

Background: Hypothalamic long-chain fatty acids (LCFA) and glucose are critical for energy balance, but it is not known if their metabolism is coupled.
Results: Glucose regulates hypothalamic metabolism of palmitate via AMP-activated kinase.
Conclusion: Glucose and LCFA metabolism is coupled in a cell-type and LCFA-dependent manner.
Significance: This is the first evidence for glucose regulation of LCFA metabolic fate in the hypothalamus.
Keywords: AMP-activated Kinase (AMPK), Astrocytes, Fatty Acid Oxidation, Glucose Metabolism, Hypothalamus, Neural Metabolism, Fatty Acid Esterification, Oleate, Palmitate
Abstract
Hypothalamic controls of energy balance rely on the detection of circulating nutrients such as glucose and long-chain fatty acids (LCFA) by the mediobasal hypothalamus (MBH). LCFA metabolism in the MBH plays a key role in the control of food intake and glucose homeostasis, yet it is not known if glucose regulates LCFA oxidation and esterification in the MBH and, if so, which hypothalamic cell type(s) and intracellular signaling mechanisms are involved. The aim of this study was to determine the impact of glucose on LCFA metabolism, assess the role of AMP-activated Kinase (AMPK), and to establish if changes in LCFA metabolism and its regulation by glucose vary as a function of the kind of LCFA, cell type, and brain region. We show that glucose inhibits palmitate oxidation via AMPK in hypothalamic neuronal cell lines, primary hypothalamic astrocyte cultures, and MBH slices ex vivo but not in cortical astrocytes and slice preparations. In contrast, oleate oxidation was not affected by glucose or AMPK inhibition in MBH slices. In addition, our results show that glucose increases palmitate, but not oleate, esterification into neutral lipids in neurons and MBH slices but not in hypothalamic astrocytes. These findings reveal for the first time the metabolic fate of different LCFA in the MBH, demonstrate AMPK-dependent glucose regulation of LCFA oxidation in both astrocytes and neurons, and establish metabolic coupling of glucose and LCFA as a distinguishing feature of hypothalamic nuclei critical for the control of energy balance.
Introduction
The hypothalamus controls energy homeostasis by integrating hormonal and nutrient signals such as long-chain fatty acids (LCFA)4 and glucose (1). The modulation of glucose homeostasis and food intake by glucose and LCFA relies on the intracellular metabolism of these nutrients. Esterification and oxidative metabolism of LCFA has been shown to mediate the effects of LCFA on glucose and energy homeostasis (2–5), and the actions of glucose in the brain mainly involve changes in the AMP/ATP ratio (6). In peripheral tissues, glucose regulates the partitioning of LCFA-CoA between oxidation and esterification, a process fundamental to glucose regulation of insulin release by the pancreatic beta cell (7). Despite this knowledge, it is not known if glucose and LCFA metabolism is coupled in the hypothalamus nor which cell types and intracellular signaling pathways are involved.
The coupling of glucose and LCFA metabolism in peripheral tissues is known to require specific glucose-derived metabolites and enzymes (8). Intracellular metabolism of glucose inhibits the activity of a key energy sensing enzyme, AMP-activated kinase (AMPK). Several studies have established the importance of AMPK in the mediobasal hypothalamus (MBH) in glucose sensing (9, 10), action (11), and the counterregulatory response to hypoglycemia (12, 13). Glucose inhibition of hypothalamic AMPK leads to the activation of acetyl-CoA carboxylase (ACC), thereby leading to the generation of malonyl-CoA from glucose-derived acetyl-CoA (14, 15). In peripheral tissues it is known that malonyl-CoA inhibits LCFA-CoA mitochondrial oxidation via inhibition of carnitine palmitoyl transferase-1a (CPT-1a), the isoform expressed in the liver, and CPT-1b, the muscle isoform (8). In the hypothalamus, CPT-1a and CPT-1c, the brain-specific isoform, are expressed, but only CPT-1a possesses the prototypical mitochondrial acyltransferase activity (16). Although inhibition of CPT-1a and LCFA-CoA oxidation by malonyl-CoA has been suggested in the brain (15, 17–20), this model is not consistent with the results of some studies showing that increased malonyl-CoA does not affect LCFA-CoA levels in the hypothalamus (21–23). However, LCFA-CoA oxidation rates in response to glucose have never been measured in the hypothalamus.
Beyond changes in oxidative flux, it is also not known whether or not glucose affects LCFA-CoA partitioning between oxidation and esterification in the hypothalamus and if this process depends on AMPK. Hypothalamic neurons (24) and glia (25–29) are both able to sense glucose, and the importance of astroglia in central nutrient sensing has been recently emphasized (30). It is unclear if glucose modulation of LCFA-CoA metabolism occurs in neurons and/or astrocytes and whether or not such a feature distinguishes hypothalamic nuclei from other brain regions. Finally, it remains to be elucidated if the type of LCFA (carbon chain length and saturation degree) differentially affects glucose-regulated LCFA metabolism. The goal of the present study was to 1) determine whether or not glucose regulates LCFA metabolism in the hypothalamus, 2) establish if oleate and palmitate metabolism are differentially modulated by glucose, 3) identify the role of AMPK in glucose-regulated LCFA metabolism, and 4) identify which hypothalamic cell type(s) is involved. To this end we have employed a combination of in vitro models consisting of hypothalamic neurons, hypothalamic, and cortical astrocytes cultures as well as ex vivo MBH and cortical slices to measure glucose metabolism, LCFA oxidation, and esterification rates in response to glucose and pharmacological AMPK manipulation.
EXPERIMENTAL PROCEDURES
Reagents
Culture media and serum were from Wisent. Radioactive tracers were from PerkinElmer Life Sciences, and all other reagents were from Sigma unless otherwise noted. The NPY RIA kit was from Phoenix Pharmaceuticals (Burlingame, CA).
Animals
Four-to-five-week-old male Wistar rats and C57Bl/6 mice were purchased from Charles River. Animals were housed 2 per cage on a 12-h light/dark cycle at 21 °C with free access to water and standard diet. All procedures were approved by the Institutional Committee for the Protection of Animals (CIPA) at the Centre Hospitalier de l'Université de Montréal.
Neuronal Cell Lines Culture
GT1-7 (a generous gift from Dr. Pamela Mellon, San Diego, CA) and N46 neurons (Cellutions Biosystems, Toronto, ON, Canada) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 25 mm glucose, and 1% penicillin/streptomycin at 37 °C in 95% O2, 5% CO2. Cells were used at an ∼70% confluence for every experiment. To assess the effect of glucose on the expression of esterification enzymes in N46 neurons, cells were starved during 12 h in DMEM containing 10% FBS at 1 mm glucose. Then cells were maintained for 24 h in DMEM containing 10% FBS with 1 or 15 mm glucose.
Primary Astrocytes Culture and Immunocytochemistry
Primary cultures of hypothalamic and cortical astrocytes were prepared from 1-day-old C57Bl/6 pups using a protocol adapted from the group of Magistretti and co-workers (31). Briefly, after decapitation, the brains were removed, and the hypothalami and cortices were dissected and transferred into 6-well plates containing 2 ml of DMEM. The tissues were dissociated by passing through syringe needles of decreasing diameter (22 gauge followed by 25 gauge) 6 times. The cells were plated in polyornithine-coated T25 flasks and maintained in DMEM containing 25 mm glucose and supplemented with 44 mm NaHCO3, 1% antibiotic-antimycotic, and 10% FBS at 37 °C in 95% O2, 5% CO2. Astrocytes were cultured for 14 to 21 days before use (∼70% confluence).
Astrocytes cultured on coverslips were fixed in 4% formalin and blocked in presence of phosphate-buffered saline (PBS) with 5% bovine serum albumin (BSA) and 0.05% Triton. Cells were then incubated with a glial-fibrillary acidic protein primary antibody (1:1000, Dako, Canada) in 5% BSA and 0.05% Triton in PBS overnight at 4 °C followed by secondary antibody incubation (1:1000, Alexa Fluor 568, A-11004, Invitrogen) in 0.25% BSA for 2 h at room temperature. The coverslips were mounted onto glass slides with Vectashield (Vector Laboratories) containing DAPI (1.5 μg/ml). Cells were observed with a Zeiss fluorescent microscope.
RNA and Real-time Quantitative PCR
N46 and GT1-7 neurons and primary astrocytes grown in 6-well plates were rinsed with ice-cold PBS before RNA extraction using the TRIzol method (Invitrogen). RNA concentration was quantified spectrophotometrically. 900 ng of total RNA was reverse-transcribed by M-MuLV reverse transcriptase (Invitrogen) with random hexamers following the manufacturer's conditions. The reaction mix was then diluted 5-fold before use.
Quantitative gene expression was measured from 1:10 cDNA dilutions. Real-time PCR was performed using the QuantiFast SYBR Green PCR kit (Qiagen) according to the manufacturer's guidelines on a Corbett Rotor-Gene 6000. Data were analyzed using the standard curve method and normalized to 18 S expression levels. The list and sequence of primers is provided in Table 1.
TABLE 1.
