LUV. We prepared DMPC LUVs with or without 25% (mole ratio) SM and measured exogenously added NBD-PS inward translocation using sodium dithionate quenching at various times, adapting a previously described method (33). Preliminary studies revealed that robust NBD-PS translocation occurred at 23 °C, the DMPC phase transition temperature, but not at 4 or 37 °C. DMPC:SM (3:1) LUVs had 1.5-fold faster initial rates (over the first 5 min) of NBD-PS inward translocation than pure DMPC LUVs (p < 0.0001, Fig. 7A). Because SM has a phase transition temperature of 42 °C, we determined the phase transition temperature (Tm) of the DMPC and DMPC:SM LUVs directly by DPH anisotropy. The DMPC and DMPC:SM LUVs had Tm values of 23.0 and 24.9 °C, respectively; and the disordered phase for the DMPC:SM LUVs had a higher plateau anisotropy value, indicative of more order in the disordered phase compared with the DMPC LUVs (Fig. 7B). At 23 °C, the DMPC LUVs were much more disordered than the DMPC:SM LUVs; thus, the faster initial rate of NBD-PS flip by the DMPC:SM LUVs cannot be attributed to increased lipid disorder.
FIGURE 7.

Effect of SM on NBD-PS translocation, anisotropy and rHDL formation in liposomes. A, DMPC and DMPC:SM (3:1 mole ratio) LUVs were incubated with 25 μm NBD-PS at RT and the rate of NBD-PS translocation was determined by sodium dithionite quenching as described under “Experimental Procedures.” Values plotted are the mean ± S.D., n = 3, *, p < 0.05 versus DMPC at each time point by two-tailed t test. B, the lipid phase transition temperature was determined by DPH anisotropy in DMPC and DMPC:SM (3:1) LUVs as described under “Experimental Procedures.” C, apoAI-mediated solubilization of DMPC (solid line) and DMPC:SM (3:1, dashed line) LUVs was determined by incubating these LUVs with apoAI (20:1 mass ratio lipid:apoAI) and recording the decrease in turbidity over time. D, β-methyl cyclodextrin-mediated cholesterol stripping from liposomes containing different amounts of PS. Cholesterol efflux to cyclodextrin was measured at 7 min at 23 °C (n = 3, *, p < 0.05 versus 0 and 1% POPS by ANOVA with a Bonferroni post test).
ApoAI incubation with ABCA1 expressing cells forms nascent HDL, but apoAI can also spontaneously interact with DMPC liposomes, solubilizing the lipids, decreasing the turbidity of the liposome suspension, and generating reconstituted HDL (44). This reaction occurs fastest at the lipid phase transition temperature, as it is thought that apoAI can best penetrate the liposome surface at phase transition boundaries. We performed apoAI-mediated MLV clearance assays at 25 °C, the phase transition temperature of the DMPC:SM MLVs, and we found that inclusion of SM in the DMPC MLVs greatly retarded MLV clearance (Fig. 7C). These results indicate that at 25 °C, where phase transition boundaries are expected to be more prevalent for the DMPC:SM MLVs than the DMPC MLVs, the presence of SM impaired apoAI-mediated lipid solubilization.
To further probe the role of exposed anionic phospholipids in enhanced cholesterol extractability from membranes, we performed cyclodextrin-mediated extraction of radiolabeled cholesterol from liposomes with varying amounts of PS. The liposomes were made with POPC:cholesterol (9:1), a tracer amount of radiolabeled cholesterol, and 0, 1, or 5% POPS. As shown in Fig. 7D, the MLVs containing 5% PS had higher levels of cholesterol extraction by cyclodextrin. Thus, membrane PS had a direct effect on the extractability of cholesterol, which was not dependent upon membrane SM.
Cellular PS Flip Is Dependent upon ATP
The reduced rate of cellular flipping of NBD phospholipids upon depletion of SM could be due to either a direct effect on the activity of ATP-dependent flippases and/or to inherent changes in properties of the plasma membrane independent of flippase function. To examine the role of flippases on NBD-PS inward translocation, we repeated the NBD-PS cellular translocation assay in cells depleted of ATP by preincubation with 2-deoxyglucose. As shown in Fig. 8A glucose deprivation by 2-deoxyglucose led to a ∼5-fold decrease in cellular ATP levels. 2-Deoxyglucose treatment also decreased NBD-PS flip by 59% versus control (p < 0.001), a larger decrease in PS flip than was observed by SMase treatment (32% versus control, p < 0.001, Fig. 8B). Combining SMase treatment with ATP depletion led to an additional small decrease in NBD-PS flip. Taken together with the liposome studies, our data indicate that the majority of anionic phospholipid cellular flippase activity is mediated by ATP-dependent flippases, and suggest that SM can promote PS translocation by two pathways: 1) by increasing the activity of cellular flippases; and 2) by a direct change in the membrane property increasing flippase-independent PS translocation.
