

Abstract
Objective:
To describe the imaging and clinical features in type II (late-onset) Alexander disease (AxD).
Methods:
We retrospectively identified all cases of type II AxD evaluated at Mayo Clinic, Rochester from January 1996 to February 2012. Clinical and neuroimaging data abstracted from the record included age at onset of symptoms, age at diagnosis, first symptom, neurologic symptoms, physical/neurologic findings on examination, genetic testing and/or biopsy (if performed), and MRI findings.
Results:
Thirteen patients with type II AxD were identified. Median age at onset was 38 years (range: 12–63). Five patients were female. Eleven of 13 patients had atrophy of the medulla while all 13 had medullary T2 hyperintensity. In 7 patients, these brainstem regions showed patchy enhancement. Five subjects had T2 signal change in the middle cerebellar peduncle, with associated contrast enhancement in 4 subjects. Eleven of 12 patients with T2 fluid-attenuated inversion recovery (FLAIR) imaging had pial FLAIR signal change in the medulla. Nine of 12 patients with spinal cord imaging had cord atrophy, and 3 of 9 of these evaluated with contrast had cervical cord enhancement.
Conclusions:
Our study confirms prior reports of atrophy and signal change of the medulla and spinal cord in late-onset AxD. We expand on previous imaging studies by identifying middle cerebellar peduncle and pial FLAIR signal changes as important diagnostic clues. Variable patchy enhancement may occur in regions of T2 hyperintensity, leading to diagnostic uncertainty. In addition, we demonstrate that previously emphasized clinical features such as palatal tremor may not be common. We affirm that age at onset predicts clinical phenotype and imaging findings.
Alexander disease (AxD)1 is a leukodystrophy that is pathologically characterized by the presence of Rosenthal fibers. Mutations in the glial fibrillary acidic protein (GFAP) gene cause AxD.2 Traditionally, AxD has been categorized into infantile, juvenile, and adult forms according to the age at onset.3 MRI criteria for the infantile variant consist of frontal white matter changes; a periventricular rim with high T1 and low T2 signal; T2 hyperintensity (herein referred to as signal abnormality) involving the basal ganglia, thalamus, and brainstem; and contrast enhancement of particular gray and white matter structures (ventricular lining, periventricular rim of tissue, white matter of the frontal lobes, optic chiasm, fornix, basal ganglia, thalamus, dentate nucleus, and brainstem).4 Imaging and clinical features of late-onset AxD have been less frequently described. Available studies suggest that the clinical and radiologic presentation differs significantly from infantile-onset cases.5–11 Adult-onset cases appear to have less cerebral white matter involvement. Medullary and spinal cord atrophy occurs in most, but often with a mixture of additional findings that may present diagnostic challenges.5,12 In a review of more than 215 cases of AxD, AxD was divided into 2 groups: type I was characterized by early onset, seizures, megalencephaly, and typical MRI features, and type II with a later age at onset characterized by brainstem features and atypical MRI findings.13 Given the paucity of neuroimaging data in type II AxD, we examined MRI findings in 13 cases of type II AxD.
METHODS
To identify all patients evaluated with AxD between January 1, 1996 and February 1, 2012, we used the Mayo Clinic Medical Records Linkage System. The computer search strategy used the text words “Alexander disease.” Given this approach, any patient with AxD mentioned in the text was evaluated (n = 132). Only patients with type II AxD were included. Patients were included if they had genetic or pathologic confirmation of AxD or if a first-degree relative with similar imaging and clinical features had genetic or pathologic evidence of AxD. Clinical data abstracted for all patients included age at onset of symptoms, age at diagnosis, first symptom, neurologic symptoms, physical/neurologic findings on examination, genetic testing and/or biopsy (if performed), and MRI findings.
MRI interpretation.
All MRIs were reviewed in detail by a board-certified, unblinded neuroradiologist (K.S.). MRIs were systematically evaluated for features described in prior reports including periventricular signal changes, brainstem atrophy, brainstem enhancement, parenchymal brainstem signal change, spinal cord atrophy, spinal cord enhancement and signal change, cerebellar enhancement/signal change, dentate signal change, “tadpole atrophy” (relative sparing of pons), periventricular rim of decreased T2 signal and increased T1 signal, garland-like feature along ventricular walls (ependymal nodularity), and thalamic/basal ganglia signal abnormality. Additional features systemically analyzed after initial review of all cases demonstrating a possible pattern included brainstem pial fluid-attenuated inversion recovery (FLAIR) signal change and middle cerebellar peduncle involvement.