Primers used for real-time quantitative PCR
POMC, pro-opiomelanocortin; GK, glucokinase; PDH, pyruvate dehydrogenase; DGAT, diacylglycerol acyltransferase 1 and 2; GPAT, glycerol-3-phosphate acyltransferase.
| Gene | Forward | Reverse |
|---|---|---|
| NPY | ctccgctctgcgacactaca | aatcagtgtctcagggctgga |
| AgRP | tgctactgccgcttcttcaa | ctttgcccaaacaacatcca |
| POMC | aggcctgacacgtggaagat | aggcaccagctccacacat |
| CPT-1a | gaggaccctgaggcatctat | gaatgctctgcgtttatgcc |
| CPT-1b | ccggaaaggtatggccactt | gaagaaaatgcctgtcgccc |
| CPT1-c | tccgatggggcagaagtaga | agtcattccagacacgccag |
| ACC1 | atgtccgcactgactgtaacc | tccatagccgacttccatagc |
| ACC2 | tctgatgaggaccctagtgc | acatgctgggcctcatagtag |
| FAS | gtgatagccggtatgtcggg | tagagcccagccttccatct |
| MCD | agaagatcagcgagtgtgagg | agtcagagccacatgcagaac |
| GK | atcttctgttccacggagagg | gatgttaaggatctgccttcg |
| GLUT2 | ccaggtccaatcccttggtt | cccaaggaagtccgcaatgt |
| PDHa1 | accagagaggatgggctcaa | aggcttcctgaccatcacac |
| PDHa2 | atcagcaacaacctcagcaat | ggttctggatcagtcgtagca |
| DGAT1 | ggatctgaggtgccatcgtc | atcagcatcaccacacacca |
| DGAT2 | catcatcgtggtgggaggtg | tgggaaccagatcagctccat |
| GPAT1 | cggaactgaactggagaagtg | gatgaattgctggtgctcctt |
| GPAT2 | aagactgagccggcatgttc | gtgacaggaccacacccttt |
| AGPAT1 | gacagagatacagccagccg | gctccattctggtcacctca |
| AGPAT2 | tacgccaaggtcggtctcta | accagctgatgatgctcatgt |
| AGPAT3 | aggaaaacacctgtccacgg | actgagaacagccgtccaag |
| AGPAT4 | gtgcttgcgagatgttgtcc | atggggatcctccgaacgta |
| AGPAT5 | accggggtccagatattgct | gcatgtccgcaacaatccag |
| AGPAT6 | ctactacgccatggtgggac | ggtggcgatctttcacctca |
| AGPAT7 | cccggcgtactcaaagttct | ttggcataaagggtggggtc |
| AGPAT8 | gcggtgtcagaatcatggtg | cggctgctaatccagcgata |
Hypothalamic and Cortical Slices
The slice protocol was adapted from a protocol previously described (32). After decapitation, the brain was rapidly removed and immersed in ice-cold cutting solution of the composition 1.25 mm NaH2PO4·H2O, 2.5 mm KCl, 7 mm MgCl2-6H2O, 0.5 mm CaCl2·2H2O, 28 mm NaHCO3, 8 mm d-glucose, 1 mm l-(+)-ascorbate, 3 mm sodium pyruvate, 5 mm Hepes, and 200 mm sucrose, pH 7.25, and osmolarity 310–320 mosm oxygenated with 95% O2, 5% CO2. Four 300-μm-thick sections containing the hypothalamus were cut from each rat using a Vibratome while being continuously immersed in ice-cold cutting solution. The MBH, which includes the arcuate nucleus plus the ventro- and dorso-median hypothalamus, and the mediolateral area of the motor and somatosensory cortex were dissected on each section as previously described (12, 32).
Slices were allowed to recover for 1 h at room temperature in artificial cerebrospinal fluid (aCSF) containing 118 mm NaCl, 3 mm KCl, 1.2 mm NaH2PO4·H2O, 1 mm MgCl2·6H2O, 1.5 mm CaCl2·2H2O, 5 mm Hepes, 25 mm NaHCO3, 5 mm d-glucose 5, and 15 mm sucrose, pH 7.25, and osmolarity 310–320 mosm, oxygenated with 95% O2, 5% CO2. Slices dissected from each hemisphere were pooled together in two distinct incubation chambers to allow two treatment conditions per animal. Slices were then preincubated at 37 °C for 30 min in aCSF containing 1 mm glucose before treatment under constant oxygenation.
For Neuropeptide Y (NPY) secretion in MBH slices, slices were incubated in aCSF containing 1 mm glucose and 0.1 mm palmitate pre-complexed to 0.1% BSA for 1 h 40 min. The media were then collected and replaced by aCSF, 1 mm glucose, 0.1 mm palmitate pre-complexed to 0.1% BSA and 50 mm KCl for 20 min after which the media were collected. NPY radioimmunoassay was performed following the manufacturer's recommendation. At the end of slice experiments, samples were washed in cold PBS, frozen in liquid nitrogen, and stored at −80 °C until subsequent homogenization for protein extraction.
Muscle and Liver Slices
The soleus muscle and liver were dissected on animals after decapitation. A 500-μm-thick section of the liver was cut using a slicing matrix. The entire soleus and the liver section were preincubated at 37 °C for 30 min in Krebs-Ringer bicarbonate HEPES buffer, pH 7.3, supplemented by 0.1% BSA and 1 mm glucose under constant oxygenation.
LCFA Oxidation in Cultured Neurons and Astrocytes
The measurement of LCFA oxidation in neurons and primary astrocytes cultures was performed using a protocol adapted from a methodology already described for cultured beta cells (33). Briefly, cells grown in T25 flasks were starved for 2 h in DMEM containing 1 mm glucose followed by a 30-min preincubation in the same media plus 0.5% fatty acid-free BSA. Cells were then incubated 2 h in the presence of different glucose concentrations (1, 8, and 15 mm), 0.3 mm palmitate or oleate pre-complexed to 0.5% BSA, 0.1 μCi/ml [1-14C]palmitate, or 0.1 μCi/ml [1-14C]oleate with or without 200 μm etomoxir, 25 μm Compound C (CpC, Calbiochem), or 1 mm 5-amino-1-β-d-ribofuranosyl-imidazole-4-carboxamide (AICAR, Toronto Research Chemicals). AICAR was dissolved in sterile water. 0.25% DMSO was used as a vehicle for CpC, and 0.25% DMSO was added in all the conditions. The flasks were sealed at the beginning of the incubation with a stopper containing a filter (Whatman GF/B paper) pre-soaked in 5% KOH. The incubation was stopped by the injection of 0.2 ml of 40% perchloric acid into each flask via a needle through the cap to acidify the medium and liberate the CO2. After overnight isotopic equilibration at room temperature, filters were removed, and the trapped 14CO2 and 14C acid-soluble products generated by the oxidation of [14C]palmitate or [14C]oleate were counted to calculate total palmitate and oleate oxidation. The results were normalized by cell protein content.
Glucose Utilization and Oxidation in Cultured Neurons and Astrocytes
Neurons and primary astrocytes grown in T25 flasks were starved in DMEM containing 1 mm glucose for 2 h and preincubated in DMEM containing 1 mm glucose containing 0.5% BSA during 30 min as described for LCFA oxidation. Cells were then incubated in the presence of different glucose concentrations (1, 8, and 15 mm), 0.3 mm palmitate pre-complexed to 0.5% BSA, d-[U-14C]glucose (0.2 μCi/ml), and d-[5-3H]glucose (0.5 μCi/ml). The reaction was stopped by the addition of 0.2 ml of 40% perchloric acid. 14CO2 generated was counted to estimate glucose oxidation, whereas 3H2O was counted to estimate glucose utilization. The results were normalized by cell protein content.
LCFA Oxidation and Glucose Utilization in Brain Slices, Muscle, and Liver
Because brain slices have to be constantly oxygenated, 14C-labeled oleate and palmitate tracers could not be used to measure LCFA oxidation via the release of 14CO2. Therefore, 3H tracers were employed to estimate the amount of 3H2O generated and released in the media. Cortical and MBH slices were preincubated in aCSF containing 1 mm glucose and 0.1% BSA for 30 min followed by a 2-h incubation with different concentrations of glucose (0.5, 1, 5, and 10 mm), 0.1 mm palmitate, or oleate pre-complexed to 0.13% BSA, [9,10(n)-3H]palmitate or [9,10(n)-3H]oleate (2 μCi/ml) or d-[5-3H]glucose (0.5 μCi/ml) with and without etomoxir (200 μm) or CpC (25 μm) at 37 °C under constant oxygenation with 95% O2, 5% CO2. 0.25% DMSO was used as a vehicle for CpC, and 0.25% DMSO was added under all conditions. Palmitate oxidation was measured in a 500-μm-thick liver section, and the entire soleus muscle was measured under similar conditions except that incubations were performed in Krebs-Ringer bicarbonate HEPES buffer. For glucose utilization measurements, slices were incubated at 1 or 10 mm glucose in the presence of 0.1 mm palmitate pre-complexed to 0.1% BSA. At the end of the incubation, the medium was collected and acidified with concentrated HCl (10% of volume). Samples were placed in scintillation vials containing cold water and incubated for 24 h at 50 °C under constant agitation. After equilibration, 3H2O was counted to calculate palmitate or oleate oxidation or glucose utilization. The results were normalized by slice protein content.
