

Abstract
HMGB1 released from necrotic cells or macrophages functions as a late inflammatory mediator, and has been shown to induce cardiovascular collapse during sepsis. Thus far, however, the effect(s) of HMGB1 in the heart are not known. We determined the effects of HMGB1 on isolated feline cardiac myocytes by measuring sarcomere shortening in contracting cardiac myocytes, intracellular Ca2+ transients using fluo-3, and L-type calcium currents using whole cell perforate configuration of the patch clamp technique. Treatment of isolated myocytes with HMGB1 (100 ng/ml) resulted in a 70% decrease in sarcomere shortening and a 50% decrease in the height of the peak Ca++ transient within 5 min (p <0.01). The immediate negative inotropic effects HMGB1 on cell contractility and calcium homeostasis were partially reversible upon washout of HMGB1. A significant inhibition of the inward L-type calcium currents also was documented by the patch clamp technique. HMGB1 induced the PKCε translocation and a PKC inhibitor significantly attenuated the negative inotropic effects of HMGB1. These studies show for the first time that HMGB1 impairs sarcomere shortening by decreasing calcium availability in cardiac myocytes through modulating membrane calcium influx, and suggest that HMGB1 maybe act as a novel myocardial depressant factor during cardiac injury.
Keywords: Inflammation, innate immunity, myocardial function, sepsis
INTRODUCTION
Myocardial dysfunction, a central component in the complex pathophysiology of sepsis, contributes to the high mortality associated with this disorder. Indeed, in patients who develop cardiovascular impairment the overall mortality rises from 20% to 70–90% (18). Lipopolysaccharide (LPS) depresses intrinsic myocardial contractility and is believed to be an important factor contributing to myocardial dysfunction during sepsis (17; 21). Although the endogenous agents that contribute to the myocardial depression observed in sepsis remain elusive, studies suggest that TNF, IL-1β, and nitric oxide (NO) play a role (1, 7, 13, 14).
HMGB1 was originally described as a nuclear DNA-binding protein involved in the transcriptional regulation and gene expression. However in 1999 Wang et al. (27) reported that activated macrophages could secrete HMGB1. HMGB1 was subsequently shown to be released systemically in murine models of endotoxemia and sepsis induced by cecal perforation (28). Interestingly, the release of HMGB1 was delayed when compared to tumor necrosis factor and remained elevated for a period of 36 hours. Further supporting its importance in the pathogenesis of sepsis was the observation that passive immunization with neutralizing anti-HMGB1 antibodies prevented organ dysfunction in septic mice (28). Recently Gibot et al.(2) have shown that as in experimental models of sepsis, HMGB1 plasma concentration correlate with the degree of organ dysfunction during septic shock. However, HMGB1 levels alone did not predict outcome in this patient cohort.
Although previous studies have primarily addressed the role of TNF, IL-1β and NO in the pathogenesis of sepsis-induced LV dysfunction, (1, 7, 13, 14), HMGB1 also is expressed in the heart following stress and therefore may contribute to the development of cardiac depression (22). In considering the cytokine properties of HMGB1 and its proposed role in sepsis-induced organ dysfunction, we sought to determine whether HMGB1 could directly depress myocardial function in vitro.
METHODS
Effects of HMGB1 on cardiac myocyte function
The methods for isolating and culturing adult feline cardiac myocytes have been described in detail elsewhere (10). Sarcomere shortening and calcium transients were assessed in isolated contracting cardiac myocytes using the IonOptix MyoCam system (IonOptix Corp, Milton, MA). Freshly isolated cells were loaded with 10 μM Fluo 3-AM (Invitrogen) in HEPES solution containing (in mM) NaCl 137, KCl 5.4, CaCl2 1.8, Dextrose 15, MgSO4 1.3, NaH2PO4 1.2, HEPES 20 (pH 7.4 with NaOH ) at 37°C in the dark for 15 minutes. Fluo-3 loaded cells were then attached to coverslips pre-coated with 10 μg/ml laminin and mounted in a perfusion chamber (RC 47 FSLP, Warner Instrument). Cells were then placed on the stage of an inverted microscope (OLYMPUS IX 70) and superfused with the same HEPES solution to remove unloaded dye. The excitation light was set at 485-nm wavelength and emission was collected at 530-nm wavelength. The stage of the microscope was illuminated with red light (640–750 nm) through bright-field illumination optics, to allow simultaneous measurements of fluorescence changes and cell shortening. The cells were paced at 0.5 Hz following a 2 minute period of stabilization (baseline), the cells were superfused with diluent or HMGB1 (1–100 ng/ml; Sigma-Aldrich, St. Louis, MO). Sarcomere motion and fluorescence changes were recorded continuously using Ionoptix acquisition software and the data were stored for an off-line analysis by IonWizard software (Ionoptix 1999, version 5). The HMGB1 (Sigma-Aldrich, St. Louis, MO) used in these studies was a full-length recombinant human protein that was expressed in Escherichia coli. In preliminary control studies we established that purified LPS (0.25 – 25 μg/ml, Invivogen, San Diego, CA) had no effect on sarcomere shortening over a 30-minute period (data not shown). In additional control studies, we treated the cells with 100 ng/ml of HMGB1 obtained from R & D Systems (Minneapolis, MN), as well HMGBiotech (Milano, Italy). These commercial preparations employed for these studies were also tested for endotoxin levels using the amebocyte lysate assay (Lonza, Switzerland).
