

Background: BMPs affect osteoclastogenesis in vitro, but the effects of BMP signaling on osteoclastogenesis in vivo are not well understood.
Results: Conditional deletion of BMPRII in osteoclasts results in reduced osteoclastogenesis, resulting in increased bone.
Conclusion: BMP signaling is required for proper bone remodeling in vivo.
Significance: Identifying factors affecting osteoclast differentiation increase understanding of bone remodeling regulation in vivo.
Keywords: Bone, Bone Morphogenetic Protein (BMP), MAP Kinases (MAPKs), Osteoclast, Signal Transduction, BMPRII, Conditional Deletion
Abstract
To investigate the role of bone morphogenetic protein (BMP) signaling in osteoclastogenesis in vivo, we eliminated BMPRII in osteoclasts by creating a BMPRIIfl/fl;lysM-Cre mouse strain. Conditional knock-out (cKO) mice are osteopetrotic when compared with WT controls due to a decrease in osteoclast activity. Bone marrow macrophages (BMMs) isolated from cKO mice are severely inhibited in their capacity to differentiate into mature osteoclasts in the presence of M-CSF and receptor activator of NF-κB (RANK) ligand. We also show that BMP noncanonical (MAPK) and canonical (SMAD) pathways are utilized at different stages of osteoclast differentiation. BMP2 induces p38 phosphorylation in pre-fusion osteoclasts and increases SMAD phosphorylation around osteoclast precursor fusion. Phosphorylation of MAPKs was decreased in differentiated BMMs from cKO animals. Treating BMMs with the SMAD inhibitor dorsomorphin confirms the requirement for the canonical pathway around the time of fusion. These results demonstrate the requirement for BMP signaling in osteoclasts for proper bone homeostasis and also explore the complex signaling mechanisms employed by BMP signaling during osteoclast differentiation.
Introduction
Bone remodeling, the replacement of old, damaged bone with new bone, is essential for maintaining structural integrity of the skeleton and mineral homeostasis. The process is regulated by communication between osteocytes, capable of sensing damage to bone, osteoclasts, cells that respond by resorbing damaged bone, and osteoblasts, cells responsible for secreting fresh osteoid, which later mineralizes to form mature bone (reviewed in Ref. 1). Proper regulation of the remodeling process is critical to bone health, whereas decoupling of bone formation and bone resorption leads to bone diseases such as osteoporosis.
Osteoclasts are multinucleated giant cells derived from the monocyte/macrophage branch of the hematopoietic lineage. Macrophage colony-stimulating factor (M-CSF) and receptor activator of NF-κB ligand (RANKL)5 are essential for initiating osteoclast differentiation. RANKL is expressed by both osteoblasts and osteocytes, although recent evidence supports osteocytes as the primary source of osteoclast-initiating RANKL during bone remodeling in vivo (2, 3).
In addition to M-CSF and RANKL, additional factors influence osteoclast differentiation. For example, T-cell cytokines can either inhibit (IFN-γ, IL-4, IL-10) or promote (TNF-α, IL-6, IL-17) osteoclast differentiation (reviewed in Ref. 4). Bone morphogenetic proteins (BMPs) are members of the TGF-β family of signaling molecules. BMPs bind to the heterotetrameric receptor complex consisting of two type I and two type II receptors, inducing phosphorylation of SMADs 1, 5, and 8, enabling them to form a trimeric complex with SMAD4 and be retained in the nucleus to regulate gene expression. In addition to signaling through the SMAD pathway, it is well known that TGF-β family receptors also mediate signaling through noncanonical pathways such as the MAP kinase pathway. TGF-β utilizes both AKT, through TAK1, and SMAD pathways to regulate cell survival in mature osteoclasts (5). BMPs also transduce their signals using the MAPK pathway in mature osteoclasts to promote resorption and cell survival (6), and BMPs have also been shown to activate MAPKs through TAK1 in chondrocytes (7, 8). Several laboratories, including ours, have shown that osteoclasts express BMP receptors and respond directly to BMPs through activation of SMAD signaling (6, 9–11). Expanding on previous studies, we showed that in the presence of BMP2, bone marrow macrophages (BMMs) require less RANKL to differentiate into osteoclasts and that the addition of BMP2 leads to the formation of larger osteoclasts than in the presence of RANKL alone. We also showed that deleting the BMP inhibitor Twisted gastrulation (TWSG1) in mice results in osteopenia due to an increase in osteoclast differentiation, suggesting a stimulatory function for BMP signaling in osteoclasts in vivo (9). Conversely, RANKL-mediated osteoclast differentiation is severely inhibited in the presence of the BMP inhibitor noggin, and shRNA knockdown of BMPRII in WT osteoclasts results in fewer and smaller osteoclasts in vitro. Together, these results demonstrate that increased BMP signaling promotes osteoclastogenesis, whereas loss of BMP signaling reduces it.
Despite abundant in vitro data showing a role for BMPs in regulating osteoclast differentiation, the skeletal effects of eliminating BMP signaling specifically in osteoclasts in vivo as well as the intracellular pathways through which BMPs transduce their signals in osteoclasts are not known. Therefore, to evaluate the effects of BMP signal deficiency in osteoclasts, we generated conditional knock-out mice where BMPRII is deleted in myeloid lineage cells, which includes osteoclasts, by crossing BMPRII-floxed mice containing loxP sites flanking exons 4 and 5 (BMPRIIfl/fl) with mice expressing Cre recombinase driven by the lysozyme M promoter (lysM-Cre). lysM-Cre mice were selected to give us the ability to examine loss of BMP signaling throughout osteoclast differentiation, including early precursors. We present data that mice lacking BMPRII in osteoclasts display increased bone mass when compared with WT littermates due to decreased osteoclast differentiation. Furthermore, we also present data characterizing BMP signaling during osteoclast differentiation and provide evidence that BMP2 utilizes both noncanonical MAPK signaling and canonical SMAD signaling at different stages of osteoclast differentiation. The results presented herein not only offer convincing evidence for a role of BMP signaling affecting osteoclasts in vivo, but also provide insight into the complex ways in which the signaling machinery utilized by BMPs changes throughout osteoclast differentiation.
