

Abstract
Objective:
To determine the extent of an inherited contribution to amyotrophic lateral sclerosis (ALS) mortality.
Methods:
Death certificates (DCs) from 1904 to 2009 were analyzed from patients with at least 3 generations recorded in the Utah Population Database, a genealogic and medical database of more than 2 million Utah residents. Among probands whose DCs listed ALS, the relative risk (RR) of death with ALS was determined among spouses and first- through fifth-degree relatives, using birth year-, sex-, and birthplace-matched cohorts.
Results:
Eight hundred seventy-three patients with ALS met the inclusion criteria. Among 3,531 deceased first-degree relatives of probands, the RR of dying with ALS was increased compared with control cohorts (RR = 4.91, 95% confidence interval 3.36, 6.94). The RR of dying with ALS was also increased among 9,386 deceased second-degree relatives (RR = 2.85, 95% confidence interval 2.06, 3.84). The RR of dying with ALS was not increased among third- through fifth-degree relatives. More affected first-degree relatives were male (p = 0.014). No cases of conjugal ALS were observed.
Conclusions:
This study is suggestive of familial clustering in excess of expected for ALS. Our results confirm the results of prior studies of familial ALS, suggesting applicability of our findings to other mixed European populations. Furthermore, this work expands on previous studies by quantifying the RR of ALS among more distant relatives. The use of mortality data obtained from DCs reduces the ascertainment and recall bias of many previous studies. Finally, the excess of ALS among second-degree relatives and lack of conjugal ALS are strongly supportive of a genetic contribution.
Amyotrophic lateral sclerosis (ALS) is a largely sporadic neurodegenerative disease, although a minority of patients have a clinically indistinguishable familial form of ALS (fALS). Despite common use of the term “fALS,” there is no universally accepted definition.1 The most frequently used definition requires a history of ALS in the proband and in at least one first- or second-degree relative.2 Prior studies examining the incidence of fALS have been limited by recall bias and were often unable to include relatives beyond the first degree.2 Thus, these studies may underestimate the true incidence of fALS and do not quantify risk in more distant relatives. The listing of ALS on death certificates (DCs) from a massive patient and genealogic database, such as the Utah Population Database (UPDB), may serve to provide a more accurate estimate of the frequency of fALS and the risk in extended relatives. Because ALS is clinically well recognized, progresses rapidly, and is inevitably fatal, accuracy of coding of ALS on DCs is estimated to be at least 70% to 90%.3 The purpose of this study was to define familial clustering of ALS mortality using the UPDB and Utah DCs. To our knowledge, this study represents the first large population-based study to use mortality data to estimate the familial relative risk (RR) in ALS beyond first-degree relatives. This study provides an improved understanding of the risk of ALS among extended family members of patients with ALS.
METHODS
Genealogic data.
The UPDB is a computerized genealogic/medical resource created from the linkage of multiple data sources. Genealogic data have been record-linked to disease data for the state, including all Utah DCs from 1904. More than 2.5 million individuals in the UPDB belong to genealogies with at least 3 generations of data; some pedigrees extend to 15 generations connecting to the Utah pioneers. The resource includes genealogic data for the original Utah pioneers (members of the Church of Jesus Christ of Latter-day Saints, or Mormons) and their descendants.4 Utah's founding pioneers were composed of a sizable, largely unrelated mixture of European population.5 This population continued to have high rates of immigration for years after its founding in 1847.5 Studies using pedigree data, migration matrices, and isonymy have all shown low levels of inbreeding in the founding population of Utah.6
Inclusion criteria.
The genetic relationships among the individuals in the UPDB whose Utah DC listed ALS as a cause of death between 1904 and 2009 have been analyzed to describe the familiality of death from ALS. The cause of death on Utah DCs was previously encoded using ICD codes, revisions 6–10. For deaths occurring before 1956, ICD-10 coding was retrospectively assigned. ICD codes were used to identify patients in the database with ALS listed as the primary or a contributing cause of death on a DC (DCs were limited to only one cause of death until 1980) with at least 3 generations of genealogy in the UPDB. Given the evolving ICD coding system, these codes include ALS as well as other motor neuron diseases, including progressive muscular atrophy, progressive bulbar palsy, and progressive lateral sclerosis, but exclude spinal muscular atrophy, which has been indicated as the most complete method to capture patients using DCs.7 The frequency of deaths by ICD code is shown in table e-1 on the Neurology® Web site at www.neurology.org. For simplicity, we use the term ALS to describe the studied condition, with the understanding that we are also referring to other, less common forms of motor neuron disease. Only patients whose age at death was greater than 14 years were included because younger deaths were more likely to represent cases of miscoded spinal muscular atrophy rather than ALS.
