

Summary
In recent years, active research using genomic, cellular and animal modeling approaches has revealed the fundamental forces driving the development of autoimmune diseases. Type I IFN (IFN) imprints unique molecular signatures in a list of autoimmune diseases. IFN is induced by diverse nucleic acid-containing complexes, which trigger innate immune activation of plasmacytoid dendritic cells (pDCs). IFN primes, activates or differentiates various leukocyte populations to promote autoimmunity. Accordingly, IFN signaling is essential for the initiation and/or progression of lupus in several experimental models. However, the heterogeneous nature of SLE requires better characterization on how IFN pathways are activated and subsequently promote the advancement of autoimmune diseases. Given the central role of type I IFN, various strategies are devised to target these cytokines or related pathways to curtail the progression of autoimmune diseases.
Keywords: Type I Interferon, Systemic lupus erythematosus, Autoimmune disease, Plasmacytoid dendritic cells, Toll-like receptor, Nucleic acids, Innate immune activation, Anti-interferon therapy, Lupus model, Interferon induction
Type I IFN pathway is broadly implicated to autoimmune diseases
Since its discovery more than half a century ago, type I interferon has been recognized as the most important immune mediator involved in anti-viral protection. In contrast to type II interferon which is encoded by single IFN_ gene, type I IFN encompasses similarly structured products of many genes in human genome, including 13 IFN-α subtypes, IFN-β, IFN-ε, IFN-κ and IFN-ω. These proteins unanimously bind to and signal through a receptor complex comprised of IFN-α/β receptor 1(IFNAR1) and IFNAR2, which activates a prominent Jak-Stat signaling pathway. The consequence of this signaling is the expression of multitude of IFN-inducible genes, some of which have direct antiviral functions. The description of IFN biology and the molecular mechanism of IFN-mediated viral control can be found in other reviews [1,2]. It is important to point out that, IFN production as an immediate early response during viral infections is of transient nature for its duration. Therefore, healthy individuals do not have sustained IFN in their system, contrary to the pathological conditions described below. As IFN also possess potent anti-tumor activity, recombinant IFN has been routinely used to treat chronic viral infections or malignant cancers in patients.
The first autoimmune condition, where type I interferon is implicated, is systemic lupus erythematosus (SLE) [3]. SLE is a systemic autoimmune disease with multiple organ involvement, characteristic antinuclear autoantibodies and formation of immune complexes [4]. Lupus patients contain circulating IFN and “IFN signature” – transcript expression of a panel of type I IFN-responsive genes - in their peripheral blood, which frequently correlate with the disease severity [5–9]. In childhood-onset systemic lupus erythematosus, the levels of serum IFNα is positively correlated with circulating anti-dsDNA autoantibodies and SLE Disease Activity index scores [10]. Not only 90% of pediatric SLE patients and more than 50% of adult patients display peripheral IFN signature, half of the biopsied glomeruli from SLE kidneys also contain IFN-inducible transcripts, suggesting a central role played by IFN-mediated pathogenesis [10,11]. As a prototypic systemic autoimmune disease, SLE has been extensively studied in recent years and significant knowledge has been acquired regarding the source of IFN, the innate immune signaling pathways underlying its induction, and genetic risk factors involved (discussed in following sections).
It turns out that abnormal IFN presence is not limited to SLE, but is rather prevalent in various autoimmune pathologies. In psoriasis, a cutaneous autoimmune inflammatory condition, IFN signature is detected in the psoriatic plaques and induced IFN production further exacerbates the spread of the lesions [12]. Sjögren syndrome is a disease of which the salivary and lacrimal glands are the targets of destructive autoimmune reactions. Similar to SLE, gene expression analysis revealed the activation of IFN pathway, an important clue for understanding the disease pathogenesis [13]. Systemic sclerosis is a complex disease with features of extensive fibrosis and circulating autoantibodies against various cellular antigens. An activated type I IFN system with detectable IFN signature and IFNα serum levels is associated with the vascular pathology and fibrotic process [14]. A severe, multisystem autoimmune disease, dermatomyositis manifests as muscle, skin and vasculature pathologies, which frequently associate with tissue calcification. Multiple recent studies revealed significant upregulation of type I IFN pathway in both adult and juvenile dermatomyositis patients [15]. Recently, a genome wide survey identified a common gene set involved in the type I IFN pathway that are unanimously upregulated in patients with SLE, rheumatoid arthritis, myositis, and systemic sclerosis [16].
Administration of IFNα to patients with malignant or viral diseases occasionally induces a lupus-like syndrome, suggesting a causative relationship between IFN and lupus [6]. Recently, a group of inheritable diseases have been linked by the prominent type I interferon presence and collective autoimmune pathogenesis [17]. Aicardi-Goutieres syndrome (AGS) is a severe inflammatory disorder mimicking congenital infection with marked IFN production and occasionally overlapping features with SLE. Mutations in three prime repair exonuclease 1 (TREX1), a major DNA exonuclease important for clearance of endogenous DNA and anti-retroviral infection, have been shown to cause AGS and familial childblain lupus [18]. Interestingly, heterozygous mutations in TREX1 represent the single most common cause of monogenic lupus [19]. Moreover, the genetic mutation responsible for spondyloenchondrodysplasia, a disease with significant overlapping manifestation with SLE, has been identified [20,21]. Interestingly, tartrate-resistant acid phosphatase mutant leads to increased phospholated osteopontin, which results in elevated IFN production and exhibition of SLE and lupus-related autoimmunity. Collectively, these studies support a dominant role of type I interferon in lupus-like autoimmunity.
Autoimmune diseases comprise numerous very diverse pathologies from organ specific to systemic autoimmune manifestations. However, despite the tissues targeted and the nature of autoantibodies involved, a group of these diseases as discussed above share a common molecular link: abnormal IFN production and upregulation of the IFN-inducible gene panel. The mode of type I interferon activation under autoimmune conditions is rather unique and distinct from the neutrophil-driven gene profile induced jointly by type I/II IFN in patients with active tuberculosis infection [22]. The mechanism for its induction and the functional consequence of type I IFN is therefore critical for understanding the pathogenesis of autoimmune diseases.
pDCs as a major cellular source of IFN
pDCs are unique innate immune leukocytes that exert specialized function as the major type I IFN producer during the early host response to viral infections. They are derived from bone marrow precursors, circulating in blood and reside in T cell rich area of secondary lymphoid organs. Upon exposure to viral particles, pDCs can produce extraordinarily high amounts of all subtypes of type I and type III (IFNλ1–3) interferon in a matter of several hours. Although the frequency of pDCs is low (< 1% of all leukocytes), these cells are responsible for the majority of IFN produced during early viral infections due to the superior IFN production on per cell basis. Mice lacking pDCs are defective to control infections by mouse hepatitis virus, murine cytomegalovirus, or persistent strain of LCMV [23,24].