EXPERIMENTAL PROCEDURES
Cell Culture
Primary BMMs from BMPRII+/+;lysM-Cre, BMPRII+/fl;lysM-Cre, or BMPRIIfl/fl;lysM-Cre in the C57Bl/6 background were harvested and cultured as described previously (11). C57Bl/6 WT cells were used for Cre-expressing adenovirus (Ad-Cre) and BMP2 treatment experiments. Osteoclasts were cultured in minimum essential media α supplemented with l-glutamine (Life Technologies), FBS, and penicillin/streptomycin. BMMs were flushed with minimum essential media α (Life Technologies) from the marrow cavity of the femur and tibia. Red blood cells were lysed with RBC lysis buffer (150 mm NH4Cl, 10 mm KHCO3, 0.1 mm EDTA, pH 7.4). BMMs were spun down, resuspended in minimum essential media α supplemented with conditioned medium containing M-CSF, and plated on 100-mm tissue culture plates overnight. The following day, the nonadherent population of cells was counted and plated for experiments. Cells were maintained in medium containing M-CSF for two more days, at which time medium was changed and RANKL (and, if applicable, additional treatment (i.e. BMP2)) was added to initiate osteoclast differentiation.
Mice
BMPRIIfl/fl mice were originally created in the C57Bl/6 background as described in Ref. 12. We obtained the mice from Dr. Michael O'Connor (University of Minnesota). Mice were crossed with the lysM-Cre-expressing mouse (The Jackson Laboratory). We generated Twsg1−/− mice as described previously (13). Use and care of the mice in this study were approved by the University of Minnesota Institutional Animal Care and Use Committee.
μCT Analysis
Microcomputed tomography (μCT) was performed at the University of Minnesota. Femora were subjected to micro-CT scanning (model XT H 225, Nikon Metrology, Inc.) at conditions of 90 kV, 90 μA, 0.5° steps with 708 ms of exposure time and pixel size at 12 μm. Three-dimensional reconstructions were made from the original images using the software CT Pro 3D (Nikon metrology). VG Studio Max 2.1 (Volume Graphics GmbH) was used for visualization and three-dimensional rendering and creation of bmp files. Then, histomorphometry analyses were performed using SkyScan CT-Analyzer Version 1.12 (Bruker micro-CT).
Immunoblotting
Cell protein lysates were harvested from osteoclasts in modified radioimmune precipitation buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% IGEPAL, 0.25% sodium deoxycholate, 1 mm EDTA) supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo Scientific). Lysates were cleared by centrifugation at 12,000 × g at 4 °C. Proteins were resolved by SDS-PAGE, transferred to PVDF membrane (Millipore), and incubated at 4 °C overnight with primary antibodies. p38, P-p38, ERK1/2, P-ERK1/2, α-tubulin, P-SMAD1/5/8, P-JNK, and JNK antibodies were obtained from Cell Signaling Technology. SMAD1/5/8 antibody was obtained from Santa Cruz Biotechnology. HRP-conjugated anti-rabbit and anti-mouse were incubated with membranes, washed, and incubated with Amersham ECL Prime Western blotting detection reagent (GE Healthcare).
ELISA
Serum was harvested from animals at 3 months of age and subjected to ELISA as per the manufacturer's protocol. Carboxyl-terminal telopeptide of collagen I (CTX) was detected using the RatLaps enzyme-linked immunosorbent assay (Immunodiagnostic Systems). Osteocalcin was detected using a mouse osteocalcin kit (Biomedical Technologies Inc.)
Quantitative Real-time PCR
Quantitative real-time PCR was performed using the MyiQ Single Color real-time PCR detection system (Bio-Rad). RNA was harvested from cells using TRIzol reagent (Ambion, Life Technologies) and quantified using UV spectroscopy. cDNA was prepared from 1 μg of RNA using the iScript cDNA synthesis kit (Bio-Rad) as per the manufacturer's protocol. Experimental genes were normalized to L4 or GAPDH as indicated in the figure legends. Primer sequences follow: tartrate-resistant acid phosphatase (TRAP) (forward) 5′-CGT CTC TGC ACA GAT TGC A; (reverse) 5′-GAG TTG CCA CAC AGC ATC AC; NFATc1 (forward) 5′-TCA TCC TGT CCA ACA CCA AA; (reverse) 5′-TCA CCC TGG TGT TCT TCC TC; cathepsin K (forward) 5′-AGG GAA GCA AGC ACT GGA TA; (reverse) 5′-GCT GGC TGG AAT CAC ATC TT; DC-STAMP (forward) 5′-GGG CAC CAG TAT TTT CCT GA; (reverse) 5′-TGG CAG GAT CCA GTA AAA GG; ATV6v0d2 (forward) 5′-TCA GAT CTC TTC AAG GCT GTG CTG; (reverse) 5′-GTG CCA AAT GAG TTC AGA GTG ATG; L4 (forward) 5′-CCT TCT CTG GAA CAA CCT TCT CG; (reverse) 5′-AAG ATG ATG AAC ACC GAC CTT AGC.
Histological Analysis
Femurs were harvested from mice and placed in Z-fix solution for 6 h. The bones were then decalcified in EDTA for 10 days, embedded in paraffin, and sectioned. Bone sections were stained with TRAP as per the manufacturer's protocol (387A, Sigma-Aldrich). ImageJ was used to quantify bone surface and osteoclast surface from TRAP-stained slides.
Dynamic Histomorphometry
50 mg/kg of tetracycline (Sigma-Aldrich) was delivered to mice by intraperitoneal injection 7 days and 2 days prior to harvest. Femora were dehydrated in gradient alcohol, cleared with xylene, and embedded in methyl methacrylate for dynamic histomorphometric analysis. Serial frontal sections (5 μm in thickness) of each sample were made using a hard tissue microtome (Polycut E, Leica Microsystems). Photographs were taken by using a digital charged-coupled device (CCD) camera (DP71, Olympus) attached to a fluorescent microscope (100×) with a long-pass filter (Model: 41012, Chroma Technology Corp.). Lengths of fluorescent labeled/unlabeled measurement and distances between doubling were done by using the software Image Pro Plus 6.1 (Media Cybernetics). Secondary spongiosa (1–3 mm below the growth plate) in the metaphysis of the distal femur was subjected to dynamic histomorphometric analyses according to the recommendations of the American Society for Bone and Mineral Research (ASBMR) Histomorphometry Nomenclature Committee (14).
TRAP Staining of Cells
BMMs were differentiated to the indicated time, fixed with 4% paraformaldehyde for 10 min, and washed with PBS. Cells were then incubated in TRAP stain (0.05 m acetate buffer; 0.03 m sodium tartrate; 100 μg/ml naphthol AS-MX phosphate; 0.01% Triton X-100; 0.3 mg/ml Fast Red Violet LB stain) for ∼10 min at 37 °C. Size and number of osteoclasts were quantified using ImageJ.
