

Abstract
Objectives:
We determined whether head trauma was associated with amyloid deposition and neurodegeneration among individuals who were cognitively normal (CN) or had mild cognitive impairment (MCI).
Methods:
Participants included 448 CN individuals and 141 individuals with MCI from the Mayo Clinic Study of Aging who underwent Pittsburgh compound B (PiB)-PET, fluorodeoxyglucose-PET, and MRI. Head trauma was defined as a self-reported brain injury with at least momentary loss of consciousness or memory. Regression models examined whether head trauma was associated with each neuroimaging variable (assessed as continuous and dichotomous measures) in both CN and MCI participants, controlling for age and sex.
Results:
Among 448 CN individuals, 74 (17%) self-reported a head trauma. There was no difference in any neuroimaging measure between CN subjects with and without head trauma. Of 141 participants with MCI, 25 (18%) self-reported a head trauma. MCI participants with a head trauma had higher amyloid levels (by an average 0.36 standardized uptake value ratio units, p = 0.002).
Conclusions:
Among individuals with MCI, but not CN individuals, self-reported head trauma with at least momentary loss of consciousness or memory was associated with greater amyloid deposition, suggesting that head trauma may be associated with Alzheimer disease–related neuropathology. Differences between CN individuals and individuals with MCI raise questions about the relevance of head injury–PET abnormality findings in those with MCI.
Several studies have suggested that a history of head trauma is associated with an increased risk of Alzheimer disease (AD) dementia and results in an earlier age at onset of dementia compared to those without a head trauma.1−7 However, there is not complete consensus as several other large studies have not found an association.8–12 Further, the mechanism by which head trauma may increase the risk of AD dementia is not well understood. Autopsy studies have reported significant amyloid-β deposition in up to 30% of persons who die acutely following a brain injury, including children.13−15 In line with greater brain amyloid-β deposition, studies also suggest that CSF levels of amyloid-β 1-42 are lower, and tau levels higher, immediately after severe traumatic brain injuries.16,17 Assessments of the long-term effects of head trauma on in vivo measures of amyloid deposition and Alzheimer-related neurodegeneration are lacking. In the present study, we sought to determine whether a history of head trauma with at least momentary loss of consciousness and/or memory was associated with in vivo neuroimaging measures of Alzheimer-related neuropathology, including amyloid PET imaging, hippocampal volume, and fluorodeoxyglucose (FDG)-PET hypometabolism. We included cognitively normal (CN) individuals and those with mild cognitive impairment (MCI) enrolled in the population-based Mayo Clinic Study on Aging (MCSA).
METHODS
Subjects.
The MCSA is a population-based study of cognitive aging among Olmsted County, MN residents that began in October 2004 and initially enrolled individuals aged 70 to 89 years. Details of the study design and participant recruitment are provided elsewhere.18,19 All MCSA subjects undergo a clinical and cognitive assessment every 15 months that includes a study coordinator interview, neurologic evaluation, and neuropsychological test battery. Beginning in 2006, both newly and previously enrolled subjects were offered the opportunity to undergo PET imaging. The present analyses consisted of 589 individuals with a self-reported history of head trauma who underwent Pittsburgh compound B (PiB)-PET, FDG-PET, and MRI.
Standard protocol approvals, registrations, and patient consents.
The study protocols were approved by the Mayo Clinic and Olmsted Medical Center Institutional Review Boards. All subjects provided signed informed consent to participate in the study and in the imaging protocols.
Neuropsychological assessments.
A psychometrist administered a neuropsychological battery that included 9 tests covering 4 domains: 1) memory (Auditory Verbal Learning Test Delayed Recall Trial,20 Wechsler Memory Scale-Revised Logical Memory & Visual Reproduction II21); 2) language (Boston Naming Test22 and Category Fluency23); 3) executive function (Trail Making Test B24 and Wechsler Adult Intelligence Scale-Revised [WAIS-R] Digit Symbol subtest25); and 4) visuospatial skills (WAIS-R Picture Completion and Block Design subtests).25
For the purpose of determining impairment for an MCI diagnosis, the raw scores on each test were age-adjusted using normative data from the Mayo's Older American Normative Studies.26 The adjusted test scores within each domain were then summed and scaled to obtain domain-specific z scores to inform a clinical MCI diagnosis.18 In the current analyses and table 1, z scores were not adjusted for age but were created by averaging and scaling the individual z-scored tests within each domain. The domain z scores were then averaged and scaled to create a global z score. The measurement of subjective memory complaints was adapted from the first 5 questions on the Blessed Memory Test. Questions 1 to 4 were given a score of 2 if subjects reported “definitely worse than when I was younger,” 1 if they reported “slightly worse,” and 0 if they reported “as good or better.” Item 5 (problems remembering appointments correctly) was scored 1 for yes and 0 for no. Questions 1 to 5 were summed for a score ranging from 0 (no concern) to 9 (highest concern).