pDCs’ extraordinary ability to produce IFN is underscored by their intrinsic expression of a signaling machinery that can effectively respond to nucleic acids associated with microbial agents. In contrast to conventional dendritic cells, human pDCs selectively express high levels of Toll-like receptor (TLR) 7 and TLR9, two important innate immune sensors that detect RNA or DNA ligands. TLR9 preferentially bind to the unmethylated CpG motifs present in the 2′ deoxyribose backbone of natural DNA, which is more prevalent in microbial over mammalian DNA [12]. In addition to TLR7/9, pDCs endogenously express high levels of IRF7, a key signaling mediator to initiate IFN transcription. Since both TLR7 and TLR9 are strategically located intracellularly and nucleic acid engagement only occurs in specific endolysosome locations, a unique membrane trafficking pathway are essential for TLR7/9 signaling and IFN production in pDCs [12]. To trigger high IFN production, nucleic acids with particulate-like property or as part of a protein complex engage TLR9 primarily in the early endosome of pDCs to trigger MyD88 signaling and subsequent IRF7 activation, which initiates the transcription of all type I and type III interferon subtypes.
TLRs are ancient innate immune receptors that recognize the pathogen-associated molecular patterns (PAMP). Human genome encodes 10 TLRs that display differential binding specificities towards ligands of lipid, protein or nucleic acid nature. Interestingly, several TLRs, TLR3, TLR7, TLR8 and TLR9, can bind different types of nucleic acids in the lumen of endosomes and induce type I IFN production. While innate immune sensors, such as TLRs, are essential for immune protection against microbial infections, the Toll hypothesis predicts that autoimmune diseases originate from imperfect innate immune discrimination of microbe from self through receptors detecting microbial DNA and RNA [25]. This idea is supported by the fact that genetic risk factors associated with SLE include a group of genes whose products are directly involved in TLR and type I IFN signaling pathways [4,26,27]. Functional polymorphism of 3’ UTR of TLR7 with increased TLR7 expression and elevated IFN signature is associated with human male SLE, consistent with the lupus manifestation in mice with increased gene dosage of TLR7 [28]. Genetic variants of IRF5, IRF7 and IRF8 are associated with increased SLE susceptibility, which is correlated with increased serum IFNα in patients [27,29].
While pDCs are activated readily by viral particles, they do not respond to naked natural DNA or RNA. Spontaneous IFN production is safeguarded by shielding TLR7/9 inside the cells and ubiquitous presence of nucleases in the tissue environment. However, several host-derived factors, by binding to self–nucleic acids and delivering them to endosomal compartments, can nevertheless trigger TLR signaling and induce type I IFN from pDCs. It is well known that many SLE patients have reduced capacity to clear apoptotic cells, which correlates with elevated levels of circulating immune complexes (IC) containing autoantibodies and nucleic acids . These ICs can stimulate autoreactive B cells by dual engagement of B cell receptor and TLRs, thus promote further lupus pathogenesis. For pDCs, nucleic acid-containing ICs can be internalized by binding to Fc receptor (FcγRIIα, CD32) and delivered to the endolysosome to activate TLRs (Figure 1). While TLR9 is activated by DNA-containing ICs, such as autoantibody complexed with nucleosomes, TLR7 is potently stimulated by RNA-containing ICs, made of autoantibody bound to U1 small nuclear RNA in pDCs that leads to IFN production [6,12]. Blocking TLR signaling activation by interfering with the endosomal delivery or competing off the active sites on TLR7/9 significantly diminishes the amount of IFN induced by ICs from pDCs, highlighting the significance of TLR activation pathway in abnormal IFN induction in SLE.
Figure 1. Human pDCs are activated by nucleic acid-containing complexes to produce type I IFN.

SLE ICs containing chromatin or ribonucleoprotein (RNP) are internalized by pDCs via binding to Fc receptor and activating TLR9 or TLR7, respectively, to induce IFN production. Other nucleic acid-containing complexes, such as LL-37 or amyloid fibrils, also activate pDCs to trigger IFN secretion. SLE Neutrophils, which are primed by pDC-produced IFN and stimulated by autoantibodies, undergo NETosis. Nucleic acids-containing NET then prompts IFN production from pDCs.
It has been recently revealed that pDCs’ IFN response to autoimmune ICs is subjected to regulation by multilayered mechanisms. A network of monocytes, NK cells, and pDCs maintains certain level of control: monocytes inhibit RNA-containing ICs-induced IFNα production via secreting TNF-α, PGE2, and reactive oxygen species; whereas significantly enhanced IFN secretion is promoted by NK cells via MIP-1β secretion and LFA-mediated cell–cell contact [30,31]. Complement component C1q deficiency is the strongest known susceptibility factor for SLE. Interestingly, majority of SLE patients have low levels of C1q, reciprocal to the increased type I IFN activity during active disease. C1q binding to ICs gears the predominant binding of these interferongenic complexes towards monocytes other than pDCs, which markedly attenuates IFN production [32,33].
While SLE patients have somewhat reduced pDC numbers in their blood, infiltrating pDCs are found abundantly in the skin lesions of cutaneous lupus patients [12]. In both dermal lesions and in noninflammatory skin, SLE patients have active pDCs correlated with positive IFNα transcript. It is apparent that IFN signature and pDCs infiltration are involved in several cutaneous autoimmune diseases, where cytotoxic attack leads to degeneration of the basal epidermal layer [34]. Gilliet et al demonstrated that pDC-derived type I IFN is essential to drive the development of psoriasis; and furthermore, LL37, an antimicrobial peptide present in the skin of psoriasis patients, binds to self-nucleic acids and activate TLR7/9, leading to the selective IFN production by pDCs [35] (Figure 1). In addition to profound IFN signature in blood, increased numbers of pDCs as well as MxA protein have been detected in both skin and muscle from patients with juvenile dermatomyositis, implicating the pDC-IFN axis in tissue inflammation [36].
Parallel with significant IFN signature, SLE blood also selectively express a panel of genes involved in granulopoiesis, which correlates with the abnormal presence of large number of immature neutrophils [5,37]. Neutrophils are innate immune cells that rapidly infiltrate infection or injury sites for host protection. Uniquely, neutrophils can undergo a peculiar form of cell death, namely NETosis, characterized by formation of NET, decondensed chromatin threads decorated with cytoplasmic proteins endorsed with anti-microbial activity [38]. It was recently shown that IFN can prime neutrophils for NETosis, a process that is strongly stimulated by autoantibodies against ribonucleoprotein complex or LL37 [39,40]. Subsequently, the generated NET stimulates pDCs to induce type I inteferon, a self-perpetuating loop to sustain the IFN production (Figure 1). High-mobility group box 1 (HMGB1), a highly abundant chromatin-binding protein, is implicated clinically in SLE and exert multiple immunological functions [41]. Consistently, HMGB1 constitutes part of NET, which functions as a potent IFN inducer [40]. HMGB1 has been shown to facilitate activation of mouse pDCs by forming a DNA-containing complex capable of engaging TLR9 [42]; however, this function does not seem to be preserved in humans [43,44].
