

Background: Apoptosis and autophagy are coordinately regulated, but the underlying mechanisms are incompletely understood.
Results: Bcl-2 specifically interacts with GABARAP via a conserved EWD motif, resulting in impaired GABARAP lipidation.
Conclusion: Sequestration of GABARAP is likely to contribute to the down-regulation of autophagy by Bcl-2.
Significance: Interfering with pro-survival functions of Bcl-2 (including its impact on autophagy) represents a promising strategy for cancer therapy.
Keywords: Apoptosis, Autophagy, Bcl-2, NMR, Protein-Protein Interactions, GABARAP, LC3, Lipidation
Abstract
Apoptosis and autophagy are fundamental homeostatic processes in eukaryotic organisms fulfilling essential roles in development and adaptation. Recently, the anti-apoptotic factor Bcl-2 has been reported to also inhibit autophagy, thus establishing a potential link between these pathways, but the mechanistic details are only beginning to emerge. Here we show that Bcl-2 directly binds to the phagophore-associated protein GABARAP. NMR experiments revealed that the interaction critically depends on a three-residue segment (EWD) of Bcl-2 adjacent to the BH4 region, which is anchored to one of the two hydrophobic pockets on the GABARAP molecule. This is at variance with the majority of GABARAP interaction partners identified previously, which occupy both hydrophobic pockets simultaneously. Bcl-2 affinity could also be detected for GEC1, but not for other mammalian Atg8 homologs. Finally, we provide evidence that overexpression of Bcl-2 inhibits lipidation of GABARAP, a key step in autophagosome formation, possibly via competition with the lipid conjugation machinery. These results support the regulatory role of Bcl-2 in autophagy and define GABARAP as a novel interaction partner involved in this intricate connection.
Introduction
Cellular integrity critically relies on the presence of degradation systems with the potential to dispose of defective biomolecules and organelles or to disintegrate cytoplasmic material for recycling of its constituents. One of these fundamental pathways is autophagy, which has been found to be deregulated in a series of human diseases, such as cancer and neurodegeneration, as well as in aging (1). During autophagy,5 cytosolic components are engulfed by a crescent-shaped double membrane termed the phagophore, which is subsequently closed to form an autophagosome. Upon fusion of the autophagosome with lysosomes, its contents are decomposed by hydrolytic enzymes (2).
In yeast, conjugation of autophagy-related protein (Atg)6 8, a member of the ubiquitin superfamily, with phosphatidylethanolamine is a critical requirement for autophagosome formation (3). Lipidated Atg8 (Atg8-II) is located on phagophore and autophagosome membranes, representing a well established marker of autophagic structures (4). Atg8 has several homologs in mammals, including three variants of microtubule-associated protein light chain 3 (LC3A, LC3B, and LC3C), GABAA receptor-associated protein (GABARAP), glandular epithelial cell protein 1 (GEC1), and Golgi-associated ATPase enhancer of 16 kDa (GATE-16). Similar to Atg8, these proteins are substrates of a lipid conjugation machinery, resulting in their membrane attachment (5). Based on sequence similarities, GABARAP, GEC1, and GATE-16 on the one hand and the LC3s on the other have been proposed to form two distinct subfamilies, which appear to exert slightly different functions in autophagosome biogenesis (6).
GABARAP is a versatile protein with a wide range of interaction partners, which are involved in various aspects of vesicle transport and fusion as well as apoptosis (7). The observation of abnormal GABARAP expression levels in cancer cells suggests a potential role of this protein during cancer development (8, 9). Structural studies using nuclear magnetic resonance (NMR) spectroscopy as well as x-ray crystallography have revealed that most interactions of GABARAP are mediated by two hydrophobic pockets on its surface (10–14), and that this binding site is also available in the lipidated form of the molecule (15). Thus, at least subsets of GABARAP binding partners are expected to compete for the interaction surface, suggesting an intricate spatio-temporal patterning of different complexes (7).
In an effort to find novel GABARAP ligands, we previously identified the signature XXWXXΦ as a minimal binding motif (Fig. 1A). In this sequence, Φ is hydrophobic, whereas X can be any residue, but at least one of these needs to be acidic (10, 11). Based on this motif, we identified Nix (Nip-like protein x) as a direct interaction partner of GABARAP (16). Nix, alternatively named Bnip3L (Bcl-2/adenovirus E1B 19-kDa interacting protein 3-like), is a pro-apoptotic protein containing a single B cell lymphoma (Bcl) 2 homology (BH) 3 region. Although in Bnip3L, the BH3 motif has been suggested to mediate interaction with Bcl-2 (17), the closely related Bnip3 appears to utilize different sites for this purpose (18). Bcl-2 is the founding member of a protein family comprising both pro- and anti-apoptotic factors. Notably, the most prominent members of the anti-apoptotic subgroup (Bcl-2, Bcl-xL, and Bcl-w) are distinguished by the presence of a well conserved BH4 sequence. Because Nix binds to GABARAP via its N-terminal WVEL region (16, 19), which conforms to the XXWXXΦ motif, simultaneous binding of Nix to GABARAP and Bcl-2 may be feasible.
FIGURE 1.
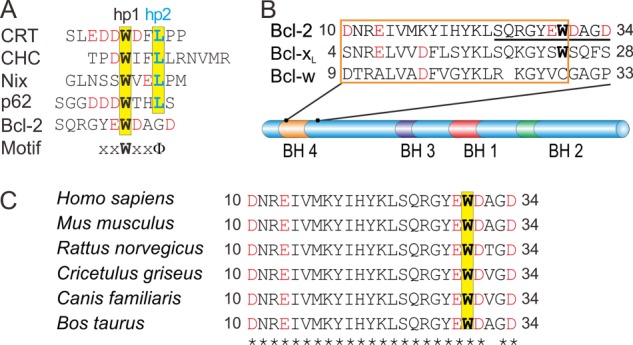
GABARAP interaction partners. A, sequence alignment of the known GABARAP ligands calreticulin (CRT, Ref. 11), clathrin heavy chain (CHC, Ref. 10), Nix (16), p62 (44), and Bcl-2 (this work). Acidic residues are highlighted in red. Amino acids that are known or expected to interact with hydrophobic pocket hp1 are shown in bold black, those bound to hp2 are highlighted in blue. B, sequence alignment of BH4 segments (boxed, following the annotations in the UniProt database), together with four subsequent residues, for human Bcl-2, Bcl-xL, and Bcl-w (colored as above). Bcl-2 residues Ser24-Asp34, which are implicated in GABARAP binding according to NMR titration experiments, are underlined. C, comparison of mammalian Bcl-2 sequences, demonstrating the strict conservation (indicated by asterisks) of BH4 residues and the GABARAP binding motif.
We therefore decided to explore the impact of Bcl-2 on the GABARAP-Nix interaction. Surprisingly, Bcl-2 turned out to also display significant affinity for the GABARAP molecule. The present work describes the results of a subsequent effort to characterize the direct Bcl-2 interaction with GABARAP and its potential role in vivo.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Plasmid construction, expression, and purification of human GABARAP in Escherichia coli BL21 DE3 have been described previously (20).
DNA fragments coding for LC3A, LC3B, GATE-16, and GEC1 were amplified by polymerase chain reaction (PCR) and cloned into pGEX-4T-2 or pET-15b for expression of glutathione S-transferase (GST) fusion proteins or His-tagged proteins, respectively. All constructs were sequenced to confirm their correctness and expressed in E. coli BL21 DE3 cells. Once cultures reached an optical density (600 nm) of about 0.6, expression was induced with 1 mm isopropyl β-d-thiogalactopyranoside and cells were harvested after 3 h of growth at 37 °C. His-tagged fusion proteins were purified from the soluble extract by nickel-nitrilotriacetic acid affinity chromatography, and GST fusion proteins were purified by glutathione-Sepharose affinity chromatography. If necessary, fusion tags were cleaved off by thrombin hydrolysis, followed by further purification by size exclusion chromatography. An analogous strategy was followed for expression and purification of soluble human Bcl-xL (residues 1–204), using a vector (21) kindly provided by Dr. Joseph T. Opferman (St. Jude Children's Research Hospital, Memphis, TN).