Adenovirus Infection of Osteoclasts
Bone marrow macrophages were isolated as described above. Prior to stimulation with RANKL, the cells were incubated with 50 multiplicity of infection of Cre recombinase or control adenovirus for 3 h at 37 °C in the presence of M-CSF. After 5 days, RNA was extracted for use in quantitative RT-PCR, protein was extracted for Western blotting, or cells were stained for TRAP.
RESULTS
BMPRII Is Required for Efficient Osteoclast Differentiation
Previously, our laboratory showed that BMP2 enhances RANKL-mediated osteoclast differentiation and that noggin, an inhibitor of BMP signaling, blocks osteoclast differentiation (11). In that study, we also observed impaired osteoclastogenesis upon shRNA-mediated knockdown of BMPRII. To further validate the requirement of BMP signaling for osteoclast formation, we compared osteoclast differentiation of BMMs from WT and BMPRIIfl/fl mice (12) following infection with Ad-Cre. As shown in Fig. 1, A–C, cultures from BMPRIIfl/fl mice infected with Ad-Cre showed a significant decrease in both size and number of multinucleated osteoclasts when compared with Ad-Cre-infected WT cultures, indicating severely inhibited osteoclast differentiation in vitro. Real-time RT-PCR confirmed that BMPRII and cathepsin K (Ctsk, a marker for osteoclast differentiation) mRNA expression is decreased in the Ad-Cre-treated BMPRIIfl/fl osteoclasts (Fig. 1, D and E). BMPRII protein expression is also reduced in Ad-Cre-infected osteoclasts (Fig. 1F). These data support our previous work indicating that BMP signaling through BMPRII is essential for efficient osteoclast differentiation in vitro (11).
FIGURE 1.

Cre expression by adenovirus in BMPRIIfl/fl BMMs inhibits osteoclast differentiation. A–C, BMMs from either WT or BMPRIIfl/fl mice were infected with adenovirus expressing Cre recombinase prior to RANKL treatment. Cells were then differentiated in the presence of RANKL and M-CSF, TRAP-stained, imaged, and quantified. Oc, osteoclast. N.Oc, number of osteoclasts. MNOC, multinucleated osteoclast. D and E, real-time RT-PCR was used to measure BMPRII and cathepsin K gene expression following infection of WT or BMPRIIfl/fl osteoclasts. F, BMPRII protein levels were analyzed by Western blot. Error bars represent S.D. * = p < 0.05; and ** = p < 0.005.
BMPRIIfl/fl;lysM-Cre Mice Have Increased Bone Mass due to Reduced Bone Resorption
The in vivo skeletal phenotype was evaluated using μCT analysis. For the remainder of the study, BMPRII+/+;lysM-Cre will be referred to as wild-type (WT), BMPRII+/fl;lysM-Cre will be referred to as heterozygotes (Het), and BMPRIIfl/fl;lysM-Cre will be referred to as conditional knock-out (cKO). μCT analysis of 1-month-old animals showed no difference in bone volume fraction between WT, Het, and cKO animals (data not shown). At 3 months of age, however, cKO mice showed significantly increased bone volume (BV/TV), bone surface (BS), trabecular thickness (Tb.Th), and trabecular number (Tb.N.) when compared with WT animals (Fig. 2, A–E). This corresponded to significantly reduced trabecular separation (Tb.Sp) in cKO mice versus WT mice (Fig. 2F). Het mice showed an intermediate phenotype that was significant when compared with WT mice for certain parameters (BS and Tb.N).
FIGURE 2.

cKO mice have increased bone volume fraction corresponding with an increase in trabecular parameters. A, μCT images of femurs from WT, Het, and cKO mice. B–F, volumetric data quantifying bone volume fraction (BV/TV), bone surface (BS), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular separation (Tb.S). Values represent mean plus S.D. Sample sizes of mice are: WT, n = 9; Het, n = 7; cKO, n = 10. Significance was calculated for WT versus Het and WT versus cKO. Error bars represent S.D. * = p < 0.05; and ** = p < 0.005.
To determine the cause of increased bone volume, we examined the status of bone formation and resorption. Osteoclast activity was decreased in cKO mice when compared with WT animals, as measured by CTX ELISA at 3 months of age (Fig. 3A). When compared with WT serum, CTX levels were reduced ∼40% (p = 0.0005) in cKO serum and ∼20% (p < 0.05) in Het serum. This decrease in osteoclast activity agrees with the increased bone mass we observe in cKO mice. We assessed the presence of osteoclasts in histological sections stained with TRAP (Fig. 3C). There was no difference in osteoclast surface per bone surface between WT and cKO mice (Fig. 3D). Next we investigated whether bone formation rate was altered in cKO mice by performing dynamic histomorphometric analyses following double tetracycline labeling (Fig. 3E). Mineralized surface/bone surface (MS/BS), mineralized apposition rate, and bone formation rate were calculated and showed no significant differences between the three genotypes (Fig. 3, F–H). As a measure of osteoblast activity, we measured serum osteocalcin levels by ELISA and found no significant difference between WT, Het, and cKO animals (Fig. 3B). Based on these results, we conclude that defective osteoclast activity rather than altered bone formation is responsible for the increased bone mass we observe in the cKO mice.
FIGURE 3.

In vivo osteoclast activity is decreased in cKO mice. A and B, serum was obtained from mice at 3 months and subjected to ELISA assays. ELISA data shown are representative experiments that have been repeated three times. * = p < 0.005; *** = p = 0.0005. OCN, osteoclast number. C, TRAP staining of histological sections from WT and cKO mice. D, osteoclast surface per bone surface (Oc.S/BS) was quantified from TRAP-stained sections: WT, n = 2; cKO, n = 9. E, representative images of tetracycline double labeling from WT, Het, and cKO. F–H, quantification of double labeling. MS/BS, mineralized surface/bone surface; MAR, mineral apposition rate; and BFR/BS, bone formation rate/bone surface. No significance between the genotypes. WT and Het, n = 5; cKO, n = 8. Error bars represent S.D.