LCFA Esterification in Cells and Brain Slices
Oleate and palmitate esterification into neutral lipids was measured using 14C-labeled tracers and thin layer chromatography (TLC). Cells (neurons and astrocytes) grown in T25 flasks were starved for 2 h in DMEM containing 1 mm glucose followed by a preincubation of 30 min in the same media plus 0.5% BSA. Cells were then incubated during 2 h in the presence of different glucose concentrations (1 and 15 mm), 0.3 mm palmitate or oleate pre-complexed to 0.5% BSA, [1-14C]palmitate or [1-14C]oleate (0.1 μCi/ml). MBH slices were preincubated in aCSF containing 1 mm glucose for 30 min and then incubated for 2 h with different concentrations of glucose (1 and 10 mm), 0.1 mm palmitate or oleate pre-complexed to 0.13% BSA and [1-14C]palmitate or [1-14C]oleate (2 μCi/ml) at 37 °C under constant bubbling with 95% O2, 5% CO2.
At the end of the incubation cells and explants were collected, washed with cold PBS, and rapidly frozen in 0.5 ml (cells) or 0.2 ml (slices) of methanol:HCl (100:1) in liquid nitrogen. Total lipids were extracted using the Folch method. Briefly, samples were homogenized using a pestle and loaded into pre-chilled glass tubes containing 2 ml of chloroform then washed with 0.5 ml of methanol and HCl. Water with 0.9% NaCl was added, and samples were vigorously vortexed for 15 s and centrifuged at 900 × g for 15 min at 4 °C. After centrifugation, the lower phase (organic) was transferred into pre-chilled glass tubes and dried under N2. Each sample was suspended in 50 μl of chloroform and loaded on the TLC plates (Whatman). The samples were delivered by small drops, and 10 μl of the esterification mix was loaded to quantify total palmitate or oleate tracer radioactivity. Total lipids were separated using a solvent for neutral lipids (petroleum ether/ether/acetic acid; 70:30:1) for separation of total phospholipids from mono-, di-, and triacylglycerols. Plates were imaged using a phosphor screen (GE Healthcare) after 9 to 10 days of exposure, and the signal was quantified using a Typhoon scanner (GE Healthcare). Results were normalized by slice and cell protein content.
Treatment for Western Blot Analysis
Western blotting was performed to examine changes in AMPK (Thr-172) and ACC (Ser-79) phosphorylation in response to glucose, AICAR, or CpC in cultured cells or brain slices. Cells were starved in DMEM containing 1 mm glucose and incubated for 15 min in DMEM containing 1 or 15 mm glucose with or without CpC (25 μm) or AICAR (1 mm). AICAR was dissolved in sterile water. 0.25% DMSO was used as a vehicle for CpC, and 0.25% DMSO was added in all the conditions. Cortical and MBH slices were preincubated in aCSF containing 1 mm glucose and incubated in aCSF containing 1 or 10 mm glucose for 15 min. After washing with cold PBS, samples were lysed in ice-cold lysis buffer containing 1 m Tris-HCl, pH 7.5, 1 m NaCl, 400 mm Na2 EDTA, 100 mm EGTA, pH 7.5, Triton X-100, sodium pyrophosphate, 100 mm β-glycerophosphate, 100 mm Na3VO4, and 10 mg/ml leupeptin supplemented with 1% protease inhibitor mixture (Cell Signaling), 1 mm PMSF, and aprotonin. Total protein (20 μg) was resolved on a 7.5% acrylamide gel and transferred to a nitrocellulose membrane (Bio-Rad) blocked with 5% nonfat milk in Tris-buffered saline containing 0.2% Tween 20 for 1 h. Blots were then incubated with a P-AMPK or P-ACC primary antibody (1:1000, Cell Signaling) overnight at 4 °C followed by secondary antibody incubation (HRP-goat anti rabbit, 1:10,000, Bio-Rad) for 1 h at room temperature. The membranes were then stripped and incubated with an AMPK or ACC primary antibody (1:1000, Cell Signaling). Chemiluminescence (Western Lightning, PerkinElmer Life Sciences) was quantified on scanned films using densitometry.
Statistics
Data are expressed as the means ± S.E. Intergroup comparisons were performed by one-way ANOVA with Bonferroni post-tests or by Student's t test when comparing two conditions. p < 0.05 was considered significant.
RESULTS
Glucose and LCFA Oxidation Are Coupled via AMPK in Hypothalamic Neurons
GT1-7 and N46 hypothalamic neuronal cell lines were chosen for intracellular metabolism studies based on previous reports demonstrating “glucose-sensing” capacities (34–36). GT1-7 neurons mainly express the agouti-related protein (AgRP) neuropeptide, whereas N46 cells express both AgRP and NPY (Fig. 1A). Expression of pro-opiomelanocortin was extremely low compared with AgRP and NPY. The glucose transporter GLUT2 and glucokinase were not detected in GT1-7 and N46 neurons. However, key enzymes involved in malonyl-CoA metabolism, including ACC, malonyl-CoA decarboxylase (MCD) (the malonyl-CoA degrading enzyme), and fatty acid synthase, which catalyzes palmitate synthesis from malonyl-CoA, as well as enzymes of LCFA (CPT-1) and glucose (pyruvate dehydrogenase) oxidation (Fig. 1B) were expressed in GT1-7 and N46 neurons. Expression levels of CPT-1a were similar in GT1-7 and N46 neurons and were about 2-fold higher than CPT-1b. CPT-1c was the most abundant CPT-1 isoform in GT1-7 cells.
FIGURE 1.
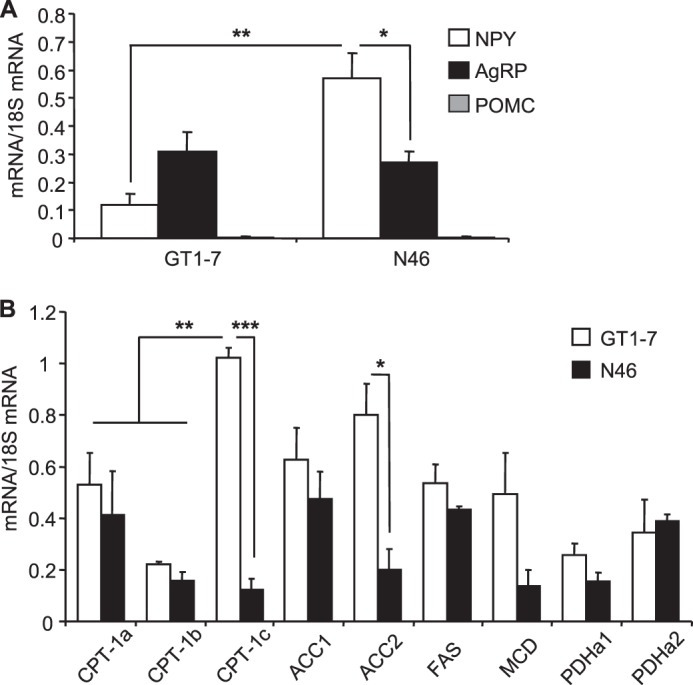
Neuropeptides and metabolic enzymes expression profiles in GT1-7 and N46 hypothalamic neurons. Neuropeptides (A) and enzymes (B) expression was determined by qPCR, and mRNA levels were normalized to 18 S levels. AgRP, NPY, pro-opiomelanocortin (POMC), CPT-1, ACC, fatty acid synthase (FAS), MCD, pyruvate dehydrogenase (PDH). Results are shown as means ± S.E. n = 3–5 performed in duplicate. Statistical analyses were performed with one-way ANOVA with Bonferroni post-tests. *, **, and ***, p < 0.05, 0.01, and 0.001, respectively.
Palmitate oxidation was measured in response to increasing glucose concentrations in both GT1-7 and N46 hypothalamic neurons. First, the CPT-1 inhibitor etomoxir significantly decreased palmitate oxidation by 38 and 70% in GT1-7 and N46 neurons, respectively (Fig. 2, A and D). Second, increasing glucose concentrations decreased palmitate oxidation in both hypothalamic cell lines (Fig. 2, A and D). However, the effect of glucose was more pronounced in N46 neurons and reached a maximum at 8 mm glucose compared with GT1-7 cells. To determine whether the effect of glucose was related to differences in glucose metabolism, its utilization and oxidation were measured in GT1-7 and N46 neurons. Glucose utilization and oxidation were increased by ∼2-fold in response to 8 and 15 mm glucose in N46 neurons (Fig. 2, E and F). Despite a similar trend, glucose utilization and oxidation were significantly increased by ∼1.6 and ∼1.4-fold, respectively, only at 15 mm glucose in GT1-7 neurons (Fig. 2, B and C). These results suggest that the higher rate of glucose metabolism in N46 neurons inhibits palmitate oxidation at lower glucose concentrations when compared with GT1-7 neurons. Altogether, these results show that palmitate oxidation is decreased in response to glucose in hypothalamic neurons.
FIGURE 2.

Glucose inhibits LCFA oxidation via AMPK in hypothalamic neuronal cell lines. Palmitate oxidation (A), glucose utilization (B), and oxidation (C) in response to increasing glucose concentrations or etomoxir (200 μm) in GT1-7 neurons is shown. Palmitate oxidation (D), glucose utilization (E), and oxidation (F) in response to increasing glucose concentrations or etomoxir (200 μm) in N46 neurons is shown. Western blot (G), quantitation of Thr-172 phospho-AMPK levels (H), and palmitate oxidation (I) in N46 neurons treated with glucose with or without CpC (25 μm) or AICAR (1 mm) is shown. Oleate oxidation (J) in response to glucose in N46 neurons is shown. Results are shown as the means ± S.E. n = 3–6 independent experiments with each condition performed in duplicate for intracellular metabolism measurements. n = 3–4 independent experiments for Western blot analyses. Statistical analyses were performed with one-way ANOVA with Bonferroni post-tests except for oleate oxidation (unpaired Student's t test). *, **, and ***, p < 0.05, 0.01, and 0.001, respectively versus 1 mm glucose. ##, p < 0.01 versus 15 mm glucose.
