

Abstract
Hyperekplexia is a neurological disorder associated primarily with mutations in the α1 subunit of glycine receptors (GlyRs) that lead to dysfunction of glycinergic inhibitory transmission. To date, most of the identified mutations result in disruption of surface expression or altered channel properties of α1-containing GlyRs. Little evidence has emerged to support an involvement of allosteric GlyR modulation in human hyperekplexia. Here, we report that recombinant human GlyRs containing α1 or α1β subunits with a missense mutation in the α1 subunit (W170S), previously identified from familial hyperekplexia, caused remarkably reduced potentiation and enhanced inhibition by Zn2+. Interestingly, mutant α1W170Sβ GlyRs displayed no significant changes in potency or maximum response to glycine, taurine, or β-alanine. By temporally separating the potentiating and the inhibitory effects of Zn2+, we found that the enhancement of Zn2+ inhibition resulted from a loss of Zn2+-mediated potentiation. The W170S mutation on the background of H107N, which was previously reported to selectively disrupt Zn2+ inhibition, showed remarkable attenuation of Zn2+-mediated potentiation and thus indicated that W170 is an important residue for the Zn2+-mediated GlyR potentiation. Moreover, overexpressing the α1W170S subunit in cultured rat neurons confirmed the results from heterologous expression. Together, our results reveal a new zinc potentiation site on α1 GlyRs and a strong link between Zn2+ modulation and human disease.
Introduction
Hyperekplexia is an inherited neuronal disorder characterized by hypertonia and exaggerated startle reflex to unexpected sensory stimuli (Bakker et al., 2006). Hyperekplexia is caused by dysfunction of glycinergic transmission that can result from diverse root causes that include loss of function of glycine receptors (GlyRs), reduced receptor clustering of GlyRs at synapses, or reduced glycine release from presynaptic terminals (Harvey et al., 2008). Most cases of identified familial hyperekplexia are associated with dominant or recessive missense mutations of the glra1 gene, which encodes the α1 subunit of GlyRs (Lynch, 2004). These mutations commonly lead to severe impairments of α1 GlyR-mediated synaptic inhibition either by reducing surface expression of synaptic GlyRs or by disrupting basic channel properties, such as agonist binding affinity, channel gating, and channel conductance (Chung et al., 2010). Complete loss of function mutations of α1 subunit has also been reported, causing hyperekplexia symptoms that are clinically indistinguishable from patients carrying missense glra1 mutations (Brune et al., 1996). However, in animal models, deletion of functional α1 subunits caused more severe symptoms and juvenile death in mutant mice, suggesting that deficits in human α1 subunit might be more efficiently compensated (Buckwalter et al., 1994).
GlyRs are activated by multiple endogenous agonists, including glycine, β-alanine, and taurine. In addition, GlyRs also exhibit significant allosteric modulation by various effectors, such as Zn2+, endocannabinoids, steroids, and alcohols (Lynch, 2004). Among these allosteric modulators, Zn2+ is enriched in presynaptic vesicles in many regions of the CNS and acts as a potent allosteric modulator of inhibitory GlyRs (Sensi et al., 2011). Low concentrations (<10 μm) of Zn2+ potentiate GlyR currents, whereas high concentrations (>10 μm) inhibit GlyR-mediated responses (Bloomenthal et al., 1994; Laube et al., 1995). Before this study, there has been no evidence from reported human mutations that hyperekplexia is caused by impaired allosteric modulation without affecting basic receptor properties of GlyRs. Although selectively removing sensitivity to Zn2+-mediated potentiation produced exaggerated startle reactions in knockin mice (Hirzel et al., 2006), the different phenotype severities between human and murine glra1 null mutations make it difficult to predict whether simply disrupting Zn2+-mediated modulation of GlyRs also causes hyperekplexia in humans.
In this study, we report that the missense mutation W170S in the GlyR α1 subunit, which was recently identified from Omani families with hyperekplexia and mild mental retardation (Al-Futaisi et al., 2012), caused almost complete loss of Zn2+-mediated potentiation and enhancement of Zn2+-mediated inhibition. Unlike previously reported missense α1 mutations from human cases of hyperekplexia, α1W170S-containing GlyRs displayed no significant alterations in agonist sensitivities, maximal current responses, or current–voltage (I-V) relations. The major alteration of α1W170S was the ablated sensitivity to Zn2+-mediated potentiation, which was determined in both a recombinant expression system and in cultured neurons. Collectively, our findings demonstrate that impaired allosteric modulation of α1 GlyRs by Zn2+ may directly lead to human hyperekplexia.
Materials and Methods
cDNA constructs and transfection.