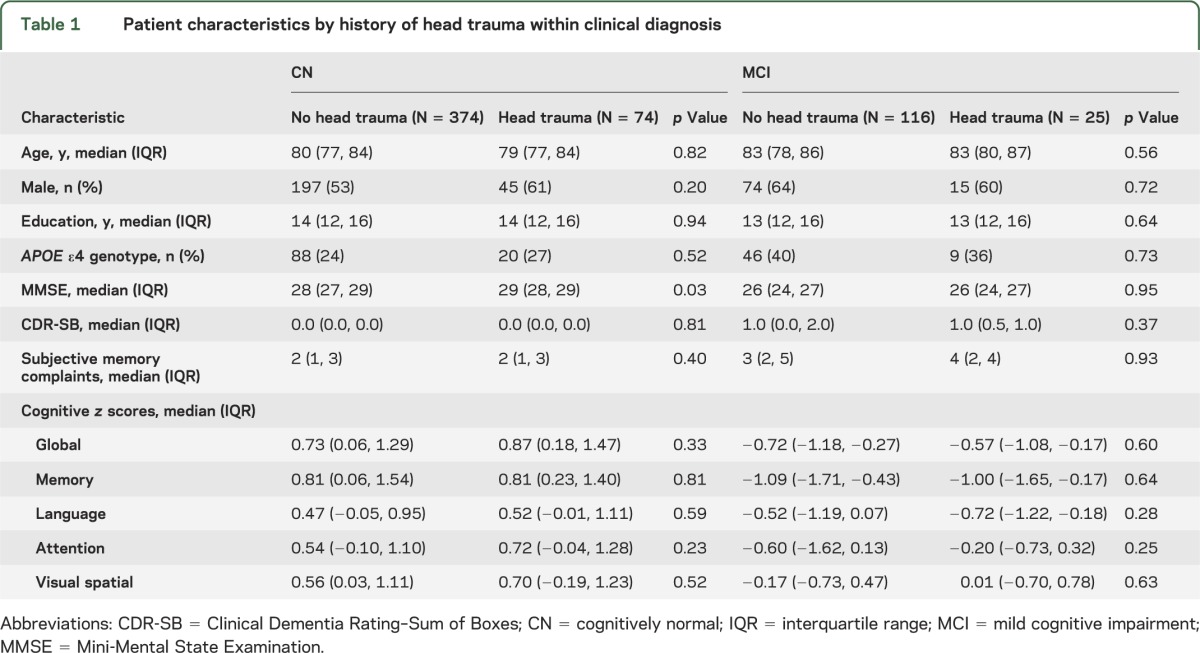
Table 1.
Patient characteristics by history of head trauma within clinical diagnosis
Diagnostic categories.
Impairment in a cognitive domain was assessed by comparing the person's domain score with the score in normal subjects from the same population. A score of ≤ −1.0 SD below the age-specific mean in the general population was considered possible cognitive impairment. A decision about impairment in a cognitive domain was not based on an algorithm but on a consensus agreement among the examining physician, study coordinator, and neuropsychologist, taking into account years of education, prior occupation, and visual or hearing deficits.18,19 MCI was diagnosed according to the following published criteria: cognitive concern by subject, informant (from Clinical Dementia Rating [CDR]), study coordinator, or physician; impairment in ≥1 of the 4 cognitive domains; essentially normal functional activities (from CDR and Functional Activities Questionnaire); and absence of dementia.27 Dementia was diagnosed according to the DSM-IV criteria.28
Assessment of head trauma.
Participants completed a medical history form in which they self-reported whether a previous head trauma had occurred. Participants were asked the following question: “Have you ever experienced any head injuries that led you to see a doctor, stay in the hospital, lose your memory, or become unconscious?” If yes, participants were asked “How many head injuries have you had?”, “How old were you at the time of the first injury?”, “Did this injury cause loss of memory or loss of consciousness?”, and “How long were you unconscious/unable to remember?” For the purposes of this study, participants were coded as having a head trauma only when it was accompanied by at least momentary loss of memory or consciousness.
Amyloid PET methods.