Amyloid fibrils are stable insoluble aggregates of terminal misfolded protein products with extensive β sheet structures, which can accumulate extracellularlly or intracellularly [45]. Multiple aberrant polypeptides are implicated in multiple human pathological conditions exemplified by Alzheimer’s’ disease [46], whereas an increasing list of functional amyloids participate in diverse cellular functions [47–49]. Amyloid dispositions in vivo are frequently heterogeneous and contain non-proteinaceous cofactors [45,50]. We first observed that amyloid precursor proteins rapidly convert to amyloid fibrils in the presence of nucleic acids or glycosaminoglycans [51]. Distinct from protein only amyloid or fibrils containing glycosaminoglycan, nucleic acid-containing amyloid fibrils are potent inducer of type I IFN from human pDCs (Figure 1). Complexed with amyloid precursor protein, self-DNA or -RNA are protected from nucleases in the environment, effectively taken up then is retained in the early endosomes of pDCs, where the prolonged TLR9 activation can promote MyD88 signaling and subsequent IRF7 activation, which initiates the transcription of type I IFN genes. Importantly, distinct from ICs or NET, nucleic acid-containing amyloid induces IFN production independent of the function of autoantibodies and FcγRIIα.
IFN promotes autoimmunity by broad effects on leukocytes
Beyond the antiviral effects, type I IFN modulates the functions of all key leukocytes involved in innate and adaptive immunity. Dendritic cells (DCs) are professional antigen-presenting cells (APCs) which, upon innate immune sensing, initiate and orchestrate adaptive immune responses. DCs are particularly important in maintaining tolerance and driving autoimmunity. Mice harboring DC-specific ablation of Blimp-1 or A20, or expressing baculovirus caspase inhibitor all develop severe lupus-like syndrome, demonstrating a powerfully pathogenic role of DCs with altered activation threshold or unrestrained presence [52–54].
It was first discovered that SLE sera differentiate human monocytes into DCs that function as potent APCs in a type I IFN-dependent manner [5]. In addition to activating T cells, SLE-DCs can strongly promote T cell-independent plasmablast differentiation [55]. IFN in tissue sustains the local recruitment of inflammatory monocytes and promotes their differentiation into macrophages [56].
Additionally, IFN profoundly modifies the functions of myeloid DCs, a subset of DCs that are distinct from pDCs. The resulting DCs display increased expression of costimulatory and MHC molecules, enhance antigen processing and presentation by both MHC I and II molecules [57,58]. These features are crucial for the subsequent stimulation of autoreactive T cells, likely leading to strong TH1 polarization of the immune response [59]. IFN signaling in DCs has been shown to enhance the primary antibody response and induces isotype switching, at least partly due to the DCs’ ability to stimulate the development of lymph node–resident T follicular helper cells, which are critical for B cell development [1,60].
Autoantibody production serves as a hallmark of various autoimmune diseases. In SLE, immune complexes containing autoantibodies contribute directly to disease pathology. Aside from their effect on monocytes and DCs that promote B cell differentiation, type I IFN directly activates B cells and T cells in vivo to promote antibody response [1,61]. Together with IL-6 produced as a result of TLR activation of pDCs, IFN induces the differentiation of mature B cells into long-lived plasma cells [5]. In lupus-prone NZB/W F1 mice, exogenous IFNα stimulates undiminished production of short-lived plasma cells [62]. What is more, IFN activates myeloid cells to secrete BAFF and APRIL, two TNF superfamily ligands that are crucial for B cell survival and activation [63].
Separately, type I IFN directly affects T lymphocytes on the development of CD4+ helper memory T cells and the proliferation and expansion of antigen-specific and long-lived central memory CD8+ T cells [61,64]. IFNα/β limits Th17 differentiation, likely via dampening IL-23 while increasing IL-27 production by macrophages and DCs [65,66]. Although IFN does not affect the recruitment of neutrophils on the inflammation sites, it primes SLE neutrophils to promote their death induced by SLE autoantibodies. As discussed earlier, NETosis generates nucleic acid-containing NETs to induce IFN production by pDCs [39,40].
IFN propels autoimmune diseases in vivo
Animal models are valuable tools to allow better understanding of disease pathogenesis, where in vitro studies are confirmed, cellular mechanisms are established, therapeutic interventions are tested, and results are correlated with human clinic diseases. While the etiology of SLE remains elusive, a recent comprehensive genetic analysis classified SLE into three subphenotypes - those having single, cumulative, and no known genetic association [67]. As a reflection, multiple lupus animal models have been created to study this complex and heterogeneous disease experimentally. Despite the strong association between type I interferon, pDCs and human SLE, the central question remains – what are the roles played by type I interferon and pDCs in vivo?
The most commonly studied lupus mice are inbred mouse strains that spontaneously develop a disease with characteristics of human SLE [68,69]. These lupus-prone rodents harbor multigenic mutations that predispose them to autoimmune development; however, they lack the prominent IFN-I signature associated with clinic lupus. Therefore, it remains unclear how endogenous type I IFN and pDCs participate in the pathogenesis of spontaneous murine lupus. Also notable is that, although administration of IFNα to patients occasionally induces lupus, excessive IFN is insufficient to break the immune tolerance in Balb/c mice [5]. In contrast, systemically induced IFNα production in young NZB/W F1 and B6.Sle123 mice significantly accelerated lupus nephritis [62,70]. Conversely, IFN receptor deficiency or TLR7/9 inhibition greatly ameliorated lupus in NZB/W F1 mice [71,72]. Interestingly, the skin inflammation in the same mice, which mimics cutaneous lupus, is dependent on pDC activation via TLR7 and TLR9, which results in a persistent type I IFN gene signature [73].