Wild-type (WT) human GlyR α1 (hGlra1) and the human GlyR β subunit (hGlrb) were subcloned into the pBK-CMV NB-200 expression vector (Liu et al., 2010). α1W170S, α1H107N, and α1W170S/H107N plasmids were constructed by site-directed mutagenesis of hGlra1. The sequences of all the plasmids were confirmed by automated DNA sequencing. Purified plasmids encoding the WT or mutant GlyR α1 alone or with β subunits (1:10; total plasmid amount 3–4.5 μg) were transfected into HEK293T cells by electroporation (NEPA21, NEPA GENE). The α1 and α1β compositions of GlyRs were confirmed by their shifted sensitivity to picrotoxin using whole-cell patch-clamp recordings. A small amount (∼0.5 μg) of pcDNA3-GFP was cotransfected along with GlyR subunits to act as a transfection marker and facilitate the visualization of transfected cells during electrophysiological experiments. Cells were replated on glass coverslips after transfection and cultured for an additional 15–24 h before patch-clamp recordings.
Neuronal culture and overexpression.
Cultured cortical neurons were prepared from the neocortex of day 18 fetal rats (from embryos of either sex) as described previously (Liu et al., 2010) and were transiently transfected with α1 subunits (1 μg) and GFP (0.3 μg) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols. Electrophysiological recordings were performed 3–7 d after plating.
Whole-cell patch-clamp recordings.
Whole-cell recordings were performed under voltage-clamp mode using an Axopatch 200B (Molecular Devices). Whole-cell currents were recorded at a holding potential of −60 mV, and signals were low-pass filtered at 2 kHz and digitized at 10 kHz (Digidata, 1440A). Recording pipettes (3–5 MΩ) were filled with intracellular solution that contained the following (in mm): 140 CsCl, 10 HEPES, 4 Mg-ATP, and 0.5 BAPTA (pH 7.20, osmolarity, 290–295 mOsm). The coverslips were continuously superfused with the extracellular solution containing the following (in mm): 140 NaCl, 5.4 KCl, 10 HEPES, 1.0 MgCl2, 1.3 CaCl2, and 20 glucose (pH 7.4, 305–315 mOsm). To evoke glycine currents, we used fast perfusion of glycine and other agonists with a computer-controlled multibarrel fast perfusion system (Warner Instruments). Maximum currents (Imax) were evoked by the highest concentration of the agonist as determined by the dose–response curves. Zn2+ was applied both in the bath and together with the agonist-containing solutions. All experiments were performed at 23–25°C.
Homology modeling.
The mature hGlyR α1 subunit was modeled on the crystal structure of the glutamate-gated chloride channel α (GluCl) (Hibbs and Gouaux, 2011) using I-TASSER server (Zhang, 2008; Roy et al., 2010, 2012). All 3D images were subsequently rendered using the UCSF Chimera package (Pettersen et al., 2004).
Data analysis.
Values are expressed as mean ± SEM. One-way ANOVA or a two-tailed Student's t test was used for statistical analysis, and p values <0.05 were considered to be statistically significant. Dose–response curves were created by fitting data to the Hill equation: I = Imax/[1 + (EC50/[A])nH], where I is the current, Imax is the maximum current, [A] is a given concentration of agonist, and nH is the Hill coefficient.
Results
The startle disease mutation W170S did not alter heteromeric GlyR agonist sensitivities or channel permeability
Glycine receptor-mediated currents were tested by whole-cell voltage-clamp recordings in HEK293T cells expressing WT or mutant GlyRs. Because the extracellular N terminus of α1 contains the GlyR agonist binding site and previously reported missense mutations at this region typically exhibit a feature of impaired sensitivity to agonist activation (Chung et al., 2010), we first examined whether agonist binding affinities were altered in α1W170S GlyRs. The dose–response curve of glycine-activated, homomeric α1W170S GlyRs currents was shifted toward the right compared with homomeric α1WT GlyRs, with the EC50 increased from 62.9 ± 5.0 μm to 127.6 ± 6.9 μm (nH = 3.38 ± 0.49 and 1.69 ± 0.14; n = 5 and n = 10, respectively; p < 0.05; Fig. 1A). The agonist sensitivity of α1W170S was also examined with the other two endogenous GlyR partial agonists, β-alanine and taurine. Homomeric α1W170S showed increased sensitivity to activation by β-alanine (EC50 = 226.9 ± 14.3 μm and 99.4 ± 8.0 μm, nH = 2.40 ± 0.03 and 1.56 ± 0.03, n = 9 and n = 9 in α1WT and α1W170S, respectively; p < 0.01; Fig. 1B) and no change in sensitivity to activation by taurine (EC50 = 256.6 ± 29.9 and 213.7 ± 19.1 μm, nH = 1.55 ± 0.05 and 1.57 ± 0.04, n = 8 and n = 8, respectively; p > 0.05; Fig. 1C). As the majority of α1-containing GlyRs in the adult CNS consist of heteromeric α1β GlyRs that may exhibit different current kinetics than homomeric receptors (Lynch, 2004), we also investigated the potential for altered agonist sensitivities in heteromeric α1W170Sβ GlyRs. Interestingly, the α1W170Sβ receptors did not show significant impairment of the sensitivity to glycine (EC50 = 116.4 ± 5.1 and 80.0 ± 5.1 μm, nH = 2.45 ± 0.02 and 1.96 ± 0.03, n = 5 and n = 6 in α1WTβ and α1W170Sβ, respectively; p > 0.05; Fig. 1D), β-alanine (EC50 = 91.0 ± 7.6 and 95.5 ± 8.0 μm, nH = 1.55 ± 0.03 and 1.83 ± 0.04, n = 9 and n = 8, respectively; p > 0.05; Fig. 1E), or taurine (EC50 = 371.1 ± 25.6 and 194.4 ± 18.9 μm, nH = 1.60 ± 0.03 and 1.45 ± 0.04, n = 8 and n = 8, respectively; p < 0.05; Fig. 1F). These data suggest that the hyperekplexia phenotype observed in humans carrying W170S alleles is not likely to be mediated by impaired agonist responsiveness of mutant GlyRs.