PET images were acquired using a PET/CT scanner (DRX, GE Healthcare). A CT image was obtained for attenuation correction. The 11C PiB-PET scan, consisting of four 5-minute dynamic frames, was acquired from 40 to 60 minutes after injection. Image analysis was done using our in-house fully automated image processing pipeline.29 An amyloid PET standardized uptake value ratio (SUVR) was formed by calculating the median uptake over voxels in the prefrontal, orbitofrontal, parietal, temporal, anterior cingulate, and posterior cingulate/precuneus regions of interest (ROIs) for each subject and dividing this meta-ROI by the median uptake over voxels in the cerebellar gray matter ROI of the atlas. We have previously shown in this cohort that 90% diagnostic sensitivity for clinically diagnosed AD dementia corresponds to a PiB-PET cutpoint of 1.5,30 and therefore included this cutpoint as our dichotomous outcome.
FDG-PET methods.
FDG-PET images were obtained on the same day 1 hour after the amyloid PET scan. FDG-PET scans were analyzed using the pipeline described above.29 The angular gyrus, posterior cingulate, and inferior temporal cortical ROIs defined an “Alzheimer signature” meta-ROI,31 which was normalized to pons and vermis.
Structural MRI methods.
All subjects underwent MRI scanning at 3T with a standardized protocol that included a 3D magnetization-prepared rapid acquisition gradient echo sequence. Hippocampal volume was measured with FreeSurfer software version 4.5.0. Each subject's raw hippocampal volume was adjusted by their total intracranial volume (TIV) such that the value represents the difference in cm3 between a subject's measured hippocampal volume and the subject's predicted volume based on the subject's TIV.30
Statistical methods.
Differences in variables between those with and without a head trauma were initially evaluated by clinical status (i.e., separately among CN and MCI participants) using Wilcoxon rank sum tests and χ2 tests. We first assessed the association between a self-reported history of head trauma and each neuroimaging measuring as a continuous outcome using linear regression controlling for age and sex. Because PiB-PET SUVR values tended to be skewed toward higher values, we analyzed these data on the log-transformed scale. To obtain estimated differences on the SUVR scale, we back-transformed the estimates and used bootstrap resampling to obtain confidence intervals (CIs). We note that results for PiB-PET varied little when we fit an untransformed model with or without bootstrap resampling.
We used logistic regression controlling for age and sex to examine whether head trauma was associated with increased odds of pathologically significant amyloid and neurodegenerative changes by dichotomizing each neuroimaging marker at the estimated 90th percentile of the AD distribution in this cohort.30 For example, this corresponds to a PiB-PET cutpoint of 1.5.
RESULTS
The characteristics of the 589 participants by clinical diagnosis and self-reported head trauma are shown in table 1. Compared to CN individuals, participants with MCI were more likely to have an APOE ε4 allele (39% vs 24%, p = <0.001), had more subjective memory complaints (median [interquartile range (IQR)]: 3 [2, 5] vs 2 [1, 3], p = <0.001), and performed significantly (p < 0.001) worse on all cognitive tests. Subjects with MCI were also older (median [IQR]: 83 [79, 86] vs 80 [77, 84], p < 0.001), and tended to be male (63% vs 54%, p = 0.06) and to have less education (median [IQR]: 13 [12, 16] vs 14 [12, 16]), p = 0.08). The median age of first self-reported head trauma did not differ by cognitive status (CN: 22 [12, 62] vs MCI: 23 [16, 70], p = 0.39) or sex (women: 32 [14, 66] vs men: 21 [14, 61], p = 0.39). Within the CN group, those with self-reported head trauma performed slightly better on the Mini-Mental State Examination (p = 0.03; see table 1). There were no other differences (all p > 0.05) between CN subjects with and without a history of head trauma in any demographic or cognitive characteristic, including the proportion with an APOE ε4 allele (table 1). There were also no differences in any factor by self-reported head trauma in the MCI group.
Frequency of self-reported head trauma in CN and MCI individuals.
The percentage of individuals self-reporting a head injury with at least momentary loss of consciousness or memory was similar between the CN and MCI groups (17% vs 18%, p = 0.74). The median number of years [IQR] between the age of the first self-reported head trauma and the neuroimaging measure also did not differ between CN and MCI participants (58 [17–67] years for CN vs 56 [14–67] years for MCI, p = 0.70).
Relationship between head trauma and neuroimaging measures among CN and MCI individuals.
Among CN individuals, there were no associations between head trauma and amyloid accumulation, hippocampal volume, or FDG-PET hypometabolism when examining the variables as either continuous or dichotomous measures (tables 2 and 3, figure).
Table 2.