Currently, there is no clear delineation between Mendelian inherited autoimmune diseases and acquired ones, albeit influencing factors such as infections or aging have been suggested as contributors in the latter category [74,75]. As significant number of autoimmune patients harbor no apparent genetic association, it is important to evaluate lupus pathogenesis within a relatively competent immune system. However, mice that spontaneously develop lupus have a multitude of intrinsic abnormalities that predispose them to autoimmune development [76], therefore other autoimmune models are necessary to be studied. In particular, an inducible experimental lupus model that re-creates the disease-initiating event would be especially informative. Immunization of non-autoimmune mice with bacterial DNA with a carrier protein induced production of anti-bacterial DNA antibody; however, the antibody failed to cross-react with mammalian DNA [77]. Injection of a large number of apoptotic human cells into wild-type mice resulted in modest and transient autoantibody production without clinical changes [78]. Purified HMGB1-nucleosome complexes induced the generation of autoantibodies against dsDNA and histone, a process dependent on the signaling of TLR2 [44]. However, the autoantibody response was limited to the immunogen administrated; in any case, lupus-like disease was not reported. Altogether, these attempted efforts revealed a powerful mechanism by which a healthy immune system maintains tolerance and avoids overzealous autoimmunity, even when the self antigens are given at high doses.
Recently we have been successful to induce a lupus-like syndrome in non-autoimmune mice after immunization with nucleic acid-containing amyloid [79](Figure 2). Consistent with their potent pDC activation function in vitro, DNA-containing amyloid fibrils induced a rapid type I IFN response correlated with selective pDC infiltration and activation in mice. Immunization of Balb/c mice with DNA-containing amyloid fibrils resulted in anti-nuclear serology against a panel of self-antigens, including ssDNA, RNA, Sm/RNP and histone. The mice deposited antibodies in the kidneys and exhibited detectable proteinuria. Intriguingly, pDC depletion selectively obstructed IFN response and abolished autoantibody generation, suggesting an essential role of pDC-IFN axis in initiation of systemic autoimmunity. This inducible lupus model for the first time allows in vivo characterization how pDCs, the primary IFN producer in SLE, and innate immune signaling pathways contribute to the pathogenesis of lupus.
Figure 2. Induction of type I IFN initiates systemic autoimmunity in vivo.
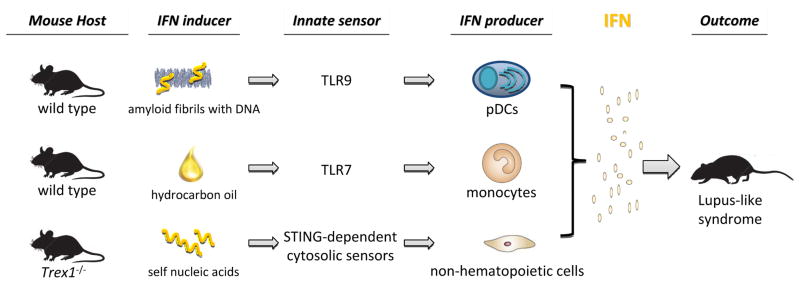
Murine experimental lupus models illustrate that activation of different innate immune pathways induce type I IFN and cause systemic lupus-like autoimmunity.
Separately, wild type mice, after i.p. injection with hydrocarbon oil pristane, manifest prolonged inflammation and eventually develop an array of autoantibodies and glomerulonephritis [80]( Figure 2). In this model, type I IFN expression was detected from a population of monocytes via TLR7 activation [81,82]. Pristane, which can be ingested in food or inhaled from exposure to oil products, imitates an environmental trigger for lupus [80]. Interestingly, prolonged oral administration of pristane reportedly results in amyloidosis in mice [83].
Numerous single-gene deletions in mice have resulted in lupus-like syndrome, which highlights the importance of individual checkpoint controls in maintaining immune tolerance [68,84]. A growing list of innate immune sensors in the cytoplasm of many cell types can respond to intracellular pathogens by inducing type I IFN, a process that does not rely on membrane-associated TLRs (see review in [85]). Largely consistent with TREX1 mutations in human, Trex1−/− mice develop a severe multi-organ autoimmune disease, that is driven by intrinsic production of type I IFN from nonhematopoietic cells in response to the accumulation of nucleic acids [86](Figure 2). As a major cellular exonuclease, Trex1, i.e. DNase III, is crucial to remove aberrant products of retrotransposon replication [87]. Notably in Trex1−/− mice, early IFN production is mediated by an adaptor molecule STING in a TLR-independent manner. Interestingly, STING also mediates the lethal inflammatory disease initiated by self DNA manifested in DNase II−/− mice [88]. MRL-lpr mice that harbor lpr mutation in Fas gene are well studied lupus-prone strain. In these mice, type II IFN signature prevails and IFN_ plays a more pathogenic role instead of type I IFN [89,90]. As multiple human in vitro studies suggest importance of neutrophils and NETs in SLE [39,40,91,92], the contribution of NETosis in murine lupus was studied recently in MRL-lpr model. MRL-lpr mice lacking NADPH oxidase gene that is essential for NETosis unexpectedly have exacerbated lupus, arguing against a significant pathogenic role of NETs in SLE [93]. The precise mechanism how NADPH oxidase inhibits lupus remains to be elucidated.
Therapeutic strategies to treat autoimmune diseases
The current treatment options for SLE patients often involve the use of immunosuppressive regimens, including glucocorticoids, cytotoxic drugs and antimalarial compounds. However, even high-dose methylprednisolone pulse therapy, an aggressive immunosuppressive approach, fails to induce the desired apoptosis of pDCs due to their ongoing TLR activation by ICs [94]. With the encouragement from the approval of Belimumab (anti-BAFF mAb), major drug development effort directly targets B cells – the leukocyte secreting autoreactive antibodies with pathological consequences (recently reviewed in [95,96]). Given the central role played by type I IFN pathway in propelling lupus in human and in experimental models, strategies to interfere with the key steps by which this central mediator influences the development of autoimmunity represent a rational design for the next generation of therapeutic remedies for lupus and beyond.
Type I interferon poses a major challenge for drug targeting as it comprises multiple subsets of closely related proteins. Potential health risk also exists because IFN is extremely important for host’s anti-viral responses. To directly repress the levels of active type I IFN in SLE patients, multiple humanized monoclonal antibodies with specificity towards IFNα or IFNα/β are currently in early phase (I or II) clinical trials, including Sifalimumab (MedImmune LLC), Rontalizumab (Genentech, Inc.), and AGS-009 (Argos Therapeutics Inc.). It is encouraging that sifalimumab is safe and elicits a dose dependent inhibition of the IFNα pathway [97]. Interestingly, SLE patients with neutralizing autoantibodies against IFNα have less active disease [98]. To induce such protective immunity, an IFNα kinoid vaccine (Neovacs S.A.) has been tested in an early phase trial with reasonable safety and efficacy results [99]. Despite this, caution needs to be taken with this approach as anti-cytokine autoantibodies are associated with life-threatening immunodeficiency, as in the case of patients with anti-type II IFN antibodies [100].