RR in relatives.
To estimate the RR of death with ALS among relatives, the observed rate of death with ALS in relatives was compared with the expected rate of death with ALS calculated internally in the UPDB, as follows. All individuals in the UPDB who belong to at least 3 generations of genealogy, and who have a coded cause of death were assigned to 1 of 132 birth year- (5 years), sex-, and birthplace-specific (Utah or not) cohorts. The rate of death with ALS for each cohort was estimated as the total number of individuals with ALS reported as a cause, or contributing cause, of death in each cohort, divided by the total number of individuals with a DC in the cohort.
The expected number of relatives dying with ALS was estimated by counting all relatives of probands who have a DC (by cohort, each relative counted only once regardless of how many times they are identified as a relative of the degree of interest), then multiplying the number of deceased relatives (per cohort) by the cohort-specific rate of death with ALS, then summing over all cohorts. R = observed/expected is an unbiased estimator of RR and can be calculated for different relationships. Two-sided probabilities were calculated under the null hypothesis RR = 1.0, under the assumption that the number of observed deaths follows a Poisson distribution with mean equal to the expected number of deaths.
Genealogical Index of Familiality.
The Genealogical Index of Familiality (GIF) statistic was developed specifically for the UPDB.8 The GIF analysis considers all genetic relationships between cases and measures the average relatedness among all pairs within a set of individuals. The relatedness measure implements the Malécot coefficient of kinship,9 defined as the probability that randomly selected homologous genes from the 2 individuals are identical by descent from a common ancestor. For example, for siblings, the coefficient is 0.25 (1/22); for grandparent/grandchild, the coefficient is 0.125 (1/23); and for first cousins, the coefficient is 0.0625 (1/24). The contribution to the GIF statistic is smaller for pairs with a greater genetic distance. The case GIF is defined as the average of the coefficients of kinship between all possible pairs of cases (×105). To test the hypothesis of no excess relatedness among the set of all deaths with ALS, the case GIF was compared with the empirical distribution of GIF statistics estimated from 1,000 sets of matched controls. Controls were randomly selected from all individuals with genealogic data and a DC, and matched to cases by birth cohort (5 years), sex, and birthplace (Utah or not). These analytical methods, including GIF analysis, have previously been applied to describe the familial and genetic contribution to mortality for multiple phenotypes including intracranial aneurysms,10 influenza,11 and asthma.12
The GIF statistic can also be estimated while ignoring all close relationships (relationships closer than first cousins); this allows a test of the hypothesis that excess relatedness has been observed among distant relatives. This test is termed the distant GIF or dGIF test and allows determination of whether the excess familial clustering observed could all be due to shared environmental effects.
Standard protocol approvals, registrations, and patient consents.
This research was limited to the analysis of unidentifiable data. There was no contact with human subjects, thus no informed consent was required.
RESULTS
Eight hundred seventy-three individuals meeting inclusion criteria were identified in the UPDB from 1904 to 2009. As shown in table e-1, the majority of patients were identified with the most recent version of ICD codes. Among all records from 1904 to 2009 with at least 3 generations of data, the lifetime risk of ALS is 1 in 800. When results are restricted to the time period between 1990 and 2009, the lifetime risk of ALS is 1 in 391. The mean age of death of patients with ALS is 66 years, with a peak in the 65 to 69 age group (n = 146). The average age of death is slightly higher in females than males; the age of death with ALS has increased over time for both sexes. The majority of the patients (all but 6) were white.
GIF analysis was used to test whether there was an excess of relatedness between cases compared with the expected relatedness in the UPDB population, as estimated from age-, sex-, and birthplace-matched sets of controls. The sample size, average case relatedness (case GIF), mean control relatedness (control GIF), and empirical significance are shown in table 1. GIF analysis demonstrated an overall excess of relatedness among probands compared with controls (p < 0.001). The results for the dGIF analysis, which ignores close relationships while testing for an excess of relatedness, are also shown in table 1; no excess distant relatedness was observed (p = 0.412).
Table 1.