Estimated difference in mean neuroimaging measure for head trauma vs no head trauma from linear regression models adjusted for age and sex
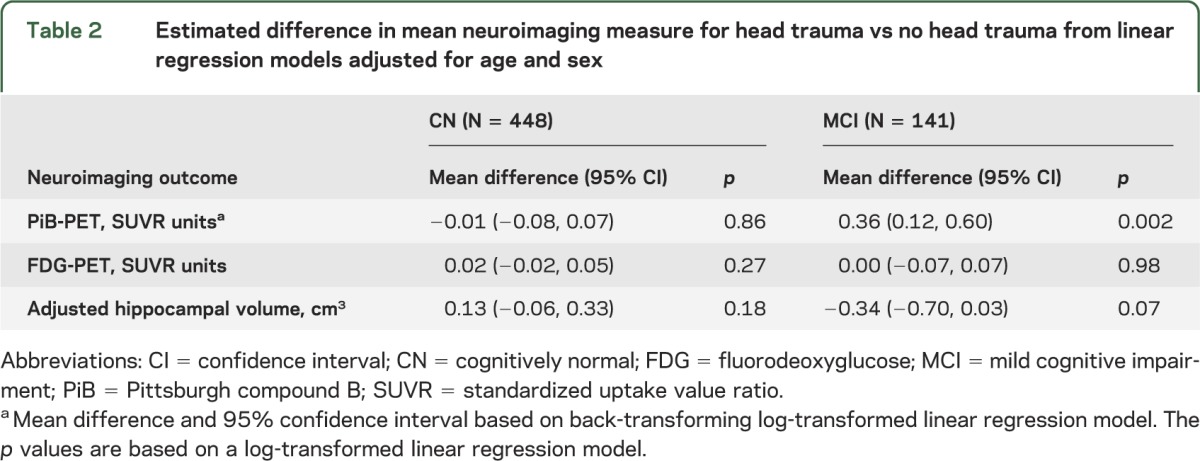
Table 3.
Odds of abnormal neuroimaging measure in CN and MCI participants for head trauma vs no head trauma based on logistic regression models adjusted for age and sex
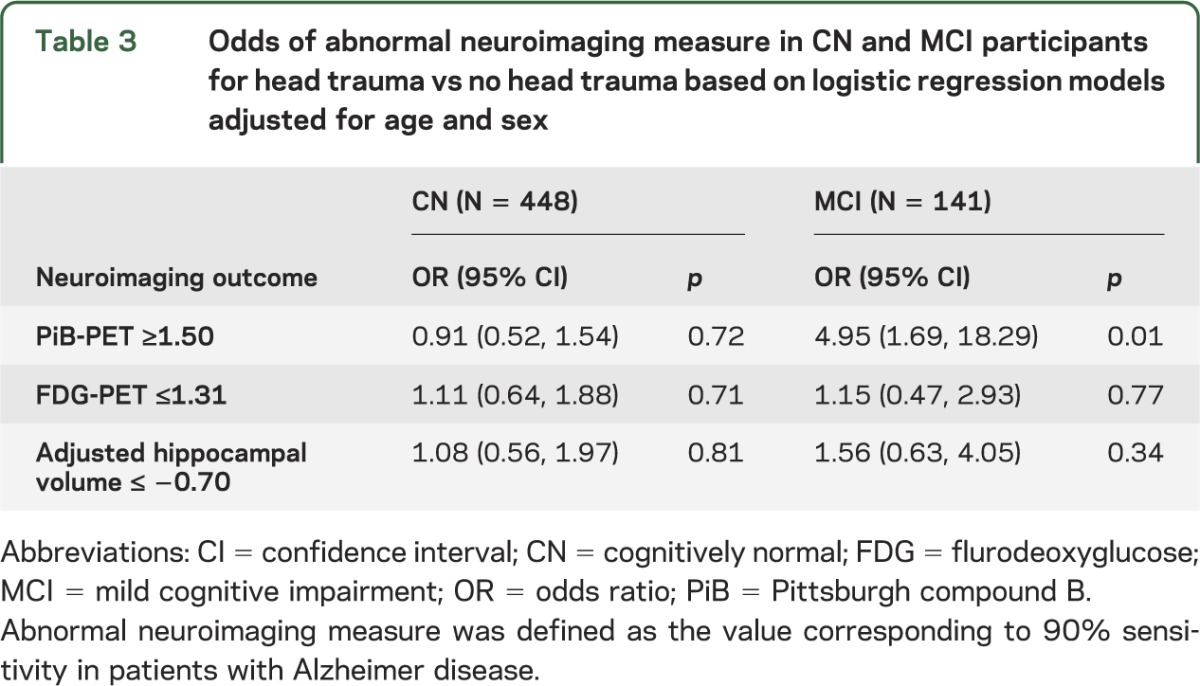
Figure. PiB-PET, FDG-PET, and adjusted hippocampal volume by cognitive status and history of head trauma.