FIGURE 8.
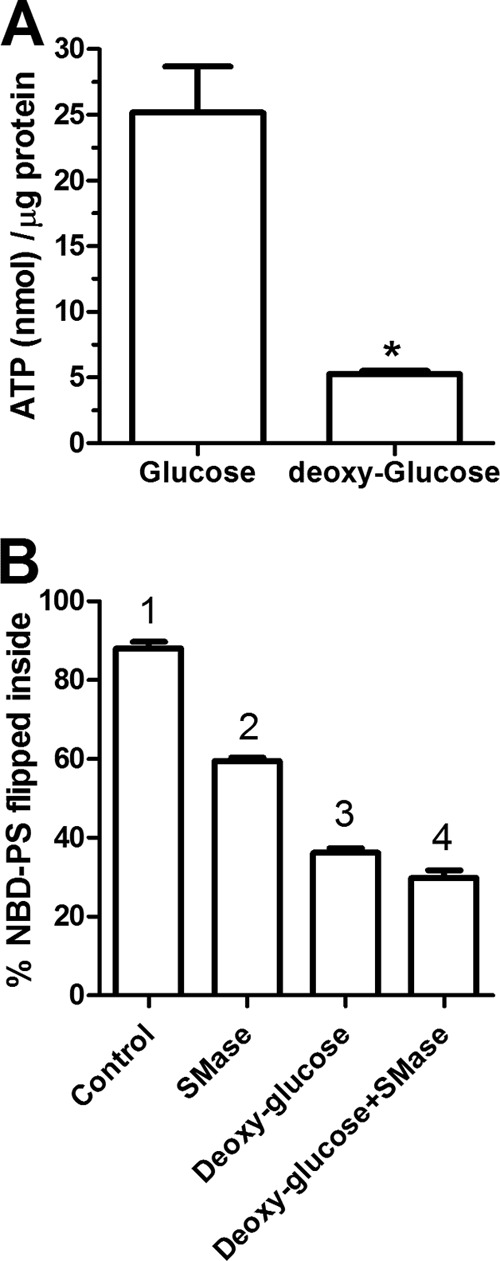
Majority of cellular NBD-PS translocation is ATP dependent. RAW264.7 macrophages were grown in the presence of glucose or 2 mm deoxy-d-glucose for 4 h. A, ATP levels were measured in freshly prepared cell extracts using the ATP determination kit from Molecular Probes (n = 3; *, p < 0.001 by two-tailed t test). B, the ATP-depleted (deoxyglucose) and control cells were treated with 3 units/ml of SMase for the final 1 h. These cells were then incubated with 25 μm NBD-PS at 37 °C for 10 min in phenol red-free DMEM. After 10 min, the media was replaced with PBS. The cells were gently scraped and then subjected to flow cytometry analysis as described in the legend to Fig. 6. Values are the % NBD-PS translocated into the cells, mean ± S.D. of the median fluorescence from 3 independent wells, different numbers above the bars show p < 0.001, by ANOVA with a Bonferroni post test.
DISCUSSION
Eukaryotic cells have evolved robust mechanisms to maintain their plasma membrane asymmetry. Anionic phospholipids such as PS and PE are highly enriched in the plasma membrane inner leaflet due to the activity of cellular PS/PE flippases belonging to class P4 type ATPases (3). In contrast, some members of the ABC gene family (reviewed in Ref. 6) have floppase activity, including ABCA1, which can mediate the flop of PS to the outer leaflet of the plasma membrane (14).
Sphingomyelin is the second most abundant lipid and major sphingolipid component of HDL and the plasma membrane, and it forms lipid rafts that are enriched in free cholesterol (45, 46). Using somatic cell genetics, CHO cells defective in ceramide transfer protein, which cannot produce endogenous SM from ceramide, were found to have increased cellular cholesterol efflux to cyclodextrin, without altering the level of cell surface-free cholesterol, but increasing the availability of plasma membrane-free cholesterol for efflux (47). A similar finding was reported in fibroblasts incubated with SMase (48). These studies demonstrate that SM plays a role in the availability of plasma membrane-free cholesterol, but not its absolute levels.