To investigate the involvement of AMPK in glucose inhibition of palmitate oxidation, N46 neurons were treated with or without the AMPK inhibitor, CpC (25 μm), at low glucose (1 mm) or in the presence or absence of AICAR, the AMPK activator (1 mm), at high glucose (15 mm). Western blot analysis revealed that AMPK phosphorylation was significantly decreased by CpC and high glucose compared with low glucose conditions, whereas AICAR blocked the effect of high glucose (Fig. 2, G and H). In line with the effect on AMPK activity, CpC decreased palmitate oxidation at low glucose, whereas AICAR prevented glucose inhibition of palmitate oxidation (Fig. 2I). These results demonstrate that the inhibition of palmitate oxidation by glucose is dependent on AMPK activity. The oxidation of the monounsaturated LCFA oleate was also measured in N46 neurons to assess potential difference between LCFA. Interestingly, basal oleate oxidation was significantly lower compared with palmitate (9.8 ± 0.6 versus 7.2 ± 0.1 nmol/mg/h, p < 0.01) but was inhibited by glucose similarly to palmitate (Fig. 2J).
Glucose Inhibits Palmitate Oxidation via AMPK in Hypothalamic but Not Cortical Astrocytes
Astrocytes are the most abundant cells in the central nervous system and are well known to oxidize LCFA (37). Therefore, we decided to investigate whether glucose regulates LCFA oxidation in astrocytes and whether there are potential differences in glucose action depending on the nature of the astrocytes. To this end we developed and validated mouse astrocyte cultures generated from cortices or hypothalami isolated from newborn mice (P1).
The purity of the culture was assessed by immunocytochemistry using glial fibrillary acidic protein as a marker for astrocytes. Our results showed that ∼88% of cells in both cortical and hypothalamic cultures were glial-fibrillary acidic protein-positive (Fig. 3A). We found that less than 1% of cells were Iba1 positive (microglial marker) and no staining for NeuN (neuronal marker) was observed (data not shown). The expression profile of key enzymes of LCFA metabolism was assessed in primary astrocytes by qPCR. GLUT2 and glucokinase mRNA were not detected in astrocyte cultures. CPT-1a and -1c were the main isoforms expressed in astrocytes with higher CPT-1c levels observed in hypothalamic astrocytes (Fig. 3B). CPT-1a, ACC1, MCD, fatty acid synthase, and pyruvate dehydrogenase (PDHa1 and -a2) expression levels were significantly higher in cortical versus hypothalamic astrocytes (Fig. 3B).
FIGURE 3.

Glial fibrillary acidic protein and metabolic enzymes expression profiles in hypothalamic and cortical astrocytes. A, Glial fibrillary acidic protein (GFAP) and DAPI immunostaining in cortical (left) and hypothalamic (right) astrocytes. B, enzyme expression was determined by qPCR, and mRNA levels were normalized to 18 S levels. CPT-1, ACC, fatty acid synthase (FAS), MCD, pyruvate dehydrogenase (PDH). Results are shown as the means ± S.E. n = 5 performed in duplicate. Statistical analyses were performed with one-way ANOVA with Bonferroni post-tests. *, **, and ***, p < 0.05, 0.01, and 0.001, respectively. ###, p < 0.001 versus CPT-1a and -1b.
Palmitate oxidation was measured in response to glucose in hypothalamic and cortical astrocytes as described for hypothalamic neurons. Oxidation rates were decreased by ∼75% in response to etomoxir in both astrocyte cultures (Fig. 4A). No differences were observed in basal palmitate oxidation between hypothalamic and cortical astrocytes. However, palmitate oxidation was inhibited by glucose specifically in hypothalamic astrocytes (Fig. 4A). Interestingly, the differential response to glucose was not related to differences in glucose utilization and oxidation in hypothalamic versus cortical astrocytes (Fig. 4, B and C). To determine the implication of AMPK in glucose inhibition of palmitate oxidation, cortical and hypothalamic astrocytes were treated with AICAR in presence of high glucose (15 mm). Western blot analysis revealed that glucose decreased P-AMPK and P-ACC in hypothalamic astrocytes compared with low glucose conditions, an effect that was prevented by AICAR (Fig. 4, D and E). In cortical astrocytes, P-AMPK and P-ACC levels remained unchanged in response to glucose. In contrast, AICAR increased both P-AMPK and P-ACC in cortical astrocytes (Fig. 4, D and E). The inhibitory effect of glucose on palmitate oxidation was prevented by AICAR treatment in hypothalamic astrocytes, whereas AICAR had no effect on palmitate oxidation in cortical astrocytes (Fig. 4F).
FIGURE 4.

Glucose decreases palmitate oxidation via AMPK in hypothalamic but not cortical astrocytes. Palmitate oxidation (A), glucose utilization (B), and oxidation (C) in response to glucose or etomoxir (200 μm) in hypothalamic and cortical astrocyte cultures are shown. Western blot (D) and quantitation of Thr-172 P-AMPK and Ser79 P-ACC levels (E) in hypothalamic and cortical astrocytes are shown. Palmitate oxidation (F) in response to glucose with or without AICAR (1 mm) in hypothalamic and cortical astrocytes is shown. Results are shown as the means ± S.E. n = 4–7 independent experiments with each condition performed in duplicate for intracellular metabolism measurements. n = 3 independent experiment for Western blot analyses. Statistical analyses were performed with one-way ANOVA with Bonferroni post-tests. *, **, and ***, p < 0.05, 0.01, and 0.001 respectively versus 1 mm glucose.
Glucose and Palmitate Oxidation Are Coupled in Hypothalamic but Not Cortical Slices
The metabolic coupling between glucose and LCFA was investigated in a physiological model of acute brain slices ex vivo. LCFA oxidation was measured in MBH (consisting of the arcuate nucleus plus the ventro- and dorso- median-hypothalamus) and cortical slices from male Wistar rats using palmitate and oleate radioactive tracers. The viability of MBH slices was tested by measuring NPY secretion in response to KCl after incubation in the oxidation media. KCl induced an ∼5-fold increase in NPY secretion, indicating that the MBH slices were viable (Fig. 5A). Notably, we found that the palmitate oxidation rate was significantly higher in the MBH compared with the cortex, liver, and soleus muscle (Fig. 5, B and C). In addition, palmitate oxidation in the MBH and cortex was significantly decreased by ∼90% in response to etomoxir, indicating that palmitate oxidation is mainly CPT-1-dependent (Fig. 5C). Interestingly, palmitate oxidation was significantly decreased in response to glucose in the MBH, whereas oxidation remained unchanged with increasing glucose concentrations in cortical slices (Fig. 5, D and E). Glucose utilization was significantly higher in MBH compared with cortical slices at low glucose (Fig. 5F). However, the -fold increase in glucose utilization in response to glucose (∼ 2-fold) was similar in both areas. In line with the effect of glucose on palmitate oxidation in the MBH, we found that phosphorylation of AMPK was significantly decreased by glucose in the MBH, whereas it remained unchanged in cortical slices (Fig. 5, G and H). We also investigated whether glucose affects oleate oxidation in MBH slices. Basal oleate oxidation at 1 mm glucose was similar to palmitate. However, in contrast to palmitate, glucose had no effect on oleate oxidation (Fig. 5I), suggesting that its oxidation is not regulated by glucose and AMPK. To address this question, palmitate and oleate oxidation were measured at low glucose with and without CpC. Although CpC significantly decreased palmitate oxidation, oleate oxidation remained unchanged (Fig. 5J), supporting the idea that oleate oxidation is independent of AMPK activity. Importantly, oleate oxidation was not altered by etomoxir, suggesting that its oxidation is CPT-1-independent (Fig. 5K).
FIGURE 5.

Glucose inhibits palmitate but not oleate oxidation in hypothalamic slices. A, basal and KCl-stimulated NPY secretion in MBH slices. B, palmitate oxidation in MBH slices and peripheral tissues. C, palmitate oxidation in MBH and cortical slices with or without etomoxir (200 μm). Palmitate oxidation in MBH (D) and cortical (E) slices in response to increasing concentrations of glucose are shown. F, glucose utilization in MBH and cortical slices. Western blot (G) and quantitation of Thr-172 phospho-AMPK levels (H) in response to glucose in MBH and cortical slices. Oleate oxidation (I) in response to glucose in MBH slices is shown. J, palmitate and oleate oxidation in MBH slices at 1 mm glucose treated or not with CpC (25 μm). K, oleate oxidation in MBH slices at 1 mm glucose ± etomoxir (200 μm). Results are shown as the means ± S.E. n = 4–5 per condition for Western blots, 5–6 per condition for glucose utilization, and 8–14 animals per condition for palmitate and oleate oxidation. Statistical analyses were performed with one-way ANOVA with Bonferroni post-tests except for oleate oxidation and quantitation of AMPK phosphorylation (unpaired Student's t test). *, **, and ***, p < 0.05, 0.01, and 0.001, respectively versus 0.5 and 1 mm glucose or vehicle. $$, p < 0.01 versus MBH.
