

Abstract
Background & Aims
Obesity is a risk factor for pancreatic ductal adenocarcinoma (PDAC), but it is not clear how obesity contributes to pancreatic carcinogenesis. The oncogenic form of KRAS is expressed during early stages of PDAC development, and is detected in almost all of these tumors. However, there is evidence that mutant KRAS requires an additional stimulus to activate its full oncogenic activity, and that this stimulus involves the inflammatory response. We investigated whether the inflammation induced by a high-fat diet, and accompanying up-regulation of cyclooxygenase-2 (COX2), increases Kras activity during pancreatic carcinogenesis in mice.
Methods
We studied mice with acinar cell-specific expression of KrasG12D (LSL-Kras/Ela-CreERT mice) alone or crossed with COX2 conditional knockout mice (COXKO/LSL-Kras/Ela-CreERT). We also studied LSL-Kras/PDX1 -Cre mice. All mice were fed isocaloric diets with different amounts of fat, and a COX2 inhibitor was administered to some LSL-Kras/Ela-CreERT mice. Pancreata were collected from mice and analyzed for Kras activity, levels of phosphorylated ERK, inflammation, fibrosis, pancreatic intraepithelial neoplasia (PanIN), and PDACs.
Results
Pancreatic tissues from LSL-Kras/Ela-CreERT mice fed high-fat diets (HFDs) had increased Kras activity, fibrotic stroma, and numbers of PanINs and PDACs than LSL-Kras/Ela-CreERT mice fed control diets; the mice fed the HFDs also had shorter survival times than mice fed control diets. Administration of a COX2 inhibitor to LSL-Kras/Ela-CreERT mice prevented these effects of HFDs. We also observed a significant reduction in survival times of mice fed HFDs. COXKO/LSL-Kras/Ela-CreERT mice fed HFDs had no evidence for increased numbers of PanIN lesions, inflammation, or fibrosis, as opposed to the increases observed in LSL-Kras/Ela-CreERT mice fed HFDs.
Conclusions
In mice, a HFD can activate oncogenic Kras via COX2, leading to pancreatic inflammation and fibrosis, and development of PanINs and PDAC. This mechanism could be involved in the association between risk for PDAC and HFDs.
Keywords: Pancreatic cancer, signal transduction, oncogene, prostaglandin
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer death in the US, with a dismal 5-year survival rate of 3-5%. 1 In 2012 it was estimated that 43,920 Americans will be diagnosed with PDAC and 37,390 of those patients will die from the disease, most within less than one year of diagnosis. 1 Several studies have demonstrated an elevated body mass index and excessive body weight (BW) to be significant risk factors for PDAC. 2-6 This is a great concern, as obesity rates are rising worldwide. 7 The number of obese individuals in the U.S. alone has doubled to 59 million over the past two decades. One of the key causes of this surge in obesity is consumption of a high-fat diet (HFD). Therefore, determining the mechanistic effect of such diet on PDAC development at the molecular level is critical.
Mutations of oncogenic KRAS occur almost universally in PDAC patients. 8-12 Ras is a guanine nucleotide-binding protein that acts as a binary switch to activate several important cellular signaling pathways. Normally, Ras is loaded with GDP and is inactive until acted upon by guanine exchange factors (GEFs) that are under the control of receptors for a variety of growth factors, cytokines, and other signaling molecules. GEFs exchange GDP to GTP allowing Ras to be active and able to stimulate downstream pathways. Downstream signaling pathways stimulated by active Ras which are important in cell growth, differentiation and cell survival include phosphoinositide 3-kinase, mitogen-activated protein kinase (MAPK), and Ral. 10-12 Normally, the effects of Ras activation are transient due to intrinsic GTP hydrolysis activity of the Ras molecule which is greatly accelerated by GTPase activating proteins (GAPs). Hydrolysis of GTP to GDP returns Ras to its inactive state. In contrast, oncogenic KRAS results from point mutations, most specifically in codon 12, which limits the ability of GAPs to accelerate GTP hydrolysis. 10-12 Thus, once loaded with GTP, oncogenic Ras displays prolonged signaling. For this reason, oncogenic Ras is often been said to be constitutively active. 13 However, a recent analysis showed that this is not the case. 14 Rather, oncogenic Kras activity requires a stimulus, but once stimulated, it is slow to return to its basal state. Furthermore, because high Ras activity can generate inflammatory mediators via activation of several mechanisms, including nuclear factor -kB, cyclooxygenase 2 (COX2), signal transducer and activator of transcription-3 and others, activation of oncogenic Kras beyond a threshold initiates a Ras-inflammation feed-forward loop in which elevated expression of inflammatory mediators activate and prolong Kras activity. 14, 15
Initiation of PDAC is accompanied by inflammation, desmoplasia and the formation of non-invasive ductal precancerous lesions, pancreatic intraepithelial neoplasia (PanINs). 16 PanINs are an initial step in the progression of PDAC. 16, 17 Investigators previously showed that consumption of a HFD induced pancreatic inflammation and accelerated PanIN development in a mouse model with developmental expression of oncogenic Kras using a progenitor cell marker (p48Cre) that spontaneously generated PanINs. 18 Therefore, we hypothesized that consumption of a HFD can be an inflammatory stimulus to trigger Kras signaling beyond the pathological threshold, initiating the development of PDAC. We tested this hypothesis using elastase-CreERT and Pdx1-Cre murine models to activate oncogenic Kras expression. In the elastase-CreERT model in which only adult acinar cells express Kras, minimal spontaneous PanIN formation is observed at early ages. 14, 19 Furthermore, we previously found that COX2 was a critical component of the Ras-inflammation feed-forward loop involved in PDAC initiation by inflammatory stimuli. 14 Therefore, in the current study we tested whether COX2 was necessary for HFD-induced pathological changes in animals expressing oncogenic Kras.