Effects of HMGB1 on L-type calcium currents
L-type channel activity was measured as described (19). Briefly, freshly isolated adult feline cardiac myocytes were studied in a chamber mounted on an inverted microscope (Nikon, Japan) and were superfused with a normal physiological salt solution containing (in mmol/L): NaCl 145, KCl 4, CsCl 5 (to block potassium currents), MgCl2 1, CaCl2 2, HEPES 5, Glucose 11 (pH 7.4) at 37°C. Low resistance (1–4 MΩ) patch pipettes were filled with a solution containing (in mmol/L): caesium glutamate 100, KCl 40, MgCl2 1, Na2-ATP 4, EGTA 0.5, Hepes 5 (pH 7.2) and nystatin (150 mg/ml). Experiments were initiated after sufficient access to the cytosol was achieved (access resistance < 15 MΩ; 10–20 minutes). Whole cell L-type Ca2+ currents were measured with 400 ms depolarizing test pulses from a holding potential of −40mV (to inactivate sodium current). Junction potential was not corrected and was less than 10 mV. The cell capacitance was measured using small hyperpolarizing test steps. Membrane potentials were controlled with an Axopatch 2B (Axon Instruments, Concord, ON) voltage-clamp amplifier using pClamp8 (Axon Instrument) software and acquired with a Digidata 1200 analog digital converter (Axon Instruments). The data were analyzed with Clampfit (Axon Instruments) and presented with Origin 6.0 (Microcal Software, Inc.). Only myocytes with minimal (<10%) rundown of ICa-L were included in the data analysis.
Effects of HMGB1 on PKC-ε translocation
Freshly isolated feline ventricular myocytes were cultured in M199 medium supplemented with 0.1 % human serum albumin (25 % USP) and 1 % insulin, transferring and selenium (ITS [Cellgro]) for 6 hours. The culture medium was then aspirated and the cells suspended in HEPES solution (described above) and were then exposed to HMGB1 (100 ng/ml) for 1, 3, and 10 minutes. The cells were lysed by adding 0.3 ml lysis buffer (50 mM NaF, 2 mM EGAT, 2 mM EDTA, 500 μM sodium orthovanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 0.5 mM phenylmethylsulfonyl fluoride, 20 mM Tris, pH 7.4). Cell lysates were centrifuged at 100,000 × g for 1 hour at 4 °C and the supernatant saved as the cytosolic fraction. The pellet was then resuspended in lysis buffer containing 1% Triton X-100, and sonicated. The resuspended pellets were incubated in a shaking ice bath for 15 min, centrifuged at 14,000 × g for 10 min, and the supernatant saved as the membrane fraction. Aliquots of the cytosolic and membrane fractions were subjected to electrophoresis and immunoblotting for PKC-ε using a mouse monoclonal antibody (BD Biosciences, 1:2000). Translocation of PKC-ε was defined as the ratio of membrane fraction to cytosolic fraction.
Effects of PKC inhibition on the functional effects of HMGB1
To determine whether PKC-ε inhibition was sufficient to attenuate the effects of HMGB1 (100 ng/mL), freshly isolated cardiac myocytes were pre-treated for 10 minutes with Ro-31-8220 (0.001-1 μM; Calbiochem, San Diego, CA), a PKC inhibitor, prior to assessing sarcomere shortening as described above.