Standard protocol approvals, registrations, and patient consents.
All patients consented to the use of their clinical records for the purpose of research, and the study was approved by the Mayo Clinic institutional review board.
RESULTS
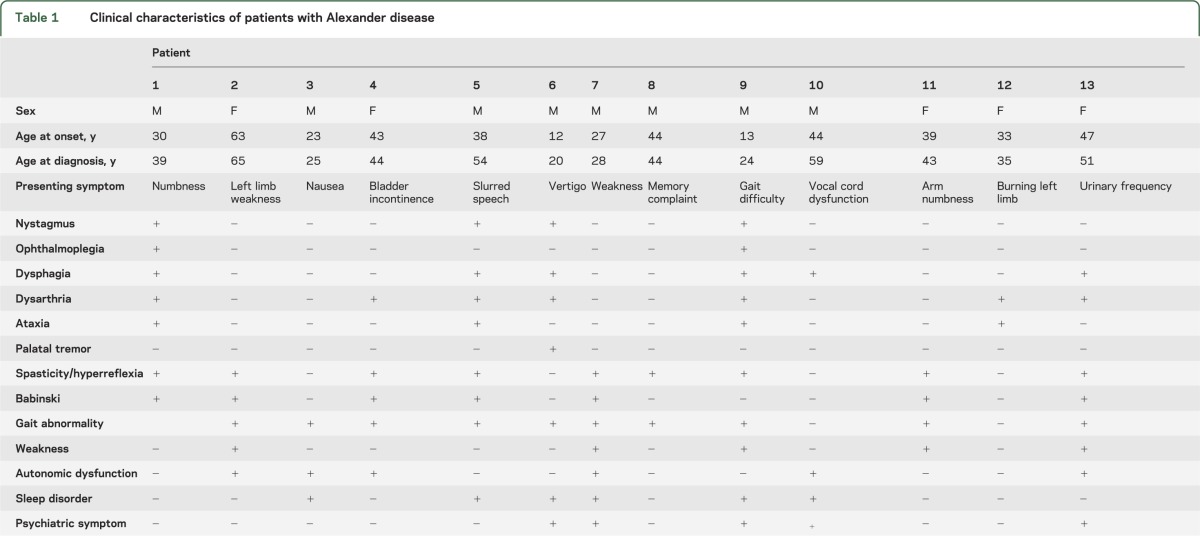
Thirteen patients were identified. Median age at onset was 38 years (range: 12–63), and 5 were female. Clinical characteristics are summarized in table 1. Eight patients had mutations in the GFAP gene (see table e-1 on the Neurology® Web site at www.neurology.org).
Table 1.
Clinical characteristics of patients with Alexander disease
Ancillary testing.
A mild peripheral neuropathy was noted on nerve conduction studies in 1 of 5 patients who underwent EMG and nerve conductions. One patient underwent a thermoregulatory sweat test, which revealed diffuse anhidrosis.
Family history.
Ten subjects had a positive family history of probable AxD. Patient 13 was the mother of patient 12. Patient 10 is the father of patient 3. Patient 8 is the son of patient 2.
Pathology.
Patient 1 underwent a biopsy of the middle cerebellar peduncle, which demonstrated excessive Rosenthal fiber deposition in the gray and white matter, consistent with AxD. Patient 2 had an autopsy, which demonstrated severe atrophy of medulla oblongata and spinal cord with Rosenthal fiber deposition. Patient 3 had a biopsy of the medulla, which demonstrated dense Rosenthal fiber deposition, mild hypercellularity, and focal chronic inflammation, initially interpreted as representing pilocytic astrocytoma. Patient 4's daughter had an autopsy consistent with AxD. Patient 8 is the son of patient 2 who had autopsy evidence of AxD.
Imaging.