The mechanism for the stimulation of ABCA1 efflux activity by SM depletion is controversial. Tamehiro et al. (29) reported that myriocin treatment increases the cell surface expression of ABCA1 without changing the overall expression of ABCA1. They found that SPT1 binds directly to ABCA1, trapping it in the ER, and that myriocin disrupts the SPT1-ABCA1 interaction, allowing ABCA1 to traffic to the plasma membrane. However, Yamauchi et al. (49) showed that cellular SM depletion by SMase digestion increases ABCA1 levels by inhibiting its turnover. In SM-depleted mutant CHO cells, ABCA1-mediated cholesterol efflux was increased without altering the cell surface or total ABCA1 levels (50). Also, a genetic lowering of macrophage SM via a different subunit of serine palmitoyltransferease (Sptlc2-hemizygous mice) was also reported to increase the ABCA1-dependent cholesterol efflux to apoAI in cholesterol-loaded cells, associated with a decrease in SPT1 protein and an increase in total ABCA1 protein levels (51). Meanwhile Ghering and Davidson (40) reported that the addition of a short chain ceramide analog to ABCA1 expressing cells increases ABCA1 levels and cholesterol efflux to apoAI, whereas physiological ceramides had no effect.
In the current study, we found that treatment of cells with myriocin, fumonisin B1, or SMase all led to increased ABCA1 activity. These three treatments all deplete SM levels, whereas the first two are predicted to deplete and the third to increase ceramide levels. Thus, the level of SM rather than any other sphingolipid species seems to be critical for increasing ABCA1 activity. In addition, we did not see any increase in ABCA1 activity upon treatment of RAW264.7 cells with C2-dihydoceramide, ruling out short-chain ceramides as the mediator for enhanced cholesterol efflux. Furthermore, we found that both myriocin and SMase treatments induced cholesterol efflux to cyclodextrin in cells without ABCA1 expression, similar to what was observed previously upon SMase treatment or in cells with defective SM synthesis (47, 48, 50). In addition, SM depletion by myriocin or SMase increased ABCA1 activity (Fig. 3A) without increasing total or cell surface ABAC1 levels (Fig. 3B). Thus, any potential effects on ABCA1 levels cannot explain the results in our studies. Our findings confirm those in mutant CHO cells, in that both ABCA1-dependent and -independent cholesterol efflux are stimulated when SM is depleted.
Overall, prior studies and our data indicate the existence of links between SM depletion and ABCA1-dependent and -independent cholesterol efflux activity, although the exact mechanism of these effects are controversial or unknown. It is possible that the ABCA1-dependent and -independent effects of SM depletion may share the same mechanism or they may occur via different mechanisms. One possibility is that SM depletion decreases lipid rafts, where SM and free cholesterol (FC) are enriched in microdomains of the plasma membrane, and depletion of rafts would redistribute plasma membrane FC to non-raft domains. This redistribution of cholesterol to a non-raft microdomain was observed in mutant CHO cells defective in endogenous SM synthesis (47). We confirmed in RAW267.4 mouse macrophages that depletion of SM by myriocin or SMase treatment decreased rafts using cell surface GM1 levels as a surrogate for rafts. Thus, the ABCA1-independent increase in free cholesterol efflux to cyclodextrin (Fig. 4A) may be in part mediated by its redistribution to a non-raft domain. Most researchers agree that ABCA1 is not located in lipid rafts and that it mediates cholesterol efflux primarily from non-raft domains (52–54). ABCA1 activity seems to destabilize lipid rafts and has been shown to promote the transfer of FC from the raft to the non-raft domain in BHK cells (55), and Abca1-deficient macrophages are reported to have more lipid rafts (56).
However, Abca1 induction has only a very minor effect on the initial rates of cholesterol efflux to cyclodextrin at 37 or 21 °C in RAW264.7 macrophages (57), indicating that ABCA1 does not necessarily have a large effect on FC redistribution in the plasma membrane in macrophages, and that there may be some degree of cell type specificity for ABCA1 effects on plasma membrane FC distribution. Thus, SM depletion may evoke a distinct mechanism in addition to FC redistribution that can increase ABCA1-mediated cholesterol efflux activity to apoAI (Fig. 1). This was also indicated by our finding that myriocin increased cholesterol efflux to NaTC in HEK cells, but even more so in cells transfected with WT and particularly the W590S mutant isoform of ABCA1 (Fig. 4B).
The first indication of what this separate mechanism might be was our finding that SM depletion increased cell surface PS in an ABCA1-independent fashion (Fig. 5). In fact, ABCA1 itself also raises cell surface PS, although this activity is greatly diminished in the W590S ABCA1 mutation that leads to Tangier disease (15, 24–26). The SM depletion-mediated increase in cell surface PS was sufficient to compensate for the defective PS translocation in the W590S-Abca1 expressing HEK cells, restoring cholesterol efflux to apoAI (Fig. 3G) and NaTC (Fig. 4B), providing direct proof that the ABCA1 PS floppase activity plays a role in ABCA1-mediated cholesterol efflux. Previously we had demonstrated that increased cell surface PS is, by itself, not sufficient to promote cholesterol efflux to apoAI, although we did not claim that the PS floppase activity of ABCA1 is not required for its cholesterol efflux activity (58).