Effects of blocking the receptor for advanced glycation end products (RAGE) and TLR4 on the functional effects of HMGB1
To determine whether the negative inotropic effects of HMGB1 were mediated by activation of Toll-like receptor 4 (TLR4) and/or the receptor for advanced glycation end-products (RAGE), freshyly isolated cardiac myocytes were pre-treated for 30 minutes with an anti-RAGE antibody (10 ug/ml; R & D System, Minneapolis, MN) or anti-TLR4 antibdoy (10 μg/ml; clone HTA125; Gene Tex, San Antonio, TX) prior to stimulation with HMGB1 (100 ng/ml) as described above.
Statistical Analysis
All data are expressed as the mean ± SEM. Statistical significance was evaluated by 2-way ANOVA. A post hoc test of least significant differences (Bonferroni or Dunnett’s) was used to determine differences among groups where appropriate. A probability value of P < 0.05 was considered to be statistically significant.
RESULTS
Effects of HMGB1 on cardiac myocyte function
Following an initial period of stabilization (2 min), freshly isolated adult feline cardiac myocytes were superfused with diluent or 100 ng/ml of HMGB1. Figures 1A and 1B depict continuous tracings of sarcomere shortening (upper panel) and calcium transients (lower panel) for diluent treated cells, whereas Figures 1C and 1D depict tracings of sarcomere shortening and calcium transients in HMGB1 treated cells. As shown, treatment had no significant effect on sarcomere motion, or the peak calcium transients during the period of observation. In contrast treatment with HMGB1 resulted in a rapid (within 5 min) decrease in sarcomere shortening that was accompanied by a decrease in the peak amplitude of the calcium transient. Importantly, the effects of HMGB1 (Figure 1B) were partially reversible following washout of HMGB1 from the superfusate Figure 2 summarizes the results of group data. Treatment with 100 ng/ml of HMGB1 resulted in a significant (p < 0.01) 70% decrease in sarcomere shortening, which was accompanied by a significant (p < 0.01) 50 % decrease in peak fluorescence brightness. The effects of HMGB1 on sarcomere shortening and peak fluorescence brightness were partially reversible during the washout phase of the experiment. Sarcomere shortening following washout of HMGB1 did not differ significantly from baseline. To determine whether the effects of HMGB1 (Sigma) on sarcomere shortening were spurious, that is secondary to either an artifact of the commercial preparation, and/or endotoxin contamination of the recombinant protein, we repeated the above experiments using 2 additional commercially available preparations. As shown in the Table, all of the preparation s of HMGB1 studied provoked a significant decrease in sarcomere shortening in isolated contracting cardiac myocytes. Furthermore, concentrations of endotoxin that overlapped those found in the HMGB1 preparations did not affect sarcomere shortening following a 10–30 minute exposure (data not shown), consistent with the repeated observation that effects of endotoxin on isolated cardiac myocytes are time dependent, and require ~ 6 hours to become manifest.
Figure 1. Effects of HMGB1 on cardiac myocyte contractility and intracellular calcium transients.

Recordings of myocyte contractility during the entire period of experimentation in individual cells treated with diluent (A) or HMGB1 (100 ng/mL; B). Vertical axis shows absolute sarcomere length in microns or fluorescence brightness (in arbitrary units). The horizontal axis represents time (in seconds). Representative pacing events recorded under baseline (C) and post treatment with HMGB1 (100 ng/mL; D). Vertical axis represents fluorescence brightness in arbitrary units.
Figure 2. Effects of HMGB1 on cardiac myocyte contractility and peak amplitude of calcium transients.
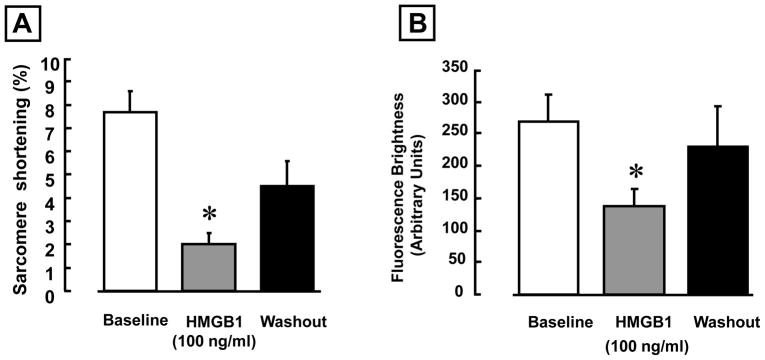
(A) Effect of 100 ng/ml HMGB1 on the sarcomere shortening. (B) Effect of 100 ng/ml HMGB1 on the peak fluorescence brightness. (* p < 0.01 vs. baseline, N = 9 cells from 4 hearts).