In Vitro Osteoclast Differentiation Is Inhibited in cKO BMMs
Based on our findings in vivo, we hypothesized that osteoclasts derived from BMMs of cKO mice would differentiate less efficiently than their WT counterparts in culture. As expected, we found significantly smaller TRAP-positive multinucleated osteoclasts in cKO mice when compared with WT mice when BMM-derived osteoclast precursors were allowed to differentiate in the presence of RANKL and M-CSF (Fig. 4, A–C). Similar to the in vivo phenotype, Het mice showed an intermediate phenotype that was significant when compared with WT mice. To our surprise, we observed an increase in the number of multinucleated osteoclasts in cKO mice. However, the total nuclei number in a given field remained unchanged (Fig. 4D), whereas the average number of nuclei per osteoclast was lower (Fig. 4E), indicating that differentiating BMMs from the cKO and Het mice resulted in more multinucleated osteoclasts containing fewer nuclei. This result is suggestive of an inhibition in fusion of mononucleated preosteoclasts into multinucleated osteoclasts. To investigate whether resorption is altered in cKO osteoclasts, BMMs from WT, Het, and cKO were differentiated on calcium phosphate-coated plates. The percentage of resorbed area was 50% less (p < 0.0001) in cultures containing cKO osteoclasts when compared with those containing WT osteoclasts (Fig. 4F).
FIGURE 4.

cKO BMMs are impaired in their ability to differentiate into mature osteoclasts. BMMs harvested from WT, Het, or cKO animals were differentiated in culture in the presence of M-CSF and RANKL, TRAP stained, imaged, and counted. Only osteoclasts with ≥3 nuclei were counted. A, representative images from each of the genotypes. B and C, quantification of osteoclast size (OC Size) and number (N.Oc). D and E, fixed cells were stained with DAPI to visualize nuclei. Total nuclei on each image and average (Avg.) nuclei per osteoclast were quantified. F, quantification of in vitro resorptive activity. Values represent mean ± S.D. * = p < 0.05; ** = p < 0.005; and **** = p < 0.0001. Error bars represent S.D.
In addition, the expression of genes associated with osteoclast differentiation was altered between WT, Het, and cKO animals (Fig. 5). Multiple markers of osteoclast differentiation, Acp5, Ctsk, ATP6v0d2, and OC-STAMP, display reduced mRNA expression by quantitative RT-PCR in Het and cKO when compared with WT controls. In addition, the expression of NFATc1, a transcription factor required for osteoclast differentiation, is reduced in Het and cKO osteoclasts. Therefore, elimination of BMPRII in osteoclasts leads to a decrease in expression of genes critical for osteoclast differentiation and fusion.
FIGURE 5.
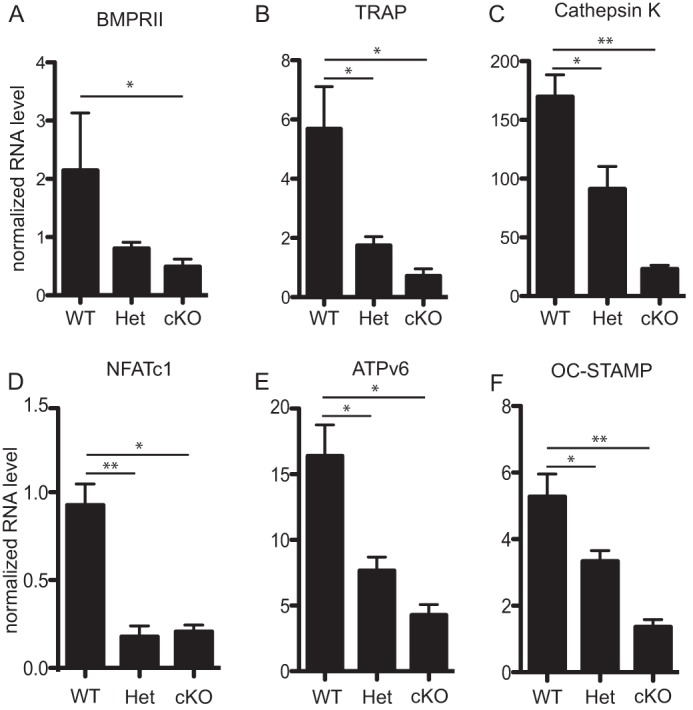
Osteoclast marker gene expression is reduced in Het and cKO osteoclasts. Real-time quantitative PCR was run on cDNA extracted from mature osteoclasts differentiated from WT, Het, and cKO BMMs. A, BMPRII mRNA expression was shown to validate gene expression knockdown in these osteoclasts. B–F, expressions of Acp5, Ctsk, NFATc1, ATP6v0d2, and OC-STAMP were included as multiple markers of osteoclast differentiation. Values represent the mean ± S.D. * = p < 0.05; and ** = p < 0.005. Error bars represent S.D.
The BMP Noncanonical (MAPK) Pathway, but Not the BMP Canonical (SMAD) Pathway, Is Affected in BMPRIIfl/fl;lysM-Cre Osteoclasts
As BMP signals can be transduced through multiple pathways, we next investigated the status of the canonical SMAD and noncanonical MAPK intracellular signaling pathways in BMPRII-deficient osteoclasts throughout differentiation. Levels of phosphorylated and nonphosphorylated forms of p38, ERK1/2, JNK, and SMAD proteins were analyzed by Western blot. Levels of phosphorylated p38 (P-p38) and phosphorylated ERK1/2 (P-ERK1/2) were decreased most notably on days 3 and 4 in cKO osteoclasts; levels of phosphorylated JNK levels were reduced on days 2–4. In differentiated WT osteoclasts, P-SMAD1/5/8 increases steadily throughout differentiation as we have shown previously (Fig. 6, P-SMAD1/5/8, lower band) (11). In contrast, P-SMAD1/5/8 is not consistently reduced in cKO osteoclasts. These results show that primarily the noncanonical pathway is affected in cKO osteoclasts and that signaling through the noncanonical MAPK arm of the BMP pathway is essential for osteoclast differentiation.
FIGURE 6.