Box plots with superimposed data points for PiB-PET (A), FDG-PET (B), and adjusted hippocampal volume (C). The p values indicate the difference between those with and without head trauma based on linear regression models adjusted for age and sex. Boxes indicate the median and quartiles, with vertical “whiskers” extending to the nearest data point within 1.5 times the value of the interquartile range. CN = cognitively normal; FDG = fluorodeoxyglucose; MCI = mild cognitive impairment; PiB = Pittsburgh compound B; SUVR = standardized update value ratio.
In contrast to CN individuals, a history of head trauma in individuals with a diagnosis of MCI was associated with higher global amyloid levels. In terms of SUVR units, the mean difference was +0.36 (95% CI 0.12‒0.60, p = 0.002; table 2 and figure). This corresponds to a mean level about 18% higher in those with vs without a head trauma. In terms of PiB positivity, defined as PiB-PET SUVR >1.5, subjects with MCI had nearly a 5-fold higher odds of elevated amyloid accumulation compared to those without a head trauma (odds ratio 4.95, 95% CI 1.69‒18.29; table 3). There were no associations (at p < 0.05) between head trauma and either FDG-PET or hippocampal volume when examined as continuous or dichotomous measures.
In additional analyses among CN individuals, there were no interactions with head trauma and APOE genotype for dichotomous or continuous measures of hippocampal volume, amyloid, or FDG-PET hypometabolism. The small number of MCI individuals with head trauma who had an APOE ε4 allele (n = 9) precluded us from examining an interactive effect within this group. However, there were no differences between the percentage of APOE ε4 allele carriers among MCI individuals with and without head trauma (36% vs 40%, p = 0.73).
DISCUSSION
In this study, we determined whether a history of head trauma with at least momentary loss of consciousness or memory was associated with in vivo measures of Alzheimer pathology in a population-based study of CN and MCI individuals aged 70 years and older. The frequency of self-reported head trauma did not differ by cognitive status (i.e., CN vs MCI). Among CN individuals, there were no associations between a history of head trauma and amyloid PET, FDG-PET hypometabolism, or hippocampal volumes. In contrast, among subjects with MCI, those with a self-reported head trauma had higher amyloid PET SUVR. Differences between the CN and MCI groups raise questions about the relevance of head trauma–PET abnormality findings in those with MCI.
Several epidemiologic studies have suggested that a history of head trauma is associated with an increased risk of AD,1−7 including a prospective study of hospitalized World War II Navy and Marine veterans with well-characterized nonpenetrating head injuries.5 In contrast, other large studies and meta-analyses have not found an increased risk of AD among those with head trauma.8–12 Our findings are in line with these latter studies in that the frequency of self-reported head trauma did not differ between CN and MCI cases. We expected to find at least a trend for more self-report in patients with MCI based on the literature and also due to the self-reported nature of the assessment and potential for recall bias; participants with cognitive problems may be more likely to seek an answer for their cognitive changes. Limiting the head trauma to at least momentary loss of consciousness or memory may have somewhat mitigated this potential recall bias. It is also possible that we did not see an association because those most susceptible to the adverse effects of head trauma developed AD and thus were not included in the present study because the MCSA initially enrolled individuals without dementia.