Table.
| Vendor | Endotoxin [ng/mg protein] | Diluent % Sarcomere Shortening | HMGB1 [100ng/ml] % Sarcomere Shortening | % Decrease from Baseline | P vs. Diluent |
|---|---|---|---|---|---|
| Sigma | 25 | 8.87 ± 0.71 | 4.40 ± 0.60 | 70 ± 3.4 | 0.0002 |
| R & D Systems | 10–100 * | 9.29 ± 0.45 | 5.28 ± .78 | 46 ± 6.0 | 0.0002 |
| HMGBiotech | <0.4 | 9.76 ± 0.64 | 6.57 ± 0.63 | 33 ± 3.1 | 0.001 |
R & D reports <1EU/μg protein. The conversion used was 10 EU/ng.
To determine whether the effects of HMGB1 were dose dependent, we treated isolated contracting cardiac myocytes with HMGB1 concentrations ranging from 1–100 ng/ml. Figure 3A and B summarize the effects of different HMGB1 concentrations on sarcomere shortening and the peak amplitude of calcium transients, respectively. As shown, at concentrations ≥ 10 ng/ml HMGB1 produced a significant (p < 0.01 compared to diluent) decrease in sarcomere shortening and peak amplitude of calcium transients. Thus, pathophysiological concentrations of HMGB1 are sufficient to produce negative inotropic effects and depress calcium transients in isolated contracting cardiac myocytes.
Figure 3. Dose-dependent effects of HMGB1 on myocyte contractility and intracellular calcium transients.
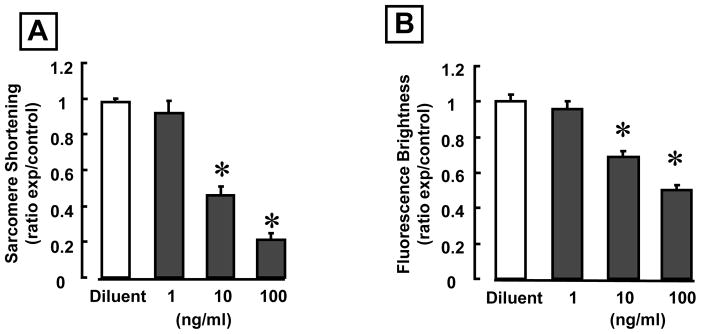
(A) Effects of increasing concentrations of HMGB1 on sarcomere shortening. (B) Effects of increasing concentrations of HMGB1 on the peak amplitude of calcium transients. (*p < 0.01 vs. diluent; N = 8 cells/condition).
Effects of HMGB1 on L-type calcium currents
To determine the mechanisms for the negative inotropic effects of HMGB1, we evaluated L-type calcium channel activity in diluent and HMGB1 treated adult cardiac myocytes. Figures 4A and 4 B depict the effect on the HMGB1 on the inward calcium current. As shown by the representative tracings in Figure 4A, exposure to HMGB1 (10 ng/ml) dramatically reduced the inward L-type calcium current. A partial recovery of channel activity was noted after washout of HMGB1. Figure 4B, which summarizes the results of group data, shows that HMGB1 (10 ng/ml) led to a significant (P <0.01) inhibition (70%) of the inward calcium current in isolated myocytes, and that this was partially reversible following washout. Time course experiments (Figure. 4C) revealed that the effect of HMGB1 occurred within 1–2 minutes and reached plateau within 5 min after challenge. These latter findings are in agreement with the effects of HMGB1 on myocyte contractility and peak amplitude of intracellular calcium transients shown in Figures 2A and 2B.
Figure 4. Effect of HMGB1 on L-type calcium currents.

(A) Inward L-type calcium currents recorded under baseline conditions, after HMGB1 treatment, and washout. (B) Relative effects of HMGB1 (10 ng/ml) on the L-type calcium currents in isolated myocytes. (C) Time course of the L-type calcium currents in the presence and after washout of HMGB1. (N = 8 cells/condition).
Effect of HMGB1 on PKC-ε translocation
Activation of PKC-ε has been linked to the inhibition of L-type calcium channel activity in mammalian cardiac myocytes (3). Accordingly, we examined the effects of HMGB1 (100 ng/mL) on PKC-ε activation in isolated feline cardiac myocytes. Figure 5A depicts representative Western blots for cytosolic and membrane PKC-ε fractions, in the presence and absence of HMGB1, whereas Figure 5B summarizes the results of group data. As shown HMGB1 induced a significant (p < 0.01) translocation of PKC-ε from the cytosol to the membrane within 10 minutes, consistent with the rapid activation of the L-calcium currents in the whole cell patch clamp studies (Figure 4C).