Glucose Increases Palmitate but Not Oleate Esterification in N46 Neurons and Slices
To determine whether inhibition of LCFA oxidation translates into increased LCFA esterification, the incorporation of LCFA into neutral lipids was measured in N46 neurons, hypothalamic astrocytes cultures, and MBH slices. We found that in the three models, palmitate and oleate were mainly esterified in triacylglycerol (TAG) and phospholipids (PL) (Fig. 6). Under basal conditions, oleate esterification in TAG and PL was significantly higher compared with palmitate, whereas its esterification in diacylglycerol (DAG) was reduced in comparison to palmitate in neurons and slices (Fig. 6, A and C). Oleate esterification was not affected by glucose, whereas palmitate esterification in DAG and TAG was increased in response to glucose (Fig. 6, A and C). Interestingly, glucose did not affect palmitate esterification in hypothalamic astrocytes (Fig. 6B).
FIGURE 6.

Glucose regulation of hypothalamic LCFA esterification into neutral lipids. LCFA esterification into monoacylglycerol (MAG), DAG, TAG, non-esterified fatty acid (NEFA), and PLs. Esterification of palmitate (Pal) and/or oleate (Ole) in N46 neurons (A), hypothalamic astrocytes (B), and MBH slices (C) in response to glucose. Results are shown as the means ± S.E. n = 4–6 independent experiments with each condition performed in duplicate for esterification in cultured cells and n = 11–14 animals per condition for esterification in slices. Statistical analyses were performed with one-way ANOVA with Bonferroni post-tests. * and ***, p < 0.05 and 0.001, respectively versus 1 mm glucose. $, $$, and $$$, p < 0.05, 0.01 and 0.001, respectively, versus palmitate.
In line with increased palmitate esterification in DAG and TAG in response to glucose in neuronal cells, we found that N46 neurons expressed different isoforms of enzymes involved in TAG biosynthesis through the glycerol phosphate pathway (38) including glycerol-3-phosphate acyltransferase 1 but not glycerol-3-phosphate acyltransferase 2, 1-acylglycerol-3-phosphate acyltransferase 1–8 (AGPAT), and diacylglycerol acyltransferase 1 and 2. Interestingly, the expression of AGPAT 1, 5, 7, and 8 and glycerol-3-phosphate acyltransferase 1 was significantly increased in response to high glucose (Fig. 7).
FIGURE 7.
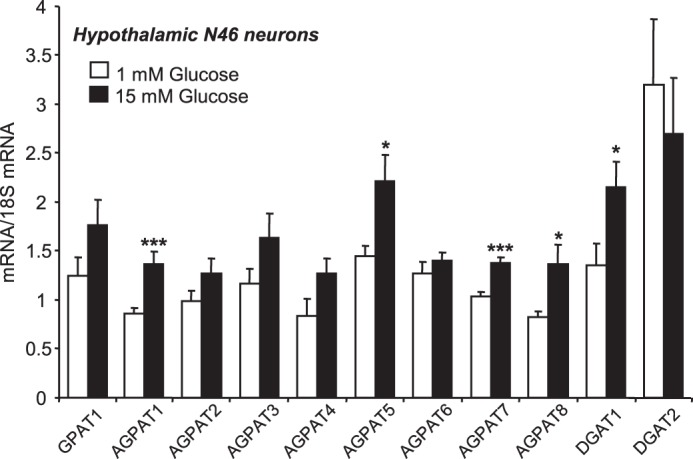
Glucose increases the expression of esterification enzymes in N46 neurons. Gene expression was determined by qPCR in N46 neurons after 24 h of exposure to 1 or 15 mm glucose. mRNA levels were normalized to 18 S levels. GPAT, glycerol-3-phosphate acyltransferase; AGPAT, 1-acylglycerol-3-phosphate acyltransferase; DGAT, diacylglycerol acyltransferase. Results presented are the means ± S.E. from three separate studies performed in duplicate. Statistical analyses were performed with an unpaired Student's t test. * and ***, p < 0.05 and 0.001, respectively, versus 1 mm glucose.
DISCUSSION
The main objectives of this study were to examine whether glucose regulates LCFA intracellular metabolism via AMPK-dependent mechanisms and assess potential differences in LCFA metabolism and its regulation by glucose in different brain areas, cell types, and for different LCFA. Using complementary models, we demonstrate for the first time that glucose and palmitate metabolism is coupled in hypothalamic neuronal cell lines, primary astrocytes, and slices. The inhibition of palmitate oxidation by glucose resulted in increased palmitate esterification into neutral lipids in neurons and MBH slices. Glucose inhibition of palmitate oxidation in hypothalamic neurons and astrocytes was prevented by AMPK activation, whereas AMPK inhibition decreased palmitate oxidation at low glucose. Taken together, these results establish the existence of an AMPK-dependent coupling between glucose and palmitate metabolism in the hypothalamus.
Importantly, the effect of glucose was specific to the hypothalamus as phosphorylation of AMPK and palmitate oxidation in cortical astrocyte cultures or slices remained unchanged in response to glucose. The absence of glucose effect on cortical P-AMPK is in agreement with previous reports showing that high glucose, hypoglycemia, or neuroglucopenia does not modulate AMPK activity in cortical areas (12, 39, 40). Interestingly, the lack of response to glucose was not related to a lower capacity to metabolize glucose as the -fold increase in glucose utilization and oxidation rates in response to glucose were similar in hypothalamic versus cortical astrocytes and in MBH versus cortical slices. This suggests a dissociation between glucose metabolism and AMPK activity that may involve differences in ATP generation and/or in the expression of the α1-AMPK isoform in cortical versus hypothalamic regions that is less sensitive to glucose compared with the α2 isoform (12, 39, 41). Although it is difficult to speculate on mRNA levels, there is also a possibility that the increased expression of fatty acid synthase and MCD in cortical astrocytes translates into lower levels of malonyl-CoA generated in response to glucose in cortical versus hypothalamic astrocytes. Additional studies aimed at measuring glucose-derived metabolites in astrocytes will be required to address this hypothesis.
To our knowledge this is the first report showing that glucose regulates P-AMPK and LCFA oxidation in primary hypothalamic but not cortical astrocytes, suggesting that hypothalamic astrocytes, similarly to hypothalamic neurons, have glucose sensing properties. These findings are in line with growing literature showing that hypothalamic glial cells are involved in central glucose sensing and action (25–29). It is important to mention that we did not observe an increase in palmitate oxidation in response to AICAR in cortical astrocytes. These results are in contrast to a study by Blázquez et al. (42) showing that AICAR increases ketogenesis in cortical astrocytes, suggesting that AMPK activation increases palmitate oxidation. The reason for this discrepancy is unclear but could involve differences in animal species (rat versus mouse in the current study), conditions of culture and treatment, and/or the possibility that palmitate oxidation is already maximal at low glucose and cannot be further enhanced by AICAR in our culture model. This hypothesis is supported by the fact that despite increased P-AMPK and P-ACC in response to AICAR, palmitate oxidation remained unchanged in cortical astrocytes.
Our study also highlights important differences in palmitate oxidation rates between hypothalamic neurons versus astrocytes as well as MBH versus cortical slices. In agreement with previous reports suggesting that LCFA oxidation in the brain mainly occurs in astrocytes (37, 43), we found that palmitate oxidation is significantly higher in astrocytes compared with GT1-7 and N46 neurons. Interestingly, despite higher CPT-1a expression levels in cortical astrocytes, the palmitate oxidation rate was similar in hypothalamic and cortical astrocytes and was decreased to the same extent by etomoxir, suggesting a similar oxidative capacity at low glucose concentrations. In agreement with the literature (16), we found that CPT-1a and -1c were the most abundant CPT-1 isoforms expressed in brain cells. However, the expression of CPT-1c in cultured astrocytes is not consistent with earlier studies showing a neuronal localization in the adult mouse brain (44, 45). This discrepancy could be related to the age of the animals used to generate astrocytes (P1) and/or the culture conditions.
In contrast to CPT-1a, the biochemical function of CPT-1c is not well defined. The initial studies concluded that CPT-1c did not have the prototypical acyltransferase activity (46, 47), but a later study identified a weak palmitoyl-CoA transferase activity in the endoplasmic reticulum of neurons (45), suggesting that CPT-1c is not involved in LCFA oxidation. This hypothesis is supported by recent results showing that CPT-1c promotes ceramide biosynthesis in the hypothalamus (48, 49). These data and the expression profile of CPT-1 isoforms in our models strongly suggest that LCFA oxidation in hypothalamic cells is mainly dependent on CPT-1a.
In tissue preparations ex vivo, palmitate oxidation was about 2- and 3-fold higher in the MBH compared with the liver and soleus muscle, respectively. These results are consistent with previous ex vivo studies showing higher palmitate oxidation rates in hypothalamic areas compared with the liver (50, 51).
In addition, in slice preparation, palmitate oxidation was about 3-fold higher in the MBH compared with cortical slices, suggesting a higher LCFA oxidative capacity. In line with this finding, we observed a similar difference in palmitate oxidation between MBH and cortical slices obtained from mouse brain (data not shown). Although the reason for this difference is unknown, this could be related to a lower number of astrocytes in cortical versus hypothalamic areas (52) and/or differences in the expression of proteins and enzymes involved in LCFA metabolism (LCFA transporters, acyl-CoA synthase, and CPT-1) that may affect LCFA uptake. Nonetheless, our results are consistent with previous reports showing regional differences in palmitate oxidation and incorporation in rat (51, 53) or human brain (54) and high levels of palmitate incorporation in the arcuate nucleus compared with non-hypothalamic areas (53) after systemic administration of radiolabeled palmitate tracers. In addition, in agreement with the former study in vivo (53), we found here that palmitate oxidation rate in MBH and cortical slices was proportional to the rate of glucose utilization.