Studies in mice and rats report a post–traumatic brain injury increase in intra-axonal amyloid-β concentrations, but these animals do not develop amyloid plaques.32–34 Mice deficient in β-amyloid–converting enzymes have significantly improved pathologic and behavioral outcomes following a head injury,35 suggesting that the amyloid-β increases, even without plaque deposition, are detrimental and result in progressive brain atrophy.36 Swine models of head rotational acceleration, which more closely mimic human pathology, result in swollen axons, increased amyloid-β levels, and diffuse plaques in both gray and white matter.37–39
Among humans, CSF studies of patients with severe head trauma also reported lower amyloid-β 1-42 and/or higher tau levels within a month of injury.16,17 However, as these individuals had severe brain trauma, it is unclear whether milder head trauma would also be associated with in vivo measures of Alzheimer-related brain pathology in later life. In the current study, we did not find a relationship between self-reported head trauma and amyloid deposition, hippocampal volume, or FDG hypometabolism utilizing an “Alzheimer signature” meta-ROI31 among CN participants. In contrast, among individuals with MCI, head trauma was associated with significantly higher amyloid PET deposition but there were no significant findings for hippocampal volume or FDG-PET hypometabolism. This result is intriguing given that we did not see a difference in the frequency of head trauma between CN and MCI participants. There are several potential explanations for these findings. First, it is possible that the higher amyloid, which head trauma may have contributed to, resulted in progression to MCI and that is why we only see an association among the MCI group. Notably, however, there were no differences in any of the cognitive tests by self-reported head trauma within the CN or MCI groups, which we might expect if head trauma was associated with AD progression. Second, we examined whether the MCI group had a longer time between the reported age of the head trauma and neuroimaging measure than the CN group. If this was the case, it is possible that the CN group simply did not have as long a time to develop the neuropathology or associated cognitive symptoms. However, there were no differences between CN and MCI participants in the number of years between the head trauma and neuroimaging measures. Third, the present study included individuals aged 70 years and older. Studies have suggested that head trauma is associated with an earlier age at onset of AD, particularly among APOE ε4 carriers.4,5 It is possible that those most affected by head trauma, due either to genetic or other susceptibility or to more severe injuries, developed cognitive impairment or dementia prior to their age of enrollment in the MCSA and therefore are not included in this study. We did not find an interaction between APOE ε4 and head trauma in CN individuals. Given the small number of MCI individuals with a head trauma and an APOE ε4 allele, we were unable to accurately assess an interaction among this group. Lastly, an alternative explanation, albeit a somewhat controversial one, is that amyloid levels may be a response to neuronal (i.e., myelin) injury. Therefore, the association between head trauma and amyloid appears only in the MCI group because amyloid is a byproduct of the myelin repair process and occurs only after a critical level of demyelination has occurred.40
Limitations of the study warrant consideration. First, information on head trauma was self-reported, which is limited by recall bias. However, as there were no differences between CN and MCI individuals, it is unlikely that recall bias was an issue. Second, we had small numbers of individuals with more than one head trauma or with more than momentary loss of consciousness/memory, which limited our ability to assess an injury severity association. Despite these limitations, there are several strengths. The present study incorporated a large number of CN and MCI individuals from a population-based study with in vivo neuroimaging. Many of the previous studies that assessed the long-term effects of head trauma in relation to AD examined only clinical onset and did not have estimates of neuropathology. Those that did were autopsy samples of selected individuals and were not population based.
Head trauma with at least momentary loss of consciousness or memory was associated with greater amyloid deposition among subjects with MCI but not CN individuals. The lack of an association in CN individuals is contradictory but may be more reliable given the much larger sample size. However, the results in MCI suggest that the etiology of cognitive impairment in MCI individuals with head trauma is more likely to be AD pathology than in MCI individuals without head trauma. This is consistent with some human and animal data and deserves additional study because it would provide in vivo evidence for a cause and effect mechanistic link between prior head trauma and AD.
ACKNOWLEDGMENT
The authors would like to thank Dr. Cynthia L. Leibson for her helpful comments and advice throughout the project.
GLOSSARY
- AD
Alzheimer disease
- CDR
Clinical Dementia Rating
- CI
confidence interval
- CN
cognitively normal
- DSM-IV
Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition
- FDG
fluorodeoxyglucose
- IQR
interquartile range
- MCI
mild cognitive impairment
- MCSA
Mayo Clinic Study of Aging
- PiB
Pittsburgh compound B
- ROI
region of interest
- SUVR
standardized uptake value ratio
- TIV
total intracranial volume
- WAIS-R
Wechsler Adult Intelligence Scale-Revised
AUTHOR CONTRIBUTIONS
Dr. Mielke: drafting/revising the manuscript, including medical writing for content, and generated the first and final drafts; study concept or design; analysis and interpretation of data. Dr. Savica: drafting/revising the manuscript, including medical writing for content; study concept or design; analysis and interpretation of data. Ms. Wiste: drafting/revising the manuscript, statistical analysis, interpretation of the data. Mr. Weigand: drafting/revising the manuscript, statistical analysis, interpretation of the data. Dr. Vemuri: drafting/revising the manuscript, analysis and interpretation of data. Dr. Knopman: drafting/revising the manuscript, study concept or design, analysis and interpretation of data. Dr. Lowe: drafting/revising the manuscript, study concept or design, study supervision. Dr. Roberts: drafting/revising the manuscript, acquisition of data. Dr. Machulda: drafting/revising the manuscript, acquisition of data. Dr. Geda: drafting/revising the manuscript, acquisition of data. Dr. Petersen: drafting/revising the manuscript, obtaining funding, acquisition and interpretation of data. Dr. Jack: drafting/revising the manuscript, obtaining funding, acquisition of data, study supervision, study concept and design, analysis and interpretation of data.