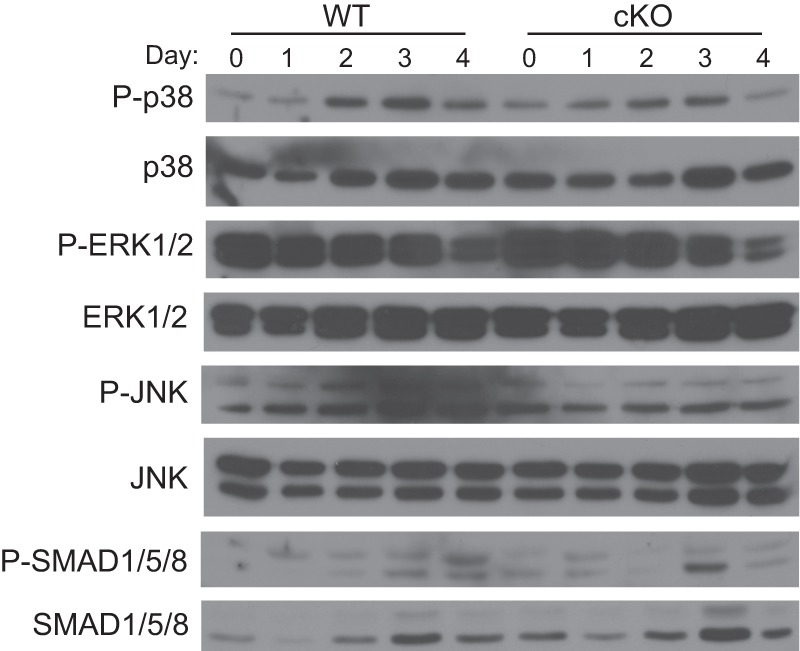
p38, JNK, and ERK1/2 phosphorylation is reduced in cKO osteoclasts. Cell protein lysates were harvested at the indicated day from BMMs differentiated in RANKL and subjected to Western blot for total and phosphorylated forms of p38, JNK, ERK1/2, and SMAD1/5/8.
BMP2 Signals through Noncanonical and Canonical Pathways during Early and Late Stages of Osteoclast Differentiation, Respectively
In our culture system, mononuclear osteoclast fusion begins on day 3 following RANKL treatment; mature osteoclasts form by day 5. In a previous study, we observed an increase in P-SMAD1/5/8 levels around day 3 of WT osteoclast differentiation, leading us to propose that the canonical BMP pathway might be critical during fusion (11). Similarly, we have found that induction of P-SMAD in osteoclastic cells by exogenous BMP is most efficient in day 3 osteoclasts (Ref. 9 and data not shown). To elucidate the temporal relationship between noncanonical and canonical BMP signaling during osteoclast differentiation, we next examined these pathways using WT BMMs that were differentiated with M-CSF and RANKL in the presence and absence of BMP2. Protein lysates were harvested each day for 5 days. In agreement with previous data (11), P-SMAD1/5/8 levels are first detectable starting at day 3 in cultures treated with only M-CSF and RANKL (Fig. 7A). In the presence of BMP2, P-SMAD1/5/8 levels are observed a day earlier (day 2) and are present at higher levels when compared with cultures without BMP2. With regard to noncanonical BMP pathways, phosphorylated p38 and ERK1/2 were observed throughout differentiation starting on day 1. Treatment with BMP2 increased phosphorylated p38 levels on days 1 and 2 of differentiation and returned to base-line levels on day 3–5 that were comparable with cultures treated with RANKL alone. In contrast to the cKO osteoclast extracts, in which P-ERK1/2 levels were reduced when compared with WT osteoclast extracts, treatment with BMP2 does not affect P-ERK1/2 levels over the course of differentiation when compared with RANKL-only cultures.
FIGURE 7.

p38 and SMADs mediate the BMP2 response at different stages of differentiation. A, WT osteoclasts were differentiated in the presence of RANKL (1 ng/ml) alone or RANKL + BMP2 (30 ng/ml), and cell protein lysates were harvested daily. Day 0 osteoclasts have only been treated with M-CSF, not RANKL nor BMP2. Protein lysates were harvested over the course of differentiation and subjected to Western blot. B, BMMs from WT and TWSG1-null mice were differentiated, and protein lysates were harvested at the indicated days following initiation of differentiation. C, BMMs were differentiated to day 3 in the presence of RANKL (60 ng/ml), serum-starved for 4 h, and then stimulated with BMP2 (30 ng/ml). Lysates were collected at the indicated times following stimulation and subjected to Western blot for total and phosphorylated p38, JNK, ERK1/2, and SMAD1/5/8.
To corroborate these findings, we utilized BMMs harvested from TWSG1-null mice, which we have previously been shown to have increased BMP signaling (9). SMAD 1/5/8 phosphorylation is increased in TWSG1-null mice as expected (Fig. 7B). Similar to treatment with BMP2, TWSG1-null BMMs undergoing differentiation display increased p38 phosphorylation at early time points, whereas levels of p38 phosphorylation return to levels of WT phosphorylation in post-fusion osteoclasts. As with BMP2 treatment, levels of phosphorylated ERK are unaffected.
We next evaluated the capacity of BMP2 to activate MAPKs following a period of serum starvation. To address this question, osteoclasts were differentiated to day 3, serum-starved for 4 h, and then stimulated with serum-free medium containing recombinant BMP2. Phosphorylation of p38 and ERK1/2 is rapidly stimulated and detected at high levels 15 min following BMP2 treatment (Fig. 7C), after which time levels of p38 phosphorylation are reduced below even unstimulated condition, whereas P-ERK1/2 levels return to pre-stimulation levels. Levels of phosphorylated JNK are reduced upon stimulation with BMP2. We observe no change in the phosphorylation of SMAD 1/5/8.
Taken together, these data indicate that BMP2 rapidly activates p38 and ERK phosphorylation following stimulation. In addition, BMPs impact both MAPKs and SMADs in a temporal specific manner during osteoclast differentiation.
Dorsomorphin, a Small-molecule SMAD Pathway Inhibitor, Inhibits Fusion of Mononuclear Osteoclasts
Because we observe P-SMAD1/5/8 activation in the absence of TWSG1 and in response to BMP2 during osteoclast differentiation, we next evaluated whether the SMAD-mediated pathway is required for osteoclast differentiation. WT BMMs were differentiated in the presence of increasing concentrations of dorsomorphin, a chemical inhibitor of type I BMP receptors that preferentially blocks phosphorylation of SMAD1/5/8. Fig. 8A demonstrates that dorsomorphin effectively inhibits SMAD phosphorylation in a dose-dependent fashion. WT BMMs were differentiated in the presence of RANKL for 3 days, pretreated with the indicated concentration of dorsomorphin or DMSO control, and stimulated with BMP2. In a separate experiment, BMMs were differentiated in the presence of dorsomorphin over either the entirety of differentiation, days 1–2 (pre-fusion), or days 3–5 (post-fusion). Dorsomorphin treatment over the course of differentiation effectively inhibited osteoclast differentiation at even lower concentrations (150 nm) (Fig. 8B). Treatment with dorsomorphin for only days 1–2 was not sufficient to inhibit osteoclast differentiation, whereas treatment over days 3–5 effectively inhibited osteoclast differentiation. Although the formation of multinucleated osteoclasts was reduced by dorsomorphin treatment, we observed an increased number of mononucleated TRAP+ osteoclasts. This observation suggests that dorsomorphin blocks osteoclastogenesis at the stage of mononuclear cell fusion. These data support our immunoblot results showing that BMP2 only utilizes the SMAD pathway around the time of osteoclast precursor fusion (Fig. 7A).