One limitation of our study is that malonyl-CoA levels were not measured in the different models in response to glucose. However, the decreased AMPK phosphorylation (in N46, hypothalamic astrocytes, and slices) and the concomitant decrease in ACC phosphorylation in GT1-7 neurons (data not shown) and hypothalamic astrocytes in response to glucose strongly support that inhibition of palmitate oxidation is mediated via inhibition of CPT-1 by malonyl-CoA.
The degree of saturation and carbon chain length of LCFA are known to affect their metabolic fate; thus, we hypothesized that oleate metabolism might be different compared with palmitate. In N46 neurons we found that oleate oxidation at low glucose was significantly lower than palmitate oxidation. However, oleate oxidation was inhibited by glucose similarly to palmitate. These findings were not replicated in MBH slices where basal oleate oxidation was similar to palmitate oxidation and remained unchanged in response to glucose, suggesting that oleate oxidation is not regulated by glucose and AMPK in MBH slices. This hypothesis was confirmed by experiments showing that palmitate but not oleate oxidation was significantly decreased by CpC in MBH slices. Importantly, oleate oxidation remained unchanged in response to CPT-1 inhibition by etomoxir. This strongly suggests that palmitate oxidation is mainly mitochondrial and CPT-1-dependent, whereas oleate is preferentially oxidized in peroxisomes where oxidation is CPT-1-independent (55) and, therefore, not altered by changes in glucose concentration or AMPK activity.
All together our results demonstrate that palmitate oxidation rate is inhibited by glucose in all the models tested, whereas oleate oxidation was not affected by glucose in slices. To get further insights on the effect of glucose on LCFA partitioning, we measured LCFA esterification into neutral lipids. Our results show that oleate esterification into neutral lipids was not significantly affected by glucose. In contrast, glucose increased palmitate esterification into DAG and TAG in both N46 neurons and slices as well as esterification into PL in cultured neurons. In agreement with these results, we found several isoforms of enzymes involved in DAG and TAG biosynthesis expressed in N46 neurons and up-regulated in response to glucose as previously described in adipocytes and hepatocytes (56, 57).
In hypothalamic astrocytes, despite a trend toward increased palmitate esterification into DAG in response to glucose (p = 0.07), monoacylglycerol, TAG, and PL remained unchanged. Interestingly, glycerol-3-phosphate acyltransferase 1 and all the AGPAT and diacylglycerol acyltransferase isoforms detected in N46 neurons were also expressed in hypothalamic astrocytes (data not shown), suggesting that the lack of glucose effect on DAG and TAG esterification in astrocytes is not related to differences in enzyme expression.
Combined with results showing high palmitate oxidation rates in astrocytes, this suggests that LCFA metabolism is mainly channeled toward mitochondrial oxidation in astrocytes. However, because it was not measured, we cannot rule out that oleate esterification and/or oxidation is differently regulated by glucose when compared with palmitate in astrocytes.
Our findings demonstrate that glucose-induced LCFA esterification is dependent on both LCFA and cell type. To our knowledge, this is the first study showing that glucose acutely increases LCFA esterification into DAG and TAG in hypothalamic neurons and slices. This suggests that chronic hyperglycemia and/or hyperlipidemia could lead to accumulation of lipids that may have deleterious effects on insulin sensitivity. This hypothesis is supported by recent studies showing that high fat feeding increases hypothalamic DAG and TAG content in mice (58) and that palmitate induces insulin resistance in the arcuate nucleus (59) and in N44 hypothalamic neurons (60). Interestingly, activation of AMPK by AICAR prevented the deleterious effect of palmitate on insulin signaling in N44 neurons (45). In light of our findings in a similar neuronal cell line, it is tempting to speculate that AMPK activation might increase palmitate oxidation while inhibiting intracellular accumulation of lipids, thereby protecting neurons from palmitate-induced lipotoxicity as already demonstrated in pancreatic beta cells (61).
An important question related to our ex vivo results in slices is whether they are representative of in vivo conditions. The range of glucose concentration was chosen based on extracellular brain glucose concentrations measured in vivo, from 0.5 mm under hypoglycemic conditions to ∼5 mm in the postprandial state (62) and on previous electrophysiology reports studying glucose sensing on brain slices (63, 64). Therefore, we believe that the results described in slices may reflect in vivo conditions. In contrast to glucose, extracellular concentrations of LCFA in the brain are unknown. Thus, LCFA concentrations were chosen within the physiological range of circulating LCFA levels (0.1 mm). Importantly, LCFA were always pre-complexed to albumin with a LCFA/albumin molar ratio of 1:5. Nonetheless, this concentration might be higher compared with central LCFA level and, therefore, have confounding effects on intracellular metabolism.
Taken together, our findings have important implications. First, they suggest that the dissociation observed between AMPK, malonyl-CoA, and LCFA-CoA levels in previous reports (21–23) might be explained by differences in LCFA-CoA metabolism in neurons versus astrocytes and/or differences in the type of LCFA. In line with this idea, it was shown that central leptin administration increases palmitoyl-CoA level without affecting oleyl-CoA level in the paraventricular nucleus of the hypothalamus (21). In addition, they suggest that the previously described effect of MCD overexpression in the MBH, the malonyl-CoA-degrading enzyme, leading to increased food intake and adiposity (17, 18) may involve modulation of malonyl-CoA and LCFA-CoA oxidation in astrocytes and/or neurons. Our data showing a high LCFA oxidative capacity in astrocyte cultures compared with neurons support the idea of a cellular coupling between astrocytes and neurons or the so called “metabolic-sensing unit” according to which changes in LCFA oxidation in astrocytes may generate metabolic signals such as ketone bodies that in turn modulate MBH neurons activity (65). Clearly, further investigations aimed at modulating MCD or CPT-1 activity specifically in neuronal subpopulations or astrocytes are required to assess their respective contribution in LCFA metabolism and action in the MBH in vivo.
Second, they support the concept that central LCFA action and sensing is dependent on the nutritional status and/or glucose concentrations. Indeed, central LCFA anorexigenic action and capacity to decrease glucose production are lost after 3 days of overfeeding (4, 66). At the cellular level, studies have established that the effect of oleate on neuronal activity is dependent on extracellular glucose concentrations in primary cultures of ventromedial nucleus neurons (67) and in arcuate neurons in brain slices (64). However, in the current study, glucose did not affect oleate oxidation and esterification in MBH slices, suggesting that the effect of glucose on oleate action is independent of a metabolic coupling between glucose and oleate. In agreement with this idea, etomoxir had a minor effect on oleate-induced calcium mobilization in ventromedial nucleus neurons (67). Thus, how glucose affects oleate-sensing in MBH neurons is still unclear, and further studies are needed to assess whether glucose modulates LCFA metabolism and action in the MBH.
Third, our results provide a metabolic basis for the different effect of centrally administered LCFA on feeding (68, 69). Indeed, oleate but not palmitate has an anorectic action making it tempting to speculate that such differences could be related to the difference between palmitate and oleate metabolism observed in our study.
To summarize, this is the first study investigating the partitioning of saturated (palmitate) and monounsaturated (oleate) LCFA between oxidation and esterification in response to glucose in the MBH. Our results demonstrate that glucose regulates palmitate metabolism via AMPK in hypothalamic but not cortical areas. In contrast, the metabolism of the monounsaturated LCFA oleate was not responsive to glucose, suggesting that the metabolic coupling is dependent on the type of LCFA. Finally, they support the role of hypothalamic astrocytes in central glucose and LCFA action. Further studies are needed to determine the functional role of the metabolic coupling between glucose and LCFA in the control of energy balance by the hypothalamus and in the etiology of obesity and type 2 diabetes.
Acknowledgments
We thank Dr. Mireille Bélanger for precious help setting up the astrocyte cultures. We thank Drs. Vincent Poitout, Marc Prentki, Murthy Madiraju, and Xavier Fioramonti for critical discussions and reading of the manuscript and Dr. Pamela Mellon for providing the GT1-7 neuronal cell line.
This work was supported by grants from the Canadian Institutes of Health Research (MOP115042), Fonds de Recherche Quebec-Sante, Societe Francophone du Diabete, Diabete Quebec, and Canadian Foundation for Innovation (to T. A.).
- LCFA
- long-chain fatty acid
- AMPK
- AMP-activated kinase
- MBH
- mediobasal hypothalamus
- ACC
- acetyl-CoA carboxylase
- CPT
- carnitine palmitoyl transferase
- aCSF
- artificial cerebrospinal fluid
- NPY
- neuropeptide Y
- CpC
- Compound C
- AICAR
- 5-amino-1-β-d-ribofuranosyl-imidazole-4-carboxamide
- MCD
- malonyl-CoA decarboxylase
- TAG
- triacylglycerol
- PL
- phospholipid
- DAG
- diacylglycerol
- ANOVA
- analysis of variance
- AgRP
- agouti-related protein
- AGPAT
- 1-acylglycerol-3-phosphate acyltransferase.