We identified that the mechanism by which SM depletion led to increased cell surface PS is via a decrease in the rate of inward translocation (flip) of anionic phospholipids, even in the absence of ABCA1 expression (Fig. 6). The effect of SM to increase PS flip was confirmed in DMPC liposomes (Fig. 7A). This increase in cell surface PS could partially restore the activity of the PS-floppase-deficient Abca1 W590S mutation (Figs. 1A and 3G). However, it is difficult to ascertain whether increased cell surface PS mediates increased cholesterol efflux independent of the SM content of the membrane. Therefore, we used liposomes again, and demonstrated that adding 5% PS to POPC:cholesterol (9:1) MLVs increased cholesterol extraction by β-methyl cyclodextrin (Fig. 7D). Thus, the combination of cellular and liposome studies illuminate the role of SM on PS flip, and the effect of PS on cholesterol efflux/extractability.
SM may play a role in regulating phospholipid movement across membranes by virtue of its ability to make phase boundary domains where gel and liquid disordered phases abut each other and membrane lattice defects occur. These molecular packing defects at the phase transition temperature have been shown to promote the rapid transbilayer movement of NBD-labeled phospholipids in liposomes (33). In agreement with this finding, we observed the rapid flip of NBD-PS in the DMPC liposomes at its phase transition temperature of 23 °C, but not at 4 or 37 °C. We found only a minor increase in the phase transition temperature of DMPC liposomes upon the addition of 25% SM (Fig. 7B). At the lipid phase transition temperature, the membrane lattice defects at phase boundaries are also thought to mediate the increased rate of microsolubilization of phospholipid liposomes by apoAI (59). However, we observed less, and not more, apoAI-mediated solubilization of the DMPC:SM (3:1) liposomes versus the DMPC only liposomes despite performing this study at 25 °C, the measured phase transition temperature for the DMPC:SM mixture. This result implies that SM may also have a direct effect on apoAI to inhibit lipid solubilization.
Recently, SM was shown to alter the translocation of a neutral lipid, diacylglycerol, although in the opposite direction of what we observed with anionic phospholipids. Ueda et al. (60) found that adding SM to liposomes decreased the spontaneous flip of NBD-labeled diacylglycerol with no effect on the flip of NBD-PC, and SM depletion was shown to increase flipping of cell surface diacylglycerol. Combining our results with those of Ueda et al. (60), SM appears to have contrasting effects on diacylglycerol and anionic phospholipid flipping, with no effect on the flip of PC.
Myriocin has been shown to decrease atherosclerosis and induce lesion regression in apoE-deficient mice, associated with its lowering of plasma cholesterol levels (28, 61–63). Cholesterol absorption is decreased in mice by myriocin treatment as well as by hemizygous deficiency in the gene encoding for SPT1, which are associated with increased expression of the intestinal sterol exporters ABCG5 and ABCG8 and decreased expression of ABCA1 (64). We speculate that SM reduction in the intestinal epithelium could also play a role in decreased cholesterol binding to the cell membrane and flipping cholesterol to the inner leaflet of the membrane, similar to our observed reduced flip of anionic phospholipid; although, cholesterol flipping is thought to be independent of ATPase/flippases, it may be decreased upon changes in phase boundaries on the surface of the cell membrane after SM depletion. Although we did not observe an effect of SM depletion on NBD-cholesterol flipping in macrophages, this negative result could be due to the disruptive effect of the NBD label on cholesterol.
Thus it appears that SM depletion may have several consequences that can act together to increase cholesterol efflux. First, SM depletion frees FC from rafts for more rapid extraction by cyclodextrin; second, SM depletion decreases the flip of anionic phospholipids leading to higher levels cell surface PS that can promote ABCA1-mediated cholesterol efflux; and third, SM depletion may promote apoAI lipid solubilization, independent of changes in the lipid phase boundaries. Further work is required to identify the SM-dependent PS flippases that play role in cholesterol efflux. These SM-dependent PS flippases may provide novel therapeutic targets for increasing cholesterol efflux from macrophages.
This work was supported, in whole or in part, by National Institutes of Health Grant PO1 HL098055 (to J. D. S. and S. L. H.).
- PS
- phosphatidylserine
- SPT1
- serine palmitoyltransferase subunit 1
- SM
- sphingomyelin
- SMase
- sphingomyelinase
- NaTC
- sodium taurocholate
- LUV
- large unilamellar vesicles
- DMPC
- dimyristylphosphatidylcholine
- DPH
- 1,6-diphenyl-1,3,5-hexatriene
- MLV
- multilamellar vesicles
- CTB
- cholera toxin B subunit
- FC
- free cholesterol
- 8-Br-cAMP
- 8-bromo-cAMP
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine
- ABC
- ATP-binding cassette
- DMSO
- dimethyl sulfoxide
- ANOVA
- analysis of variance
- PE
- phosphatidylethanolamine
- DAOS
- N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3,5-dimethoxyaniline.