FIGURE 8.
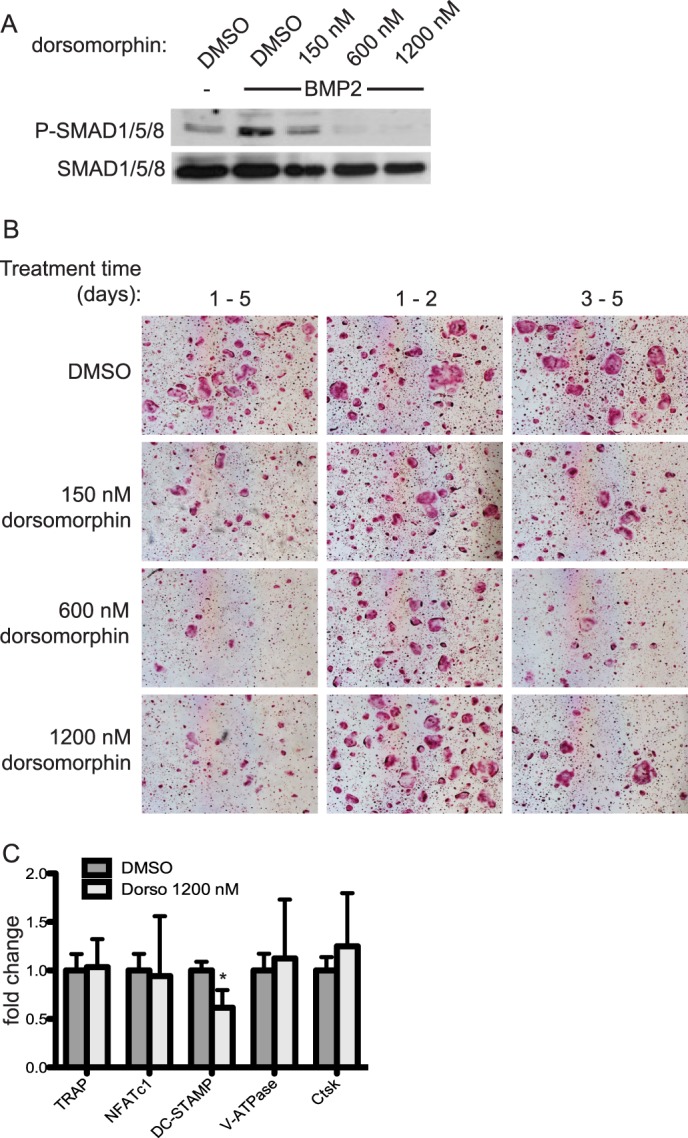
Dorsomorphin treatment inhibits differentiation on days 3–5 of osteoclast differentiation. A, efficacy of dorsomorphin was measured by its ability to block BMP2-induced SMAD phosphorylation by Western blot. Fusion-staged osteoclasts were preincubated with DMSO or dorsomorphin for 30 min and then stimulated with BMP2 for 30 min. B, osteoclasts were differentiated in the presence of RANKL (60 ng/ml) and dorsomorphin or DMSO vehicle control for days 1–5, days 1–2, or days 3–5. Cells were TRAP-stained and imaged. C, cDNA from day 5 osteoclasts was subjected to quantitative real-time PCR for various osteoclast marker genes. Expression of each gene is graphed as -fold change relative to its expression in DMSO-treated control cells. Dorso, dorsomorphin. Numbers represent mean ± S.D. * = p < 0.05. Error bars represent S.D.
To gain further insight into the mechanism by which dorsomorphin affected osteoclastogenesis, we performed quantitative RT-PCR on RNA from osteoclasts differentiated with or without dorsomorphin. Gene expression was measured for Acp5, NFATc1, DC-STAMP, V-ATPase, and Ctsk. Of these, only mRNA expression of DC-STAMP, a protein critical for mononuclear osteoclast fusion, was down-regulated. These data support the observation that cell fusion is blocked and suggest regulation of DC-STAMP at least at the transcriptional level to be one possible mechanism.
DISCUSSION
In this study, we show that BMP signaling is required for normal in vivo osteoclastogenesis. BMMs harvested from BMPRIIfl/fl mice and infected with Cre-expressing adenovirus differentiated less efficiently into mature osteoclasts than did WT cells infected with Cre-expressing adenovirus. Defective osteoclastogenesis in BMPRIIfl/fl;lysM-Cre mice interferes with proper bone remodeling, leading to increased bone volume fraction and bone surface when compared with WT mice. These results correspond to an increase in trabecular number and thickness and a decrease in trabecular separation at 3 months of age. μCT analysis of 1-month-old animals failed to reveal any difference between WT and cKO mice, suggesting that the defect we observe is due to impaired remodeling rather than improper bone development during embryogenesis (data not shown). We also observe a decrease in osteoclast activity in vivo as indicated by CTX ELISA. In support of the in vivo data, BMMs harvested from Het and cKO mice and cultured in the presence of M-CSF and RANKL have impaired differentiation when compared with those from WT mice. However, we observe no change in osteoclast surface per bone surface, measured by TRAP-stained histological sections, between WT and cKO animals. Furthermore, BMMs harvested from Het and cKO animals and differentiated on osteo-assay plates displayed less resorptive activity than differentiated WT osteoclasts. These data strongly support the hypothesis that BMPRII is essential for proper osteoclast differentiation and show that loss of BMPRII in vivo leads to osteopetrosis caused by reduced bone resorption.
The findings herein are consistent with current understanding of how BMPs regulate osteoclasts. Initial studies showed that BMP2 positively affected osteoclast differentiation as Kanatani et al. (15) showed an increase in osteoclast formation and function. Later studies confirmed the stimulatory effect of BMP2 on osteoclasts, including data that the osteoclast marker Ctsk was up-regulated in response to BMP2 treatment (10, 16). These studies also confirmed that BMPRIa, BMPRII, SMAD1, and SMAD5 are expressed in osteoclasts. Another group recently deleted Bmpr1a either in osteoblasts, using col1a1-Cre, or in osteoclasts, using cathepsin K-Cre (17). In contrast to our findings, they found that deleting Bmpr1a in osteoclasts led to an increase in bone formation due in part to an increase in osteoblast activity in these mice. Although they observed a decrease in osteoclast number in tibia bone sections in Bmpr1aΔOC/ΔOC mice, spleen-derived osteoclasts differentiated normally in culture. They ascribe this to BMPR1A regulating osteoblast-osteoclast coupling. We observed no change in osteoblast activity in our mouse model, demonstrated by unchanged osteocalcin serum levels and bone formation rate between WT and cKO mice.