A synthetic cDNA (obtained from Geneart) coding for a soluble human Bcl-2 (isoform 2)/Bcl-xL chimera (Ref. 22, hereafter denoted Bcl-2#) was cloned into a modified pET-15b vector using BamHI and XhoI restriction sites introduced by PCR. The protein was expressed in E. coli BL21 DE3 cells and purified with nickel-nitrilotriacetic acid-agarose, followed by tobacco etch virus protease digestion and size exclusion chromatography.
Peptides corresponding to the interaction region of Bcl-2 were custom synthesized with acetylated N termini and amidated C termini and purified to >95% (JPT Peptide Technologies). The wild-type peptide contained Bcl-2 residues 24–34 (SQRGYEWDAGD); in addition, peptides carrying single point mutations (E29Q, W30A, and D31N) were used.
Cell Lines
Rat embryonic fibroblast (REF) cells expressing mouse Bcl-2 (23) or transfected with a blank vector were a kind gift of Christoph Borner (University of Freiburg, Germany) to A.M. and were grown in high-glucose Dulbecco's modified Eagle's medium (DMEM, PAA Laboratories) containing 10% fetal calf serum (FCS, Biochrom) as well as 1% penicillin/streptomycin (Invitrogen). Jurkat cells were grown in RPMI 1640 medium (PAA Laboratories) containing 10% FCS. Mouse embryonic fibroblasts (MEFs) were prepared by crushing E12.5–13.5 embryos in sterile cell culture dishes in the presence of culture medium and passaged as soon as they reached confluence, until they were immortalized based on morphology. At this stage, cells were cultured in high-glucose DMEM (Invitrogen) supplemented with 10% FCS, 100 μg/ml of streptomycin, and 100 units/ml of penicillin. To obtain stably transfected cell lines, wild-type and mutated Bcl-2 were inserted into the pPB-CAG-EBNXN vector (obtained via the Sanger Institute) containing PiggyBac transposon sites, which facilitate stable insertion into the genome of a target cell when co-expressed with a transposase (23). The cDNAs were N terminally tagged with a hemagglutinin epitope and C terminally connected to an internal ribosomal entry site followed by the yellow fluorescent protein Venus. These constructs were co-transfected with a plasmid encoding the transposase in a ratio of 1:4; 48 h after transfection, Venus-positive cells were selected on a MoFlo XDP (Beckman-Coulter) cell sorter. After three to four rounds of sorting, cells were ∼99% positive for Venus fluorescence and thus considered stably transfected. Successful expression was confirmed by Western blotting. For ongoing culture, cells were grown in the absence of antibiotics.
Determination of the Bcl-2 Binding Interface on GABARAP
NMR spectra were recorded with a cryogenically cooled Z-pulse field gradient 1H{13C,15N} probe at 25 °C on a Varian NMR spectrometer at a proton frequency of 800 MHz. The initial sample contained 75 μm 15N-GABARAP in 20 mm Tris-HCl, pH 7.8, 5 mm dithiothreitol (DTT), and 7% (v/v) deuterium oxide (D2O). Successive addition of unlabeled Bcl-2# (or Bcl-xL) finally resulted in a sample containing equal concentrations (65 μm) of both proteins. 1H-15N-HSQC (heteronuclear single quantum coherence) spectra were collected with up to 200 complex points in the 15N time domain, up to 64 scans per t1 point, and a 1.5-s recycle delay. Data were processed with NMRPipe (24) and analyzed with NMRView (25).
NMR Titration of 15N-GABARAP with Bcl-2-derived Peptides
NMR spectra were recorded at 25 °C on a Varian NMR spectrometer at a proton frequency of 600 MHz. The initial sample contained 200 μm 15N-GABARAP in 25 mm NaH2PO4/Na2HPO4, pH 6.9, 100 mm KCl, 100 mm NaCl, 50 μm EDTA, and 5% (v/v) D2O. Successive addition of unlabeled Bcl-2 peptides (wild-type, E29Q, W30A, and D31N, respectively) finally resulted in a sample containing 132 μm GABARAP and 265 μm Bcl-2 peptide. Again, 1H-15N-HSQC spectra were collected during the titration. Data were processed with NMRPipe and evaluated with CcpNmr Analysis (26).
Backbone Resonance Assignment of Bcl-2
Uniformly 13C- and 15N-labeled samples of 300 μm Bcl-2# were prepared using 20 mm Tris-HCl, pH 7.8, 5 mm DTT, and 7% (v/v) D2O. NMR experiments were performed at 25 °C on Varian NMR spectrometers, equipped with cryogenically cooled Z-pulse field gradient probes or room temperature XYZ-pulse field gradient probes at proton frequencies of 900, 800, and 600 MHz. To obtain backbone resonance assignments of the affected residues, two-dimensional 1H-15N-HSQC, three-dimensional BEST-HNCA (27), three-dimensional CBCACONH (28), and 6–10 ppm band selective (1H-1H-15N)-NOESY-HSQC spectra were recorded. Data were processed with NMRPipe and analyzed with CARA (29) and CcpNmr Analysis. The resulting assignment (covering 126 of 162 residues) has been deposited in the Biological Magnetic Resonance Data Bank (code 19559).
Determination of the GABARAP Binding Interface on Bcl-2
1H-15N-HSQC spectra were recorded at a proton frequency of 900 MHz. The initial sample contained 120 μm 15N-Bcl-2#. Successive addition of unlabeled GABARAP finally resulted in a sample containing equal concentrations (70 μm) of Bcl-2# and GABARAP.
Modeling of the GABARAP·Bcl-2 Complex
A model of the GABARAP·Bcl-2 complex was calculated using HADDOCK, version 2.0 (30, 31). For GABARAP, the crystal structure of the complex with a calreticulin-derived peptide (PDB code 3DOW, Ref. 13) was chosen as starting model. In the case of Bcl-2, we used the solution structure of the Bcl-2/Bcl-xL chimera (PDB code 1GJH, Ref. 22) to generate variants with different side chain orientations of Trp30 using the basin-hopping parallel tempering algorithm (32, 33). In this procedure, we applied random Cartesian moves to the atoms of residues 27–33 only, followed by energy minimization of the entire model. To allow structures of higher energy to be accepted by the basin-hopping algorithm, we used temperatures between 5,000 and 50,000 K for the 16 replicas. For each of these, 200 basin-hopping steps were performed and the 10 structures with lowest energy were saved. The resulting 160 structures were examined visually, and the one with the side chain of Trp30 pointing mostly outward was selected for subsequent docking. For evaluation of energies we used the CHARMM22 potential (34). The basin-hopping parallel tempering algorithm is implemented in the GMIN software.
For in silico docking of the two molecules using HADDOCK, 35 ambiguous interaction restraints were defined based on the changes in the 1H-15N-HSQC spectra observed upon complex formation. Additionally, the Tyr28-Ala34 region of Bcl-2 was defined as fully flexible segment. HADDOCK was then carried out using the web server expert interface with default program parameters. Analysis of the final 200 water-refined models of the GABARAP·Bcl-2 complex resulted in the identification of two clusters; a statistical evaluation is provided in Table 1. The first cluster comprised more than 75% of all obtained models as well as the model with the lowest energy. The latter was chosen for graphic representation of the GABARAP·Bcl-2 complex, after removal of the Bcl-xL insertion.
TABLE 1.