Figure 1.

The W170S mutation in α1β GlyRs showed no significant changes in agonist sensitivities, maximal current responses, or I-V relationship. A–C, Dose–response curves for α1WT and α1W170S homomeric GlyRs normalized to maximal currents induced by glycine (A), β-alanine (B), and taurine (C). D–F, Dose–response curves for α1WTβ and α1W170Sβ heteromeric GlyRs normalized to maximal currents induced by glycine (D), β-alanine (E), and taurine (F). G, Maximal responses (Imax) of glycine-, β-alanine-, and taurine-induced currents in α1WTβ and α1W170Sβ GlyRs. H, The β-alanine- and taurine-mediated Imax were normalized to glycine-mediated Imax from the same cell. I, Representative traces of 100 μm glycine-induced currents at different holding potentials from −60 mV to 60 mV. J, Peak I-V relations (normalized to the current recorded at −60 mV) of α1WTβ and α1W170Sβ GlyRs. n.s., Not significant.
Missense mutations in other regions of α1 have been previously characterized that result in only moderate changes in GlyR agonist affinity yet also a remarkable reduction of maximal whole-cell currents (Saul et al., 1999). We found that, in α1W170Sβ receptors, the maximal currents (Imax) evoked by saturating agonist concentrations, including 3 mm glycine (α1WTβ, Imax = 9.4 ± 1.2 nA, n = 7; α1W170Sβ, Imax = 7.5 ± 0.9 nA, n = 11; p > 0.05), β-alanine (α1WTβ, Imax = 8.4 ± 1.2 nA, n = 7; α1W170Sβ, Imax = 7.3 ± 1.0 nA, n = 11; p > 0.05), or 5 mm taurine (α1WTβ, Imax = 7.3 ± 1.4 nA, n = 7; α1W170Sβ, Imax = 6.6 ± 0.9 nA, n = 11; p > 0.05) were not significantly changed compared with the α1WTβ receptors (Fig. 1G). The Imax induced by β-alanine (α1WTβ, Iala/Igly = 90.7 ± 8.6%, n = 7; α1W170Sβ, Iala/Igly = 95.7 ± 4.5%, n = 11; p > 0.05) or taurine (α1WTβ, Itau/Igly = 90.8 ± 17.5%, n = 7; α1W170Sβ, Itau/Igly = 85.8 ± 5.2%, n = 11; p > 0.05) as a percentage of the glycine-induced Imax from the same cell was also indistinguishable between α1WTβ and α1W170Sβ GlyRs (Fig. 1H). In addition, the I-V relationship of 100 μm glycine-induced responses in α1W170Sβ receptors showed no difference compared with α1WTβ receptors, indicating that there were no changes in Cl− ion permeability (Fig. 1I,J). Together, these results suggest that the basic channel properties of recombinant α1W170Sβ GlyRs were not significantly different from that of α1WTβ GlyRs.
Impaired Zn2+-mediated modulation in α1W170S-containing GlyRs
To investigate how the W170S mutation affects glycine α1 receptors, we generated a homology model of α1 GlyR based upon the template of the crystal structure of glutamate-gated chloride channels, which share 45% identical and 62% positive sequences with the GlyR α1 subunit (Hibbs and Gouaux, 2011). The homology model showed that W170 was located at loop F in the outer face of the N terminus domain. It is structurally close to the previously reported Zn2+ potentiation site (Miller et al., 2005b), with particular proximity (<4 Å) to D194, a key residue for Zn2+ binding (Fig. 2A). Therefore, we investigated the possibility that the W170S mutation might affect the sensitivity of GlyRs to Zn2+ modulation. Low concentrations (0.1–1 μm) of Zn2+ remarkably enhanced homomeric α1WT currents evoked by 30 μm glycine (EC10), whereas high concentrations (>10 μm) of Zn2+ inhibited responses of α1WT GlyRs (Fig. 2B top, C). Interestingly, Zn2+-mediated potentiation of GlyRs was substantially attenuated in α1W170S GlyRs (Fig. 2B, bottom; C, top). Coexpression of the β subunit did not rescue the deficit of Zn2+ potentiation in α1W170S, suggesting that the ablated Zn2+ potentiation was preserved in α1W170Sβ GlyRs (Fig. 2C, bottom).