REFERENCES
- 1. Pomorski T., Menon A. K. (2006) Lipid flippases and their biological functions. Cell. Mol. Life Sci. 63, 2908–2921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pomorski T., Holthuis J. C., Herrmann A., van Meer G. (2004) Tracking down lipid flippases and their biological functions. J. Cell Sci. 117, 805–813 [DOI] [PubMed] [Google Scholar]
- 3. van der Mark V. A., Elferink R. P., Paulusma C. C. (2013) P4 ATPases. Flippases in health and disease. Int. J. Mol. Sci. 14, 7897–7922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tarling E. J., de Aguiar Vallim T. Q., Edwards P. A. (2013) Role of ABC transporters in lipid transport and human disease. Trends Endocrinol. Metab. 24, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nagao K., Kimura Y., Mastuo M., Ueda K. (2010) Lipid outward translocation by ABC proteins. FEBS Lett. 584, 2717–2723 [DOI] [PubMed] [Google Scholar]
- 6. Quazi F., Molday R. S. (2011) Lipid transport by mammalian ABC proteins. Essays Biochem. 50, 265–290 [DOI] [PubMed] [Google Scholar]
- 7. Davidson W. S., Hazlett T., Mantulin W. W., Jonas A. (1996) The role of apolipoprotein AI domains in lipid binding. Proc. Natl. Acad. Sci. U.S.A. 93, 13605–13610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oram J. F. (2003) HDL apolipoproteins and ABCA1. Partners in the removal of excess cellular cholesterol. Arterioscler. Thromb. Vasc. Biol. 23, 720–727 [DOI] [PubMed] [Google Scholar]
- 9. Oram J. F., Vaughan A. M. (2000) ABCA1-mediated transport of cellular cholesterol and phospholipids to HDL apolipoproteins. Curr. Opin. Lipidol. 11, 253–260 [DOI] [PubMed] [Google Scholar]
- 10. Chambenoit O., Hamon Y., Marguet D., Rigneault H., Rosseneu M., Chimini G. (2001) Specific docking of apolipoprotein A-I at the cell surface requires a functional ABCA1 transporter. J. Biol. Chem. 276, 9955–9960 [DOI] [PubMed] [Google Scholar]
- 11. Rigot V., Hamon Y., Chambenoit O., Alibert M., Duverger N., Chimini G. (2002) Distinct sites on ABCA1 control distinct steps required for cellular release of phospholipids. J. Lipid Res. 43, 2077–2086 [DOI] [PubMed] [Google Scholar]
- 12. Tall A. R. (2003) Role of ABCA1 in cellular cholesterol efflux and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 23, 710–711 [DOI] [PubMed] [Google Scholar]
- 13. Wang N., Tall A. R. (2003) Regulation and mechanisms of ATP-binding cassette transporter A1-mediated cellular cholesterol efflux. Arterioscler. Thromb. Vasc. Biol. 23, 1178–1184 [DOI] [PubMed] [Google Scholar]
- 14. Alder-Baerens N., Müller P., Pohl A., Korte T., Hamon Y., Chimini G., Pomorski T., Herrmann A. (2005) Headgroup-specific exposure of phospholipids in ABCA1-expressing cells. J. Biol. Chem. 280, 26321–26329 [DOI] [PubMed] [Google Scholar]
- 15. Wang S., Gulshan K., Brubaker G., Hazen S. L., Smith J. D. (2013) ABCA1 Mediates unfolding of apolipoprotein AI N terminus on the cell surface before lipidation and release of nascent high-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 33, 1197–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oram J. F., Vaughan A. M. (2006) ATP-Binding cassette cholesterol transporters and cardiovascular disease. Circ. Res. 99, 1031–1043 [DOI] [PubMed] [Google Scholar]
- 17. Pagler T. A., Wang M., Mondal M., Murphy A. J., Westerterp M., Moore K. J., Maxfield F. R., Tall A. R. (2011) Deletion of ABCA1 and ABCG1 impairs macrophage migration because of increased Rac1 signaling. Circ. Res. 108, 194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang X., Collins H. L., Ranalletta M., Fuki I. V., Billheimer J. T., Rothblat G. H., Tall A. R., Rader D. J. (2007) Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J. Clin. Invest. 117, 2216–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Westerterp M., Murphy A. J., Wang M., Pagler T. A., Vengrenyuk Y., Kappus M. S., Gorman D. J., Nagareddy P. R., Zhu X., Abramowicz S., Parks J. S., Welch C., Fisher E. A., Wang N., Yvan-Charvet L., Tall A. R. (2013) Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ. Res. 112, 1456–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yvan-Charvet L., Ranalletta M., Wang N., Han S., Terasaka N., Li R., Welch C., Tall A. R. (2007) Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Invest. 