Cluster statistics for GABARAP·Bcl-2# complex models obtained with HADDOCK
| Cluster 1 | Cluster 2 | |
|---|---|---|
| HADDOCK scorea | −137.0 ± 7.6 | −78.6 ± 6.2 |
| Cluster size | 154 | 42 |
| Root mean square deviation-Emin (Å) | 0.7 ± 0.4 | 7.6 ± 0.2 |
| Evdw (kcal/mol) | −43.5 ± 8.2 | −39.0 ± 5.6 |
| Eelec (kcal/mol) | −808.7 ± 61.3 | −522.1 ± 27.7 |
| Edes (kcal/mol) | 52.3 ± 3.4 | 34.8 ± 4.4 |
| ERV (kcal/mol) | 159.9 ± 50.7 | 299.8 ± 35.2 |
| BSA (Å2) | 2078.3 ± 60.4 | 1460.1 ± 69.1 |
a The following abbreviations were used: Emin, backbone root mean square distance from the lowest-energy structure; Evdw, Van der Waals energy; Eelec, electrostatic energy; Edes, desolvation energy; ERV, restraints violation energy; and BSA, buried surface area.
Surface Plasmon Resonance (SPR) Spectroscopy
SPR studies were carried out on a BiacoreX optical biosensor (GE Healthcare). Following standard procedures for amine coupling, 1 μm recombinant Bcl-2# without fusion tag in 10 mm sodium acetate, pH 4.0, was used for coupling to the carboxymethylated dextran matrix of a CM5 sensor chip surface. A reference surface was treated identically, but not exposed to Bcl-2# for immobilization. Recombinant GABARAP, GEC1, GATE-16, LC3A, and LC3B were dissolved in running buffer (10 mm Hepes, pH 7.4, 150 mm NaCl, 3 mm EDTA, 1 mm β-mercaptoethanol, and 0.005% surfactant P20) at concentrations between 1 and 60 μm and injected at 30 μl/min and 25 °C. Response units were recorded as a function of time. The averaged signal intensities between 90 and 120 s after injection were used for evaluation. The dissociation constant (Kd) was obtained for each analyte by nonlinear regression according to the steady state affinity model, using BIAevaluation software.
Pulldown Analysis
Immobilization of GABARAP and pulldown with Jurkat cell extracts or competitive pulldown with REF or Bcl-2-overexpressing REF cell extracts were performed as described previously (11). Eluted proteins from the pulldown procedure were analyzed by Western blot analysis. The primary antibodies were mouse anti-Bcl2 IgG (Acris Antibodies) and rabbit anti-Atg4B IgG (Santa Cruz Biotechnology). Blots were stained by chemiluminescence (SuperSignal West Pico chemiluminescent substrate, Pierce) and documented using a ChemiDoc system (Bio-Rad).
Co-immunoprecipitation Assay
Jurkat cells were sonicated in lysis buffer (10 mm Na2HPO4, 1.76 mm KH2PO4, pH 7.4, 137 mm NaCl, 2.7 mm KCl, 1% Nonidet P-40, 0.5% sodium cholate, protease inhibitor mixture) and centrifuged at 10,000 × g for 30 min at 4 °C. The supernatant was incubated with rabbit anti-human Bcl-2 polyclonal antibody (Cell Signaling Technology) or without antibody as a control at 4 °C overnight with gentle mixing. Immobilized recombinant protein A-agarose (Pierce) was added and the mixture was incubated with gentle mixing for 2 h at room temperature. The agarose was washed three times with a buffer containing 25 mm Tris-HCl, pH 7.2, and 150 mm NaCl. Subsequently, bound protein was eluted by 100 mm glycine, pH 2.5, and analyzed by SDS-PAGE and Western blotting. GABARAP immunoreactivity was probed by rabbit anti-human GABARAP polyclonal IgG (Pineda) and HRP-coupled goat anti-rabbit IgG (Pierce). Bcl-2 immunoreactivity was detected by mouse anti-Bcl-2 monoclonal antibody (Acris Antibodies) and HRP-coupled goat anti-mouse IgG (Pierce).
For immunoprecipitation of Bcl-2 or GABARAP from MEF cell lysates, antibodies were covalently coupled to an amine-reactive agarose (Pierce Co-IP Kit, Thermo Scientific), following the manufacturer's instructions. Cells were incubated with ice-cold lysis buffer (25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 5% glycerol) without sonication, followed by centrifugation at 13,000 × g for 10 min at 4 °C. In each experiment, 500 μl of supernatant diluted to a total protein concentration of 1 mg/ml was applied to the appropriate antibody-coupled resin in a spin column. After washing three times with 200 μl of lysis buffer, bound protein was recovered after incubation with 60 μl of elution buffer, pH 2.8, at room temperature. Following precipitation with methanol/chloroform, the eluate was analyzed by SDS-PAGE and Western blotting, using antibodies as outlined above.
Cell Death Assays
Cell death was determined using Sytox green staining (Invitrogen) in the presence of different staurosporine concentrations. After incubation with staurosporine for 48 h, the cells were analyzed by high-content imaging using a BD Pathway 855 microscope. Data were obtained from 4 independent experiments using 3 replicates per experiment. Viabilities were normalized to the untreated cells, which were set as 100%.
Immunocytochemistry
The cells were fixed, permeabilized, and stained as previously described (16). Detection of GABARAP was performed with polyclonal rabbit anti-GABARAP antibody (Pineda) and rhodamine-labeled bovine anti-rabbit IgG (Santa Cruz Biotechnology). For staining of nuclear DNA, we used 4′,6-diamidino-2-phenylindole (DAPI). Immunofluorescence was observed with an LSM 710 confocal laser scanning system (Carl Zeiss MicroImaging) using an EC-Plan-Neofluar ×40/1.30 oil DIC objective. GABARAP positive particles were quantified in individual frames using the ImageJ software (NIH), using parameters as outlined in Ref. 36.
Statistical Analysis
All numerical results are given as mean ± S.E., and represent data from a minimum of three independent experiments. We determined the statistical significance of differences between experimental groups by the two-tailed unpaired Student's t test using SigmaPlot (Systat Software).
RESULTS
GABARAP Directly Binds to Bcl-2
After our accidental discovery of GABARAP binding to Bcl-2, we first determined the dissociation constant of this complex using SPR spectroscopy, for comparison with the GABARAP-Nix interaction. As recombinant Bcl-2 is known to be poorly soluble, we used a soluble and functional chimera (Bcl-2#) in which the loop connecting the BH4 and BH3 regions (residues 35–91) of Bcl-2 (isoform 2) was replaced by residues 35–50 of Bcl-xL, and which lacked the C-terminal membrane anchor (22). We found that GABARAP directly binds to Bcl-2# with stronger affinity (Kd = 26 μm, Fig. 2A) than to Nix (Kd = 100 μm, Ref. 16). As GABARAP shares significant similarities in sequence and function with other members of the Atg8 family, we additionally examined the interaction between Bcl-2 and several GABARAP homologs by SPR spectroscopy and found that GEC1 bound Bcl-2# with a Kd of 72 μm (Fig. 2B). In contrast, GATE-16, LC3A, and LC3B yielded SPR signals that indicate Kd values larger by at least 1 order of magnitude.
FIGURE 2.
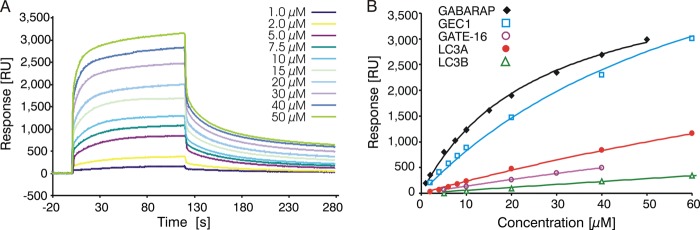
Binding of GABARAP homologs to Bcl-2. A, SPR sensorgrams resulting from injections of GABARAP at different concentrations, with Bcl-2# immobilized on the chip as described under “Experimental Procedures.” B, steady state binding curves of GABARAP, GEC1, GATE-16, LC3A, and LC3B interacting with immobilized Bcl-2#. Nonlinear regression analysis yielded the Kd for GABARAP-Bcl-2# to be 25.9 ± 1.3 μm, compared with 71.5 ± 5.3 μm for the GEC1-Bcl-2# interaction. Binding of GATE-16, LC3A, and LC3B to Bcl-2# was very weak, suggesting Kd values significantly exceeding 200 μm.