Our laboratory has previously studied modulation of osteoclast differentiation in the context of altered BMP signaling. Loss of the BMP inhibitor, TWSG1, or treatment with BMP2 increases osteoclast differentiation and results in osteopenia in Twsg1−/− mice (9, 11, 18). Osteoclast differentiation was enhanced in part by an increase in DC-STAMP RNA expression in Twsg1−/− osteoclasts, which agrees with the data presented above (Fig. 8C) showing that DC-STAMP expression is repressed in the presence of dorsomorphin. We also showed previously that BMP2 can enhance differentiation of osteoclasts cultured in the presence of suboptimal RANKL (11) and that treatment of cultures with the BMP2 antagonist, noggin, effectively blocked osteoclast differentiation. Interestingly, noggin treatment up until day 3 was effective at blocking osteoclast differentiation, whereas it had little inhibitory effect if noggin treatment was begun on day 3 or later. These results are in contrast to our findings in this study that show that BMP receptor inhibition with dorsomorphin blocks osteoclast differentiation only at days 3–5. The exact reason for this discrepancy is not known; however, one could speculate that noggin treatment may be preferentially blocking noncanonical BMP signaling at early time points.
Investigating the signaling pathways altered in our mouse model, we show that the impaired osteoclastogenesis we observe in cKO mice corresponds to defective MAPK signaling as p38 JNK, and ERK1/2 phosphorylation were all reduced. Throughout differentiation of cKO BMMs, SMADs remained capable of being activated. One possible explanation for this is that a truncated form of BMPRII may be expressed in BMPRIIfl/fl;lysM-Cre mice as the floxed region removes exons 4 and 5 and introduces a premature stop codon (12). Previously, a truncated product, similar to that which could be expressed in our system, was shown to function as a dominant negative affecting p38 but not SMAD activation when transfected into C2C12 cells (19). We have not, however, tested whether a truncated protein product is expressed in our system. Alternatively, the persistence of SMAD1/5/8 activation could be explained by findings showing that in the absence of BMPRII, ActRIIa is able to compensate and facilitate BMP signaling (20). Furthermore, TGFβ1 has also been shown to activate SMAD1/5/8 in certain contexts (21). Therefore, compensation by other TGFβ superfamily signaling components is a plausible explanation for the continued activation of SMAD1/5/8 that we observe. Similar observations have been made in other systems; for example, a recent study has shown that shRNA knockdown of BMPRII interferes with ERK activation but not SMAD activation in mature osteoclasts (6). We further explored the signaling profile of differentiating WT osteoclasts in the presence and absence of BMP signaling. We found that BMPs transduce their signals through both noncanonical and canonical pathways dependent on the stage of differentiation; at early stages (pre-fusion), BMPs signal through MAPKs to mediate their effects, whereas around the time of cell fusion, there is a switch to signaling through the canonical SMAD pathway.
Experiments using the SMAD inhibitor dorsomorphin support our model, showing that SMAD signaling is important by day 3 of osteoclast differentiation but dispensable prior to day 3. Quantitative RT-PCR data show that expression of DC-STAMP, which is involved in cell fusion, is repressed in the presence of dorsomorphin. Therefore, one mechanism by which SMADs affect osteoclast fusion could be through regulation of DC-STAMP transcription.
Testing the requirement of p38 for mediating BMP signals during early osteoclast differentiation is complicated as p38 has an essential role during osteoclast differentiation through multiple pathways, including RANKL signaling (22, 23). As it is unclear how BMP signaling is transduced exclusively through either canonical or noncanonical pathway, it is currently not possible to disrupt MAPK signaling mediated specifically through BMPs. Further research is required to determine the signaling bridge between BMPRII and MAPKs in osteoclasts. One likely candidate is TGF-β-activated kinase 1 (TAK1), which is known to mediate BMP activation of p38 in chondrocytes (7, 8, 24). However, designing experiments knocking down TAK1 to study BMP-mediated signals is again complicated in osteoclasts as RANKL is also known to activate MAPKs through a complex containing TAK1 (25). Despite these challenges in elucidating the importance of BMP-induced MAPK signaling for osteoclast differentiation in vitro, the BMPRII cKO mice show a change in MAPK signaling but not SMAD signaling, suggesting that eliminating the noncanonical pathway alone in response to BMP signaling is sufficient to cause bone defects due to impaired osteoclastogenesis.
In summary, the current study confirms that BMP signaling is critical for osteoclast differentiation in vivo. Beyond that, we find the BMP-related intracellular signaling events throughout osteoclast differentiation to be rather complex. BMPs modulate osteoclast differentiation at multiple steps, but how those signals are transduced and the targets of those signals entirely depend on the stage of differentiation. Perhaps the most interesting question is: how does the switch from noncanonical to canonical signaling occur? Ongoing work in the laboratory continues to investigate the mechanisms by which BMP signaling impacts the signaling milieu throughout osteoclast differentiation.
This work was supported, in whole or in part, by National Institutes of Health Grants R01AR056642 (to R. G.); R01AR061352 (to K. M. and E. D. J.); and R01DE016601 (to A. P.).