117, 3900–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brooks-Wilson A., Marcil M., Clee S. M., Zhang L. H., Roomp K., van Dam M., Yu L., Brewer C., Collins J. A., Molhuizen H. O., Loubser O., Ouelette B. F., Fichter K., Ashbourne-Excoffon K. J., Sensen C. W., Scherer S., Mott S., Denis M., Martindale D., Frohlich J., Morgan K., Koop B., Pimstone S., Kastelein J. J., Genest J., Jr., Hayden M. R. (1999) Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 22, 336–345 [DOI] [PubMed] [Google Scholar]
- 22. Bodzioch M., Orsó E., Klucken J., Langmann T., Böttcher A., Diederich W., Drobnik W., Barlage S., Büchler C., Porsch-Ozcürümez M., Kaminski W. E., Hahmann H. W., Oette K., Rothe G., Aslanidis C., Lackner K. J., Schmitz G. (1999) The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 22, 347–351 [DOI] [PubMed] [Google Scholar]
- 23. Rust S., Rosier M., Funke H., Real J., Amoura Z., Piette J. C., Deleuze J. F., Brewer H. B., Duverger N., Denèfle P., Assmann G. (1999) Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet. 22, 352–355 [DOI] [PubMed] [Google Scholar]
- 24. Fitzgerald M. L., Morris A. L., Rhee J. S., Andersson L. P., Mendez A. J., Freeman M. W. (2002) Naturally occurring mutations in the largest extracellular loops of ABCA1 can disrupt its direct interaction with apolipoprotein A-I. J. Biol. Chem. 277, 33178–33187 [DOI] [PubMed] [Google Scholar]
- 25. Nagao K., Zhao Y., Takahashi K., Kimura Y., Ueda K. (2009) Sodium taurocholate-dependent lipid efflux by ABCA1. Effects of W590S mutation on lipid translocation and apolipoprotein A-I dissociation. J. Lipid Res. 50, 1165–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tanaka A. R., Abe-Dohmae S., Ohnishi T., Aoki R., Morinaga G., Okuhira K., Ikeda Y., Kano F., Matsuo M., Kioka N., Amachi T., Murata M., Yokoyama S., Ueda K. (2003) Effects of mutations of ABCA1 in the first extracellular domain on subcellular trafficking and ATP binding/hydrolysis. J. Biol. Chem. 278, 8815–8819 [DOI] [PubMed] [Google Scholar]
- 27. Worgall T. S. (2011) Sphingolipid synthetic pathways are major regulators of lipid homeostasis. Adv. Exp. Med. Biol. 721, 139–148 [DOI] [PubMed] [Google Scholar]
- 28. Park T. S., Panek R. L., Rekhter M. D., Mueller S. B., Rosebury W. S., Robertson A., Hanselman J. C., Kindt E., Homan R., Karathanasis S. K. (2006) Modulation of lipoprotein metabolism by inhibition of sphingomyelin synthesis in ApoE knockout mice. Atherosclerosis 189, 264–272 [DOI] [PubMed] [Google Scholar]
- 29. Tamehiro N., Zhou S., Okuhira K., Benita Y., Brown C. E., Zhuang D. Z., Latz E., Hornemann T., von Eckardstein A., Xavier R. J., Freeman M. W., Fitzgerald M. L. (2008) SPTLC1 binds ABCA1 to negatively regulate trafficking and cholesterol efflux activity of the transporter. Biochemistry 47, 6138–6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Glaros E. N., Kim W. S., Wu B. J., Suarna C., Quinn C. M., Rye K. A., Stocker R., Jessup W., Garner B. (2007) Inhibition of atherosclerosis by the serine palmitoyl transferase inhibitor myriocin is associated with reduced plasma glycosphingolipid concentration. Biochem. Pharmacol. 73, 1340–1346 [DOI] [PubMed] [Google Scholar]
- 31. Hojjati M. R., Li Z., Zhou H., Tang S., Huan C., Ooi E., Lu S., Jiang X. C. (2005) Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoE-deficient mice. J. Biol. Chem. 280, 10284–10289 [DOI] [PubMed] [Google Scholar]
- 32. Wu Z., Gogonea V., Lee X., May R. P., Pipich V., Wagner M. A., Undurti A., Tallant T. C., Baleanu-Gogonea C., Charlton F., Ioffe A., DiDonato J. A., Rye K. A., Hazen S. L. (2011) The low resolution structure of ApoA1 in spherical high density lipoprotein revealed by small angle neutron scattering. J. Biol. Chem. 286, 12495–12508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. John K., Schreiber S., Kubelt J., Herrmann A., Müller P. (2002) Transbilayer movement of phospholipids at the main phase transition of lipid membranes. Implications for rapid flip-flop in biological membranes. Biophys. J. 83, 3315–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hojjati M. R., Jiang X. C. (2006) Rapid, specific, and sensitive measurements of plasma sphingomyelin and phosphatidylcholine. J. Lipid Res. 47, 673–676 [DOI] [PubMed] [Google Scholar]