All 13 patients underwent brain MRI. Twelve of 13 patients underwent spinal cord MRI. Imaging characteristics are summarized in table 2 and illustrated in figures 1 to 3.
Table 2.
Neuroimaging features
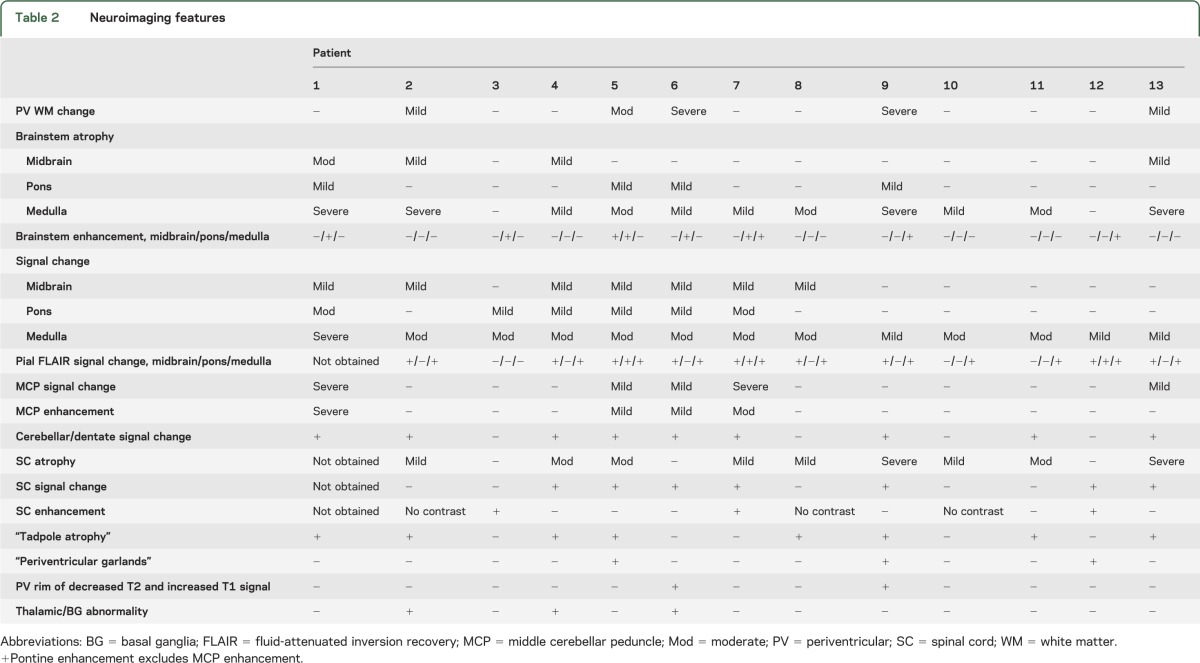
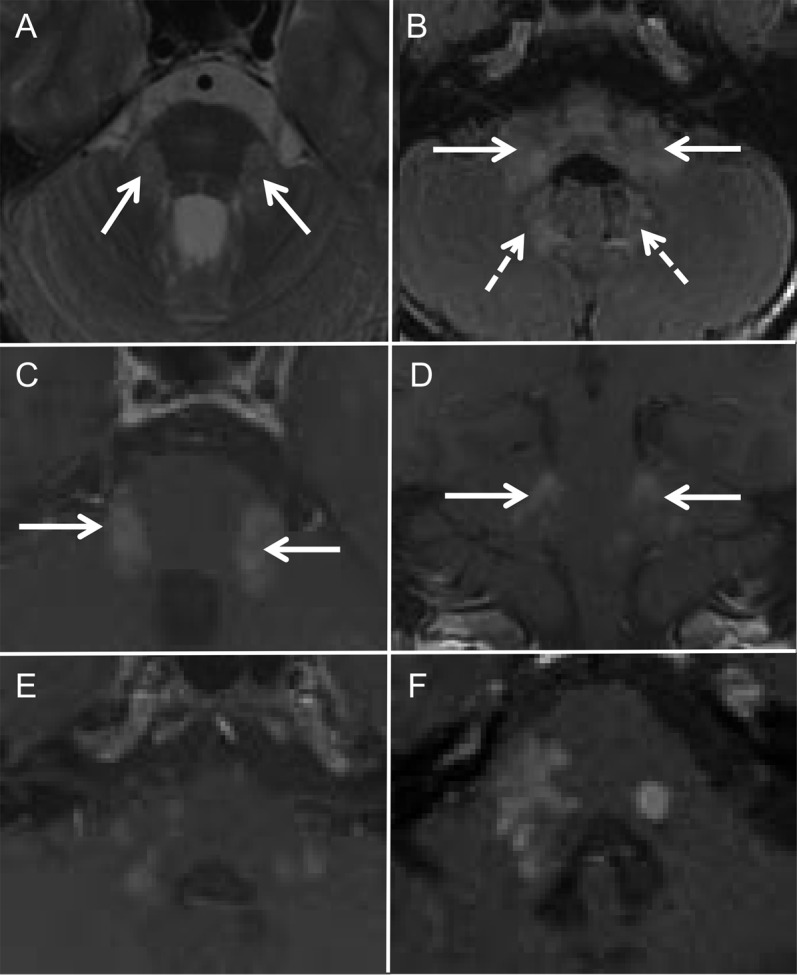
Figure 1. MRI features in Alexander disease: Brainstem and spinal cord features.