Figure 2.

W170S mutation impaired Zn2+-mediated potentiation and enhanced Zn2+-mediated inhibition of GlyR currents activated by different agonists. A, Left, The homology model of the GlyR α1 subunit based on the GluCl viewed from the outer face. Inset, Plan view of the GlyR pentamer. The arrow indicates the viewing angle. Right, The expanded illustration of the amino acid residues that affect Zn2+ potentiation. B, Representative traces represent biphasic modulation of Zn2+ on homomeric α1WT (top) or α1W170S (bottom) GlyR currents activated by glycine (EC10). C, Averaged Zn2+ concentration–response curves for the modulation of EC10 responses to glycine-activated currents in homomeric (top; α1WT, n = 7; α1W170S, n = 10) and heteromeric α1W170Sβ GlyRs (bottom; α1WT, n = 7; α1W170S, n = 10). D, Averaged Zn2+ concentration–response curves for the modulation of EC10 responses to β-alanine-activated currents in homomeric (top; α1WT, n = 5; α1W170S, n = 5) and heteromeric α1W170Sβ GlyRs (bottom; α1WT, n = 6; α1W170S, n = 5). E, Averaged Zn2+ concentration–response curves for the modulation of EC10 responses to taurine-activated currents in homomeric (top; α1WT, n = 5; α1W170S, n = 8) and heteromeric α1W170Sβ GlyRs (bottom; α1WT, n = 7; α1W170S, n = 5).
Zn2+ potentiation of GlyR current responses evoked from partial agonists, such as taurine, might behave differently from the potentiation effect of glycine-evoked responses (Lynch et al., 1998). Therefore, we tested the effects of Zn2+ on α1W170S and α1W170Sβ GlyRs-mediated currents induced by β-alanine or taurine at EC10, respectively. We found that W170S similarly abolished Zn2+-mediated potentiation of responses induced by β-alanine or taurine in both homomeric and heteromeric receptors (Fig. 2D,E). The Zn2+ inhibition was also increased under these conditions, suggesting that W170S mutations cause either attenuated potentiation or enhanced inhibition of Zn2+ in α1-containing GlyRs.
Altered Zn2+ modulation in W170S mutation was mainly the result of abolished potentiation
At Zn2+ concentrations ranging from 0.1 to 1000 μm, the biphasic Zn2+-mediated modulation of α1WT likely results from the superimposition of both positive and negative modulatory effects. Prior studies have shown that, if the GlyR has already been activated by the agonist, the positive and negative modulations of Zn2+ can be temporally separated by showing an initial potentiation followed by prolonged inhibition of GlyR currents (Lynch et al., 1998). By using this strategy, we further elucidated whether the altered responses of α1W170S to Zn2+ were the result of an ablation of Zn2+ potentiation or an increase of Zn2+ inhibition. In homomeric α1WT receptors, after 30 μm glycine was applied to induce a GlyR-mediated current, subsequent Zn2+ application caused an initial potentiation at concentrations of 0.1 to 50 μm (Fig. 3A). The Zn2+-mediated inhibition became more apparent when Zn2+ was washed out but glycine remained present (Lynch et al., 1998). In α1W170S GlyRs, however, the initial potentiation was completely absent (Fig. 3B,C), and the inhibition by Zn2+ was consequently enhanced compared with α1WT GlyRs (Fig. 3D). These data demonstrated that Zn2+-mediated positive allosteric modulation was ablated in the α1W170S.
Figure 3.