GIF and dGIF analysis

RRs of dying with ALS were estimated in relatives of probands and are shown in table 2. The RR of death with ALS is significantly increased in the 3,531 deceased first-degree relatives of individuals dying with ALS (RR = 4.91, 95% confidence interval [CI] 3.36, 6.94; p < 0.0001). When analysis of first-degree relatives is restricted to the more recent time period between 1990 and 2009, the estimated RR = 7.23 (95% CI 4.21, 11.59; p < 0.0001). RR is also significantly increased among 9,386 deceased second-degree relatives of probands (RR = 2.85, 95% CI 2.06, 3.84; p = 0.0001). RR of dying with ALS did not significantly differ from 1.0 among third-, fourth-, or fifth-degree relatives of probands. When fALS is defined as at least one first-degree relative affected, the rate of fALS cases is 3.7%. When the definition is expanded to include affected first- or second-degree relatives, the rate of fALS cases is 8.6%.
Table 2.
Estimated RR of death with ALS among relatives of individuals dying with ALS
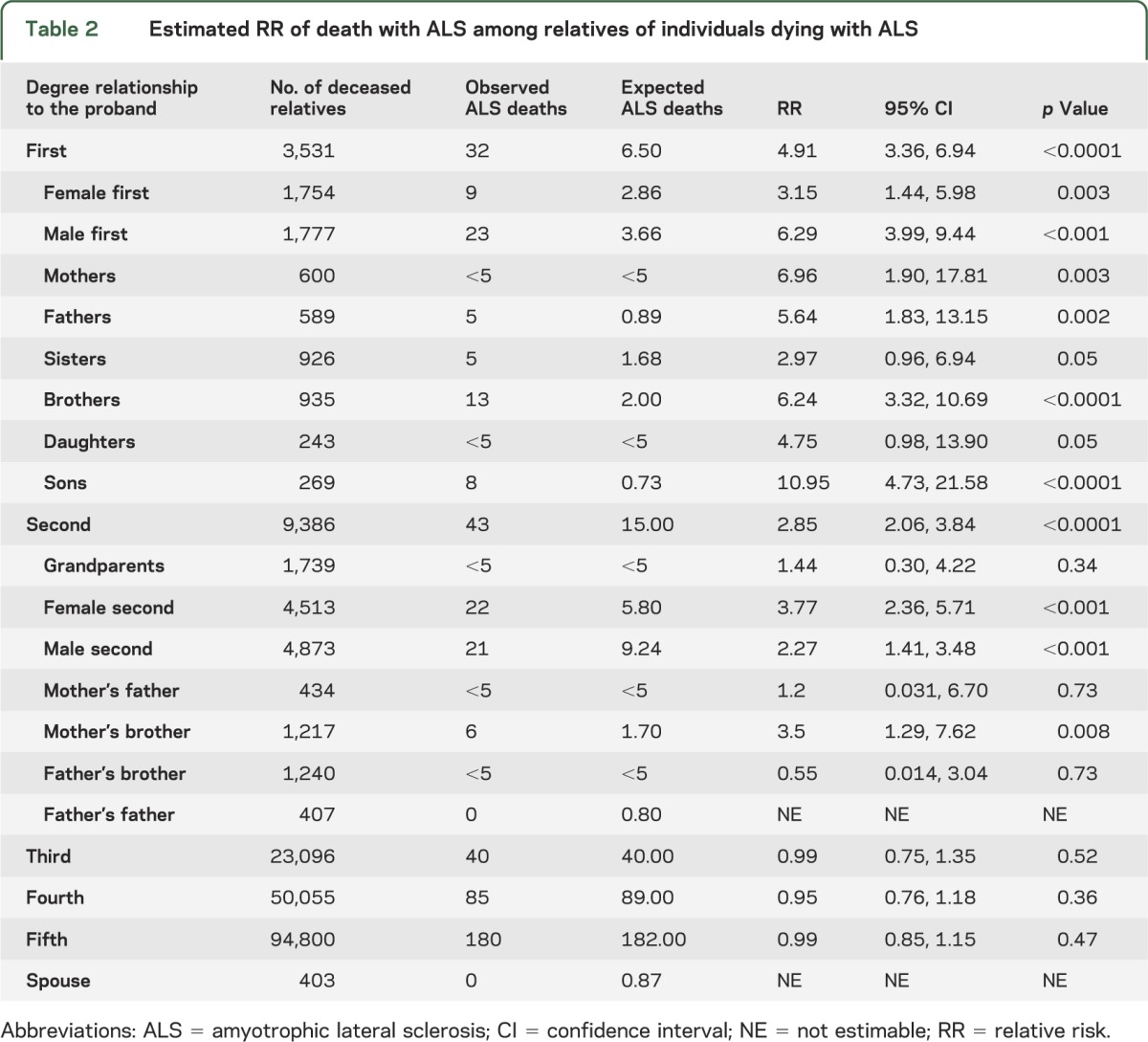
Estimated RRs by sex are shown in table 2 and figure 1. There is a male predominance (male n = 495/female n = 378) of death with ALS of 1.31 that is relatively continuous throughout 1904 to 2010, as shown in figure e-1. The RR for first-degree male relatives is higher than the RR for female first-degree relatives (RR = 6.29, 95% CI 3.99, 9.44; p < 0.001; and RR = 3.15, 95% CI 1.44, 5.98; p = 0.003, respectively). The likelihood of dying with ALS is significantly increased in the 1,777 deceased first-degree male relatives (observed = 23) vs the 1,754 deceased first-degree female relatives (observed = 9) (p = 0.014 by χ2 test).