We found that consumption of a HFD in mice with oncogenic Kras expression increased PanIN formation, fibrosis, inflammation, and PDAC leading to decreased survival. Control mice without Kras expression fed a HFD and Kras expressing mice fed a control diet (CD) showed minimal pancreas pathological alterations. Thus, this model recapitulates the risk posed by a HFD in humans expressing oncogenic Kras in the pancreas. Feeding a HFD elevated levels of Kras activity and downstream signaling activity. COX2 activity was critical to the HFD-induced pancreatic changes. These observations support Kras activation and COX2 expression as key elements of PDAC initiation and provide mechanistic insight into the connection between obesity and PDAC development, suggesting exciting possibilities for interventions to prevent PDAC.
Methods
Genetically engineered transgenic mice
LSL-KrasG12D mice 20 expressing conditional knock-in mutant KrasGI2D were attained from the Mouse Models of Human Cancer Consortium Repository NIH Bethesda, MD). A full-length elastase (Ela) gene promoter was used to drive the expression of tamoxifen-regulated CreERT specifically in adult pancreatic acinar cells in mice (Ela-CreERT) as described previously. 19 LSL-KrasG12D mice were bred with Ela-CreERT (BAC) mice to generate LSL-Kras/Ela-CreERT (LSL/BAC) double transgenic mice. COX2 conditional knockout mice (COX2 KO) mice 22 were bred with LSL/BAC mice to generate triple transgenic mice (COXKO/LSL/BAC) with additional targeted deletion of COX2 in pancreatic acinar cells after Cre activation with tamoxifen. In addition, LSL-KrasG12D mice were crossed with pancreatic-specific Cre (Pdx-1-Cre) mice 21 to generate LSL/Pdx-1 mice.
Treatments in Animals
All animal experiments were reviewed and approved by the University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee. LSL/BAC and BAC mice were given tamoxifen orally for 3 days to initiate oncogenic Kras expression in acinar cells starting at the age of 40 days. Mice were fed either a CD (Test Diet DIO 58Y2; Lab Supply) in which 10% of energy was derived from fat (LSL/BAC, n=21; BAC, n=20; LSL/Pdx-1, n=19; Pdx-1, n=9; COXKO/BAC, n=5; COXKO/LSL/BAC, n=5) or a HFD (Test Diet DIO 58Y1 van Heek Series; Lab Supply, Fort Worth, TX), in which 60% of energy was derived from fat (LSL/BAC, n=21; BAC, n=13; LSL/Pdx-1, n=10; Pdx-1, n=6; COXKO/BAC, n=5; COXKO/LSL/BAC, n=10). All mice were fed for at least 30 days and the BW of each animal was measured weekly. In addition, mice were fed a HFD for 38 days and simultaneously administered either Saline (BAC, n=4; LSL/BAC, n=15) or Celecoxib at 41.1 mg/kg (Sigma-Aldrich, St. Louis, MO) (BAC, n=4; LSL/BAC, n=16) orally. After treatments, mice were sacrificed and their pancreases harvested for histological and protein analysis.
Ras activity assay
Levels of GTP-occupied Ras from mice pancreas lysates were estimated using a Raf pull-down assay kit as recommended by the manufacturer (Millipore, Billerica, MA). Briefly, snap-frozen pancreas tissue were homogenized and sonicated on ice in a lysis buffer containing 25 mM HEPES, pH 7.5, 1% IGEPAL CA-630, 150 mM NaCl, 0.25% sodium deoxycholate, 10% glycerol, 25 mM NaF, 10 mM MgCl2, 1 mM EDTA acid, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM Na3VO4. Cellular debris was removed by centrifuging at 15,000g for 20 minutes at 4°C. Lysates were then incubated for 45 minutes at 4°C with agarose beads coated with the Raf-Ras binding domain. Beads were washed three times in lysis buffer, loading buffer added, and then vortexed, centrifuged, heated and analyzed via western blot protocol. Densities of active and total Ras protein bands were quantified using an Odyssey imaging system (version 3.0, LICOR Biosciences, Lincoln, NE) then standardized, averaged and plotted using Graphpad Prism 5.