Mapping the Binding Surfaces of Bcl-2 and GABARAP
To identify Bcl-2 residues that are affected by GABARAP binding, 1H-15N-HSQC spectra of 15N-Bcl-2# were recorded during titration with unlabeled GABAPAP. Addition of GABARAP caused several Bcl-2# resonances to be broadened beyond detection, confirming direct interaction of both proteins (Fig. 3A). Note that, due to the intermediate time scale of chemical exchange, these data did not allow direct calculation of an equilibrium constant. The affected resonances mainly correspond to residues Ser24-Asp34 (SQRGYEWDAGD) of Bcl-2. This region comprises the end of helix α1 and the beginning of the α1-α2 loop, i.e. the C-terminal end of the BH4 segment and the following four residues (Fig. 1B). Notably, the sequence is centered on a tryptophan residue, but does not fully match the XXWXXΦ signature.
FIGURE 3.

Structural investigation of the GABARAP-Bcl-2 interaction. A, superimposed 1H-15N-HSQC spectra of Bcl-2# in the absence (blue) and presence of GABARAP (yellow). The resonances most strongly affected upon GABARAP binding are labeled, and corresponding residues are highlighted in yellow on the Bcl-2# surface (top left, using PDB code 1GJH). B, superimposed 1H-15N-HSQC spectra of GABARAP in the absence (yellow) and presence of Bcl-2# (35 μm, blue; 65 μm, violet). Resonance assignments were taken from a previous report (20). The signals most strongly affected upon Bcl-2# binding are labeled, and corresponding residues highlighted on the GABARAP surface (top right, using PDB code 1KOT). Residues broadening upon addition of 35 μm Bcl-2# are colored blue, those affected in the presence of 65 μm Bcl-2# are colored violet. C, an analogous experiment, using Bcl-xL instead of Bcl-2#.
In a complementary experiment, 1H-15N-HSQC spectra of 15N-GABARAP were recorded during titration with unlabeled Bcl-2#. The spectrum without ligand exhibited the typical signals of natively folded GABARAP (37). Upon addition of Bcl-2#, a lot of resonances broadened beyond detection (Fig. 3B). Signals of Leu50 and Val51 broadened already at a Bcl-2#:GABARAP ratio of 0.5. Resonances of other residues, which were mainly located within one continuous surface region of GABARAP, disappeared or shifted in the HSQC spectra with increasing amounts of Bcl-2#, thus defining the binding area involved on the GABARAP surface. This region includes the first hydrophobic pocket (hp1) of GABARAP, whereas hp2 is much less affected. This observation is in good agreement with the binding sequence identified in Bcl-2 (YEWDAG, see above), which contains a glycine instead of the second hydrophobic residue (Φ) in the XXWXXΦ signature. (Fig. 1A). We conclude that hp1 on GABARAP is most likely to interact with Bcl-2 residue Trp30, in accordance with previous studies identifying a tryptophan side chain as a critical determinant for GABARAP binding via this hydrophobic pocket (7, 38). Notably, titration of 15N-GABARAP with Bcl-xL (Fig. 3C) did not reveal significant effects in the 1H-15N-HSQC spectrum. Although the tryptophan residue of the proposed GABARAP binding site is conserved in Bcl-xL, it is not flanked by acidic side chains. These observations suggest that ionic interactions may play an important role in the formation of a GABARAP·Bcl-2 complex.
Finally, the significance of individual Bcl-2 side chains for GABARAP binding was investigated by 1H-15N-HSQC titration experiments using 15N-GABARAP and Bcl-2-derived peptides. Whereas the wild-type peptide (Ser24-Asp34) was clearly able to interact with GABARAP (Fig. 4, A and B), no binding was detected using a mutated version in which Trp30 was replaced by alanine (Fig. 4D). Likewise, the acidic residues Glu29 and Asp31 appear to be required for complex formation. After replacing these side chains individually with their non-charged amides (yielding E29Q and D31N, respectively), the interaction was greatly reduced (Fig. 4, C and E).
FIGURE 4.

Interaction of Bcl-2-derived peptides with GABARAP. A, superposition of 1H-15N-HSQC spectra of 15N-labeled GABARAP in the presence of Bcl-2 peptide Ser24-Asp34, recorded in the course of a titration experiment. The contour plots correspond to 0 (red), 62 (orange), 89 (yellow), 125 (green), 160 (cyan), 217 (blue), and 265 μm (violet) peptide. The frame in A marks the spectral region enlarged in B-E (note the different x-y scaling). With respect to the wild type, peptides in experiments C-E were modified as indicated in the figure.
Model of the GABARAP·Bcl-2 Complex
To gain further insight into the structure of the complex, in silico docking experiments were carried out. Unfortunately, the original solution structure of Bcl-2# turned out not to be suitable for this approach because it features the Trp30 side chain as part of a hydrophobic cluster, i.e. in a non-exposed conformation. To overcome this problem, we applied a global optimization methodology termed basin-hopping to generate alternative conformations of the respective segment. The basin-hopping algorithm can be viewed as a generalization of the Monte Carlo plus energy minimization procedure (39). Moves are proposed by perturbing the current geometry, and are accepted or rejected according to the Metropolis criterion with the energy difference between the local minima obtained following minimization of the two configurations. Although basin-hopping has been successfully employed to find the global minimum of peptides and proteins, in the current work it served to generate structures with modified conformations surrounding residue Trp30. As expected, the local minima generated from the perturbed models were all higher in energy than the local minimum of the original structure because they had the tryptophan side chain in a non-preferred environment. For the actual docking, the HADDOCK system was used because it offers the possibility to incorporate prior knowledge (such as experimental data) about residues involved in the interaction, and also allows for flexibility of the local structure at the binding site. The resulting model is shown in Fig. 5 (for the proposed conformational change in the α1-α2 loop of Bcl-2, please refer to supplemental Fig. S1). Notably, the bulky tryptophan side chain (Trp30) of Bcl-2 is buried deeply in hp1 on the GABARAP surface (Fig. 5B), whereas hp2 is not occupied. Furthermore, the complex is stabilized by several hydrogen bonds and salt bridges involving, among others, Glu29 and Asp31 of Bcl-2, which chiefly interact with Lys13 and Lys46 of GABARAP (Fig. 5D).
FIGURE 5.

Model of the GABARAP·Bcl-2 complex (lacking Bcl-2 residues 35–91, for which no structural information is available). A, surface representation of GABARAP with the docked Bcl-2 molecule shown in ribbon mode. For ease of orientation, the BH segments are labeled and color-coded as described in the legend to Fig. 1. B, close-up view of the region marked in A. Trp30 of Bcl-2, shown as stick model, is inserted into hp1 of GABARAP. C, ribbon diagram of Bcl-2 bound to GABARAP. D, close-up view of the region marked in C, illustrating the proposed GABARAP-Bcl-2 interface. Key residues participating in hydrophobic interactions and salt bridges are shown; for visual clarity the former are drawn in lighter color in the case of GABARAP. Several additional contacts, particularly hydrogen bonds involving main chain atoms, are omitted from this figure. The asterisk denotes Ser24 of Bcl-2, a potential phosphorylation site (see text for details).
Interaction of GABARAP and Bcl-2 in Cells
Pulldown and co-immunoprecipitation experiments were performed to investigate whether GABARAP binds to endogenous non-chimeric Bcl-2. Immobilized GABARAP was exposed to Jurkat cell extracts and associated proteins were eluted, separated by SDS-PAGE, and probed with an anti-Bcl-2 antibody. Bcl-2 was identified among the GABARAP-associated proteins, but not in the control without immobilized GABARAP (Fig. 6A). Immunoprecipitation of Bcl-2 using anti-Bcl-2 antibody-coupled agarose with subsequent SDS-PAGE analysis and Western blotting yielded GABARAP immunoreactivity at the expected molecular mass (Fig. 6B), which further confirms the cellular interaction of Bcl-2 with GABARAP.
FIGURE 6.