Figure 1. RR of death with ALS divided into sex and relationship from the proband.

Square = male; circle = female; triangle = not sex-specific; red = significant; blue = nonsignificant or not estimable; black = proband; n = number of relatives. ALS = amyotrophic lateral sclerosis; CI = confidence interval; OD = observed death; RR = relative risk.
The RR was further examined for precise relationships of probands (table 2 and figure 1). The RR for brothers of probands is significantly increased (RR = 6.24, 95% CI 3.32, 10.69; p < 0.0001), and the RR for sisters of probands trends toward increase; however, probably because of sample size, these data did not reach statistical significance (RR = 2.97, 95% CI 0.96, 6.94; p = 0.05). The RR of dying with ALS among sons of probands is significantly increased (RR = 10.95, 95% CI 4.73, 21.58; p < 0.0001), and the RR of dying with ALS among daughters of probands trends toward increase; however, probably because of sample size, these data did not reach statistical significance (RR = 4.75, 95% CI 0.98, 13.90; p = 0.05). The RR of dying with ALS for parents of probands is significantly increased for both mothers and fathers (6.96 for mothers and 5.64 for fathers). The RR of dying with ALS is significantly increased among maternal uncles (RR = 3.50, 95% CI 1.29, 7.62; p = 0.008) but is not increased among paternal uncles (RR = 0.55, 95% CI 0.02, 3.04; p = 0.73). There were no identified spouses of probands who also died with ALS.
DISCUSSION
We have shown familial clustering in excess of expected for ALS using mortality data and a large population-based database. These data, including the mean age of death, overall familial risk, and increased male predominance, are in agreement with previous studies. A recent meta-analysis suggested that the frequency of fALS is approximately 5%, and our rate of 8.6% is likely higher because of the inclusion of second-degree relatives (the rate is 3.6% when limited to affected first-degree relatives) and the reduction of ascertainment and recall bias. Given that our results confirm prior findings, this serves to validate our data as representative of the mixed European populations that have predominately been studied. We extend these prior studies by determining that there is no increased risk of ALS among third- through fifth-degree relatives. While there is no uniform agreement of the definition of fALS, it is usually defined as ALS in either a first- or second-degree relative of a proband.1 Our study provides clear support for the use of this definition.
These data also have implications for genetic counselors, neurologists, and other clinicians involved in the care and monitoring of patients with ALS and their family members. However, the conclusions of this study have to be applied to the individual patient with caution. It is our hope that these data, when regarded in the context of other ALS studies, will serve to enable informative and appropriate conversations between patients with ALS and their caregivers.
This study also provides insights that may catalyze additional research in the field of ALS. The increase in familial clustering, especially among second-degree relatives, as well as a lack of conjugal ALS in our study population, most strongly supports the role of genetic factors in the etiology of ALS. However, we recognize that our study does not exclude an environmental contribution to fALS. The male predominance in ALS is widely reported, and a recent meta-analysis of population studies showed a male predominance of 1.3-fold, consistent with our estimate.13 Less frequently reported is the male predominance in fALS, which is most likely attributable to lack of power in most fALS studies. We report a higher risk in first-degree male relatives (approximately 6) than in first-degree female relatives (and RRs are not significantly increased in female offspring and siblings). Furthermore, our study suggests that sons of patients dying with ALS have a more than 10-fold RR of dying with ALS. We believe that the significant excess of RR among males in our study has implications for understanding genetic and/or environmental aspects of the disease. Our study reconfirms and extends similar findings over the past 2 decades.14–17 There are multiple explanations for the male predominance in fALS and while this discussion focuses on genetic causes for this increase, environmental interactions more common to males, such as occupational hazards, could also account for these differences.