STUDY FUNDING
This study was supported in part by NIH grants P50 AG016574, U01 AG006786, U01 AG037526, K01 MH068351, RO1 AG011378, and K01 AG028573. This study was also supported by the Robert Wood Johnson Foundation, the Alexander Family Alzheimer's Disease Research Professorship, GE Healthcare, the Elsie and Marvin Dekelboum Family Foundation, the MN Partnership for Biotechnology and Medical Genomics, and the Robert H. and Clarice Smith and Abigail van Buren Alzheimer's Disease Research Program, and was made possible by the Rochester Epidemiology Project (R01 AG034676).
DISCLOSURE
M. Mielke served as a consultant to Eli Lilly and receives research support from the NIH/NIA and the Alzheimer Drug Discovery Foundation. R. Savica, H. Wiste, and S. Weigand report no disclosures. P. Vemuri receives support from the NIH/NIA and Alzheimer's Association. D. Knopman serves as Deputy Editor for Neurology®; served on a Data Safety Monitoring Board for Lilly Pharmaceuticals; served as a consultant to TauRx Pharmaceuticals; was an investigator in clinical trials sponsored by Baxter, Elan Pharmaceuticals, and Forest Pharmaceuticals in the past 2 years; and receives research support from the NIH. V. Lowe serves on scientific advisory boards for Bayer Schering Pharma and GE Healthcare and receives research support from GE Healthcare, Siemens Molecular Imaging, the NIH (NIA, NCI), the MN Partnership for Biotechnology and Medical Genomics, and the Leukemia & Lymphoma Society. R. Roberts receives research support from the NIH/NIA and from Abbvie Health Economics and Outcomes Research. M. Machulda and Y. Geda report no disclosures. R. Petersen serves on scientific advisory boards for Pfizer, Inc., Janssen Alzheimer Immunotherapy, Elan Pharmaceuticals, and GE Healthcare; receives royalties from the publication of Mild Cognitive Impairment (Oxford University Press, 2003); and receives research support from the NIH/NIA. C. Jack provides consulting services for Siemens Healthcare. He receives research funding from the NIH (R01 AG011378, RO1 AG041851, RO1 AG037551, U01 HL096917, U01 AG032438, U01 AG024904) and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Graves AB, White E, Koepsell TD, et al. The association between head trauma and Alzheimer's disease. Am J Epidemiol 1990;131:491–501 [DOI] [PubMed] [Google Scholar]
- 2.Mortimer JA, van Duijn CM, Chandra V, et al. Head trauma as a risk factor for Alzheimer's disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 1991;20(suppl 2):S28–S35 [DOI] [PubMed] [Google Scholar]
- 3.O'Meara ES, Kukull WA, Sheppard L, et al. Head injury and risk of Alzheimer's disease by apolipoprotein E genotype. Am J Epidemiol 1997;146:373–384 [DOI] [PubMed] [Google Scholar]
- 4.Nemetz PN, Leibson C, Naessens JM, et al. Traumatic brain injury and time to onset of Alzheimer's disease: a population-based study. Am J Epidemiol 1999;149:32–40 [DOI] [PubMed] [Google Scholar]
- 5.Plassman BL, Havlik RJ, Steffens DC, et al. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 2000;55:1158–1166 [DOI] [PubMed] [Google Scholar]
- 6.Guo Z, Cupples LA, Kurz A, et al. Head injury and the risk of AD in the MIRAGE study. Neurology 2000;54:1316–1323 [DOI] [PubMed] [Google Scholar]
- 7.Wang HK, Lin SH, Sung PS, et al. Population based study on patients with traumatic brain injury suggests increased risk of dementia. J Neurol Neurosurg Psychiatry 2012;83:1080–1085 [DOI] [PubMed] [Google Scholar]
- 8.Mehta KM, Ott A, Kalmijn S, et al. Head trauma and risk of dementia and Alzheimer's disease: The Rotterdam Study. Neurology 1999;53:1959–1962 [DOI] [PubMed] [Google Scholar]
- 9.Launer LJ, Andersen K, Dewey ME, et al. Rates and risk factors for dementia and Alzheimer's disease: results from EURODEM pooled analyses. EURODEM Incidence Research Group and Work Groups. European Studies of Dementia. Neurology 1999;52:78–84 [DOI] [PubMed] [Google Scholar]
- 10.Katzman R, Aronson M, Fuld P, et al. Development of dementing illnesses in an 80-year-old volunteer cohort. Ann Neurol 1989;25:317–324 [DOI] [PubMed] [Google Scholar]
- 11.Dams-O'Connor K, Gibbons LE, Bowen JD, McCurry SM, Larson EB, Crane PK. Risk for late-life re-injury, dementia and death among individuals with traumatic brain injury: a population-based study. J Neurol Neurosurg Psychiatry 2013;84:177–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savica R, Parisi JE, Wold LE, Josephs KA, Ahlskog JE. High school football and risk of neurodegeneration: a community-based study. Mayo Clin Proc 2012;87:335–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts GW, Gentleman SM, Lynch A, Graham DI. beta A4 amyloid protein deposition in brain after head trauma. Lancet 1991;338:1422–1423 [DOI] [PubMed] [Google Scholar]