(A–C) Axial fluid-attenuated inversion recovery (FLAIR) MRIs from case 7 showing the pial FLAIR signal change (arrows) along the midbrain (A), pons (B), and medulla (C). Sagittal (D) and axial (E) T2-weighted MRIs from case 5 showing an area of intramedullary signal change and moderate cord atrophy. Sagittal T1 with contrast cervical spine MRI (F) from case 7 showing a dorsally located region of subpial enhancement in the upper cervical cord. Sagittal (G) and axial (H) cervical cord MRIs from case 9 showing cord atrophy (G, H) and intramedullary signal change (H).
Figure 2. MRI features in Alexander disease: Middle cerebellar peduncle.
Axial T2-weighted (A) and axial fluid-attenuated inversion recovery (B) MRIs from case 7 showing increased signal involving the middle cerebellar peduncle (A), inferior cerebellar peduncle (B, arrows), and dentate (B, dashed arrows). Axial (C) and coronal (D) T1 with contrast MRIs from case 7 showing enhancement of the middle cerebellar peduncle. Axial T1 with contrast MRIs from case 7 (E) and case 1 (F) showing patchy asymmetric enhancement of the lateral pons and middle cerebellar peduncles.
Figure 3. MRI features in Alexander disease: Periventricular and basal ganglia findings.

Axial fluid-attenuated inversion recovery (FLAIR) (A, B) and T2-weighted (C, D) MRIs from case 9 showing periventricular hyperintensity. Also shown is increased dentate signal (arrows in B) and periventricular T2 hypointensity (arrows in D). Sagittal T1-weighted MRI from case 9 (E) showing the periventricular T1 hyperintensity. Axial FLAIR MRI from case 12 (F) showing periventricular garlands. Axial T2 MRIs from case 2 (G) and case 4 (H) showing symmetrically increased signal along the medial and lateral borders of the lentiform nuclei (G) and globus pallidi (H), respectively.
Cerebral white matter.
Five patients had involvement of cerebral white matter with the most severe changes occurring at younger ages (figure 3).
Brainstem atrophy, enhancement, and signal change.
Eleven of 13 patients had atrophy of the medulla, 4 had pontine atrophy, and 4 had midbrain atrophy. Seven patients had brainstem enhancement, and enhancement of the pons was the most common (seen in 5 cases). Thirteen of 13 patients had parenchymal signal change in the medulla. Six had pontine signal change and 7 had midbrain signal change (figure 1).
Brainstem pial FLAIR signal change.
Twelve patients underwent FLAIR imaging. Eleven of 12 had pial FLAIR signal change in the medulla. Three of these had coexisting pial FLAIR change in pons and 9 had coexisting pial FLAIR signal change in the midbrain (figure 1).
Middle cerebellar peduncle involvement.
Five subjects had T2 signal change in the middle cerebellar peduncle. Associated enhancement was present in 4 of 5 patients (figure 2).
Cerebellar/dentate findings.