It is estimated that mutations in genes known to cause or contribute to ALS account for approximately 65% of fALS cases.18 Because the Utah population is of mixed European background, it is likely that a similar percentage applies to the studied population. With the exception of the rare X-linked mutation in UBQLN2 and rare case reports of SOD1 having a reduced penetrance in females, none of the known genes have any major sexual dimorphism.19–22 Thus, some of the approximately 35% of undiscovered genetic contributions may account for these differences. In addition to X-linked inheritance, sex-specific imprinting and sex-specific effects on autosomal genes are of interest.23 Another possible cause of the noted sexual dimorphism is mitochondrial involvement, which has long been implicated to have a role in ALS.24 Our data strongly support additional efforts investigating sexual dimorphism in fALS, as these may also help to identify additional involved genes and help explain the overall male predominance in ALS.
One of the inherent limitations of this study over such an extended period of time is the deficiency of ALS cases in the early years studied. In the period between 1904 and 2009, the lifetime risk of ALS in our study is lower than expected (1:800).25 When our data are restricted to only include deaths from 1990 to 2009, the lifetime risk of ALS is 1:391, which is consistent with typically described risks for ALS in a mixed European population.25 Prior DC-based studies show similar findings of increasing number of ALS cases, which was attributed to underdiagnosis of ALS as well as decreased life expectancy, and not to an increase in the incidence of the disease.26,27 While this likely explains the majority of the discrepancy in incidence, another consideration specific to our population is the lack of a long-established referral center for patients with ALS within the state (started in 1990), which may have led to an underdiagnosis of ALS. Other limitations include potential inaccuracies in DC coding, missing or misrepresented genealogic data, and censoring of cause of death information for individuals dying outside of Utah. Most of these limitations lower the likelihood of identifying probands or their relatives, which suggests that the true familial risks of ALS may even be higher than estimated.
Despite these limitations, this is one of the most comprehensive studies of the risk of ALS among relatives with the disease. Our study provides further evidence of an increased risk of ALS among close family members (especially males), a rejection of the increased risk of ALS among distant relatives, a starting point for conversations among patients with ALS, families, and caregivers, and information that may help to direct research in the field.
Supplementary Material
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- CI
confidence interval
- DC
death certificate
- dGIF
distant Genealogical Index of Familiality
- fALS
familial amyotrophic lateral sclerosis
- GIF
Genealogical Index of Familiality
- ICD
International Classification of Diseases
- RR
relative risk
- UPDB
Utah Population Database
Footnotes
Editorial, page 13
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Dr. Gibson: study design, acquisition of data, and writing of manuscript. Ms. Figueroa: study supervision. Dr. Bromberg: analysis, interpretation, and critical revision of the manuscript for important intellectual content. Dr. Pulst: study concept, supervision, and critical revision of the manuscript for important intellectual content. Dr. Cannon-Albright: study design, acquisition of data, study supervision, and critical revision of the manuscript for important intellectual content.
STUDY FUNDING
Partial support for all datasets within the Utah Population Database was provided by Huntsman Cancer Institute, University of Utah, and the Huntsman Cancer Institute's Cancer Center Support grant, P30 CA42014, from the National Cancer Institute.