Western blot analysis
Pancreas tissues from each group were homogenized using RIPA buffer, and lysates were separated using SDS-PAGE. Proteins were transferred to nitrocellulose membranes and blocked with 5% nonfat milk in phosphate-buffered saline (PBS) containing 0.05% Tween 20. Membranes were incubated with primary antibodies: Rabbit Anti-GAPDH G9545 (1:1000; Sigma-Aldrich), Mouse Anti-Ras 05-516 Clone 10 (1:1000; Millipore), p-p44/42 MAPK (T202/Y204) p-ERK 9106S (1:1000; Cell Signaling Technology, Danvers, MA), and p44/42 MAPK (total ERK) 9102 (1:1000; Cell Signaling Technology). Immunodetection of proteins was performed using Alexa-Fluor goat antimouse 800 or goat anti-rabbit 680-conjugated secondary antibodies for detection with an Odyssey Infrared Imaging System (LI-COR Biosciences). Bands densities were analyzed as described.
Histology
The pancreases of all mice were removed, washed with PBS, and placed in formalin overnight. After incubation, formalin-fixed samples were embedded in paraffin and sectioned onto slides at 5 μm thick and then stained with hematoxylin and eosin (H&E). Histological assessment was performed by a pathologist and slides were scored 1-3 on the level of inflammation (1: occasional scattered leukocytes, non-aggregating; 2: intermediate level of leukocytes; 3: aggregates of leukocytes within the pancreatic parenchyma in a follicle-like formation) based on the predominant type of inflammatory infiltrate, fibrosis (1: occasional periductular or scant perilobular fibrosis, 2: intermediate level, 3: definite extensive fibrosis, involving 50% of parenchyma with resultant parenchymal (acinar) atrophy, and frequency of PanIN lesions were analyzed by counting all foci of PanIN lesions (Supplementary Figure 5). Each lobule affected, was scored as 1, irrespective of the number of duct profiles.
Immunohistochemistry
Unstained paraffin embedded tissue were stained for: Rabbit anti-α smooth muscle actin (α-SMA) ab5694 (1:1000; Abcam, Cambridge, MA), Rabbit anti-phospho-ERK 4370 (1:100; Cell Signaling Technology) and anti-COX2 antibody (1:300; Cayman Chemicals, Ann Arbor, MI). Slides were deparaffinized using the standard procedure of two washes with Xylene, 100% Ethanol, 95% Ethanol, and one wash of 80% Ethanol. Antigen retrieval was performed using 1x DAKO target retrieval solution (10x concentrate) (DAKO, Carpinteria, CA) in a steamer for 20 minutes at 98°C followed by endogenous blocking with H2O2. Slides were then blocked using 8% fish gelatin and a primary antibody was applied overnight at 4°C. Washes were performed with PBS containing 0.05% Tween 20. Sections were incubated with rabbit on rodent horseradish peroxidase-labeled polymer RMR622H (Biocare Medical, Concord, CA) and positive labeling was detected with DAB+ Substrate system (DAKO). Gill no. 3 hematoxylin solution was used to counterstain each section. Immunohistochemical (IHC) staining for macrophages, rat anti-F4/80 (1:20; eBioscience, San Diego, CA), was performed after deparaffinization and antigen retrieval using methods described above, and nonspecific binding was removed by protein blocking with 8% fish gelatin. After washing, biotin goat anti-rat secondary was applied to the tissue sections followed by incubation with streptavidin.
Immunofluorescence
Unstained OCT-embedded frozen pancreatic tissue sections were fixed with cold acetone for 10 minutes and then washed with PBS. Slides were then blocked with 8% fish gelatin for 20 minutes at RT. Primary anti-COX2 antibody (1:300; Cayman Chemicals) in 8% fish gelatin was incubated. Slides were washed in PBS, blocked and secondary Alexa 594 Goat Anti-rabbit (1:800; Life Technologies) was applied and incubated at RT, then washed, counterstained with Hoechst 33342 (1:10000) and mounted. Slides were visualized under an epifluorescence microscope (Carl Zeiss Microscopy, Thornwood, NY) and the total fluorescence area measured using Simple PCI software program (Hamamatsu Corporation, Sewickley, PA). The total COX2 staining area/total nuclear area was calculated for each sample and normalized to the average of all LSL/BAC mice in CD.