Figure 5. HMGB1 induces PKC-ε translocation in cardiac myoctyes.

(A) Representative western blots for cytosolic and membrane PKC-ε. HMGB1 (100 ng/ml: R&D Systems) induced PKC-ε translocation from the cytosol to the membrane fraction. (B) Time course of PKCε translocation in cardiac myocytes after exposure to HMGB1 (100 ng/ml). (*p < 0.01 vs Time 0; n = 4 isolations) (C) Ro-31-8220 (Ro) significantly attenuated the effects of HMGB1 (100 ng/mL; R&D Systems) in a dose dependent manner compared to Diluent (Dil). N= 10–15 cells/condition. (*p < 0.01 vs. Diluent; ‡ p < 0.05 vs. HMGB1 alone). N= 10–15 cells/condition
To determine whether inhibition of PKC translocation was sufficient to prevent the effects of HMGB1 in cardiac myocytes, we pre-treated the cells with varying concentrations of the PKC inhibitor Ro-31-8220 (0.001-1 μM). Figure 5C summarizes the effects of Ro-31-8220 on sarcomere shortening. Ro-31-8220 had no effect on sarcomere shortening at baseline but did significantly attenuate the deleterious effects of HMGB1 on sarcomere shortening in a dose dependent manner (p<0.01 compared to HMGB1 diluent-treated cells).
RAGE and TLR4 mediate the negative inotropic effects of HMGB
Previously we have shown that the negative inotropic effects of HMGB1 were not mediated via Toll-like 2 receptor (22). HMGB1 has been reported to bind to and signal through the multivalent immunoglobulin receptor RAGE and TLR4 (12) (16). To determine whether the RAGE and or TLR4 receptors mediated the negative inotropic effects of HMGB1 we performed studies using inhibitory antibodies against RAGE and TLR4. As shown in Figure 6A, a neutralizing anti-RAGE antibody significantly attenuated the negative inotropic effects of HMGB1 in isolated cardiac myocytes. Specifically, pretreatment with a RAGE antibody resulted in a 32% increase in the extent of sarcomere shortening in HMGB1 treated cells, when compared to cells that had been pretreated with diluent alone. Figure 6B shows that the TLR4 antibody also had a significant effect on sarcomere shortening following exposure to HMGB1. Pretreatment with TLR4 antibody resulted in a significant 22% improvement in sarcomere shortening compared to HMGB1 stimulated cells that had been pretreated with diluent alone. Taken together, these data suggest that the negative inotropic effect of HMGB1 on cardiomyocytes is, at least in part, mediated by binding to both RAGE and TLR4.
Figure 6. Effects of anti-RAGE and TLR4 antibodies on HMGB1-induced negative inotropic effects in cardiac myocytes.

Cells were pre-incubated (30 min) with each antibody prior to exposure to HMGB1 (100 ng/mL). (A) RAGE blockade improved sarcomere shortening by 32% (B) TLR4 blockade improved sarcomere shortening by 22% (*p < 0.01 vs. Diluent; ‡ p < 0.05 vs. HMGB1 alone). N= 10-15 cells/condition
DISCUSSION
The results of this study show for the first time that pathophysiologically relevant concentrations of HMGB1 are sufficient to produce negative inotropic effects in isolated contracting cardiac myocytes through a mechanism that involves inhibition of the L-type calcium channel. Three lines of evidence support this statement. First, treatment with HMGB1 produced a rapid decrease in sarcomere shortening that was accompanied by a decrease in the peak amplitude of the intracellular calcium transient in isolated contracting cardiac myocytes (Figures 1 and 2). The effects of HMGB1 on sarcomere shortening and calcium homeostasis were dose dependent (Figure 3), and were partially reversible following washout of HMGB1 (Figure 2). It is unlikely that the observed negative inotropic effects of HMGB1 were secondary to LPS contamination, insofar as the HMGB1 used in this study contained 25 ng LPS per 1000 ng HMGB1, which is not sufficient to provoke negative inotropic effects within the time course that we observed negative inotropic effects with HMGB1. Indeed, in preliminary control studies we determined that purified LPS (0.25 – 25 μg/ml) had no effect on cell shortening, nor the peak amplitude of intracellular calcium transients over a 30-minute period (data not shown), consistent with prior observations (6, 8, 11, 15, 20, 24, 25). Furthermore, when we used 2 additional commercially available preparations of HMGB1, each of which had lower concentrations of contaminating endotoxin (0.4 – 10 ug.ml), we still observed significant decreases in sarcomere shortening. Of note, the magnitude of the effect of HMGB1 on sarcomere shortening exceeded the magnitude of the decrease in amplitude of the peak calcium transient, suggesting that HMGB1 may impair sarcomere shortening through calcium independent mechanisms. Alternatively, HMGB1 may provoke decreased calcium sensitivity in cardiac myocytes. Additional studies will be required to address