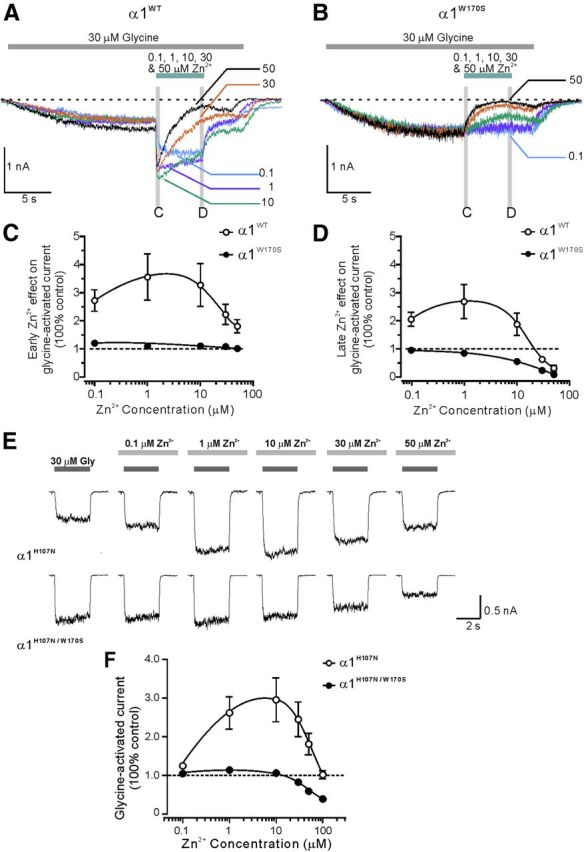
Separation of Zn2+-mediated potentiation and inhibition indicated that the W170S mutation mainly impaired Zn2+-mediated potentiation of GlyRs. A, Examples of α1WT-mediated currents showing that, after GlyR activation by glycine (EC10), subsequent Zn2+ application (5 s) at higher concentrations (30 and 50 μm) evoked an initial potentiation followed by inhibition. B, In α1W170Sβ GlyRs, the same application of Zn2+ only evoked inhibitory effects. Zn2+-mediated early effect was measured as the maximal potentiation of glycine-activated currents during the first 300 ms of Zn2+ perfusion (C). Zn2+-mediated late effect was measured as the maximal inhibition of glycine-activated currents during the last 300 ms before Zn2+ was washed out (D). C, D, Averaged dose–responses for Zn2+-mediated early and late effects on glycine-activated currents in homomeric α1WT (n = 6) and α1W170S receptors (n = 4). E, Top, Using the low Zn2+ potentiation background H107N, Zn2+ application induced only potentiation but no significant inhibition of glycine-activated currents in the homomeric α1H170N receptors. Bottom, Zn2+-mediated potentiation was ablated in the α1H170N/W170S double-mutant receptors. F, Averaged dose–responses for Zn2+-mediated modulations in α1H170N (n = 5) and α1H170N/W170S receptors (n = 16).
We further examined the effects of W170S on Zn2+-mediated potentiation by diminishing the influence of the inhibitory effects of Zn2+. The α1 mutation, H107N, has been reported to be at least 150-fold less sensitive than the α1WT to Zn2+-mediated inhibition without significantly affecting Zn2+-mediated potentiation (Miller et al., 2005a, b). Therefore, we generated the double-mutation α1H107N/W170S on the background of α1H107N (α1H107N, EC50 = 41.6 ± 3.1 μm, n = 2; α1H107N/W170S, EC50 = 136.9 ± 6.5 μm, n = 7). In α1H107N GlyRs, Zn2+ produced significant potentiation of glycine-induced responses without showing obvious inhibition at concentrations up to 100 μm (Fig. 3E,F). In contrast, Zn2+-mediated potentiation was abolished in α1H107N/W170S receptors. At Zn2+ concentrations of 50–100 μm, the W170S mutation even partially recovered the sensitivity of α1H107N to Zn2+-mediated inhibition (Fig. 3F). Together, these data revealed that W170 residue is likely a required site for Zn2+-mediated potentiation in α1 GlyRs.
Neuronal expressed α1W170S GlyRs were insensitive to Zn2+-mediated potentiation
The above results have shown that α1W170S-containing GlyRs lacked sensitivity to Zn2+-mediated potentiation in a recombinant expression system. However, the differential protein expression profiles between HEK293T cells and neurons might result in altered receptor functional characteristics in these two systems. To confirm that α1W170S-containing GlyRs are also insensitive to Zn2+-mediated potentiation when expressed in nerve cells, we overexpressed α1WT or α1W170S subunits in cultured cortical neurons as an endogenous expression system. Young cortical neurons mainly express α2 subunits, which exhibit substantially lower sensitivity to Zn2+ potentiation than that of α1 (Lynch, 2004; Miller et al., 2005b). In addition, α1- and α2-containing GlyRs have very distinct activation kinetics that can facilitate their biophysical isolation and thus also allow unambiguous confirmation of successful α1WT or α1W170S overexpression in neurons (Mangin et al., 2003; Mohammadi et al., 2003). To establish the accuracy of this strategy, we first examined the activation kinetics of α1WT and α1W170S and compared them with the kinetics of α2-containing GlyRs expressed in HEK293T cells. α1WT and α1W170S GlyRs exhibit similar 10–50% rise time (α1WT, 86.7 ± 6.9 ms, n = 8; α1W170S; 83.6 ± 7.1 ms, n = 8; p > 0.05) after application of 3 s pulses of 30 μm glycine, whereas α2 receptors expressed in HEK293T cells exhibited substantially slower activation kinetics (10–50% rise time: 243.9 ± 17.3 ms, n = 8, p < 0.01 compared with α1WT or α1W170S; Fig. 4A–C). In nontransfected cortical neurons, the same glycine application induced currents with activation kinetics similar to that of α2 receptors expressed in HEK293T cells (206.7 ± 21.6 ms, n = 7, p > 0.05 compared with α2 in HEK293T). Neurons overexpressing α1WT or α1W170S receptors showed GlyR activation kinetics with rise times ranging from 50 to 290 ms, indicating variable GlyR expression profiles. Therefore, only neurons that exhibited glycine currents with rise times <100 ms were included in our analysis and were considered to predominantly express α1WT or α1W170S GlyRs.