Collagen detection
Collagen was detected in paraffin embedded pancreatic tissue using Trichrome staining (Sigma-Aldrich). Deparaffinization was performed as described above and sections incubated in preheated Bouin solution, washed with water and stained with Weigert's Iron Hematoxylin solution, followed by staining in Biebrich Scarlet-Acid Fuchsin solution, Phosphotungstic/Phosphomolybdic Acid Solution, and Aniline Blue Solution. The slides were then washed with 1% acetic acid, and dehydrated and mounted.
Statistical Analysis
Data were analyzed using the Prism 5 software program (GraphPad Software San Diego, CA). Results are expressed as the mean ± standard error of the mean. A t-test or one-way analysis of variance was performed and P levels less the 0.05 were considered significant.
Results
A HFD increased Mouse Adiposity
To analyze the effects of a HFD on BW we compared weight gains in LSL/BAC and BAC mice fed either the CD or the HFD for thirty days beginning after tamoxifen Cre activation at 40 days of age (Figure 1A). After 30 days on the HFD, all of the mice gained significant BW (Figure 1B) and had large fatty deposits in the abdominal region regardless of the presence of oncogenic Kras. In contrast, mice on the CD had only a minor increase in weight. A difference was noted in pancreatic weight (PW) between BAC controls and LSL.BAC animals. Mice expressing oncogenic Kras and fed the HFD had significantly higher PW than did mice fed the CD (Figure 1B).
Figure 1. HFD Increased BW and PW and the initiation of the early stages of PDAC development.

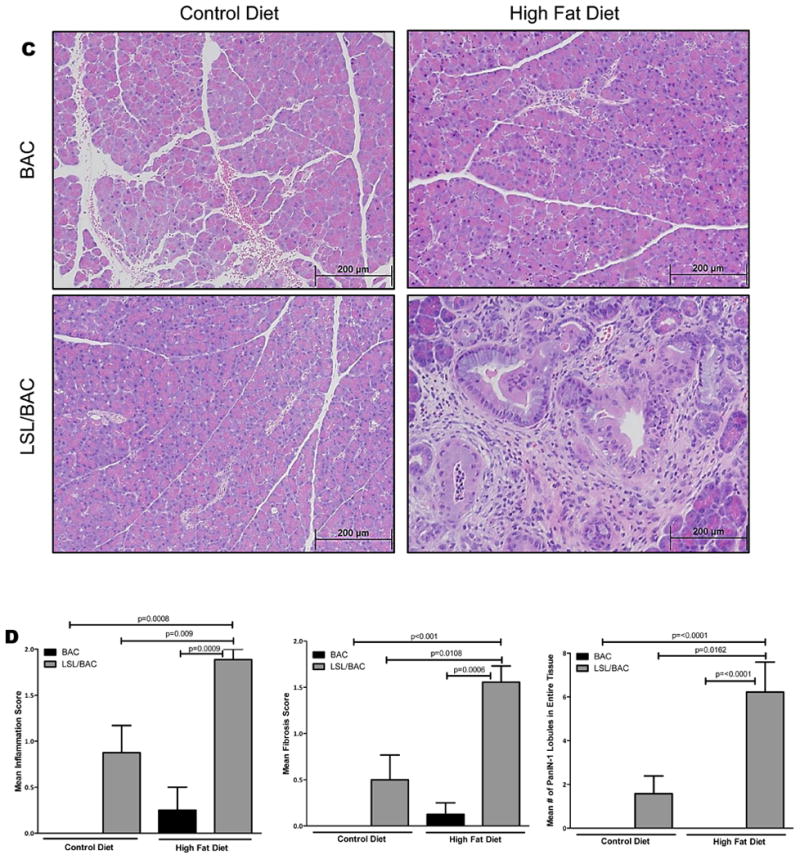
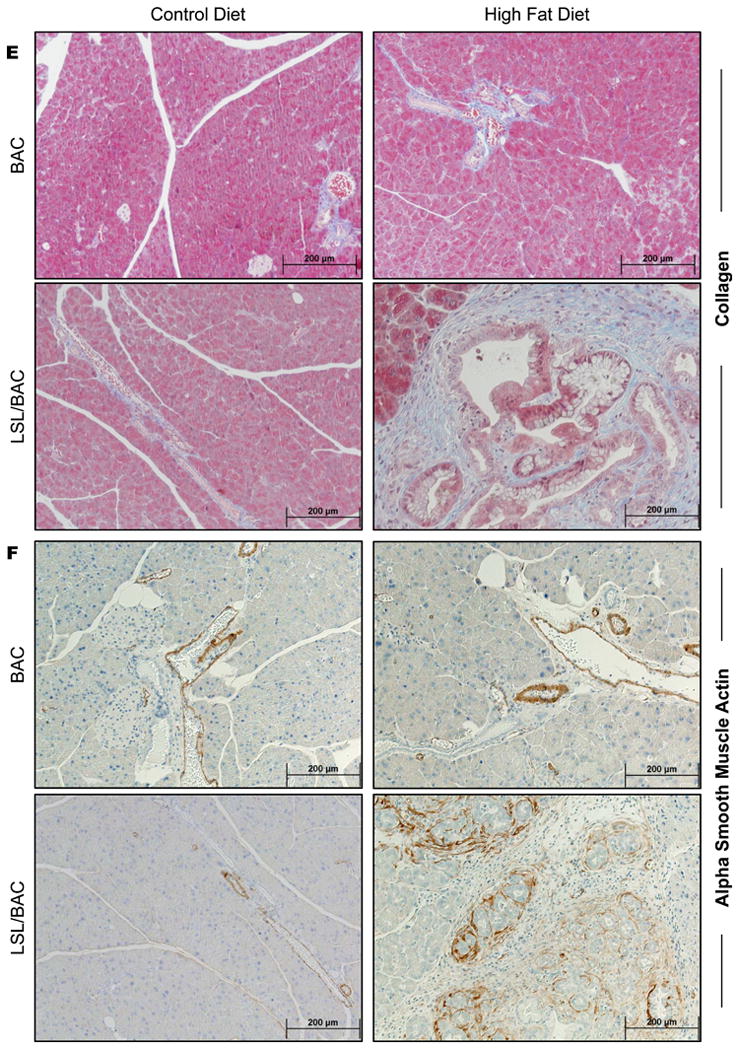
(A) At 40 days of age BAC and LSL/BAC mice were induced with tamoxifen for 3 consecutive days for Cre activation and placed on CD (BAC; n=5 and LSL/BAC; n=8) or HFD (BAC; n=9 and LSL/BAC; n=10) for thirty days. (B) BW of each mouse was measured weekly and the percent change from week 1 to week 5 of treatment was calculated. In addition, PW of each mouse was measured and the percent PW/BW was graphed for each group. (C) Representative H&E stains of BAC and LSL/BAC mice on CD and HFD are shown. (D) Each tissue sample was scored according to its level of inflammation, fibrosis, and number of PanIN-1 lesions seen in the entire tissue. (E) Collagen staining was performed for BAC and LSL/BAC animals on the CD and the HFD. (F) IHC for α--SMA expression was also performed for all groups.