Intracellular association of Bcl-2 with GABARAP. A, pulldown assays using Jurkat cell extracts and either GABARAP-coupled (+GABARAP) or unmodified Sepharose (−GABARAP). GABARAP-associated proteins were immunoblotted with anti-Bcl-2 antibody. B, endogenous proteins were immunoprecipitated from Jurkat cell extracts using an anti-Bcl-2 antibody immobilized on protein A-agarose (+Bcl-2 antibody) and non-coupled agarose as control (negative control). Bound components were immunoblotted with anti-Bcl-2 or anti-GABARAP antibodies. The right lane contains 10% of the cell lysate used for immunoprecipitation. C, co-immunoprecipitation experiments with MEF cells stably overexpressing either wild-type Bcl-2 (EWD) or the QAN mutant. Left panel, after precipitation of GABARAP from cell lysates with an immobilized antibody, both flow-through and eluate were analyzed by SDS-PAGE and Western blotting using a Bcl-2 antibody. Control cells have been transfected with the empty vector. Right panel, immunoblot showing the protein levels of overexpressed Bcl-2 as well as endogenous tubulin (loading control), prepared with 10% of the lysate used for immunoprecipitation. D, overexpression of Bcl-2 in MEF cells leads to increased viability under staurosporin treatment (see text for details).
To scrutinize the in vivo significance of the EWD motif, we performed co-immunoprecipitation experiments with MEF cells. Taking advantage of the very low levels of endogenous Bcl-2 in these cells, we generated stable lines overexpressing either wild-type human Bcl-2 or a triple mutant in which the EWD sequence was replaced by QAN. Note that the BH4 regions of mouse and human Bcl-2 (residues 10–30, Fig. 1C) as well as the GABARAP molecules share 100% sequence identity. Confocal laser scanning microscopy revealed that in both stable cell lines, Bcl-2 displayed a subcellular localization in accordance with previous reports (23, 40, 41), suggesting that the triple mutation did not disturb sorting of the protein. After immunoprecipitation with a GABARAP antibody, Bcl-2 could be detected in the eluate only for cells expressing the wild-type protein, whereas in case of the QAN mutant, immunoreactivity was found exclusively in the flow-through (Fig. 6C). As expected, the endogenous Bcl-2 was not detectable in the empty vector control cells. The inverse experiment yielded complementary results. Although endogenous GABARAP co-precipitates with overexpressed wild-type Bcl-2, it is not associated with the triple mutant (not shown). Together, these observations underscore the critical role of the acidic/hydrophobic binding motif identified in this study. We also investigated the susceptibility of these cell lines to apoptotic cell death by challenging them with staurosporin and recording the fraction of live cells as a function of staurosporin concentration using high-content imaging (Fig. 6D). As expected, overexpression of wild-type Bcl-2 increased viability, compared with control cells. Interestingly, the QAN mutant also conferred significant resistance to cell death, indicating that the GABARAP binding function of Bcl-2 is largely independent of its anti-apoptotic activity.
Bcl-2 Regulates GABARAP Lipidation
To explore the potential impact of Bcl-2 on GABARAP function, the relative expression levels of Bcl-2, GABARAP, and LC3 in control and Bcl-2-overexpressing REF cells were investigated by Western blot analysis. In Bcl-2-overexpressing cells, the ratio of non-lipidated to lipidated GABARAP (GABARAP:GABARAP-II) was much higher than in control cells (Fig. 7, A and B). In contrast, the ratio of LC3 to LC3-II did not change significantly in response to Bcl-2 overexpression (Fig. 7A). Note that for both GABARAP and LC3, the non-lipidated fractions comprise unmodified (full-length) as well as C terminally cleaved protein (form I), which are not separated due to marginal differences in molecular mass. Analysis of the subcellular distribution revealed that GABARAP-positive puncta were fewer and smaller in Bcl-2-overexpressing cells compared with control cells (Fig. 7, C–E). As GABARAP can be conjugated to phospholipids, resulting in membrane localization (5), the amount and size of the GABARAP positive dots should correlate with the lipidation level of GABARAP. We therefore conclude that overexpression of Bcl-2 results in a decrease of GABARAP lipidation, which is consistent with the Western blotting results.
FIGURE 7.

Bcl-2 overexpression reduces lipid conjugation of GABARAP. A, Western blot analysis of Bcl-2, GABARAP, LC3, and tubulin in Bcl-2-overexpressing and control REF cells. B, GABARAP:GABARAP-II ratio obtained from densitometric evaluation. Bars represent the mean ± S.E. from three independent experiments (*, p < 0.05). Subcellular localization of GABARAP in control (C) and Bcl-2-overexpressing REF cells (D) was investigated using rabbit anti-GABARAP antibody followed by rhodamine-conjugated bovine anti-rabbit antibody (red). Nuclei were stained with DAPI (blue). Scale bar is 10 μm. E, quantitation of GABARAP positive dots in control and Bcl-2-overexpressing cells. Results represent the mean ± S.E. for combined data from four independent experiments (**, p < 0.01).
Finally, we investigated whether Bcl-2 is able to compete with Atg4B for the interaction with GABARAP. For this purpose, we performed pulldown experiments with immobilized GABARAP, using lysates from REF cells with or without Bcl-2 overexpression, and tested the eluate for bound Atg4B. We found that Atg4B signals significantly decreased under conditions of Bcl-2 overexpression (Fig. 8), indicating that Bcl-2 prevents GABARAP from interacting with the conjugation machinery.
FIGURE 8.

Bcl-2 overexpression affects binding of Atg4B to GABARAP. Competitive pulldown experiments using REF cells with or without Bcl-2 overexpression and either GABARAP-coupled or unmodified Sepharose. Bound components were immunoblotted with anti-Bcl-2 or anti-Atg4B antibodies. The input panel shows 10% of the cell lysate used in the pulldown experiments, probed with anti-Bcl-2, anti-Atg4B, and anti-tubulin antibodies, respectively.
DISCUSSION
Autophagy and apoptosis are strictly regulated biological processes essential for maintenance of cell and tissue homeostasis during development as well as in response to stress. About two decades ago, proteins of the Bcl-2 family have been recognized as essential regulators of apoptotic cell death; their role in autophagy, however, has only recently been appreciated (reviewed in Ref. 42). Specifically, Pattingre et al. (43) found that Bcl-2 negatively regulates autophagy via its interaction with Beclin 1. Beclin 1 acts as an obligatory activator of Vps34, a class III phosphatidylinositol 3-kinase essential for initiation of phagophore assembly. Current evidence indicates that Bcl-2 and Bcl-xL interact with the BH3 region of Beclin 1 via a conserved hydrophobic groove formed by their own BH1, BH2, and BH3 segments. When engaged in this complex, Beclin 1 is ineffective in promoting Vps34 activity. In the presence of autophagic stimuli such as starvation, however, Beclin 1 is released from Bcl-2, resulting in activation of the phosphatidylinositol 3-kinase and hence phagophore formation.
In this work, we provide data in support of GABARAP representing a novel target of Bcl-2 in autophagy. Following the incidental discovery of this interaction in our laboratory, we applied immunoprecipitation and immunofluorescence techniques to verify its significance in living cells. Complementary HSQC NMR titrations with recombinant proteins revealed an undecapeptide centered on the C-terminal end of the Bcl-2 BH4 region to be involved in association with the conserved hydrophobic surface of the GABARAP molecule. Furthermore, we could demonstrate that the expression level of Bcl-2 affects GABARAP lipidation, suggesting a potential molecular mechanism of its inhibitory role in autophagy. Together, these results support the conclusion that the Bcl-2-GABARAP interaction contributes to the crosstalk between apoptosis and autophagy.