TLR-mediated activation of pDCs represent the major source of type I interferon in lupus, thus means to inhibit TLR signaling would significantly impact the disease progression. Several nucleotide antagonists with duo specificity for TLR7 and TLR9 have been developed and are tested in early phase trials. These include DV1179 (Dynavax Technologies), IMO-3100 (Idera Pharmaceuticals) and CpG-52364 (Pfizer Inc.). One such inhibitor was shown to be effective to reduce autoantibodies and ameliorate disease symptoms in murine lupus models [71,73,94].
In addition to these approaches, it would be plausible to selectively target pDCs or signaling molecules downstream of TLR activation that are essential for IFN induction. Human pDCs express a list of specific surface receptors, such as BDCA2 and ILT7, as well as intracellular signaling adaptors, such as IRF7 and PACSIN-1, which can serve as unique targets for monoclonal antibodies or small molecule drugs [12]. Compounds that interfere generic TLR or IFNAR signaling, such as IRAKs or JAKs, would also be potentially useful to curb TLR and IFN activation under autoimmune conditions.
Although this review focuses on the pathological association and functional significance of IFNα/β, type I interferon by no means represents the universal cytokine target for autoimmune diseases. Human autoimmune diseases encompass a large number of different diseases, of which the degree of heterogeneity and complexity even in a single disease cannot be understated. When considering anti-interferon therapies, one should not ignore either the regulatory functions of type I IFN, such as limiting TNFα production, inhibiting inflammasome activation or dampening Th-17 development [65,66,101–103], as these affected pathways are critical for diseases such as rheumatoid arthritis, various autoinflammatory diseases, or multiple sclerosis. To successfully translate the findings from basic research to clinic requires careful selection of the patients with a disease that has concrete link to the targeted molecular pathway.
Expert commentary.
Autoimmune diseases inflict 5% of general population and cause tremendous suffering in patients. The precise etiology of these chronic pathologies is unknown, but significant progress in the last decade has revealed a general molecular theme that may underlie their pathogenesis. SLE and several other autoimmune diseases display a unique signature of type I interferon activation, which generally correlates with disease severity. Now it is clear that a panel of molecular compounds containing nucleic acids can activate TLR7 or TLR9 in pDCs, which consequently leads to prominent type I IFN production. A powerful pluripotent cytokine, IFN exhibits wide range effects on leukocytes, including DCs, monocyte, B cells, T cells and neutrophils, to promote autoimmune responses. Studies of lupus animal models reveal an essential role of type I IFN in inducing autoimmunity and exacerbating the disease symptoms. Altogether, these observations strongly advocate the need for an IFN-centered, targeted therapeutic approach to treat autoimmune diseases. The current therapeutic options for lupus and others are largely non-specific and have many undesired long term side effects for patients. A specific nucleic acid-TLR-pDC-IFN pathway that prompts the autoimmune development serves as an ideal map for selecting rational drug targets. Multiple strategies to tackle this particular pathway are being developed or under assessment, which bring great hope for the arrival of next generation disease specific treatment for autoimmune diseases.
Five-year view.
The current dogma on autoimmune diseases centers on TLR activation and type I interferon, which are supported by ample in vitro and in vivo experimental evidence. There still remain several important questions regarding the basic mechanism of autoimmunity that would help guide future targeted drug development. First of all, what are the triggers of autoimmune responses? As nucleic acids are absolutely central in activating the innate immune responses preceding IFN production, can they, in any complex form, truly function as the etiological agent to initiate the immune cascade leading to autoimmunity? Second, what is the contribution by other nucleic acid sensors in supporting autoimmune development? Multiple cytosolic receptors have been identified recently that are capable of binding to nucleic acids and trigger type I IFN production in diverse cell types [85]. Although Trex1−/− mice demonstrate the significant role played by a TLR-independent pathway, mice overexpressing MDA5, a cytosolic sensor for dsRNA, failed to develop autoimmune phenotype, a striking contrast to TLR7 transgenic animals [84,104]. Therefore it remains to be elucidated whether all nucleic acid-sensing pathways contribute to autoimmunity equally or differentially. Moreover, it is highly likely that further immune activation has to occur in autoimmune diseases as excessive IFN alone is insufficient to initiate lupus [6,104]. Therefore, what are the other key immune mediators that complement type I IFN to drive disease progression? On the other hand, the functional similarity between type I and type III interferon invites a closer look at this group of cytokines to examine if they are able to replace IFN to operate under certain disease conditions.
Key issues.
A large body of studies in humans increasingly implicates type I interferon pathway in a list of autoimmune diseases, suggesting a common underlying mechanism of their pathogenesis;
Plasmacytoid dendritic cells represent a major cellular source of type I interferon that are implicated in clinical autoimmune pathologies. These cells are activated by nucleic acid-containing complexes that selectively activate endosomal TLR7 or TLR9, which leads to prominent IFN production;
Type I interferon has wide-range effects on multiple immune cell populations. By stimulating different arms of the immune system, IFN is tremendously powerful to promote autoimmune responses;
Type I IFN signaling proves to be essential in multiple lupus models. It functions prior to the onset of autoimmunity or exacerbate pre-existing lupus propensity. In a newly established model, pDCs are found to represent an essential IFN producer in vivo. Novel animal models will allow in-depth mechanistic elucidation to correlate with human disease pathogenesis.
Type I IFN pathway represents an important therapeutic target for autoimmune diseases in general. Diverse treatment strategies are being examined to downregulate IFN and dampen the progress of autoimmune diseases.
Acknowledgments
This work is supported by National Institutes of Health Grant AI074809 and grants from The University of Texas M. D. Anderson Cancer Center Institutional Research Grant Program (to W.C.).
Footnotes
Financial disclosure
The authors have no financial conflict.