A HFD Increased PanIN Lesions, Pancreatic Inflammation, and Fibrosis in Mice Expressing Oncogenic Kras
To determine whether the HFD increased the rate of PanIN lesion initiation, we fed BAC and LSL/BAC mice either the CD or the HFD and analyzed the pancreases histologically (Figure 1C). BAC mice fed either diet had normal pancreases that were indistinguishable from each other (Figure 1C). LSL/BAC mice fed the CD rarely developed fibrosis or PanINs (Figure 1C). In contrast, LSL/BAC mice fed the HFD had six-fold more PanIN lobules with extensive fibrosis as well as more inflammation than did LSL/BAC animals on the CD (Figure 1C-D). PDAC is a fibrotic tumor in which cancer cells make up as little as 10-20% of the tumor mass. Pancreatic fibrosis is thought to be controlled by pancreas-specific myofibroblasts called pancreatic stellate cells (PSCs). 23 Activated PSCs express αSMA which can be used as a semi-specific marker for these cells. 23 We hypothesized that mice with expression of oncogenic Kras and fed the HFD would have PSCs activation and increased pancreatic stroma. BAC mice on either diet displayed normal collagen distribution which was primarily around the blood vessels in the pancreas as indicated by Trichrome staining (Figure 1E). LSL/BAC mice on the CD showed a similar level of collagen disposition. In contrast, consistent with tumor development, LSL/BAC mice on the HFD showed dramatic increases in stromal collagen, often covering the majority of the pancreas (Figure 1E). Similarly, αSMA staining displayed higher levels of activated stellate cells in the pancreases of LSL/BAC mice on the HFD compared to controls (Figure 1F).
Mice Fed a HFD had Higher Levels of Activated Kras and Downstream Signaling in the Pancreas of Mice Expressing Oncogenic Kras
To determine whether Kras activity is elevated in mice with oncogenic Kras consuming a HFD, we measured activity levels in the pancreases (Figure 2A; Supplementary Figure 1A and 1C). LSL/BAC mice on the HFD had significantly higher levels of Ras activity than did those on the CD. In addition, we measured the levels of downstream targets of Ras and observed elevated expression of phospho-ERK, in LSL/BAC animals fed the HFD (Figure 2B; Supplementary Figure 1B). Phospho-ERK expression did not increase significantly in LSL/BAC mice fed the CD (Figure 2B). This observation was further confirmed through IHC and is consistent with tumor development (Figure 2C).