DISCLOSURE
S. Gibson, K. Figueroa, and M. Bromberg report no disclosures. Partial funding support for S.-M. Pulst was provided by the Noorda Foundation and grants R01NS033123 and RC4NS073009 from the NIH. Data collection for this publication was supported by R01, National Library of Medicine grant LM009331 (to L. Cannon-Albright). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Byrne S, Elamin M, Bede P, Hardiman O. Absence of consensus in diagnostic criteria for familial neurodegenerative diseases. J Neurol Neurosurg Psychiatry 2012;83:365–367 [DOI] [PubMed] [Google Scholar]
- 2.Byrne S, Walsh C, Lynch C, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2011;82:623–627 [DOI] [PubMed] [Google Scholar]
- 3.Chio A, Magnani C, Oddenino E, Tolardo G, Schiffer D. Accuracy of death certificate diagnosis of amyotrophic lateral sclerosis. J Epidemiol Community Health 1992;46:517–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skolnick MH. The Utah genealogical database: a resource for genetic epidemiology. In: Cairns J, Lyon JH, Skolnick MH, editors. Banbury Report No. 4: Cancer Incidence in Defined Populations. New York: Cold Spring Harbor Laboratory; 1980:285–297 [Google Scholar]
- 5.McLellan T, Jorde LB, Skolnick MH. Genetic distances between the Utah Mormons and related populations. Am J Hum Genet 1984;36:836–857 [PMC free article] [PubMed] [Google Scholar]
- 6.Jorde LB. Inbreeding in the Utah Mormons: an evaluation of estimates based on pedigrees, isonymy, and migration matrices. Ann Hum Genet 1989;53:339–355 [DOI] [PubMed] [Google Scholar]
- 7.Marin B, Couratier P, Preux PM, Logroscino G. Can mortality data be used to estimate amyotrophic lateral sclerosis incidence? Neuroepidemiology 2011;36:29–38 [DOI] [PubMed] [Google Scholar]
- 8.Hill JR. A survey of cancer sites by kinship in the Utah Mormon population. In: Cairns J, Lyon JH, Skolnick MH, editors. Banbury Report No. 4: Cancer Incidence in Defined Populations. New York: Cold Spring Harbor Laboratory; 1980:299–318 [Google Scholar]
- 9.Malécot G. Les Mathematiques de L'heredite. Paris: Masson & Cie; 1948 [Google Scholar]
- 10.Cannon Albright LA, Camp NJ, Farnham JM, MacDonald J, Abtin K, Rowe KG. A genealogical assessment of heritable predisposition to aneurysms. J Neurosurg 2003;99:637–643 [DOI] [PubMed] [Google Scholar]
- 11.Albright FS, Orlando P, Pavia AT, Jackson GG, Cannon Albright LA. Evidence for a heritable predisposition to death due to influenza. J Infect Dis 2008;197:18–24 [DOI] [PubMed] [Google Scholar]
- 12.Teerlink CC, Camp NJ, Bansal A, et al. Significant evidence for linkage to chromosome 5q13 in a genome-wide scan for asthma in an extended pedigree resource. Eur J Hum Genet 2009;17:636–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCombe PA, Henderson RD. Effects of gender in amyotrophic lateral sclerosis. Gend Med 2010;7:557–570 [DOI] [PubMed] [Google Scholar]
- 14.Leone M. Parental sex effect in familial amyotrophic lateral sclerosis. Neurology 1991;41:1292–1294 [DOI] [PubMed] [Google Scholar]
- 15.Leone M, De Angelis MS, Giordano M, Mutani R. Influence of ancestral gender on transmission of familial amyotrophic lateral sclerosis. Lancet 1994;344:1639. [DOI] [PubMed] [Google Scholar]
- 16.Fang F, Kamel F, Lichtenstein P, et al. Familial aggregation of amyotrophic lateral sclerosis. Ann Neurol 2009;66:94–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanby MF, Scott KM, Scotton W, et al. The risk to relatives of patients with sporadic amyotrophic lateral sclerosis. Brain 2011;134:3454–3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11:323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011;477:211–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim HY, Ki CS, Koh SH, Park KH, Sunwoo IN, Kim SH. Clinical characteristics of familial amyotrophic lateral sclerosis with a Phe20Cys mutation in the SOD1 gene in a Korean family. Amyotroph Lateral Scler 2007;8:73–78 [DOI] [PubMed] [Google Scholar]
- 21.Aoki M, Abe K, Houi K, et al. Variance of age at onset in a Japanese family with amyotrophic lateral sclerosis associated with a novel Cu/Zn superoxide dismutase mutation. Ann Neurol 1995;37:676–679 [DOI] [PubMed] [Google Scholar]
- 22.Murakami T, Warita H, Hayashi T, et al. A novel SOD1 gene mutation in familial ALS with low penetrance in females. J Neurol Sci 2001;189:45–47 [DOI] [PubMed] [Google Scholar]
- 23.Wijchers PJ, Festenstein RJ. Epigenetic regulation of autosomal gene expression by sex chromosomes. Trends Genet 2011;27:132–140 [DOI] [PubMed] [Google Scholar]
- 24.Martin LJ. Biology of mitochondria in neurodegenerative diseases. Prog Mol Biol Transl Sci 2012;107:355–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 2011;7:639–649 [DOI] [PubMed] [Google Scholar]
- 26.Chio A, Magnani C, Schiffer D. Amyotrophic lateral sclerosis mortality in Italy, 1958 to 1987: a cross-sectional and cohort study. Neurology 1993;43:927–930 [DOI] [PubMed] [Google Scholar]
- 27.Dean G, Quigley M, Goldacre M. Motor neuron disease in a defined English population: estimates of incidence and mortality. J Neurol Neurosurg Psychiatry 1994;57:450–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.