this important question. Second, treatment with HMGB1 led to a significant decrease in the inward L-type calcium current (Figure 4). Importantly, time course experiments (Figure 4C) revealed that the effect of HMGB1 was manifest within 1–2 minutes, and reached plateau by 5 minutes after HMGB1 provocation, consistent with the rapid time course of the effects of HMGB1 on the peak inward calcium transient. Taken together, these studies show that HMGB1 exerts dose dependent negative inotropic effects in cardiac myocytes by decreasing the amount of activator calcium from the inward calcium that is necessary for calcium triggered calcium release, and suggest that HMGB1 maybe act as a novel myocardial depressant factor during cardiac injury. Although the scope of this study was not intended to delineate the full spectrum of mechanisms for the deleterious negative inotropic effects of HMGB1, our results do suggest that HMGB1 induced activation of PKCε is responsible, at least in part, for the negative inotropic effects of HMGB1 in cardiac myocytes. That is, HMGB1 induced the rapid translocation of PKCε from the cytosol with a time course that was consistent with the inhibition of the L-type calcium channel, as well as the decrease in sarcomere shortening and peak intracellular calcium transients. Indeed, Hu et al.(3) have shown that activation of PKCε by a specific agonist (εV1-7) inhibits the L-type calcium channel activity in adult rat ventricular myocytes. Moreover, the negative inotropic effects of HMGB1 were sensitive to Ro-31-8220, a PKC inhibitor (Figure 6C).
HMGB1 and Tissue Injury
Although HMGB1 was originally described as a late mediator of inflammation and endotoxin lethality (27), more recent studies have shown that HMGB1 plays an important role as an early inflammatory mediator following acute tissue injury. For example, Kim et al.(4) demonstrated increased HMGB1 expression in the lung within 4 hours of inducing hemorrhagic shock. These authors showed that administration of anti-HMGB1 antibody reduced the accumulation of neutrophils in the lung as well as lung permeability. Similarly, HMGB1 levels were increased within 1 hour in ischemia-reperfusion (I/R) injury of the liver. Pertinent to the present discussion, administration of recombinant HMGB1 to mice that were undergoing partial hepatic I/R led to worsening outcomes. Moreover, a neutralizing HMGB1 antibody decreased hepatic damage following I/R injury (26). Although the present results are consistent with prior observations that the acute effects of HMGB1 may be deleterious, it is interesting to note that the exogenous administration of HMGB1 has been reported to be beneficial in a mouse model of myocardial infarction by inducing myocardial regeneration and improving cardiac function (9). Interestingly, HMBG1 has also been linked to increased angiogenesis (23). Taken together, these latter findings raise the interesting possibility that in a more chronic model of inflammation HMGB1 may play an important role in promoting cardiac repair and/or cardiac regeneration.
Previous studies have shown that mice with defective TLR4 signaling are protected against HMGB1-induced hepatic I/R injury suggesting that TLR4 serves as a receptor for HMGB1 (26). However, other studies have shown that HMGB1 signaling can also be mediated via TLR2 and/or RAGE (5, 16). Previously we have shown the HMGB1 provoked negative inotropic effects in cardiac myocytes isolated from wild type and Toll like receptor 2 (TLR2) deficient mice, suggesting that TLR2 is not necessary for mediating the negative inotropic effects of HMGB1 (22). Here we show for the first time that the deleterious effects of HMGB1 on sarcomere shortening are attributable, at least in part, to the activation of RAGE and TLR4 (Figure 6). That is, the effects of HMGB1 were partially inhibited by pretreatment with neutralizing antibodies against RAGE or TLR4. We and others have shown that lipopolysaccharide (LPS), the canonical TLR4 ligand, produces negative inotropic effects after 1–6 hours of stimulation, (6, 8, 11, 15, 20, 24, 25) suggesting that LPS-induced negative inotropic effects require de novo synthesis of one or more different biological mediators. In contrast, in the present study the effects of HMBG1 on sarcomere motion and calcium homeostasis were apparent within minutes. At present, it is not apparent why HMGB1 and LPS, both of which signal through TLR4 have different time courses for producing negative inotropic effects in cardiac myocytes. One possibility, albeit speculative is that there may be cross-talk between the RAGE and TLR4 receptors, which might explain the more rapid onset on negative inotropic effects with HMGB1. Further studies will be necessary to address this interesting question.