REFERENCES
- 1. Morton G. J., Cummings D. E., Baskin D. G., Barsh G. S., Schwartz M. W. (2006) Central nervous system control of food intake and body weight. Nature 443, 289–295 [DOI] [PubMed] [Google Scholar]
- 2. Cruciani-Guglielmacci C., Hervalet A., Douared L., Sanders N. M., Levin B. E., Ktorza A., Magnan C. (2004) Beta oxidation in the brain is required for the effects of non-esterified fatty acids on glucose-induced insulin secretion in rats. Diabetologia 47, 2032–2038 [DOI] [PubMed] [Google Scholar]
- 3. Lam T. K., Pocai A., Gutierrez-Juarez R., Obici S., Bryan J., Aguilar-Bryan L., Schwartz G. J., Rossetti L. (2005) Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat. Med. 11, 320–327 [DOI] [PubMed] [Google Scholar]
- 4. Pocai A., Lam T. K., Obici S., Gutierrez-Juarez R., Muse E. D., Arduini A., Rossetti L. (2006) Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J. Clin. Invest. 116, 1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benani A., Troy S., Carmona M. C., Fioramonti X., Lorsignol A., Leloup C., Casteilla L., Pénicaud L. (2007) Role for mitochondrial reactive oxygen species in brain lipid sensing. Redox regulation of food intake. Diabetes 56, 152–160 [DOI] [PubMed] [Google Scholar]
- 6. Jordan S. D., Könner A. C., Brüning J. C. (2010) Sensing the fuels. Glucose and lipid signaling in the CNS controlling energy homeostasis. Cell. Mol. Life Sci. 67, 3255–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Prentki M., Matschinsky F. M., Madiraju S. R. (2013) Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 18, 162–185 [DOI] [PubMed] [Google Scholar]
- 8. Ruderman N. B., Saha A. K., Kraegen E. W. (2003) Minireview. Malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology 144, 5166–5171 [DOI] [PubMed] [Google Scholar]
- 9. Claret M., Smith M. A., Batterham R. L., Selman C., Choudhury A. I., Fryer L. G., Clements M., Al-Qassab H., Heffron H., Xu A. W., Speakman J. R., Barsh G. S., Viollet B., Vaulont S., Ashford M. L., Carling D., Withers D. J. (2007) AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J. Clin. Invest. 117, 2325–2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murphy B. A., Fakira K. A., Song Z., Beuve A., Routh V. H. (2009) AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am. J. Physiol. Cell Physiol. 297, C750–C758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang C. S., Lam C. K., Chari M., Cheung G. W., Kokorovic A., Gao S., Leclerc I., Rutter G. A., Lam T. K. (2010) Hypothalamic AMP-activated protein kinase regulates glucose production. Diabetes 59, 2435–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alquier T., Kawashima J., Tsuji Y., Kahn B. B. (2007) Role of hypothalamic adenosine 5′-monophosphate-activated protein kinase in the impaired counterregulatory response induced by repetitive neuroglucopenia. Endocrinology 148, 1367–1375 [DOI] [PubMed] [Google Scholar]
- 13. McCrimmon R. J., Shaw M., Fan X., Cheng H., Ding Y., Vella M. C., Zhou L., McNay E. C., Sherwin R. S. (2008) Key role for AMP-activated protein kinase in the ventromedial hypothalamus in regulating counterregulatory hormone responses to acute hypoglycemia. Diabetes 57, 444–450 [DOI] [PubMed] [Google Scholar]
- 14. Hu Z., Cha S. H., Chohnan S., Lane M. D. (2003) Hypothalamic malonyl-CoA as a mediator of feeding behavior. Proc. Natl. Acad. Sci. U.S.A. 100, 12624–12629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wolfgang M. J., Cha S. H., Sidhaye A., Chohnan S., Cline G., Shulman G. I., Lane M. D. (2007) Regulation of hypothalamic malonyl-CoA by central glucose and leptin. Proc. Natl. Acad. Sci. U.S.A. 104, 19285–19290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao S., Moran T. H., Lopaschuk G. D., Butler A. A. (2013) Hypothalamic malonyl-CoA and the control of food intake. Physiol. Behav. 122C, 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu Z., Dai Y., Prentki M., Chohnan S., Lane M. D. (2005) A role for hypothalamic malonyl-CoA in the control of food intake. J. Biol. Chem. 280, 39681–39683 [DOI] [PubMed] [Google Scholar]
- 18. He W., Lam T. K., Obici S., Rossetti L. (2006) Molecular disruption of hypothalamic nutrient sensing induces obesity. Nat. Neurosci. 9, 227–233 [DOI] [PubMed] [Google Scholar]
- 19. Andrews Z. B., Liu Z. W., Walllingford N., Erion D. M., Borok E., Friedman J. M., Tschöp M. H., Shanabrough M., Cline G., Shulman G. I., Coppola A., Gao X. B., Horvath T. L., Diano S. (2008) UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature 454, 846–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. López M., Lage R., Saha A. K., Pérez-Tilve D., Vázquez M. J., Varela L., Sangiao-Alvarellos S., Tovar S., Raghay K., Rodríguez-Cuenca S., Deoliveira R. M., Castañeda T., Datta R., Dong J. Z., Culler M., Sleeman M. W., Alvarez C. V., Gallego R., Lelliott C. J., Carling D., Tschöp M. H., Diéguez C., Vidal-Puig A. (2008) Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab. 7, 389–399 [DOI] [PubMed] [Google Scholar]
- 21. Gao S., Kinzig K. P., Aja S., Scott K. A., Keung W., Kelly S., Strynadka K., Chohnan S., Smith W. W., Tamashiro K. L., Ladenheim E. E., Ronnett G. V., Tu Y., Birnbaum M. J., Lopaschuk G. D., Moran T. H. (2007) Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc. Natl. Acad. Sci. U.S.A. 104, 17358–17363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gao S., Keung W., Serra D., Wang W., Carrasco P., Casals N., Hegardt F. G., Moran T. H., Lopaschuk G. D. (2011) Malonyl-CoA mediates leptin hypothalamic control of feeding independent of inhibition of CPT-1a. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R209–R217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao S., Casals N., Keung W., Moran T. H., Lopaschuk G. D. (2013) Differential effects of central ghrelin on fatty acid metabolism in hypothalamic ventral medial and arcuate nuclei. Physiol. Behav. 118, 165–170 [DOI] [PubMed] [Google Scholar]
- 24. Mountjoy P. D., Rutter G. A. (2007) Glucose sensing by hypothalamic neurones and pancreatic islet cells. AMPle evidence for common mechanisms? Exp. Physiol. 92, 311–319 [DOI] [PubMed] [Google Scholar]
- 25. Guillod-Maximin E., Lorsignol A., Alquier T., Pénicaud L. (2004) Acute intracarotid glucose injection towards the brain induces specific c-fos activation in hypothalamic nuclei. Involvement of astrocytes in cerebral glucose-sensing in rats. J. Neuroendocrinol. 16, 464–471 [DOI] [PubMed] [Google Scholar]
- 26. Lam T. K., Gutierrez-Juarez R., Pocai A., Rossetti L. (2005) Regulation of blood glucose by hypothalamic pyruvate metabolism. Science 309, 943–947 [DOI] [PubMed] [Google Scholar]
- 27. Marty N., Dallaporta M., Foretz M., Emery M., Tarussio D., Bady I., Binnert C., Beermann F., Thorens B. (2005) Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J. Clin. Invest. 115, 3545–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Orellana J. A., Sáez P. J., Cortés-Campos C., Elizondo R. J., Shoji K. F., Contreras-Duarte S., Figueroa V., Velarde V., Jiang J. X., Nualart F., Sáez J. C., García M. A. (2012) Glucose increases intracellular free Ca2+ in tanycytes via ATP released through connexin 43 hemichannels. Glia 60, 53–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lanfray D., Arthaud S., Ouellet J., Compère V., Do Rego J. L., Leprince J., Lefranc B., Castel H., Bouchard C., Monge-Roffarello B., Richard D., Pelletier G., Vaudry H., Tonon M. C., Morin F. (2013) Gliotransmission and brain glucose sensing. Critical role of endozepines. Diabetes 62, 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yi C. X., Habegger K. M., Chowen J. A., Stern J., Tschöp M. H. (2011) A role for astrocytes in the central control of metabolism. Neuroendocrinology 93, 143–149 [DOI] [PubMed] [Google Scholar]
- 31. Gavillet M., Allaman I., Magistretti P. J. (2008) Modulation of astrocytic metabolic phenotype by proinflammatory cytokines. Glia 56, 975–989 [DOI] [PubMed] [Google Scholar]
- 32. Kawashima J., Alquier T., Tsuji Y., Peroni O. D., Kahn B. B. (2012) Ca2+/calmodulin-dependent protein kinase kinase is not involved in hypothalamic AMP-activated protein kinase activation by neuroglucopenia. PloS ONE 7, e36335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roduit R., Nolan C., Alarcon C., Moore P., Barbeau A., Delghingaro-Augusto V., Przybykowski E., Morin J., Massé F., Massie B., Ruderman N., Rhodes C., Poitout V., Prentki M. (2004) A role for the malonyl-CoA/long-chain acyl-CoA pathway of lipid signaling in the regulation of insulin secretion in response to both fuel and nonfuel stimuli. Diabetes 53, 1007–1019 [DOI] [PubMed] [Google Scholar]