Figure 2. Significantly increased activation of Ras and its downstream targets with elevated COX2 and F4/80+ macrophages.
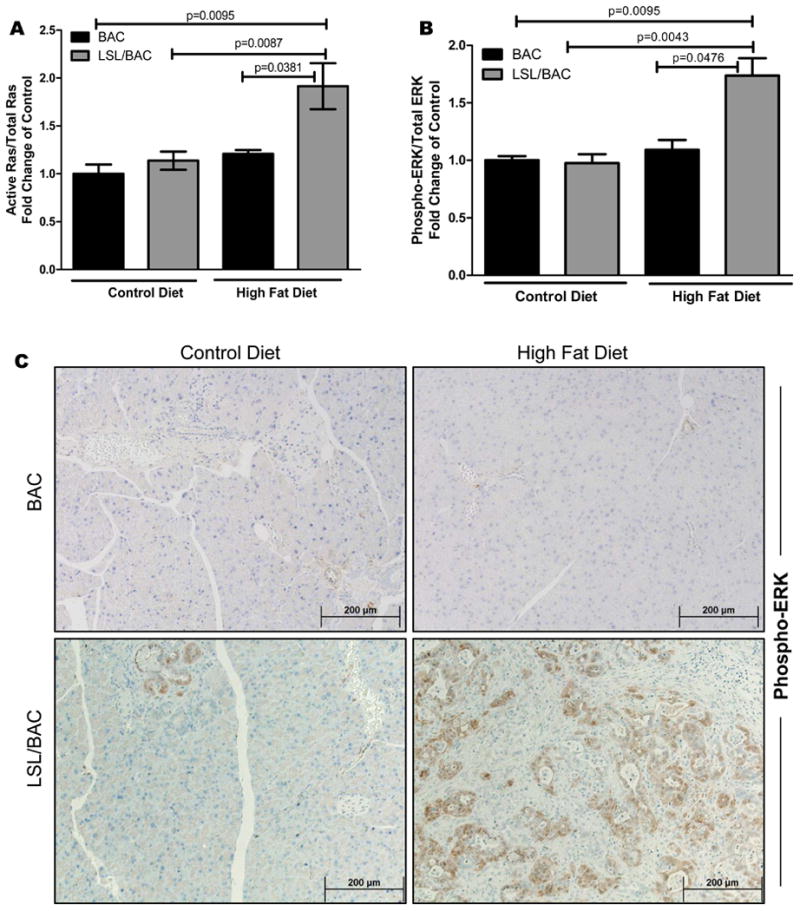
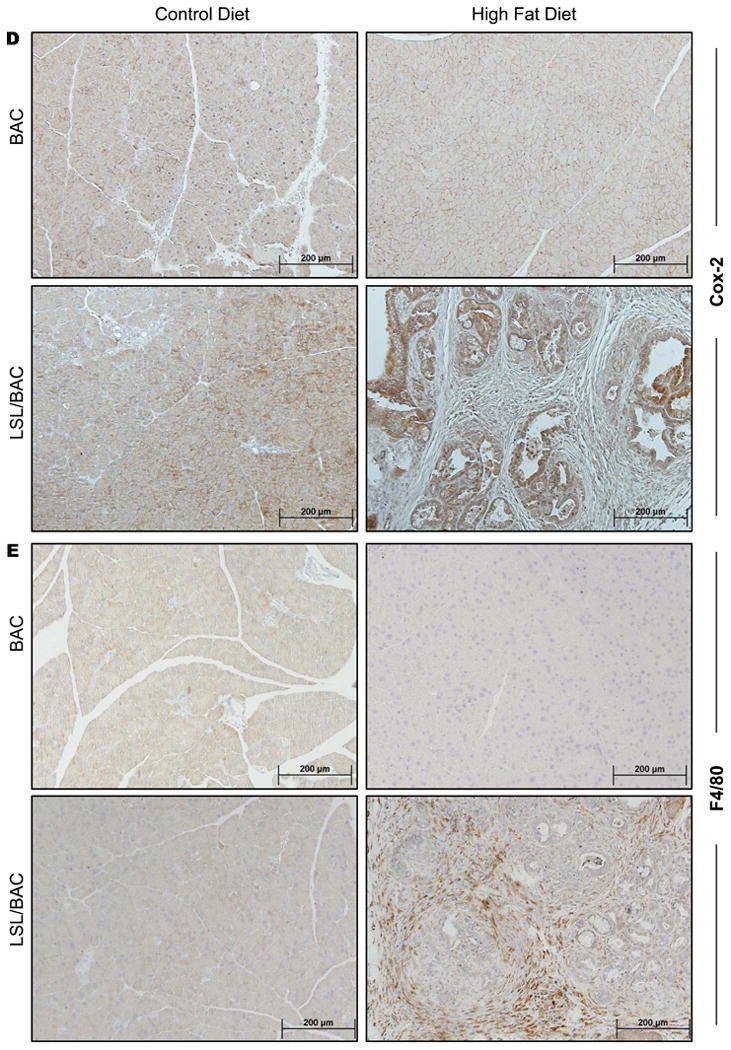
(A) Ras activity was measured by a Raf-pull down assay and was correlated with Phospho-ERK expression levels. (B) Western blot analysis for phospho-ERK expression for BAC and LSL/BAC animals on both diets was performed. (C) Additionally, IHC for phospho-ERK from all groups of mice are shown. (D & E) Pancreas tissue sections from each mouse were stained for COX2 and F4/80.