As outlined above, ligands of GABARAP often contain the sequence motif XXWXXΦ as an essential binding determinant. This signature is similar to the LC3-interacting region (44) and the Atg8 family interacting motif (45) described previously. The results of the present study suggest that, in addition to apolar side chains occupying the hydrophobic pocket(s), GABARAP-ligand interactions are stabilized by electrostatic forces, thus rationalizing the frequent occurrence of acidic residues in the XXWXXΦ motif. Indeed, part of the hydrophobic surface of GABARAP is lined by basic amino acids, which are likely to participate in salt bridges with ligand side chains. Considering these requirements, we note that the GABARAP binding motif found in Bcl-2 is not conserved in other anti-apoptotic members of the Bcl-2 family. Although the central tryptophan is present in Bcl-xL, the neighboring acidic residues are not (Fig. 1B), which is consistent with the lack of interaction with GABARAP (Fig. 3C). A distinctive property of the GABARAP·Bcl-2 complex is that binding involves only the first of the two canonical hydrophobic pockets on the GABARAP surface. To better understand possible implications of this feature, we have performed docking experiments using a modified Bcl-2# structure. The resulting model (Fig. 5) nicely reflects this type of interaction. In addition to the classical knob-and-hole tether formed by the tryptophan side chain, the interface encompasses several salt bridges and hydrogen bonds. Importantly, our model suggests that, after binding of GABARAP to Bcl-2, hp2 will still be available for interaction with other proteins, possibly establishing ternary complexes. We have recently proposed a similar binding mode for the association of N-ethylmaleimide-sensitive factor with GABARAP, where hp1 also accommodates a large hydrophobic residue (a tyrosine in this case), whereas hp2 is largely accessible (46).
While this work was under review, the crystal structure of Bcl-2 bound to a small-molecule inhibitor (PDB code 4LXD, Ref. 47) was published. Remarkably, in this structure the side chain of Trp30 is pointing away from the protein core because it is involved in a lattice contact mediated by the ligand. This observation confirms that the segment surrounding Trp30 provides sufficient flexibility to allow for re-orientation of this side chain, in accordance with our experimental data and modeling results.
To appreciate the functional significance of the GABARAP-Bcl-2 interaction, the subcellular localization of these proteins has to be taken into account. Bcl-2 is believed to largely occur on endoplasmic reticulum (ER) membranes and less on mitochondrial membranes, whereas the opposite seems to be true for Bcl-xL (48). Notably, only Bcl-2 targeted to the ER is capable of protecting against cell death, so Bcl-2 at other membranes may have different roles (49). GABARAP, in contrast, is associated with membranes of Golgi cisternae, transport vesicles, and autophagosomes. Although immunofluorescence experiments with Bcl-2-overexpressing MEF cells indicated partial colocalization of both proteins in punctate structures, this pattern did not change significantly upon mutation of the critical EWD motif (not shown). However, our co-immunoprecipitation data clearly demonstrate that GABARAP binding is abolished for the QAN mutant of Bcl-2 (Fig. 6C), which is consistent with our peptide titration results.
In response to autophagic stimuli, GABARAP and other Atg8 family proteins are subject to a ubiquitin-like conjugation process: following C-terminal cleavage by Atg4 (50), they are conjugated to phosphatidylethanolamine by the actions of Atg7, Atg3, and the Atg16L complex (51) and thus localize to autophagosomal membranes (5). Therefore, the lipidated, membrane-attached derivative (form II) is considered the functionally active species promoting autophagy. Considering these data, different mechanisms can be envisaged to explain how overexpression of Bcl-2 may influence the lipidation level of GABARAP. One such mechanism would be a direct one, where binding of GABARAP to Bcl-2 precludes its simultaneous interaction with one of the modifying proteins, the most obvious candidate being Atg4. Atg4 is a cysteine protease with four paralogs in humans, which have different binding preferences for GABARAP-related proteins (52). Atg4B, for instance, is able to cleave GATE-16, GABARAP, and LC3 (53). A recent crystallographic study featuring the Atg4B·LC3B complex revealed contact between the N-terminal YDTL sequence (a canonical LC3-interacting region) of Atg4B and the hydrophobic pockets of a symmetry-equivalent LC3B molecule (54). Similarly, Atg7 is very likely to bind Atg8 proteins via its C-terminal tail (55). We therefore propose a model in which Bcl-2 on the one hand and Atg4B (or a different component of the modification cascade) on the other compete for the hydrophobic binding pockets on the GABARAP molecule. In support of this hypothesis, our pulldown experiments demonstrate that overexpression of Bcl-2 indeed prevents Atg4B from interacting with GABARAP, thus abolishing the first step of the modification process. As expected, Bcl-2 overexpression did not significantly affect the lipidation of LC3 because the affinity of Bcl-2 for LC3 is much lower than for GABARAP.
Of course, the involvement of more indirect mechanisms, e.g. via Beclin 1, cannot be ruled out. Moreover, the attenuating effect of Bcl-2 on autophagy may not exclusively rely on its interactions with Beclin 1 and GABARAP. For instance, there is evidence that Bcl-2 can reduce autophagy by inhibition of inositol 1,4,5-trisphosphate receptors, the major Ca2+ release channels of the ER (56).
Within the signaling network of mammalian cells, Bcl-2 is now considered an integrating node in control of both autophagy and apoptosis (57). In essence, it serves to attenuate both processes by sequestration of activating molecules, death-promoting BH3 proteins in the case of apoptosis, and Beclin 1 in the case of autophagy. The results presented here add GABARAP to the latter category. In recent years, multisite phosphorylation of ER-resident Bcl-2, e.g. by JNK-1, in its largely unstructured α1-α2 loop has emerged as a master switch governing its interactions with BH3-containing proteins (58, 59). The precise mechanism by which these phosphorylation events (at Thr69, Ser70, and Ser87) will suppress binding of BH3 peptides on the opposite face of the molecule is unclear at present, but may involve a reorientation of the helical bundle. In contrast, the key residues of the proposed GABARAP binding site are located in the N-terminal part of the α1-α2 loop (Glu29-Asp31), i.e. even closer to the phosphorylation sites. Therefore, it is tempting to speculate that GABARAP inactivation by Bcl-2 might also be reversed by phosphorylation. According to this hypothesis, phosphorylation of Bcl-2 attached to the ER membrane would lead to a concerted release of two pro-autophagic proteins, Beclin 1 and GABARAP, close to their site of action, thus efficiently promoting autophagosome formation. On the other hand, protein-protein interactions by several ubiquitin-like modifiers were found to be enhanced by phosphorylation of serine residues adjacent to the hydrophobic LC3-interacting region or SUMO interacting motif sequences of their binding partners (60–62). It is interesting to note that in Bcl-2, a serine (Ser24) is located in close proximity to the GABARAP binding site (Fig. 5D). Indeed, phosphorylation of Ser24 has been implicated in the regulation of anti-apoptotic Bcl-2 functions (63); whether it also plays a role in GABARAP binding (and thus modulation of autophagy) remains to be elucidated.
Elevated Bcl-2 expression is a common finding in many types of cancer cells. Recent evidence indicates that the oncogenic potential of Bcl-2 is, to an appreciable extent, due to its effects on autophagy (64). Intriguingly, deletion of helix α1 (residues 1–22) results in a Bcl-2 variant that is devoid of anti-apoptotic activity, whereas it is still able to inhibit autophagy. The latter function is only lost after an additional deletion of residues 23–36 (35), pointing to a critical role of this stretch in autophagy regulation. The results of the current study strongly argue for an involvement of GABARAP in this effect. Indeed, our observation that the QAN mutant, whereas defective in GABARAP binding, still confers resistance to cell death (Fig. 6D) supports the notion that the inhibitory effects of Bcl-2 on apoptosis and autophagy are mediated by distinct regions of the molecule. It is important to note that the anti-autophagic activity of N terminally truncated Bcl-2 has been reported to correlate with its ability to co-immunoprecipitate with Beclin 1 from cell lysates (35, 64). Whether sequestration of Beclin 1 and GABARAP by Bcl-2 represent alternative (maybe mutually exclusive) pathways to inhibit autophagy, or whether both proteins can be bound simultaneously, will be a subject of future studies.
Acknowledgments
We thank Dr. Joseph T. Opferman for the kind gift of the pGEX4T3-Bcl-xL vector, Dr. Christoph Borner for providing stable REF cell lines, and Esther Jonas for excellent technical assistance.