In conclusion, the findings of the present study suggest that HMGB1 may act as a novel myocardial depressant factor that is released by resident myocardial cells following tissue injury. Although substances that produce negative inotropic effects have traditionally been viewed as deleterious, it is possible that locally released HMGB1 may decrease energy utilization in ischemic tissue, thereby preventing injured myocytes from worsening ATP depletion that eventuate in necrosis. This point of view is consistent with the aforementioned possibility that sustained HMGB1 signaling may play an important role in cardiac repair and/or regeneration (9, 23). This statement notwithstanding, it is likely that excessive release of HMGB1 acutely may be overtly deleterious to the heart by contributing to excessive and/or sustained inflammation and/or profound myocardial depression and myocardial collapse.
Acknowledgments
This research was supported by grants HL-58081, HL-42250, HL-073017 (to DLM) and GM62474 (to JGV) from the National Institutes of Health.
Footnotes
Conflict of Interest
The authors have no conflicts to disclose.
References
- 1.Cain BS, Meldrum DR, Dinarello CA, Meng X, Joo KS, Banerjee A, Harken AH. Tumor necrosis factor-alpha and interleukin-1beta synergistically depress human myocardial function. Crit Care Med. 1999;27:1309–1318. doi: 10.1097/00003246-199907000-00018. [DOI] [PubMed] [Google Scholar]
- 2.Gibot S, Massin F, Cravoisy A, Barraud D, Nace L, Levy B, Bollaert PE. High-mobility group box 1 protein plasma concentrations during septic shock. Intensive Care Med. 2007;33:1347–1353. doi: 10.1007/s00134-007-0691-2. [DOI] [PubMed] [Google Scholar]
- 3.Hu K, Mochly-Rosen D, Boutjdir M. Evidence for functional role of epsilonPKC isozyme in the regulation of cardiac Ca(2+) channels. Am J Physiol Heart Circ Physiol. 2000;279:H2658–H2664. doi: 10.1152/ajpheart.2000.279.6.H2658. [DOI] [PubMed] [Google Scholar]
- 4.Kim JY, Park JS, Strassheim D, Douglas I, Diaz d V, Asehnoune K, Mitra S, Kwak SH, Yamada S, Maruyama I, Ishizaka A, Abraham E. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol. 2005;288:L958–L965. doi: 10.1152/ajplung.00359.2004. [DOI] [PubMed] [Google Scholar]
- 5.Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, Nawroth P, Andersson U, Harris RA, Harris HE. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 6.Kumar A, Brar R, Wang P, Dee L, Skorupa G, Khadour F, Schulz R, Parrillo JE. Role of nitric oxide and cGMP in human septic serum-induced depression of cardiac myocyte contractility. Am J Physiol. 1999;276:R265–R276. doi: 10.1152/ajpregu.1999.276.1.R265. [DOI] [PubMed] [Google Scholar]
- 7.Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor α and interleukin 1β are responsible for in vitro myocardial cell depression induced by human shock serum. J Exp Med. 1996;183:949–958. doi: 10.1084/jem.183.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lew WY, Lee M, Yasuda S, Bayna E. Depyrogenation of digestive enzymes reduces lipopolysaccharide tolerance in isolated cardiac myocytes. J Mol Cell Cardiol. 1997;29:1985–1990. doi: 10.1006/jmcc.1997.0432. [DOI] [PubMed] [Google Scholar]
- 9.Limana F, Germani A, Zacheo A, Kajstura J, Di Carlo A, Borsellino G, Leoni O, Palumbo R, Battistini L, Rastaldo R, Muller S, Pompilio G, Anversa P, Bianchi ME, Capogrossi MC. Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res. 2005;97:73–83. doi: 10.1161/01.RES.0000186276.06104.04. [DOI] [PubMed] [Google Scholar]
- 10.Mann DL, Kent RL, Parsons B, Cooper G., IV Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation. 1992;85:790–804. doi: 10.1161/01.cir.85.2.790. [DOI] [PubMed] [Google Scholar]