Figure 4.
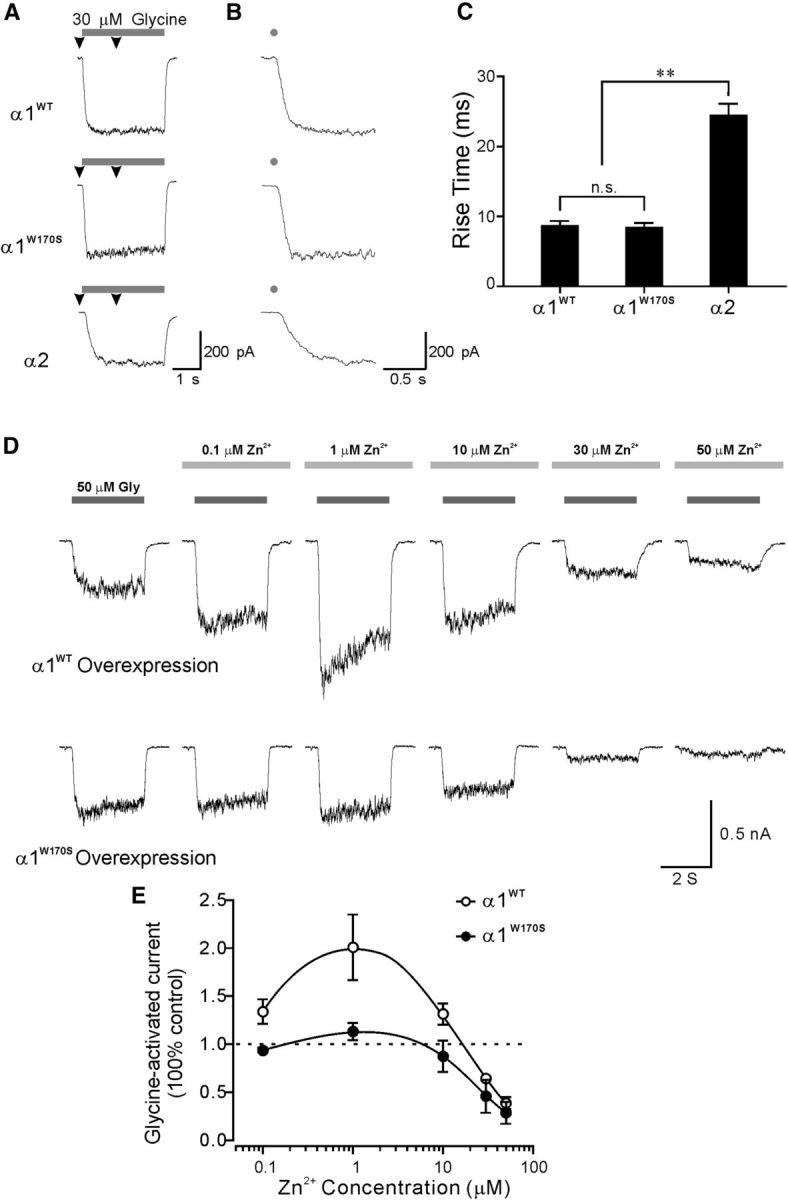
Neuronal expression of α1W170S ablated Zn2+-mediated potentiation of glycine-activated currents. A, B, Representative traces showing different activation kinetics of recombinant α1 and α2 receptors expressed in HEK293T cells. A, Arrowheads indicate regions of the traces that are expanded in B. B, Gray dots indicate application of glycine. C, Averaged 10–50 rise time of α1WT (n = 8), α1W170S (n = 8), and α2 receptors (n = 8) expressed in HEK293T cells. D, Top, Whole-cell recordings from a cortical neuron overexpressing α1WT that exhibited sensitivities to Zn2+-mediated biphasic modulation. Bottom, Whole-cell recordings from a cortical neuron overexpressing α1W170S and not exhibiting sensitivity to Zn2+ potentiation. E, Averaged dose–responses for Zn2+-mediated modulation of glycine (50 μm)-activated currents in α1WT (n = 4) and α1W170S (n = 5) overexpressed neurons.
Neurons overexpressed with α1WT showed remarkable increase in glycine-mediated currents by 0.1–10 μm Zn2+ (Fig. 4D,E; mean 10–50% rise time: 66.4 ± 8.9 ms, n = 4). In contrast, Zn2+ did not enhance glycine-mediated currents in neurons overexpressed with α1W170S (Fig. 4D,E; mean 10–50% rise time: 62.9 ± 11.6 ms, n = 5). These results confirm our findings, using a recombinant expression system, that the W170S mutation also abolished sensitivity of α1 GlyRs to Zn2+ potentiation in neurons, and this may be a major causal factor of the symptoms of hyperekplexia in humans carrying this mutant allele.