This work was supported by a fellowship of the Deutscher Akademischer Austauschdienst (DAAD) (to P. M.), a fellowship of the International Helmholtz Research School BioSoft (to M. Schwarten), and Deutsche Forschungsgemeinschaft (DFG) Grants Me 1922/9-1 (to A. M.) and Wi 1472/5 (to D. W.).
The resulting assignment (covering 126 of 162 residues) has been deposited in the Biological Magnetic Resonance Data Bank (www.bmrb.wisc.edu) with code 19559.

This article contains supplemental Fig. S1.
Within this paper, the term autophagy refers to the canonical pathway, also known as macroautophagy.
- Atg
- autophagy-related protein
- Bcl
- B-cell lymphoma
- BH
- Bcl-2 homology
- Bnip3L
- Bcl-2/adenovirus E1B 19-kDa interacting protein 3-like
- ER
- endoplasmic reticulum
- GABA
- γ-aminobutyric acid
- GABARAP
- GABAA receptor-associated protein
- GATE-16
- Golgi-associated ATPase enhancer of 16 kDa
- GEC1
- glandular epithelial cell protein 1
- GST
- glutathione S-transferase
- HSQC
- heteronuclear single quantum coherence
- hp
- hydrophobic pocket
- LC3
- light chain 3 of microtubule-associated protein
- MEF
- mouse embryonic fibroblast
- Nix
- Nip-like protein x
- REF
- rat embryonic fibroblast
- SPR
- surface plasmon resonance
- SUMO
- small ubiquitin-like modifier.
REFERENCES
- 1. Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. (2008) Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tanida I., Ueno T., Kominami E. (2004) LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ichimura Y., Kirisako T., Takao T., Satomi Y., Shimonishi Y., Ishihara N., Mizushima N., Tanida I., Kominami E., Ohsumi M., Noda T., Ohsumi Y. (2000) A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492 [DOI] [PubMed] [Google Scholar]
- 4. Kirisako T., Baba M., Ishihara N., Miyazawa K., Ohsumi M., Yoshimori T., Noda T., Ohsumi Y. (1999) Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 147, 435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kabeya Y., Mizushima N., Yamamoto A., Oshitani-Okamoto S., Ohsumi Y., Yoshimori T. (2004) LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117, 2805–2812 [DOI] [PubMed] [Google Scholar]
- 6. Weidberg H., Shvets E., Shpilka T., Shimron F., Shinder V., Elazar Z. (2010) LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 29, 1792–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mohrlüder J., Schwarten M., Willbold D. (2009) Structure and potential function of γ-aminobutyrate type A receptor-associated protein. FEBS J. 276, 4989–5005 [DOI] [PubMed] [Google Scholar]
- 8. Klebig C., Seitz S., Arnold W., Deutschmann N., Pacyna-Gengelbach M., Scherneck S., Petersen I. (2005) Characterization of γ-aminobutyric acid type A receptor-associated protein, a novel tumor suppressor, showing reduced expression in breast cancer. Cancer Res. 65, 394–400 [PubMed] [Google Scholar]
- 9. Roberts S. S., Mendonça-Torres M. C., Jensen K., Francis G. L., Vasko V. (2009) GABA receptor expression in benign and malignant thyroid tumors. Pathol. Oncol. Res. 15, 645–650 [DOI] [PubMed] [Google Scholar]
- 10. Mohrlüder J., Hoffmann Y., Stangler T., Hänel K., Willbold D. (2007) Identification of clathrin heavy chain as a direct interaction partner for the γ-aminobutyric acid type A receptor associated protein. Biochemistry 46, 14537–14543 [DOI] [PubMed] [Google Scholar]
- 11. Mohrlüder J., Stangler T., Hoffmann Y., Wiesehan K., Mataruga A., Willbold D. (2007) Identification of calreticulin as a ligand of GABARAP by phage display screening of a peptide library. FEBS J. 274, 5543–5555 [DOI] [PubMed] [Google Scholar]
- 12. Weiergräber O. H., Stangler T., Thielmann Y., Mohrlüder J., Wiesehan K., Willbold D. (2008) Ligand binding mode of GABAA receptor-associated protein. J. Mol. Biol. 381, 1320–1331 [DOI] [PubMed] [Google Scholar]
- 13. Thielmann Y., Weiergräber O. H., Mohrlüder J., Willbold D. (2009) Structural framework of the GABARAP-calreticulin interface. Implications for substrate binding to endoplasmic reticulum chaperones. FEBS J. 276, 1140–1152 [DOI] [PubMed] [Google Scholar]
- 14. Thielmann Y., Weiergräber O. H., Mohrlüder J., Willbold D. (2009) Structural characterization of GABARAP-ligand interactions. Mol. Biosyst. 5, 575–579 [DOI] [PubMed] [Google Scholar]
- 15. Ma P., Mohrlüder J., Schwarten M., Stoldt M., Singh S. K., Hartmann R., Pacheco V., Willbold D. (2010) Preparation of a functional GABARAP-lipid conjugate in nanodiscs and its investigation by solution NMR spectroscopy. ChemBioChem 11, 1967–1970 [DOI] [PubMed] [Google Scholar]
- 16. Schwarten M., Mohrlüder J., Ma P., Stoldt M., Thielmann Y., Stangler T., Hersch N., Hoffmann B., Merkel R., Willbold D. (2009) Nix directly binds to GABARAP. As possible crosstalk between apoptosis and autophagy. Autophagy 5, 690–698 [DOI] [PubMed] [Google Scholar]
- 17. Imazu T., Shimizu S., Tagami S., Matsushima M., Nakamura Y., Miki T., Okuyama A., Tsujimoto Y. (1999) Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene 18, 4523–4529 [DOI] [PubMed] [Google Scholar]
- 18. Ray R., Chen G., Vande Velde C., Cizeau J., Park J. H., Reed J. C., Gietz R. D., Greenberg A. H. (2000) BNIP3 heterodimerizes with Bcl-2/Bcl-XL and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J. Biol. Chem. 275, 1439–1448 [DOI] [PubMed] [Google Scholar]
- 19. Novak I., Kirkin V., McEwan D. G., Zhang J., Wild P., Rozenknop A., Rogov V., Löhr F., Popovic D., Occhipinti A., Reichert A. S., Terzic J., Dötsch V., Ney P. A., Dikic I. (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stangler T., Mayr L. M., Dingley A. J., Luge C., Willbold D. (2001) Sequence-specific 1H, 13C and 15N resonance assignments of human GABA receptor associated protein. J. Biomol. NMR 21, 183–184 [DOI] [PubMed] [Google Scholar]
- 21. Opferman J. T., Letai A., Beard C., Sorcinelli M. D., Ong C. C., Korsmeyer S. J. (2003) Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 426, 671–676 [DOI] [PubMed] [Google Scholar]
- 22. Petros A. M., Medek A., Nettesheim D. G., Kim D. H., Yoon H. S., Swift K., Matayoshi E. D., Oltersdorf T., Fesik S. W. (2001) Solution structure of the antiapoptotic protein bcl-2. Proc. Natl. Acad. Sci. U.S.A. 98, 3012–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Conus S., Kaufmann T., Fellay I., Otter I., Rossé T., Borner C. (2000) Bcl-2 is a monomeric protein. Prevention of homodimerization by structural constraints. EMBO J. 19, 1534–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 25. Johnson B. A., Blevins R. A. (1994) NMR view. A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 26. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) The CCPN data model for NMR spectroscopy. Development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 27. Lescop E., Schanda P., Brutscher B. (2007) A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J. Magn. Reson. 187, 163–169 [DOI] [PubMed] [Google Scholar]
- 28. Grzesiek S., Bax A. (1992) Improved 3D triple resonance NMR techniques applied to a 31 kDa protein. J. Magn. Reson. 96, 432–440 [Google Scholar]
- 29. Keller R. (2004) The Computer-Aided Resonance Assignment Tutorial, CANTINA Verlag, Goldau [Google Scholar]
- 30. Dominguez C., Boelens R., Bonvin A. M. (2003) HADDOCK. A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 31. de Vries S. J., van Dijk A. D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., Bonvin A. M. (2007) HADDOCK versus HADDOCK. New features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 69, 726–733 [DOI] [PubMed] [Google Scholar]