Dentate signal change (n = 9) was more common than cerebellar signal change outside the dentate (n = 1). The patient with signal change outside the dentate also had dentate signal change. Three patients had significant cerebellar atrophy.
Spinal cord.
Twelve patients had spinal cord MRI. Nine patients had contrast administered with spinal cord MRI. Nine patients had spinal cord atrophy, 7 patients had spinal cord signal change, and 3 patients had spinal cord enhancement. One patient with spinal cord enhancement had no spinal cord atrophy or signal change. All 12 patients had enhancement, atrophy, or signal change. Three subjects had only cervical cord imaging. Seven patients had spinal cord abnormalities in both the cervical and thoracic spinal cord. Thoracic abnormalities were limited to atrophy. Two subjects had spinal cord abnormality limited to the cervical spine (figure 1).
Tadpole atrophy (atrophy of the medulla and cervical spinal cord with relative sparing of pons).
Eight subjects had evidence of tadpole atrophy.14
Periventricular rim of decreased T2 signal and increased T1 signal.
Two patients had periventricular rim of decreased T2 signal and increased T1 signal. These were the youngest subjects in the cohort.
Garland-like feature along the ventricular wall (ependymal nodularity).
Three patients had irregularity of the ependymal surfaces of the lateral ventricles, although to a lesser degree than what has been reported in infantile AxD (figure 3).
Thalamic/basal ganglia abnormality.
Three patients had thalamic or basal ganglia signal abnormality. One patient had mild peripheral increased T2 signal in the inferior lentiform nucleus. One patient had increased T2 signal in the inferior lentiform nuclei and globus pallidi. A third patient had abnormal increased T2 signal in the caudate heads and medial thalami (figure 3).
Patterns of contrast enhancement.
Seven patients had brainstem enhancement to a varying degree. Patient 3 was imaged serially over 8 years, demonstrating that enhancement may diminish over time as atrophy evolves.
Genotype considerations.
We could not discern any specific correlation between specific GFAP mutation genotype and either MRI or pattern of clinical involvement in this series of patients with type II AxD.
Illustrative cases.
Patient 3 presented at age 23 years with nausea and headache. Brain MRI revealed diffuse patchy enhancement and expansion of the dorsal medulla. The imaging abnormality was biopsied. Pathology demonstrated Rosenthal fibers and was initially interpreted as pilocytic astrocytoma. Seven years later, without further intervention, medullary enhancement had markedly diminished with the appearance of mild atrophy and persistent signal change. This was clinically associated with evolution of cardiac and respiratory dysfunction, sleep apnea, and dysautonomia. Subsequent pathologic review with genetic analysis in this patient's father (patient 10) showed c.1154C>G on exon 7 producing p.Ser385Cys confirming the diagnosis of AxD. At last follow-up, 9 years after presentation, his neurologic examination was notable for the absence of brainstem findings including palatal tremor, and nystagmus. Autonomic symptoms continued to be disabling.
Patient 2 presented at age 65 years with 2 years of left limb weakness. Before evaluation at age 65, she developed gait unsteadiness. Neurologic evaluation was notable for hyperreflexia in the lower extremities, presence of bilateral Babinski reflex, and mild left-sided weakness. Brain MRI revealed severe atrophy of the medulla with associated T2 hyperintensity and pial FLAIR abnormality. She had mild atrophy of the cervical and thoracic spinal cord. Repeat neurologic examination 6 months later was stable. The patient died at age 67 of a gynecologic cancer.
DISCUSSION
Our findings of significant atrophy of the medulla and spinal cord support the findings of prior studies of adult-onset or type II AxD.5,7,15 We also confirmed frequent involvement of the dentate with less frequent coexisting cerebellar white matter change.5 Compared with infantile or type I AxD, involvement of the periventricular white matter was sparse.4
Pial FLAIR signal abnormality and middle cerebellar peduncle signal change or enhancement are 2 previously undescribed imaging features associated with type II AxD. Eleven of 12 patients who underwent FLAIR imaging demonstrated pial FLAIR signal abnormality most often concentrated in the medulla. The unique location of FLAIR abnormality may help differentiate from other pathologies involving the brainstem, which may have a different anatomical distribution.