- 14.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry 1994;57:419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 2012;22:142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franz G, Beer R, Kampfl A, et al. Amyloid beta 1-42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology 2003;60:1457–1461 [DOI] [PubMed] [Google Scholar]
- 17.Magnoni S, Esparza TJ, Conte V, et al. Tau elevations in the brain extracellular space correlate with reduced amyloid-beta levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain 2012;135:1268–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 2008;30:58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petersen RC, Roberts RO, Knopman DS, et al. Prevalence of mild cognitive impairment is higher in men: the Mayo Clinic Study of Aging. Neurology 2010;75:889–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rey A. L'examen Clinique en Psychologie. Paris: Presses Universitaires de France; 1964 [Google Scholar]
- 21.Wechsler D. Manual for the Wechsler Memory Scale-revised. San Antonio: The Psychological Corporation; 1987 [Google Scholar]
- 22.Kaplan E, Goodglass H, Weintraub S. The Boston Naming Test. Philadelphia: Lea & Febiger; 1983 [Google Scholar]
- 23.Strauss E, Sherman EMS, Spreen O. A Compendium of Neuropsychological Tests. New York: Oxford University Press; 2006 [Google Scholar]
- 24.Reitan R. Validity of the trail making test as an indicator of organic brain damage. Percept Mot Skills 1958;8:271–276 [Google Scholar]
- 25.Wechsler D. Wechsler Adult Intelligence Scale-Revised [Manual]. San Antonio: Psychological Corporation; 1981 [Google Scholar]
- 26.Ivnik RJ, Malec JF, Smith GE, et al. Mayo's Older Americans Normative Studies: WAIS-R, WMS-R and AVLT norms for ages 56 through 97. Clin Neuropsychol 1992;6(suppl 1):1–104 [Google Scholar]
- 27.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004;256:183–194 [DOI] [PubMed] [Google Scholar]
- 28.American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders (DSM-IV). 4th ed Washington, DC: American Psychiatric Association; 1994 [Google Scholar]
- 29.Jack CR, Jr, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008;131:665–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jack CR, Jr, Knopman DS, Weigand SD, et al. An operational approach to National Institute on Aging-Alzheimer's Association criteria for preclinical Alzheimer disease. Ann Neurol 2012;71:765–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landau SM, Harvey D, Madison CM, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 2010;75:230–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murai H, Pierce JE, Raghupathi R, et al. Twofold overexpression of human beta-amyloid precursor proteins in transgenic mice does not affect the neuromotor, cognitive, or neurodegenerative sequelae following experimental brain injury. J Comp Neurol 1998;392:428–438 [DOI] [PubMed] [Google Scholar]
- 33.Smith DH, Nakamura M, McIntosh TK, et al. Brain trauma induces massive hippocampal neuron death linked to a surge in beta-amyloid levels in mice overexpressing mutant amyloid precursor protein. Am J Pathol 1998;153:1005–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakagawa Y, Nakamura M, McIntosh TK, et al. Traumatic brain injury in young, amyloid-beta peptide overexpressing transgenic mice induces marked ipsilateral hippocampal atrophy and diminished Abeta deposition during aging. J Comp Neurol 1999;411:390–398 [PubMed] [Google Scholar]
- 35.Loane DJ, Pocivavsek A, Moussa CE, et al. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med 2009;15:377–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith DH, Chen XH, Pierce JE, et al. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma 1997;14:715–727 [DOI] [PubMed] [Google Scholar]
- 37.Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol 2004;165:357–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer's disease? Nat Rev Neurosci 2010;11:361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith DH, Chen XH, Xu BN, McIntosh TK, Gennarelli TA, Meaney DF. Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. J Neuropathol Exp Neurol 1997;56:822–834 [PubMed] [Google Scholar]
- 40.Bartzokis G. Alzheimer's disease as homeostatic responses to age-related myelin breakdown. Neurobiol Aging 2011;32:1341–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]