References
- 1.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 2.Yan N, Chen Z. Intrinsic antiviral immunity. Nat Immunol. 2012;13(3):214–222. doi: 10.1038/ni.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune Interferon in the Circulation of Patients with Autoimmune Disease. New Eng J Med. 1979;301(1):5–8. doi: 10.1056/NEJM197907053010102. [DOI] [PubMed] [Google Scholar]
- 4.Tsokos GC. Systemic Lupus Erythematosus. New Eng J Med. 2011;365(22):2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 5.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25(3):383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 6.Rönnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. LUPUS. 2008;17(5):394–399. doi: 10.1177/0961203308090020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaussabel D, Quinn C, Shen J, et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity. 2008;29(1):150–164. doi: 10.1016/j.immuni.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lugar PL, Love C, Grammer AC, Dave SS, Lipsky PE. Molecular Characterization of Circulating Plasma Cells in Patients with Active Systemic Lupus Erythematosus. PLoS ONE. 2012;7(9):e44362. doi: 10.1371/journal.pone.0044362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohl K, Tenbrock K. Inflammatory cytokines in systemic lupus erythematosus. Journal of biomedicine & biotechnology. 2011;2011:432595. doi: 10.1155/2011/432595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Postal M, Sinicato N, Peliçari K, Marini R, Lavras Costallat L, Appenzeller S. Clinical and serological manifestations associated with interferon-α levels in childhood-onset systemic lupus erythematosus. Clinics (São Paulo, Brazil) 2012;67(2):157–162. doi: 10.6061/clinics/2012(02)11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterson KS, Huang J-F, Zhu J, et al. Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. The Journal of Clinical Investigation. 2004;113(12):1722–1733. doi: 10.1172/JCI19139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8(8):594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 13.Gottenberg JE, Cagnard N, Lucchesi C, et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren's syndrome. Proc Natl Acad Sci U S A. 2006;103(8):2770–2775. doi: 10.1073/pnas.0510837103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eloranta ML, Franck-Larsson K, Lovgren T, et al. Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis. 2010;69(7):1396–1402. doi: 10.1136/ard.2009.121400. [DOI] [PubMed] [Google Scholar]
- 15.Baechler E, Bilgic H, Reed A. Type I interferon pathway in adult and juvenile dermatomyositis. Arthrit Res Ther. 2011;13(6):249. doi: 10.1186/ar3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higgs BW, Liu Z, White B, et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis. 2012;70(11):2029–2036. doi: 10.1136/ard.2011.150326. [DOI] [PubMed] [Google Scholar]
- 17.Yanick JC. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. 2011;1238 doi: 10.1111/j.1749-6632.2011.06220.x. [DOI] [PubMed] [Google Scholar]
- 18.Crow YJ, Hayward BE, Parmar R, et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38(8):917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 19.Lee-Kirsch MA, Gong M, Chowdhury D, et al. Mutations in the gene encoding the 3[prime]-5[prime] DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39(9):1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 20.Briggs T, Rice G, Daly S, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet. 2011;43(2):127–131. doi: 10.1038/ng.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lausch E, Janecke A, Bros M, et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat Genet. 2011;43(2):132–137. doi: 10.1038/ng.749. [DOI] [PubMed] [Google Scholar]
- 22.Berry MP, Graham CM, Mcnab FW, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466(7309):973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity. 2010;33(6):955–966. doi: 10.1016/j.immuni.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cervantes-Barragan L, Lewis KL, Firner S, et al. Plasmacytoid dendritic cells control T-cell response to chronic viral infection. Proc Natl Acad Sci U S A. 2012;109(8):3012–3017. doi: 10.1073/pnas.1117359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodnow CC. Immunology Discriminating microbe from self suffers a double toll. Science. 2006;312(5780):1606–1608. doi: 10.1126/science.1129797. [DOI] [PubMed] [Google Scholar]
- 26.Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med. 2011;365(17):1612–1623. doi: 10.1056/NEJMra1100030. [DOI] [PubMed] [Google Scholar]
- 27.Niewold T. Interferon alpha as a primary pathogenic factor in human lupus. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2011;31(12):887–892. doi: 10.1089/jir.2011.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen N, Fu Q, Deng Y, et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci USA. 2010;107(36):15838–15843. doi: 10.1073/pnas.1001337107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salloum R, Niewold TB. Interferon regulatory factors in human lupus pathogenesis. Translational Research. 2011;157(6):326–331. doi: 10.1016/j.trsl.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hagberg N, Berggren O, Leonard D, et al. IFN-α production by plasmacytoid dendritic cells stimulated with RNA-containing immune complexes is promoted by NK cells via MIP-1β and LFA-1. J Immunol. 2011;186(9):5085–5094. doi: 10.4049/jimmunol.1003349. [DOI] [PubMed] [Google Scholar]
- 31.Eloranta M-L, Lövgren T, Finke D, et al. Regulation of the interferon-alpha production induced by RNA-containing immune complexes in plasmacytoid dendritic cells. Arthritis and rheumatism. 2009;60(8):2418–2427. doi: 10.1002/art.24686. [DOI] [PubMed] [Google Scholar]
- 32.Santer DM, Wiedeman AE, Teal TH, Ghosh P, Elkon KB. Plasmacytoid dendritic cells and C1q differentially regulate inflammatory gene induction by lupus immune complexes. J Immunol. 2012;188(2):902–915. doi: 10.4049/jimmunol.1102797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santer D, Hall B, George T, et al. C1q deficiency leads to the defective suppression of IFN-alpha in response to nucleoprotein containing immune complexes. J Immunol. 2010;185(8):4738–4749. doi: 10.4049/jimmunol.1001731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wenzel J, Tuting T. An IFN-associated cytotoxic cellular immune response against viral, self-, or tumor antigens is a common pathogenetic feature in "interface dermatitis''. J Invest Dematol. 2008;128(10):2392–2402. doi: 10.1038/jid.2008.96. [DOI] [PubMed] [Google Scholar]
- 35.Lande R, Gregorio J, Facchinetti V, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449(7162):564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 36.Shrestha S, Wershil B, Sarwark JF, Niewold TB, Philipp T, Pachman LM. Lesional and nonlesional skin from patients with untreated juvenile dermatomyositis displays increased numbers of mast cells and mature plasmacytoid dendritic cells. Arthritis Rheum. 2010;62(9):2813–2822. doi: 10.1002/art.27529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Denny MF, Yalavarthi S, Zhao W, et al. A Distinct Subset of Proinflammatory Neutrophils Isolated from Patients with Systemic Lupus Erythematosus Induces Vascular Damage and Synthesizes Type I IFNs. J Immunol. 2010;184(6):3284–3297. doi: 10.4049/jimmunol.0902199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30(1):459–489. doi: 10.1146/annurev-immunol-020711-074942. [DOI] [PubMed] [Google Scholar]