COX2 Activity was Required for the Pathological Effects of a HFD on the Pancreases of Mice Expressing Oncogenic Kras
To further characterize the mechanism behind the effects of consuming a HFD on the pancreas, we examined pancreatic COX2 expression levels using IHC and immunofluorescence (Figure 2D; Supplementary Figure 2). In BAC mice on either diet, COX2 expression levels were low compared to LSL/BAC mice fed the HFD which had highly increased expression (Figure 2D; Supplementary Figure 2). This effect required both the HFD and expression of oncogenic Kras, as LSL/BAC mice on the CD did not had elevated expression of COX2 (Figure 2D; Supplementary Figure 2). Mice expressing oncogenic Kras fed the HFD also had increased recruitment of F4/80+ macrophages into the pancreas, as indicated using IHC and immunofluorescence (Figure 2E and Supplementary Figure 2). Double staining of COX2 and F4/80+ using immunofluorescence showed co-localization in some, but not all areas (Supplementary Figure 2B).
To further analyze the role of COX2 expression in HFD-induced pancreatic effects, we crossed LSL/BAC mice with COX2 conditional knockout mice to generate COXKO/LSL/BAC mice. After thirty days on the HFD, these animals had no evidence of increased numbers of PanIN lesions, inflammation, or fibrosis (Figure 3A; Supplementary Figure 3A). Further, COXKO/LSL/BAC mice on either diet had low Ras activity (Figure 3B; Supplementary Figure 1C), αSMA, Phospho-ERK, and COX2, and lacked the presence of F4/80+ macrophages (Supplementary Figure 4A-D) compared to LSL/BAC mice.
Figure 3. Essential role of COX2 in Kras-induced fibrosis and inflammation.
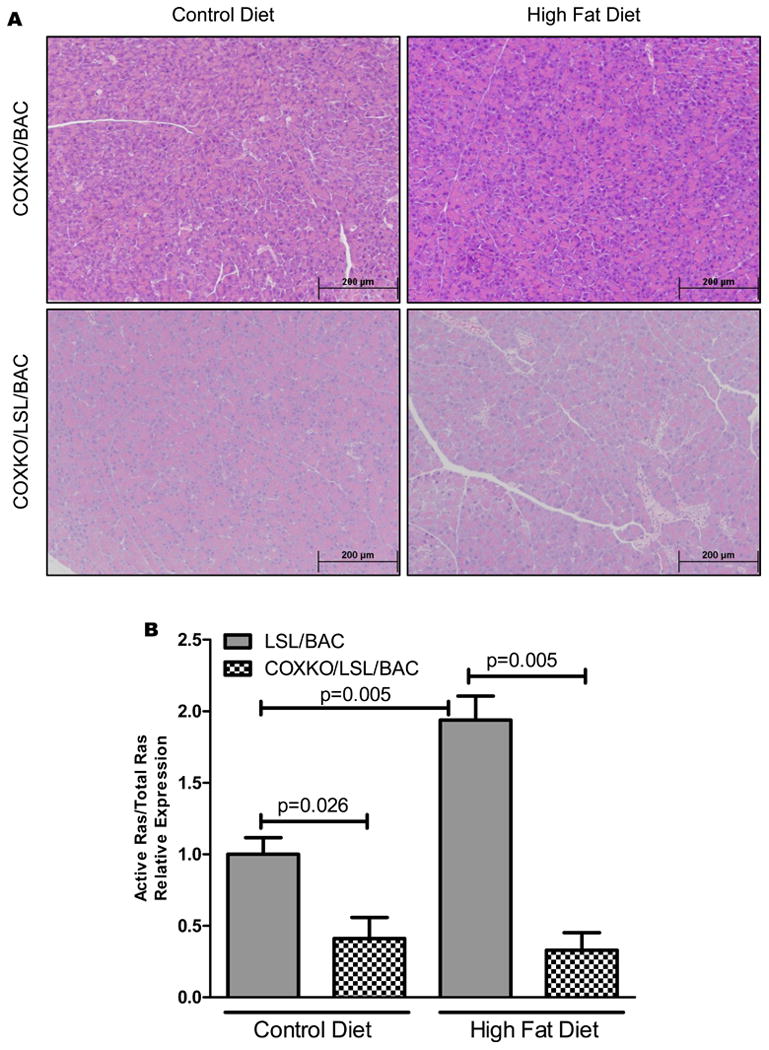
(A) Histology of COXKO/LSL/BAC mice treated with CD (COXKOBAC; n=5 and COXKO/LSL/BAC; n=5) and HFD (COXKOBAC; n=5 and COXKO/LSL/BAC; n=10) is shown. (B) Ras activity was measured through Raf-pull down assay comparing LSL/BAC animals to COXKO/LSL/BAC animals on CD and HFD.