- RANKL
- receptor activator of NF-κB ligand
- BMP
- bone morphogenetic protein
- BMM
- bone marrow macrophage
- cKO
- conditional knock-out
- μCT
- microcomputed tomography
- TRAP
- tartrate-resistant acid phosphatase
- CTX
- carboxyl-terminal telopeptide of collagen I
- Ad-Cre
- Cre-expressing adenovirus
- Het
- heterozygotes
- BS
- bone surface
- Tb
- trabecular
- P
- phosphorylated
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Henriksen K., Neutzsky-Wulff A. V., Bonewald L. F., Karsdal M. A. (2009) Local communication on and within bone controls bone remodeling. Bone 44, 1026–1033 [DOI] [PubMed] [Google Scholar]
- 2. Nakashima T., Hayashi M., Fukunaga T., Kurata K., Oh-Hora M., Feng J. Q., Bonewald L. F., Kodama T., Wutz A., Wagner E. F., Penninger J. M., Takayanagi H. (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 [DOI] [PubMed] [Google Scholar]
- 3. Xiong J., Onal M., Jilka R. L., Weinstein R. S., Manolagas S. C., O'Brien C. A. (2011) Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Takayanagi H. (2010) New immune connections in osteoclast formation. Ann. N.Y. Acad. Sci. 1192, 117–123 [DOI] [PubMed] [Google Scholar]
- 5. Gingery A., Bradley E. W., Pederson L., Ruan M., Horwood N. J., Oursler M. J. (2008) TGF-β coordinately activates TAK1/MEK/AKT/NFκB and SMAD pathways to promote osteoclast survival. Exp. Cell Res. 314, 2725–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fong D., Bisson M., Laberge G., McManus S., Grenier G., Faucheux N., Roux S. (2013) Bone morphogenetic protein-9 activates Smad and ERK pathways and supports human osteoclast function and survival in vitro. Cell. Signal. 25, 717–728 [DOI] [PubMed] [Google Scholar]
- 7. Greenblatt M. B., Shim J. H., Glimcher L. H. (2010) TAK1 mediates BMP signaling in cartilage. Ann. N.Y. Acad. Sci. 1192, 385–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shim J. H., Greenblatt M. B., Xie M., Schneider M. D., Zou W., Zhai B., Gygi S., Glimcher L. H. (2009) TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J. 28, 2028–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sotillo Rodriguez J. E., Mansky K. C., Jensen E. D., Carlson A. E., Schwarz T., Pham L., MacKenzie B., Prasad H., Rohrer M. D., Petryk A., Gopalakrishnan R. (2009) Enhanced osteoclastogenesis causes osteopenia in Twisted gastrulation-deficient mice through increased BMP signaling. J Bone Miner Res 24, 1917–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Itoh K., Udagawa N., Katagiri T., Iemura S., Ueno N., Yasuda H., Higashio K., Quinn J. M., Gillespie M. T., Martin T. J., Suda T., Takahashi N. (2001) Bone morphogenetic protein 2 stimulates osteoclast differentiation and survival supported by receptor activator of nuclear factor-κB ligand. Endocrinology 142, 3656–3662 [DOI] [PubMed] [Google Scholar]
- 11. Jensen E. D., Pham L., Billington C. J., Jr., Espe K., Carlson A. E., Westendorf J. J., Petryk A., Gopalakrishnan R., Mansky K. (2010) Bone morphogenic protein 2 directly enhances differentiation of murine osteoclast precursors. J. Cell. Biochem. 109, 672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beppu H., Lei H., Bloch K. D., Li E. (2005) Generation of a floxed allele of the mouse BMP type II receptor gene. Genesis 41, 133–137 [DOI] [PubMed] [Google Scholar]
- 13. Petryk A., Anderson R. M., Jarcho M. P., Leaf I., Carlson C. S., Klingensmith J., Shawlot W., O'Connor M. B. (2004) The mammalian Twisted gastrulation gene functions in foregut and craniofacial development. Dev. Biol. 267, 374–386 [DOI] [PubMed] [Google Scholar]
- 14. Dempster D. W., Compston J. E., Drezner M. K., Glorieux F. H., Kanis J. A., Malluche H., Meunier P. J., Ott S. M., Recker R. R., Parfitt A. M. (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 28, 2–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kanatani M., Sugimoto T., Kaji H., Kobayashi T., Nishiyama K., Fukase M., Kumegawa M., Chihara K. (1995) Stimulatory effect of bone morphogenetic protein-2 on osteoclast-like cell formation and bone-resorbing activity. J. Bone Miner. Res. 10, 1681–1690 [DOI] [PubMed] [Google Scholar]
- 16. Kaneko H., Arakawa T., Mano H., Kaneda T., Ogasawara A., Nakagawa M., Toyama Y., Yabe Y., Kumegawa M., Hakeda Y. (2000) Direct stimulation of osteoclastic bone resorption by bone morphogenetic protein (BMP)-2 and expression of BMP receptors in mature osteoclasts. Bone 27, 479–486 [DOI] [PubMed] [Google Scholar]
- 17. Okamoto M., Murai J., Imai Y., Ikegami D., Kamiya N., Kato S., Mishina Y., Yoshikawa H., Tsumaki N. (2011) Conditional deletion of Bmpr1a in differentiated osteoclasts increases osteoblastic bone formation, increasing volume of remodeling bone in mice. J. Bone Miner. Res. 26, 2511–2522 [DOI] [PubMed] [Google Scholar]
- 18. Pham L., Beyer K., Jensen E. D., Rodriguez J. S., Davydova J., Yamamoto M., Petryk A., Gopalakrishnan R., Mansky K. C. (2011) Bone morphogenetic protein 2 signaling in osteoclasts is negatively regulated by the BMP antagonist, Twisted gastrulation. J. Cell. Biochem. 112, 793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nohe A., Hassel S., Ehrlich M., Neubauer F., Sebald W., Henis Y. I., Knaus P. (2002) The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J. Biol. Chem. 277, 5330–5338 [DOI] [PubMed] [Google Scholar]
- 20. Yu P. B., Beppu H., Kawai N., Li E., Bloch K. D. (2005) Bone morphogenetic protein (BMP) type II receptor deletion reveals BMP ligand-specific gain of signaling in pulmonary artery smooth muscle cells. J. Biol. Chem. 280, 24443–24450 [DOI] [PubMed] [Google Scholar]
- 21. Oh S. P., Seki T., Goss K. A., Imamura T., Yi Y., Donahoe P. K., Li L., Miyazono K., ten Dijke P., Kim S., Li E. (2000) Activin receptor-like kinase 1 modulates transforming growth factor-β 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 97, 2626–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li X., Udagawa N., Itoh K., Suda K., Murase Y., Nishihara T., Suda T., Takahashi N. (2002) p38 MAPK-mediated signals are required for inducing osteoclast differentiation but not for osteoclast function. Endocrinology 143, 3105–3113 [DOI] [PubMed] [Google Scholar]
- 23. Lee S. E., Woo K. M., Kim S. Y., Kim H. M., Kwack K., Lee Z. H., Kim H. H. (2002) The phosphatidylinositol 3-kinase, p38, and extracellular signal-regulated kinase pathways are involved in osteoclast differentiation. Bone 30, 71–77 [DOI] [PubMed] [Google Scholar]
- 24. Gunnell L. M., Jonason J. H., Loiselle A. E., Kohn A., Schwarz E. M., Hilton M. J., O'Keefe R. J. (2010) TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J Bone Miner Res. 25, 1784–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mizukami J., Takaesu G., Akatsuka H., Sakurai H., Ninomiya-Tsuji J., Matsumoto K., Sakurai N. (2002) Receptor activator of NF-κB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol. Cell. Biol. 22, 992–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]