- 35. Hidaka H., Yamauchi K., Ohta H., Akamatsu T., Honda T., Katsuyama T. (2008) Specific, rapid, and sensitive enzymatic measurement of sphingomyelin, phosphatidylcholine and lysophosphatidylcholine in serum and lipid extracts. Clin. Biochem. 41, 1211–1217 [DOI] [PubMed] [Google Scholar]
- 36. Le Goff W., Zheng P., Brubaker G., Smith J. D. (2006) Identification of the cAMP-responsive enhancer of the murine ABCA1 gene. Requirement for CREB1 and STAT3/4 elements. Arterioscler. Thromb. Vasc. Biol. 26, 527–533 [DOI] [PubMed] [Google Scholar]
- 37. Oram J. F., Lawn R. M., Garvin M. R., Wade D. P. (2000) ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J. Biol. Chem. 275, 34508–34511 [DOI] [PubMed] [Google Scholar]
- 38. Gelderblom W. C., Jaskiewicz K., Marasas W. F., Thiel P. G., Horak R. M., Vleggaar R., Kriek N. P. (1988) Fumonisins. Novel mycotoxins with cancer-promoting activity produced by Fusarium moniliforme. Appl. Environ. Microbiol. 54, 1806–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang E., Norred W. P., Bacon C. W., Riley R. T., Merrill A. H., Jr. (1991) Inhibition of sphingolipid biosynthesis by fumonisins. Implications for diseases associated with Fusarium moniliforme. J. Biol. Chem. 266, 14486–14490 [PubMed] [Google Scholar]
- 40. Ghering A. B., Davidson W. S. (2006) Ceramide structural features required to stimulate ABCA1-mediated cholesterol efflux to apolipoprotein A-I. J. Lipid Res. 47, 2781–2788 [DOI] [PubMed] [Google Scholar]
- 41. Christian A. E., Haynes M. P., Phillips M. C., Rothblat G. H. (1997) Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 38, 2264–2272 [PubMed] [Google Scholar]
- 42. Hanada K., Pagano R. E. (1995) A Chinese hamster ovary cell mutant defective in the non-endocytic uptake of fluorescent analogs of phosphatidylserine. Isolation using a cytosol acidification protocol. J. Cell Biol. 128, 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grant A. M., Hanson P. K., Malone L., Nichols J. W. (2001) NBD-labeled phosphatidylcholine and phosphatidylethanolamine are internalized by transbilayer transport across the yeast plasma membrane. Traffic 2, 37–50 [DOI] [PubMed] [Google Scholar]
- 44. Durbin D. M., Jonas A. (1999) Lipid-free apolipoproteins A-I and A-II promote remodeling of reconstituted high density lipoproteins and alter their reactivity with lecithin:cholesterol acyltransferase. J. Lipid Res. 40, 2293–2302 [PubMed] [Google Scholar]
- 45. Chiantia S., London E. (2013) Sphingolipids and membrane domains. Recent advances. Handb. Exp. Pharmacol. 215, 33–55 [DOI] [PubMed] [Google Scholar]
- 46. Cary L. A., Cooper J. A. (2000) Molecular switches in lipid rafts. Nature 404, 945–947 [DOI] [PubMed] [Google Scholar]
- 47. Fukasawa M., Nishijima M., Itabe H., Takano T., Hanada K. (2000) Reduction of sphingomyelin level without accumulation of ceramide in Chinese hamster ovary cells affects detergent-resistant membrane domains and enhances cellular cholesterol efflux to methyl-β-cyclodextrin. J. Biol. Chem. 275, 34028–34034 [DOI] [PubMed] [Google Scholar]
- 48. Ohvo H., Olsio C., Slotte J. P. (1997) Effects of sphingomyelin and phosphatidylcholine degradation on cyclodextrin-mediated cholesterol efflux in cultured fibroblasts. Biochim. Biophys. Acta 1349, 131–141 [DOI] [PubMed] [Google Scholar]
- 49. Yamauchi Y., Hayashi M., Abe-Dohmae S., Yokoyama S. (2003) Apolipoprotein A-I activates protein kinase Cα signaling to phosphorylate and stabilize ATP binding cassette transporter A1 for the high density lipoprotein assembly. J. Biol. Chem. 278, 47890–47897 [DOI] [PubMed] [Google Scholar]