Discussion
In the present study, we found that W170 was a novel Zn2+ potentiation site at the N-terminal domain of the GlyR α1 subunit. This site was identified from patients with an autosomal recessive form of hyperekplexia exhibiting homozygote W170S missense mutation (Al-Futaisi et al., 2012). We found the W170S mutation to cause complete ablation of sensitivity of α1 GlyRs to Zn2+ potentiation. In contrast to previously identified missense glra1 mutations, it did not affect other basic electrophysiological properties of α1 GlyRs. After recombinant expression, the W170S mutation removed Zn2+ potentiation and increased the sensitivity to Zn2+ inhibition of current responses activated by glycine, β-alanine, or taurine. This substantial reduction of Zn2+-mediated potentiation was further revealed by temporally separating the positive and negative modulatory phases and by examining the W170S mutation using the H107N mutation background, which exhibits low sensitivity to Zn2+ inhibition. Furthermore, abolishment of Zn2+ potentiation was also observed in α1W170S GlyRs expressed in neuronal cultures, indicating that the alteration of Zn2+ modulation can also be observed in a native neuronal environment and might be a major causal factor of hyperekplexia in humans carrying W170S alleles.
Typically, missense mutations of glra1 identified from hyperekplexia might reduce agonist binding sensitivity, affect channel conductance, or disrupt receptor surface expression (Saul et al., 1999; Harvey et al., 2008; Chung et al., 2010). We found that homomeric α1W170S receptors had a twofold decrease in sensitivity to glycine compared with the WT receptors. However, in α1W170Sβ GlyRs, the W170S mutation exhibited no reduction in sensitivity to all tested agonists, no changes in the maximal current responses, and no change in I-V relationships, suggesting that there were no defects in the intrinsic channel properties of the GlyR and that abnormalities in these properties were unlikely to be the major cause of pathogenesis in hyperekplexia patients carrying the α1170S mutation. Instead, our results demonstrated that the major alteration of α1W170 was the ablated sensitivity to Zn2+-mediated potentiation and the consequently increased sensitivity to Zn2+ inhibition. To our knowledge, the present study is the first demonstration of disrupted allosteric modulation of GlyRs by Zn2+ as an important factor in human hyperekplexia symptoms.
Endogenous free Zn2+ is concentrated in synaptic vesicles at certain synapses and is speculated to regulate synaptic transmission (Sensi et al., 2011). The physiological role of Zn2+ in synaptic transmission has been demonstrated by studies using Zn2+ chelators or exogenous application of Zn2+ to affect the duration and amplitude of glycinergic IPSCs (Suwa et al., 2001; Eto et al., 2007). Selectively ablating sensitivity of α1 to Zn2+ potentiation led to hyperekplexia-like phenotypes in transgenic mice carrying α1D80A (Hirzel et al., 2006). In line with these studies, our findings suggest that synaptic Zn2+ plays a crucial role in glycinergic synaptic transmission and efficacy in human CNS.
Footnotes
This work was supported by Taiwan National Science Council (Grants NSC 101-2320-B-039-057, NSC 100-2632-B-039-001-MY3, NSC 102-2320-B-039-038-MY3, and NSC 102-2320-B-039-035) and China Medical University Hospital (Grant DMR-101-119). Molecular graphics and analyses were performed with the UCSF Chimera package developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). We thank Drs. Y.T. Wang and A.W. Chan for comments on the manuscript and Ms. M.L. Shen for preparation of neuronal cultures.
The authors declare no competing financial interests.