Five subjects in our series had signal change and enhancement of the middle cerebellar peduncle, a finding not emphasized in a review of 40 adult cases of AxD.7 Bilateral involvement of the middle cerebellar peduncle is useful in narrowing the differential diagnosis. In a series of 27 cases of bilateral middle cerebellar peduncle involvement on MRI, multiple system atrophy, spinocerebellar ataxia, Wilson disease, liver cirrhosis, adrenoleukodystrophy, posterior reversible encephalopathy syndrome, and neoplasms were the most frequent causes, but AxD was not listed as a potential cause.16 Therefore, AxD should be included in the differential of diseases with middle cerebellar peduncle abnormalities seen on MRI.
Palatal tremor (previously known as palatal myoclonus) has been reported to be strongly suggestive of adult-onset AxD.8,9 In our series, palatal tremor was present in only one patient. While the presence of palatal tremor may be a clue for AxD, its absence should not preclude the diagnosis from being made.
Myelopathy was a common clinical presentation for patients with type II AxD. During the evaluation of myelopathy, if medullary signal change, atrophy, or enhancement is encountered, the diagnosis of AxD should be considered.
Patterns of enhancement in AxD may lead to diagnostic uncertainty, particularly for neoplastic or inflammatory disorders. In particular, brainstem periventricular patterns in teens and younger adults raise suspicion for pilocytic astrocytoma, and pathologically, the abundance of Rosenthal fibers may further confuse the picture. The difficulty in distinguishing these disorders is highlighted in the description of patient 3. While the majority of patients present with bulbar symptoms and/or myelopathy with imaging abnormalities corresponding to the brainstem and spinal cord, patient 3 highlights the clinical imaging mismatch that can occur. The patient had a large brainstem lesion with an essentially normal neurologic examination at presentation. Therefore, the additional imaging features described here, supported by the high probability of diagnostic confirmation from analysis of the GFAP gene, are important considerations for clinicians involved in caring for these patients.
The clinical features of type II AxD are consistent with the predominant brainstem involvement. These features may vary from patient to patient, and may not be consistent within families, but account for the major portion of disability encountered. Similar to other studies, the patients with AxD from the same family in our study had varied age at onset and phenotype.15 Sleep and autonomic dysfunction and, rarely, cardiac and respiratory pacemaker dysfunction, may be particularly troubling. It is important to consider that type II AxD is a chronic disease and that imaging and clinical features may evolve over time. Additional reports of patients studied serially over an extended period would be of interest.
The 2 youngest patients in our series (age at onset 12 and 13 years, respectively) appeared to have features of both infantile- and adult-onset AxD. Both patients had atrophy of the medulla and either atrophy or signal change in the spinal cord consistent with adult-onset AxD, but they also had more extensive supratentorial periventricular white matter change. They were the only 2 patients with a periventricular rim of decreased T2 signal and increased T1 signal, a finding most often seen in infantile cases. Therefore, at least in our cases, there appears to be a group of patients with features of both infantile- and adult-onset AxD. A systematic review of known cases of AxD supports the imaging and clinical presentation of “juvenile AxD” as intermediate form with features of both infantile- and adult-onset disease.17 One case would have been categorized as juvenile and the other as adult based on prior criteria. The consistent observation of an intermediate form of AxD led one group to propose a classification system consisting of 3 groups: cerebral AxD (type I), bulbospinal AxD (type II), and intermediate form (type III).18
The retrospective nature of the study is a limitation; therefore, the clinical and radiologic features may be broader than presented because milder cases may not be clinically diagnosed.
A recent study suggested that the imaging features in type II AxD are “atypical.”13 Our study confirms that the imaging features of type II AxD are significantly different from type I AxD, but the consistent posterior spread of imaging abnormalities with age appears typical for type II AxD. The clinician should consider type II AxD in any patient who presents with brainstem dysfunction or myelopathy in conjunction with signal change, atrophy, or enhancement in the medulla or cervical cord. Pial T2 FLAIR hyperintensity in the brainstem or middle cerebellar peduncle involvement serve as additional clues to the diagnosis.