- 39.Lande R, Ganguly D, Facchinetti V, et al. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA–Peptide Complexes in Systemic Lupus Erythematosus. Sci Transl Med. 2011;3(73):73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40**.Garcia-Romo GS, Caielli S, Vega B, et al. Netting Neutrophils Are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci Transl Med. 2011;3(73):73ra20. doi: 10.1126/scitranslmed.3001201. These two studies jointly revealed an important feed-forward pathogenic loop in SLE, where autoantibody, IFN, neutrophils and pDCs corroborate to exacerbate the disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andersson U, Tracey K. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian J, Avalos AM, Mao S-Y, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 43.Popovic PJ, Demarco R, Lotze MT, et al. High Mobility Group B1 Protein Suppresses the Human Plasmacytoid Dendritic Cell Response to TLR9 Agonists. J Immunol. 2006;177(12):8701–8707. doi: 10.4049/jimmunol.177.12.8701. [DOI] [PubMed] [Google Scholar]
- 44.Urbonaviciute V, Fürnrohr BG, Meister S, et al. Induction of inflammatory and immune responses by HMGB1–nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205(13):3007–3018. doi: 10.1084/jem.20081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sideras K, Gertz M. Amyloidosis. Adv Clin Chem. 2009;47:1–44. [PubMed] [Google Scholar]
- 46.Schnabel J. Protein folding: The dark side of proteins. Nature. 2010;464:828–829. doi: 10.1038/464828a. [DOI] [PubMed] [Google Scholar]
- 47.Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid - from bacteria to humans. Trends Biochem Sci. 2007;32(5):217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 48.Badtke MP, Hammer ND, Chapman MR. Functional Amyloids Signal Their Arrival. Sci Signal. 2009;2(80):pe43. doi: 10.1126/scisignal.280pe43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maji SK, Perrin MH, Sawaya MR, et al. Functional Amyloids As Natural Storage of Peptide Hormones in Pituitary Secretory Granules. Science. 2009;325(5938):328–332. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiménez JS. Protein-DNA Interaction at the Origin of Neurological Diseases: A Hypothesis. J Alzheimer's Disease. 2010;22(2):375–391. doi: 10.3233/JAD-2010-100189. [DOI] [PubMed] [Google Scholar]
- 51.Di Domizio J, Zhang R, Stagg LJ, et al. Binding with nucleic acids or glycosaminoglycans converts soluble protein oligomers to amyloid. J Biol Chem. 2012;287(1):736–747. doi: 10.1074/jbc.M111.238477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen M, Wang Y-H, Wang Y, et al. Dendritic Cell Apoptosis in the Maintenance of Immune Tolerance. Science. 2006;311(5764):1160–1164. doi: 10.1126/science.1122545. [DOI] [PubMed] [Google Scholar]
- 53.Kim SJ, Zou YR, Goldstein J, Reizis B, Diamond B. Tolerogenic function of Blimp-1 in dendritic cells. J Exp Med. 2011;208(11):2193–2199. doi: 10.1084/jem.20110658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kool M, Van loo G, Waelput W, et al. The Ubiquitin-Editing Protein A20 Prevents Dendritic Cell Activation, Recognition of Apoptotic Cells, and Systemic Autoimmunity. Immunity. 2011;35(1):82–96. doi: 10.1016/j.immuni.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 55.Joo H, Coquery C, Xue Y, et al. Serum from patients with SLE instructs monocytes to promote IgG and IgA plasmablast differentiation. J Exp Med. 2012;209(7):1335–1348. doi: 10.1084/jem.20111644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee PY, Li Y, Kumagai Y, et al. Type I Interferon Modulates Monocyte Recruitment and Maturation in Chronic Inflammation. Am J Pathol. 2009;175(5):2023–2033. doi: 10.2353/ajpath.2009.090328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spadaro F, Lapenta C, Donati S, et al. IFN-α enhances cross-presentation in human dendritic cells by modulating antigen survival, endocytic routing, and processing. Blood. 2012;119(6):1407–1417. doi: 10.1182/blood-2011-06-363564. [DOI] [PubMed] [Google Scholar]
- 58.Simmons DP, Wearsch PA, Canaday DH, et al. Type I IFN Drives a Distinctive Dendritic Cell Maturation Phenotype That Allows Continued Class II MHC Synthesis and Antigen Processing. J Immunol. 2012 doi: 10.4049/jimmunol.1101313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Longhi MP, Trumpfheller C, Idoyaga J, et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med. 2009;206(7):1589–1602. doi: 10.1084/jem.20090247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cucak H, Yrlid U, Reizis B, Kalinke U, Johansson-Lindbom B. Type I interferon signaling in dendritic cells stimulates the development of lymph-node-resident T follicular helper cells. Immunity. 2009;31(3):491–501. doi: 10.1016/j.immuni.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 61.Tough DF. Modulation of T-cell function by type I interferon. Immunol Cell Biol. 2012;90:492–497. doi: 10.1038/icb.2012.7. [DOI] [PubMed] [Google Scholar]
- 62.Mathian A, Gallegos M, Pascual V, Banchereau J, Koutouzov S. Interferon-α induces unabated production of short-lived plasma cells in pre-autoimmune lupus-prone (NZB×NZW)F1 mice but not in BALB/c mice. Eur J Immunol. 2011;41(3):863–872. doi: 10.1002/eji.201040649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. 2009;9(7):491–502. doi: 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 64.Ronnblom L, Alm GV, Eloranta ML. Type I interferon and lupus. Curr Opin Rheumatol. 2009;21(5):471–477. doi: 10.1097/BOR.0b013e32832e089e. [DOI] [PubMed] [Google Scholar]
- 65.Guo B, Chang E, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. The Journal of Clinical Investigation. 2008;118(5):1680–1690. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moschen AR, Geiger S, Krehan I, Kaser A, Tilg H. Interferon-alpha controls IL-17 expression in vitro and in vivo. Immunobiology. 2008;213(9–10):779–787. doi: 10.1016/j.imbio.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 67**.Taylor KE, Chung SA, Graham RR, et al. Risk alleles for systemic lupus erythematosus in a large case-control collection and associations with clinical subphenotypes. PLoS Genet. 2011;7(2):e1001311. doi: 10.1371/journal.pgen.1001311. This comprehensive study elegantly correlated clinical SLE subphenotypes with genetic risk allele assocation, which signifies differential molecular mechanisms underlying the heterogeneous autoimmune pathogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morel L. Genetics of SLE: evidence from mouse models. Nat Rev Rheumatol. 2010;6(6):348–357. doi: 10.1038/nrrheum.2010.63. [DOI] [PubMed] [Google Scholar]
- 69.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 70.Fairhurst AM, Mathian A, Connolly JE, et al. Systemic IFN-alpha drives kidney nephritis in B6.Sle123 mice. Eur J Immunol. 2008;38(7):1948–1960. doi: 10.1002/eji.200837925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007;37(12):3582–3586. doi: 10.1002/eji.200737815. [DOI] [PubMed] [Google Scholar]