References
- Al-Futaisi AM, Al-Kindi MN, Al-Mawali AM, Koul RL, Al-Adawi S, Al-Yahyaee SA. Novel mutation of GLRA1 in Omani families with hyperekplexia and mild mental retardation. Pediatr Neurol. 2012;46:89–93. doi: 10.1016/j.pediatrneurol.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Bakker MJ, van Dijk JG, van den Maagdenberg AM, Tijssen MA. Startle syndromes. Lancet Neurol. 2006;5:513–524. doi: 10.1016/S1474-4422(06)70470-7. [DOI] [PubMed] [Google Scholar]
- Bloomenthal AB, Goldwater E, Pritchett DB, Harrison NL. Biphasic modulation of the strychnine-sensitive glycine receptor by Zn2+ Mol Pharmacol. 1994;46:1156–1159. [PubMed] [Google Scholar]
- Brune W, Weber RG, Saul B, von Knebel Doeberitz M, Grond-Ginsbach C, Kellerman K, Meinck HM, Becker CM. A GLRA1 null mutation in recessive hyperekplexia challenges the functional role of glycine receptors. Am J Hum Genet. 1996;58:989–997. [PMC free article] [PubMed] [Google Scholar]
- Buckwalter MS, Cook SA, Davisson MT, White WF, Camper SA. A frameshift mutation in the mouse α 1 glycine receptor gene (Glra1) results in progressive neurological symptoms and juvenile death. Hum Mol Genet. 1994;3:2025–2030. doi: 10.1093/hmg/3.11.2025. [DOI] [PubMed] [Google Scholar]
- Chung SK, Vanbellinghen JF, Mullins JG, Robinson A, Hantke J, Hammond CL, Gilbert DF, Freilinger M, Ryan M, Kruer MC, Masri A, Gurses C, Ferrie C, Harvey K, Shiang R, Christodoulou J, Andermann F, Andermann E, Thomas RH, Harvey RJ, et al. Pathophysiological mechanisms of dominant and recessive GLRA1 mutations in hyperekplexia. J Neurosci. 2010;30:9612–9620. doi: 10.1523/JNEUROSCI.1763-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto K, Arimura Y, Nabekura J, Noda M, Ishibashi H. The effect of zinc on glycinergic inhibitory postsynaptic currents in rat spinal dorsal horn neurons. Brain Res. 2007;1161:11–20. doi: 10.1016/j.brainres.2007.05.060. [DOI] [PubMed] [Google Scholar]
- Harvey RJ, Topf M, Harvey K, Rees MI. The genetics of hyperekplexia: more than startle! Trends Genet. 2008;24:439–447. doi: 10.1016/j.tig.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirzel K, Müller U, Latal AT, Hülsmann S, Grudzinska J, Seeliger MW, Betz H, Laube B. Hyperekplexia phenotype of glycine receptor α1 subunit mutant mice identifies Zn(2+) as an essential endogenous modulator of glycinergic neurotransmission. Neuron. 2006;52:679–690. doi: 10.1016/j.neuron.2006.09.035. [DOI] [PubMed] [Google Scholar]
- Laube B, Kuhse J, Rundström N, Kirsch J, Schmieden V, Betz H. Modulation by zinc ions of native rat and recombinant human inhibitory glycine receptors. J Physiol. 1995;483:613–619. doi: 10.1113/jphysiol.1995.sp020610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wu DC, Wang YT. Allosteric potentiation of glycine receptor chloride currents by glutamate. Nat Neurosci. 2010;13:1225–1232. doi: 10.1038/nn.2633. [DOI] [PubMed] [Google Scholar]
- Lynch JW. Molecular structure and function of the glycine receptor chloride channel. Physiol Rev. 2004;84:1051–1095. doi: 10.1152/physrev.00042.2003. [DOI] [PubMed] [Google Scholar]
- Lynch JW, Jacques P, Pierce KD, Schofield PR. Zinc potentiation of the glycine receptor chloride channel is mediated by allosteric pathways. J Neurochem. 1998;71:2159–2168. doi: 10.1046/j.1471-4159.1998.71052159.x. [DOI] [PubMed] [Google Scholar]
- Mangin JM, Baloul M, Prado De Carvalho L, Rogister B, Rigo JM, Legendre P. Kinetic properties of the α2 homo-oligomeric glycine receptor impairs a proper synaptic functioning. J Physiol. 2003;553:369–386. doi: 10.1113/jphysiol.2003.052142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS, Beato M, Harvey RJ, Smart TG. Molecular determinants of glycine receptor αβ subunit sensitivities to Zn2+-mediated inhibition. J Physiol. 2005a;566:657–670. doi: 10.1113/jphysiol.2005.088575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS, Da Silva HM, Smart TG. Molecular basis for zinc potentiation at strychnine-sensitive glycine receptors. J Biol Chem. 2005b;280:37877–37884. doi: 10.1074/jbc.M508303200. [DOI] [PubMed] [Google Scholar]
- Mohammadi B, Krampfl K, Cetinkaya C, Moschref H, Grosskreutz J, Dengler R, Bufler J. Kinetic analysis of recombinant mammalian α(1) and α(1)β glycine receptor channels. Eur Biophys J. 2003;32:529–536. doi: 10.1007/s00249-003-0286-y. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera: a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Yang J, Zhang Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012;40:W471–W477. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul B, Kuner T, Sobetzko D, Brune W, Hanefeld F, Meinck HM, Becker CM. Novel GLRA1 missense mutation (P250T) in dominant hyperekplexia defines an intracellular determinant of glycine receptor channel gating. J Neurosci. 1999;19:869–877. doi: 10.1523/JNEUROSCI.19-03-00869.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Paoletti P, Koh JY, Aizenman E, Bush AI, Hershfinkel M. The neurophysiology and pathology of brain zinc. J Neurosci. 2011;31:16076–16085. doi: 10.1523/JNEUROSCI.3454-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwa H, Saint-Amant L, Triller A, Drapeau P, Legendre P. High-affinity zinc potentiation of inhibitory postsynaptic glycinergic currents in the zebrafish hindbrain. J Neurophysiol. 2001;85:912–925. doi: 10.1152/jn.2001.85.2.912. [DOI] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]