- 34. Sanz C., Roncero I., Vázquez P., Navas M. A., Blázquez E. (2007) Effects of glucose and insulin on glucokinase activity in rat hypothalamus. J. Endocrinol. 193, 259–267 [DOI] [PubMed] [Google Scholar]
- 35. Madadi G., Dalvi P. S., Belsham D. D. (2008) Regulation of brain insulin mRNA by glucose and glucagon-like peptide 1. Biochem. Biophys. Res. Commun. 376, 694–699 [DOI] [PubMed] [Google Scholar]
- 36. Beall C., Hamilton D. L., Gallagher J., Logie L., Wright K., Soutar M. P., Dadak S., Ashford F. B., Haythorne E., Du Q., Jovanović A., McCrimmon R. J., Ashford M. L. (2012) Mouse hypothalamic GT1-7 cells demonstrate AMP-dependent intrinsic glucose-sensing behaviour. Diabetologia 55, 2432–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Edmond J. (1992) Energy metabolism in developing brain cells. Can. J. Physiol. Pharmacol. 70, S118–S129 [DOI] [PubMed] [Google Scholar]
- 38. Takeuchi K., Reue K. (2009) Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am. J. Physiol. Endocrinol. Metab. 296, E1195–E1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Minokoshi Y., Alquier T., Furukawa N., Kim Y. B., Lee A., Xue B., Mu J., Foufelle F., Ferré P., Birnbaum M. J., Stuck B. J., Kahn B. B. (2004) AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428, 569–574 [DOI] [PubMed] [Google Scholar]
- 40. Han S. M., Namkoong C., Jang P. G., Park I. S., Hong S. W., Katakami H., Chun S., Kim S. W., Park J. Y., Lee K. U., Kim M. S. (2005) Hypothalamic AMP-activated protein kinase mediates counter-regulatory responses to hypoglycaemia in rats. Diabetologia 48, 2170–2178 [DOI] [PubMed] [Google Scholar]
- 41. Kim M. S., Park J. Y., Namkoong C., Jang P. G., Ryu J. W., Song H. S., Yun J. Y., Namgoong I. S., Ha J., Park I. S., Lee I. K., Viollet B., Youn J. H., Lee H. K., Lee K. U. (2004) Anti-obesity effects of α-lipoic acid mediated by suppression of hypothalamic AMP-activated protein kinase. Nat. Med. 10, 727–733 [DOI] [PubMed] [Google Scholar]
- 42. Blázquez C., Woods A., de Ceballos M. L., Carling D., Guzmán M. (1999) The AMP-activated protein kinase is involved in the regulation of ketone body production by astrocytes. J. Neurochem. 73, 1674–1682 [DOI] [PubMed] [Google Scholar]
- 43. Edmond J., Robbins R. A., Bergstrom J. D., Cole R. A., de Vellis J. (1987) Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 18, 551–561 [DOI] [PubMed] [Google Scholar]
- 44. Dai Y., Wolfgang M. J., Cha S. H., Lane M. D. (2007) Localization and effect of ectopic expression of CPT1c in CNS feeding centers. Biochem. Biophys. Res. Commun. 359, 469–474 [DOI] [PubMed] [Google Scholar]
- 45. Sierra A. Y., Gratacós E., Carrasco P., Clotet J., Ureña J., Serra D., Asins G., Hegardt F. G., Casals N. (2008) CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J. Biol. Chem. 283, 6878–6885 [DOI] [PubMed] [Google Scholar]
- 46. Price N., van der Leij F., Jackson V., Corstorphine C., Thomson R., Sorensen A., Zammit V. (2002) A novel brain-expressed protein related to carnitine palmitoyltransferase I. Genomics 80, 433–442 [DOI] [PubMed] [Google Scholar]
- 47. Wolfgang M. J., Kurama T., Dai Y., Suwa A., Asaumi M., Matsumoto S., Cha S. H., Shimokawa T., Lane M. D. (2006) The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 103, 7282–7287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gao S., Zhu G., Gao X., Wu D., Carrasco P., Casals N., Hegardt F. G., Moran T. H., Lopaschuk G. D. (2011) Important roles of brain-specific carnitine palmitoyltransferase and ceramide metabolism in leptin hypothalamic control of feeding. Proc. Natl. Acad. Sci. U.S.A. 108, 9691–9696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramírez S., Martins L., Jacas J., Carrasco P., Pozo M., Clotet J., Serra D., Hegardt F. G., Diéguez C., López M., Casals N. (2013) Hypothalamic ceramide levels regulated by CPT1C mediate the orexigenic effect of ghrelin. Diabetes 62, 2329–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kasser T. R., Harris R. B., Martin R. J. (1985) Level of satiety. Fatty acid and glucose metabolism in three brain sites associated with feeding. Am. J. Physiol. 248, R447–R452 [DOI] [PubMed] [Google Scholar]
- 51. Beverly J. L., Martin R. J. (1991) Influence of fatty acid oxidation in lateral hypothalamus on food intake and body composition. Am. J. Physiol. 261, R339–R343 [DOI] [PubMed] [Google Scholar]
- 52. Emsley J. G., Macklis J. D. (2006) Astroglial heterogeneity closely reflects the neuronal-defined anatomy of the adult murine CNS. Neuron Glia Biol. 2, 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kimes A. S., Sweeney D., London E. D., Rapoport S. I. (1983) Palmitate incorporation into different brain regions in the awake rat. Brain Res. 274, 291–301 [DOI] [PubMed] [Google Scholar]
- 54. Karmi A., Iozzo P., Viljanen A., Hirvonen J., Fielding B. A., Virtanen K., Oikonen V., Kemppainen J., Viljanen T., Guiducci L., Haaparanta-Solin M., Någren K., Solin O., Nuutila P. (2010) Increased brain fatty acid uptake in metabolic syndrome. Diabetes 59, 2171–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Islinger M., Grille S., Fahimi H. D., Schrader M. (2012) The peroxisome. An update on mysteries. Histochem. Cell Biol. 137, 547–574 [DOI] [PubMed] [Google Scholar]
- 56. Meegalla R. L., Billheimer J. T., Cheng D. (2002) Concerted elevation of acyl-coenzyme A:diacylglycerol acyltransferase (DGAT) activity through independent stimulation of mRNA expression of DGAT1 and DGAT2 by carbohydrate and insulin. Biochem. Biophys. Res. Commun. 298, 317–323 [DOI] [PubMed] [Google Scholar]
- 57. Coleman R. A., Lee D. P. (2004) Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 43, 134–176 [DOI] [PubMed] [Google Scholar]
- 58. Borg M. L., Omran S. F., Weir J., Meikle P. J., Watt M. J. (2012) Consumption of a high-fat diet, but not regular endurance exercise training, regulates hypothalamic lipid accumulation in mice. J. Physiol. 590, 4377–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Benoit S. C., Kemp C. J., Elias C. F., Abplanalp W., Herman J. P., Migrenne S., Lefevre A. L., Cruciani-Guglielmacci C., Magnan C., Yu F., Niswender K., Irani B. G., Holland W. L., Clegg D. J. (2009) Palmitic acid mediates hypothalamic insulin resistance by altering PKC-θ subcellular localization in rodents. J. Clin. Invest. 119, 2577–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mayer C. M., Belsham D. D. (2010) Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons. Rescue of resistance and apoptosis through adenosine 5′ monophosphate-activated protein kinase activation. Endocrinology 151, 576–585 [DOI] [PubMed] [Google Scholar]
- 61. El-Assaad W., Buteau J., Peyot M. L., Nolan C., Roduit R., Hardy S., Joly E., Dbaibo G., Rosenberg L., Prentki M. (2003) Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 144, 4154–4163 [DOI] [PubMed] [Google Scholar]
- 62. Routh V. H. (2002) Glucose-sensing neurons. Are they physiologically relevant? Physiol. Behav. 76, 403–413 [DOI] [PubMed] [Google Scholar]
- 63. Fioramonti X., Lorsignol A., Taupignon A., Pénicaud L. (2004) A new ATP-sensitive K+ channel-independent mechanism is involved in glucose-excited neurons of mouse arcuate nucleus. Diabetes 53, 2767–2775 [DOI] [PubMed] [Google Scholar]
- 64. Wang R., Cruciani-Guglielmacci C., Migrenne S., Magnan C., Cotero V. E., Routh V. H. (2006) Effects of oleic acid on distinct populations of neurons in the hypothalamic arcuate nucleus are dependent on extracellular glucose levels. J. Neurophysiol. 95, 1491–1498 [DOI] [PubMed] [Google Scholar]
- 65. Levin B. E., Magnan C., Dunn-Meynell A., Le Foll C. (2011) Metabolic sensing and the brain. Who, what, where, and how? Endocrinology 152, 2552–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Morgan K., Obici S., Rossetti L. (2004) Hypothalamic responses to long-chain fatty acids are nutritionally regulated. J. Biol. Chem. 279, 31139–31148 [DOI] [PubMed] [Google Scholar]
- 67. Le Foll C., Irani B. G., Magnan C., Dunn-Meynell A. A., Levin B. E. (2009) Characteristics and mechanisms of hypothalamic neuronal fatty acid sensing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R655–R664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Obici S., Feng Z., Morgan K., Stein D., Karkanias G., Rossetti L. (2002) Central administration of oleic acid inhibits glucose production and food intake. Diabetes 51, 271–275 [DOI] [PubMed] [Google Scholar]
- 69. Schwinkendorf D. R., Tsatsos N. G., Gosnell B. A., Mashek D. G. (2011) Effects of central administration of distinct fatty acids on hypothalamic neuropeptide expression and energy metabolism. Int. J. Obes. (Lond) 35, 336–344 [DOI] [PubMed] [Google Scholar]