Supplementary Material
GLOSSARY
- AxD
Alexander disease
- FLAIR
fluid-attenuated inversion recovery
- GFAP
glial fibrillary acidic protein
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Dr. Graff-Radford: drafting/revising the manuscript, acquisition of data, statistical analysis, analysis or interpretation of data. Dr. Schwartz: drafting/revising the manuscript, acquisition of data. Dr. Gavrilova: drafting/revising the manuscript, analysis or interpretation of data. Dr. Lachance: drafting/revising the manuscript, study supervision. Dr. Kumar: study concept or design, drafting/revising the manuscript, analysis or interpretation of data, study supervision.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 1949;72:373–381 [DOI] [PubMed] [Google Scholar]
- 2.Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 2001;27:117–120 [DOI] [PubMed] [Google Scholar]
- 3.Li R, Johnson AB, Salomons G, et al. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann Neurol 2005;57:310–326 [DOI] [PubMed] [Google Scholar]
- 4.van der Knaap MS, Naidu S, Breiter SN, et al. Alexander disease: diagnosis with MR imaging. AJNR Am J Neuroradiol 2001;22:541–552 [PMC free article] [PubMed] [Google Scholar]
- 5.Farina L, Pareyson D, Minati L, et al. Can MR imaging diagnose adult-onset Alexander disease? AJNR Am J Neuroradiol 2008;29:1190–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martidis A, Yee RD, Azzarelli B, Biller J. Neuro-ophthalmic, radiographic, and pathologic manifestations of adult-onset Alexander disease. Arch Ophthalmol 1999;117:265–267 [DOI] [PubMed] [Google Scholar]
- 7.Namekawa M, Takiyama Y, Honda J, Shimazaki H, Sakoe K, Nakano I. Adult-onset Alexander disease with typical “tadpole” brainstem atrophy and unusual bilateral basal ganglia involvement: a case report and review of the literature. BMC Neurol 2010;10:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pareyson D, Fancellu R, Mariotti C, et al. Adult-onset Alexander disease: a series of eleven unrelated cases with review of the literature. Brain 2008;131:2321–2331 [DOI] [PubMed] [Google Scholar]
- 9.Schwankhaus JD, Parisi JE, Gulledge WR, Chin L, Currier RD. Hereditary adult-onset Alexander's disease with palatal myoclonus, spastic paraparesis, and cerebellar ataxia. Neurology 1995;45:2266–2271 [DOI] [PubMed] [Google Scholar]
- 10.van der Knaap MS, Salomons GS, Li R, et al. Unusual variants of Alexander's disease. Ann Neurol 2005;57:327–338 [DOI] [PubMed] [Google Scholar]
- 11.Probst EN, Hagel C, Weisz V, et al. Atypical focal MRI lesions in a case of juvenile Alexander's disease. Ann Neurol 2003;53:118–120 [DOI] [PubMed] [Google Scholar]
- 12.Salvi F, Aoki Y, Della Nave R, et al. Adult Alexander's disease without leukoencephalopathy. Ann Neurol 2005;58:813–814 [DOI] [PubMed] [Google Scholar]
- 13.Prust M, Wang J, Morizono H, et al. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 2011;77:1287–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Namekawa M, Takiyama Y, Aoki Y, et al. Identification of GFAP gene mutation in hereditary adult-onset Alexander's disease. Ann Neurol 2002;52:779–785 [DOI] [PubMed] [Google Scholar]
- 15.Stumpf E, Masson H, Duquette A, et al. Adult Alexander disease with autosomal dominant transmission: a distinct entity caused by mutation in the glial fibrillary acid protein gene. Arch Neurol 2003;60:1307–1312 [DOI] [PubMed] [Google Scholar]
- 16.Okamoto K, Tokiguchi S, Furusawa T, et al. MR features of diseases involving bilateral middle cerebellar peduncles. AJNR Am J Neuroradiol 2003;24:1946–1954 [PMC free article] [PubMed] [Google Scholar]
- 17.Balbi P, Salvini S, Fundaro C, et al. The clinical spectrum of late-onset Alexander disease: a systematic literature review. J Neurol 2010;257:1955–1962 [DOI] [PubMed] [Google Scholar]
- 18.Yoshida T, Sasaki M, Yoshida M, et al. Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J Neurol 2011;258:1998–2008 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.