- 32. Wales D. J., Doye J. P. K. (1997) Global optimization by basin-hopping and the lowest energy structures of Lennard-Jones clusters containing up to 110 atoms. J. Phys. Chem. A 101, 5111–5116 [Google Scholar]
- 33. Wales D. J. (2003) Energy Landscapes, Cambridge University Press, Cambridge [Google Scholar]
- 34. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiorkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 35. Trisciuoglio D., De Luca T., Desideri M., Passeri D., Gabellini C., Scarpino S., Liang C., Orlandi A., Del Bufalo D. (2013) Removal of the BH4 domain from Bcl-2 protein triggers an autophagic process that impairs tumor growth. Neoplasia 15, 315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A. M., Czaja M. J. (2009) Autophagy regulates lipid metabolism. Nature 458, 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stangler T., Mayr L. M., Willbold D. (2002) Solution structure of human GABAA receptor-associated protein GABARAP. Implications for biological function and its regulation. J. Biol. Chem. 277, 13363–13366 [DOI] [PubMed] [Google Scholar]
- 38. Thielmann Y., Mohrlüder J., Koenig B. W., Stangler T., Hartmann R., Becker K., Höltje H. D., Willbold D. (2008) An indole-binding site is a major determinant of the ligand specificity of the GABA type A receptor-associated protein GABARAP. ChemBioChem 9, 1767–1775 [DOI] [PubMed] [Google Scholar]
- 39. Li Z., Scheraga H. A. (1987) Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. U.S.A. 84, 6611–6615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gotow T., Shibata M., Kanamori S., Tokuno O., Ohsawa Y., Sato N., Isahara K., Yayoi Y., Watanabe T., Leterrier J. F., Linden M., Kominami E., Uchiyama Y. (2000) Selective localization of Bcl-2 to the inner mitochondrial and smooth endoplasmic reticulum membranes in mammalian cells. Cell Death Differ. 7, 666–674 [DOI] [PubMed] [Google Scholar]
- 41. Otter I., Conus S., Ravn U., Rager M., Olivier R., Monney L., Fabbro D., Borner C. (1998) The binding properties and biological activities of Bcl-2 and Bax in cells exposed to apoptotic stimuli. J. Biol. Chem. 273, 6110–6120 [DOI] [PubMed] [Google Scholar]
- 42. Levine B., Sinha S., Kroemer G. (2008) Bcl-2 family members. Dual regulators of apoptosis and autophagy. Autophagy 4, 600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pattingre S., Tassa A., Qu X., Garuti R., Liang X. H., Mizushima N., Packer M., Schneider M. D., Levine B. (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939 [DOI] [PubMed] [Google Scholar]
- 44. Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 [DOI] [PubMed] [Google Scholar]
- 45. Noda N. N., Ohsumi Y., Inagaki F. (2010) Atg8-family interacting motif crucial for selective autophagy. FEBS Lett. 584, 1379–1385 [DOI] [PubMed] [Google Scholar]
- 46. Thielmann Y., Weiergräber O. H., Ma P., Schwarten M., Mohrlüder J., Willbold D. (2009) Comparative modeling of human NSF reveals a possible binding mode of GABARAP and GATE-16. Proteins 77, 637–646 [DOI] [PubMed] [Google Scholar]
- 47. Souers A. J., Leverson J. D., Boghaert E. R., Ackler S. L., Catron N. D., Chen J., Dayton B. D., Ding H., Enschede S. H., Fairbrother W. J., Huang D. C., Hymowitz S. G., Jin S., Khaw S. L., Kovar P. J., Lam L. T., Lee J., Maecker H. L., Marsh K. C., Mason K. D., Mitten M. J., Nimmer P. M., Oleksijew A., Park C. H., Park C. M., Phillips D. C., Roberts A. W., Sampath D., Seymour J. F., Smith M. L., Sullivan G. M., Tahir S. K., Tse C., Wendt M. D., Xiao Y., Xue J. C., Zhang H., Humerickhouse R. A., Rosenberg S. H., Elmore S. W. (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 [DOI] [PubMed] [Google Scholar]
- 48. Kaufmann T., Schlipf S., Sanz J., Neubert K., Stein R., Borner C. (2003) Characterization of the signal that directs Bcl-xL, but not Bcl-2, to the mitochondrial outer membrane. J. Cell Biol. 160, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang N. S., Unkila M. T., Reineks E. Z., Distelhorst C. W. (2001) Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against Bax-induced cell death. J. Biol. Chem. 276, 44117–44128 [DOI] [PubMed] [Google Scholar]
- 50. Tanida I., Sou Y. S., Ezaki J., Minematsu-Ikeguchi N., Ueno T., Kominami E. (2004) HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J. Biol. Chem. 279, 36268–36276 [DOI] [PubMed] [Google Scholar]
- 51. Noda T., Fujita N., Yoshimori T. (2009) The late stages of autophagy. How does the end begin? Cell Death Differ. 16, 984–990 [DOI] [PubMed] [Google Scholar]
- 52. Li M., Hou Y., Wang J., Chen X., Shao Z. M., Yin X. M. (2011) Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J. Biol. Chem. 286, 7327–7338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hemelaar J., Lelyveld V. S., Kessler B. M., Ploegh H. L. (2003) A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. J. Biol. Chem. 278, 51841–51850 [DOI] [PubMed] [Google Scholar]
- 54. Satoo K., Noda N. N., Kumeta H., Fujioka Y., Mizushima N., Ohsumi Y., Inagaki F. (2009) The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 28, 1341–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Noda N. N., Satoo K., Fujioka Y., Kumeta H., Ogura K., Nakatogawa H., Ohsumi Y., Inagaki F. (2011) Structural basis of Atg8 activation by a homodimeric E1, Atg7. Mol. Cell 44, 462–475 [DOI] [PubMed] [Google Scholar]
- 56. Criollo A., Maiuri M. C., Tasdemir E., Vitale I., Fiebig A. A., Andrews D., Molgó J., Díaz J., Lavandero S., Harper F., Pierron G., di Stefano D., Rizzuto R., Szabadkai G., Kroemer G. (2007) Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 14, 1029–1039 [DOI] [PubMed] [Google Scholar]
- 57. Wei Y., Sinha S., Levine B. (2008) Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4, 949–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bassik M. C., Scorrano L., Oakes S. A., Pozzan T., Korsmeyer S. J. (2004) Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 23, 1207–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wei Y., Pattingre S., Sinha S., Bassik M., Levine B. (2008) JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30, 678–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wild P., Farhan H., McEwan D. G., Wagner S., Rogov V. V., Brady N. R., Richter B., Korac J., Waidmann O., Choudhary C., Dötsch V., Bumann D., Dikic I. (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhu Y., Massen S., Terenzio M., Lang V., Chen-Lindner S., Eils R., Novak I., Dikic I., Hamacher-Brady A., Brady N. R. (2013) Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 288, 1099–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stehmeier P., Muller S. (2009) Phospho-regulated SUMO interaction modules connect the SUMO system to CK2 signaling. Mol. Cell 33, 400–409 [DOI] [PubMed] [Google Scholar]
- 63. Hallin U., Kondo E., Ozaki Y., Hagberg H., Shibasaki F., Blomgren K. (2006) Bcl-2 phosphorylation in the BH4 domain precedes caspase-3 activation and cell death after neonatal cerebral hypoxic-ischemic injury. Neurobiol. Dis. 21, 478–486 [DOI] [PubMed] [Google Scholar]
- 64. Oh S., Xiaofei E., Ni D., Pirooz S. D., Lee J. Y., Lee D., Zhao Z., Lee S., Lee H., Ku B., Kowalik T., Martin S. E., Oh B. H., Jung J. U., Liang C. (2011) Downregulation of autophagy by Bcl-2 promotes MCF7 breast cancer cell growth independent of its inhibition of apoptosis. Cell Death Differ. 18, 452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]