- 11.McKenna TM, Li S, Tao S. PKC mediates LPS- and phorbol-induced cardiac cell nitric oxide synthase activity and hypocontractility. Am J Physiol. 1995;269:H1891–H1898. doi: 10.1152/ajpheart.1995.269.6.H1891. [DOI] [PubMed] [Google Scholar]
- 12.Nam YJ, Mani K, Wu L, Peng CF, Calvert JW, Foo RS, Krishnamurthy B, Miao W, Ashton AW, Lefer DJ, Kitsis RN. The apoptosis inhibitor ARC undergoes ubiquitin-proteasomal-mediated degradation in response to death stimuli: identification of a degradation-resistant mutant. J Biol Chem. 2007;282:5522–5528. doi: 10.1074/jbc.M609186200. [DOI] [PubMed] [Google Scholar]
- 13.Natanson C, Danner RL, Elin RJ, Hosseini JM, Peart KW, Banks SM, MacVittie TJ, Walker RI, Parrillo JE. Role of endotoxemia in cardiovascular dysfunction and mortality. Escherichia coli and staphylococcus aureus challenges in a canine model of human septic shock. J Clin Invest. 1989;83:243–251. doi: 10.1172/JCI113866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pagani FD, Baker LS, Knox MA, Cheng H, Fink MP, Visner MS. Load-insensitive assessment of myocardial performance after tumor necrosis factor-α in dogs. Surgery. 1992;111:683–693. [PubMed] [Google Scholar]
- 15.Panas D, Khadour FH, Szabo C, Schulz R. Proinflammatory cytokines depress cardiac efficiency by a nitric oxide-dependent mechanism. Am J Physiol. 1998;275:H1016–H1023. doi: 10.1152/ajpheart.1998.275.3.H1016. [DOI] [PubMed] [Google Scholar]
- 16.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 17.Parker JL, Adams HR. Isolated cardiac preparations: models of intrinsic myocardial dysfunction in circulatory shock. Circ Shock. 1985;15:227–245. [PubMed] [Google Scholar]
- 18.Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, Ognibene FP. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990;113:227–242. doi: 10.7326/0003-4819-113-3-227. [DOI] [PubMed] [Google Scholar]
- 19.Piacentino V, III, Dipla K, Gaughan JP, Houser SR. Voltage-dependent Ca2+ release from the SR of feline ventricular myocytes is explained by Ca2+-induced Ca2+ release. J Physiol. 2000;523(Pt 3):533–548. doi: 10.1111/j.1469-7793.2000.t01-1-00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramabadran RS, Chancy A, Vallejo J, Barger P, Sivasubramanian N, Mann DL. Targeted gene silencing of tumor necrosis factor attenuates the negative inoptropic effects of lipopolysaccharide in isolating contracting cardiac myocytes. Tex Heart Inst J. 2008 In press. [PMC free article] [PubMed] [Google Scholar]
- 21.Romanosky AJ, Giaimo ME, Shepherd RE, Burns AH. The effect of in vivo endotoxin on myocardial function in vitro. Circ Shock. 1986;19:1–12. [PubMed] [Google Scholar]
- 22.Sakata Y, Dong JW, Vallejo JG, Huang CH, Baker JS, Tracey KJ, Tacheuchi O, Akira S, Mann DL. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;292:H503–H509. doi: 10.1152/ajpheart.00642.2006. [DOI] [PubMed] [Google Scholar]
- 23.Schlueter C, Weber H, Meyer B, Rogalla P, Roser K, Hauke S, Bullerdiek J. Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol. 2005;166:1259–1263. doi: 10.1016/S0002-9440(10)62344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stein B, Frank P, Schmitz W, Scholz H, Thoenes M. Endotoxin and cytokines induce direct cardiodepressive effects in mammalian cardiomyocytes via induction of nitric oxide synthase. J Mol Cell Cardiol. 1996;28:1631–1639. doi: 10.1006/jmcc.1996.0153. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki N, Sakamoto A, Ogawa R. Effect of L-canavanine, an Inhibitor of inducible nitric oxide synthase, on myocardial dysfunction during septic shock. J Nippon Med Sch. 2002;69:13–18. doi: 10.1272/jnms.69.13. [DOI] [PubMed] [Google Scholar]
- 26.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 28.Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Wang H, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]