- 72.Santiago-Raber M-L, Baccala R, Haraldsson KM, et al. Type-I Interferon Receptor Deficiency Reduces Lupus-like Disease in NZB Mice. J Exp Med. 2003;197(6):777–788. doi: 10.1084/jem.20021996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73*.Guiducci C, Tripodo C, Gong M, et al. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J Exp Med. 2010;207(13):2931–2942. doi: 10.1084/jem.20101048. This study identified TLR-activated pDCs as major IFN producer invovled in prolonged skin inflammation, directly implicating the pathogenic role of pDCs in autoimmune conditions such as cutanneous lupus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hohensinner P, Goronzy J, Weyand C. Telomere dysfunction, autoimmunity and aging. Aging and disease. 2011;2(6):524–537. [PMC free article] [PubMed] [Google Scholar]
- 75.Cusick M, Libbey J, Fujinami R. Molecular Mimicry as a Mechanism of Autoimmune Disease. Clinical Reviews in Allergy and Immunology. 2012;42(1):102–111. doi: 10.1007/s12016-011-8294-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pathak S, Mohan C. Cellular and molecular pathogenesis of systemic lupus erythematosus: lessons from animal models. Arthritis Res Therapy. 2011;13(5):241. doi: 10.1186/ar3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pisetsky DS. Immune Activation by Bacterial DNA: A New Genetic Code. Immunity. 1996;5(4):303–310. doi: 10.1016/s1074-7613(00)80256-3. [DOI] [PubMed] [Google Scholar]
- 78.Mevorach D, Zhou JL, Song X, Elkon KB. Systemic Exposure to Irradiated Apoptotic Cells Induces Autoantibody Production. J Exp Med. 1998;188(2):387–392. doi: 10.1084/jem.188.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79**.Di Domizio J, Dorta-Estremera S, Gagea M, et al. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc Natl Acad Sci U S A. 2012;109(36):14550–14555. doi: 10.1073/pnas.1206923109. This study identified a new class of nucleic acid-containing protein complexes that are potent IFN inducers that selectively activate pDCs. In a pDC-dependent manner, these coumpounds are capable to initiate lupus in non-autoimmune mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30(9):455–464. doi: 10.1016/j.it.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee PY, Weinstein JS, Nacionales DC, et al. A Novel Type I IFN-Producing Cell Subset in Murine Lupus. J Immunol. 2008;180(7):5101–5108. doi: 10.4049/jimmunol.180.7.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee PY, Kumagai Y, Li Y, et al. TLR7-dependent and FcγR-independent production of type I interferon in experimental mouse lupus. J Exp Med. 2008;205(13):2995–3006. doi: 10.1084/jem.20080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ho F, Fu K. A new model of AA-amyloidosis induced by oral pristane in BALB/c mice. Br J Exp Pathol. 1987;68:413–420. [PMC free article] [PubMed] [Google Scholar]
- 84.Cheung YH, Loh C, Pau E, Kim J, Wither J. Insights into the genetic basis and immunopathogenesis of systemic lupus erythematosus from the study of mouse models. Semin Immunol. 2009;21(6):372–382. doi: 10.1016/j.smim.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 85.Desmet CJ, Ishii KJ. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat Rev Immunol. 2012;12(7):479–491. doi: 10.1038/nri3247. [DOI] [PubMed] [Google Scholar]
- 86.Gall A, Treuting P, Elkon KB, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36(1):120–131. doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87**.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134(4):587–598. doi: 10.1016/j.cell.2008.06.032. (86 &87) In-depth studies demonstrate that upstream IFN production mediated by a TLR-independent pathway can lead to sytemic autoimmunity, an insightful mechanism for a rare genetic risk factor in SLE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ahn J, Gutman D, Saijo S, Barber G. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci USA. 2012 doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hron JD, Peng SL. Type I IFN Protects Against Murine Lupus. J Immunol. 2004;173(3):2134–2142. doi: 10.4049/jimmunol.173.3.2134. [DOI] [PubMed] [Google Scholar]
- 90.Liu J, Karypis G, Hippen KL, et al. Genomic view of systemic autoimmunity in MRLlpr mice. Genes Immun. 2006;7(2):156–168. doi: 10.1038/sj.gene.6364286. [DOI] [PubMed] [Google Scholar]
- 91.Sangaletti S, Tripodo C, Chiodoni C, et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood. 2012;120(15):3007–3018. doi: 10.1182/blood-2012-03-416156. [DOI] [PubMed] [Google Scholar]
- 92.Knight JS, Kaplan MJ. Lupus neutrophils: 'NET' gain in understanding lupus pathogenesis. Curr Opin Rheumatol. 2012;24(5):441–450. doi: 10.1097/BOR.0b013e3283546703. [DOI] [PubMed] [Google Scholar]
- 93.Campbell AM, Kashgarian M, Shlomchik MJ. NADPH Oxidase Inhibits the Pathogenesis of Systemic Lupus Erythematosus. Science Translational Medicine. 2012;4(157):157ra141. doi: 10.1126/scitranslmed.3004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guiducci C, Gong M, Xu Z, et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature. 2010;465(7300):937–941. doi: 10.1038/nature09102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gregersen JW, Jayne DRW. B-cell depletion in the treatment of lupus nephritis. Nat Rev Nephrol. 2012 doi: 10.1038/nrneph.2012.141. advance online publication. [DOI] [PubMed] [Google Scholar]
- 96.Pisetsky DS, Grammer AC, Ning TC, Lipsky PE. Are autoantibodies the targets of B-cell-directed therapy? Nat Rev Rheumatol. 2011;7(9):551–556. doi: 10.1038/nrrheum.2011.108. [DOI] [PubMed] [Google Scholar]
- 97.Merrill JT, Wallace DJ, Petri M, et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon α monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann Rheum Dis. 2011;70(11):1905–1913. doi: 10.1136/ard.2010.144485. [DOI] [PubMed] [Google Scholar]
- 98.Morimoto AM, Flesher DT, Yang J, et al. Association of endogenous anti-interferon-alpha autoantibodies with decreased interferon-pathway and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2011;63(8):2407–2415. doi: 10.1002/art.30399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Houssiau FA, Rashkov R, Hachulla E, et al. Active immunization against IFNα with IFN-kinoid in SLE patients is safe, immunogenic and induces down-regulation of IFN-mediated genes. Arthritis Rheum. 2011;63:S963. [Google Scholar]
- 100.Browne S, Holland S. Immunodeficiency secondary to anticytokine autoantibodies. Curr Opin Allergy Clin Immunol. 2010;10(6):534–541. doi: 10.1097/ACI.0b013e3283402b41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12(2):125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Crow MK. Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Res Ther. 2010;12 (Suppl 1):S5. doi: 10.1186/ar2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Palucka A, Blanck J, Bennett L, Pascual V, Banchereau J. Cross-regulation of TNF and IFN-alpha in autoimmune diseases. Proc Natl Acad Sci USA. 2005;102:3372– 3377. doi: 10.1073/pnas.0408506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Crampton SP, Deane JA, Feigenbaum L, Bolland S. Ifih1 gene dose effect reveals MDA5-mediated chronic type I IFN gene signature, viral resistance, and accelerated autoimmunity. J Immunol. 2012;188(3):1451–1459. doi: 10.4049/jimmunol.1102705. [DOI] [PMC free article] [PubMed] [Google Scholar]