- 50. Nagao K., Takahashi K., Hanada K., Kioka N., Matsuo M., Ueda K. (2007) Enhanced apoA-I-dependent cholesterol efflux by ABCA1 from sphingomyelin-deficient Chinese hamster ovary cells. J. Biol. Chem. 282, 14868–14874 [DOI] [PubMed] [Google Scholar]
- 51. Chakraborty M., Lou C., Huan C., Kuo M. S., Park T. S., Cao G., Jiang X. C. (2013) Myeloid cell-specific serine palmitoyltransferase subunit 2 haploinsufficiency reduces murine atherosclerosis. J. Clin. Invest. 123, 1784–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Drobnik W., Borsukova H., Böttcher A., Pfeiffer A., Liebisch G., Schütz G. J., Schindler H., Schmitz G. (2002) ApoAI/ABCA1-dependent and HDL3-mediated lipid efflux from compositionally distinct cholesterol-based microdomains. Traffic 3, 268–278 [DOI] [PubMed] [Google Scholar]
- 53. Mendez A. J., Lin G., Wade D. P., Lawn R. M., Oram J. F. (2001) Membrane lipid domains distinct from cholesterol/sphingomyelin-rich rafts are involved in the ABCA1-mediated lipid secretory pathway. J. Biol. Chem. 276, 3158–3166 [DOI] [PubMed] [Google Scholar]
- 54. Fessler M. B., Parks J. S. (2011) Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J. Immunol. 187, 1529–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Landry Y. D., Denis M., Nandi S., Bell S., Vaughan A. M., Zha X. (2006) ATP-binding cassette transporter A1 expression disrupts raft membrane microdomains through its ATPase-related functions. J. Biol. Chem. 281, 36091–36101 [DOI] [PubMed] [Google Scholar]
- 56. Koseki M., Hirano K., Masuda D., Ikegami C., Tanaka M., Ota A., Sandoval J. C., Nakagawa-Toyama Y., Sato S. B., Kobayashi T., Shimada Y., Ohno-Iwashita Y., Matsuura F., Shimomura I., Yamashita S. (2007) Increased lipid rafts and accelerated lipopolysaccharide-induced tumor necrosis factor-α secretion in Abca1-deficient macrophages. J. Lipid Res. 48, 299–306 [DOI] [PubMed] [Google Scholar]
- 57. Smith J. D., Le Goff W., Settle M., Brubaker G., Waelde C., Horwitz A., Oda M. N. (2004) ABCA1 mediates concurrent cholesterol and phospholipid efflux to apolipoprotein A-I. J. Lipid Res. 45, 635–644 [DOI] [PubMed] [Google Scholar]
- 58. Smith J. D., Waelde C., Horwitz A., Zheng P. (2002) Evaluation of the role of phosphatidylserine translocase activity in ABCA1-mediated lipid efflux. J. Biol. Chem. 277, 17797–17803 [DOI] [PubMed] [Google Scholar]
- 59. Pownall H., Pao Q., Hickson D., Sparrow J. T., Kusserow S. K., Massey J. B. (1981) Kinetics and mechanism of association of human plasma apolipoproteins with dimyristoylphosphatidylcholine. Effect of protein structure and lipid clusters on reaction rates. Biochemistry 20, 6630–6635 [DOI] [PubMed] [Google Scholar]
- 60. Ueda Y., Makino A., Murase-Tamada K., Sakai S., Inaba T., Hullin-Matsuda F., Kobayashi T. (2013) Sphingomyelin regulates the transbilayer movement of diacylglycerol in the plasma membrane of Madin-Darby canine kidney cells. FASEB J. 27, 3284–3297 [DOI] [PubMed] [Google Scholar]
- 61. Glaros E. N., Kim W. S., Quinn C. M., Jessup W., Rye K. A., Garner B. (2008) Myriocin slows the progression of established atherosclerotic lesions in apolipoprotein E gene knockout mice. J. Lipid Res. 49, 324–331 [DOI] [PubMed] [Google Scholar]
- 62. Park T. S., Panek R. L., Mueller S. B., Hanselman J. C., Rosebury W. S., Robertson A. W., Kindt E. K., Homan R., Karathanasis S. K., Rekhter M. D. (2004) Inhibition of sphingomyelin synthesis reduces atherogenesis in apolipoprotein E-knockout mice. Circulation 110, 3465–3471 [DOI] [PubMed] [Google Scholar]
- 63. Park T. S., Rosebury W., Kindt E. K., Kowala M. C., Panek R. L. (2008) Serine palmitoyltransferase inhibitor myriocin induces the regression of atherosclerotic plaques in hyperlipidemic ApoE-deficient mice. Pharmacol. Res. 58, 45–51 [DOI] [PubMed] [Google Scholar]
- 64. Li Z., Park T. S., Li Y., Pan X., Iqbal J., Lu D., Tang W., Yu L., Goldberg I. J., Hussain M. M., Jiang X. C. (2009) Serine palmitoyltransferase (SPT)-deficient mice absorb less cholesterol. Biochim. Biophys. Acta 1791, 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]