As a further indication of the role of COX2 in the HFD induction of Kras generated pathology, we tested the ability of Celecoxib, a selective COX2 inhibitor, to prevent these effects. BAC and LSL/BAC mice on the HFD were treated either with saline or Celecoxib. After 38 days of treatment, LSL/BAC mice treated with Celecoxib had significantly lower levels of inflammation, fibrosis, and PanIN development compared to saline-treated mice (Figure 4).
Figure 4. Celecoxib delays HFD-induced effects.
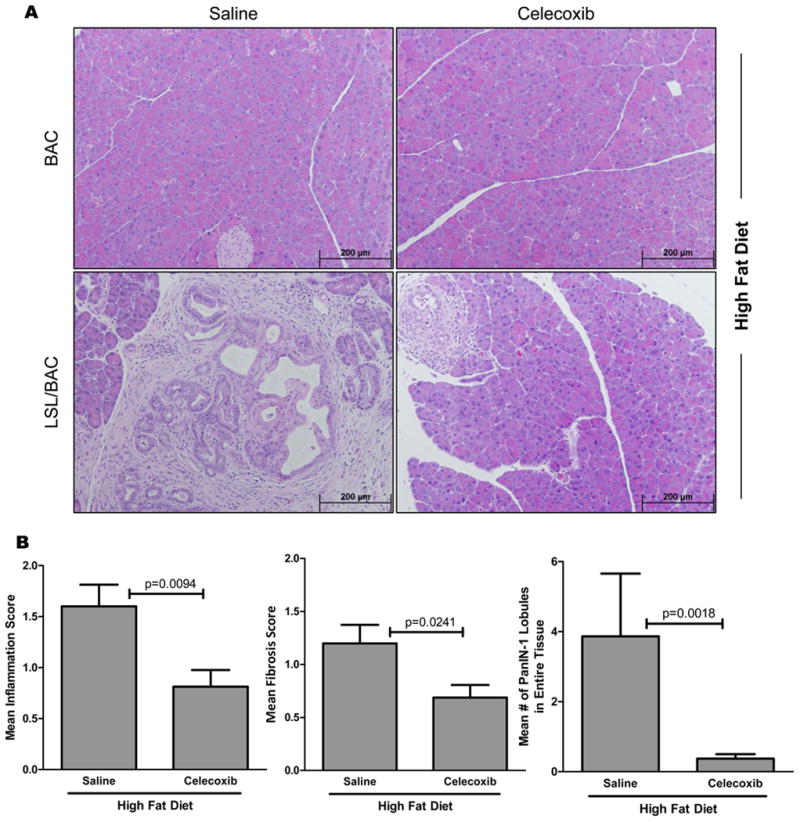
(A) Representative H&E stains of BAC and LSL/BAC animals on HFD treated with either Saline or Celecoxib (Saline; BAC, n=4; LSL/BAC, n=15 and Celecoxib; BAC, n=4; LSL/BAC, n=16) is shown. (B) LSL/BAC animals in each treatment group were histologically scored for inflammation, fibrosis, and number of PanIN lesions.
HFD Decreased Survival and Accelerated Tumor Progression
To determine how long-term consumption of HFD would affect overall survival, mice were fed CD or HFD for longer times. LSL/BAC mice on the HFD had a significant decrease in survival rate (∼250 days), whereas those on CD remain healthy (Figure 5A). Similarly, LSL/Pdx-1 mice fed the HFD had a reduced survival (∼145 days), while mice on CD survived much longer (∼220 days) (Figure 5A).
Figure 5. Decreased overall survival and promotion of tumor growth with continuous consumption of HFD.
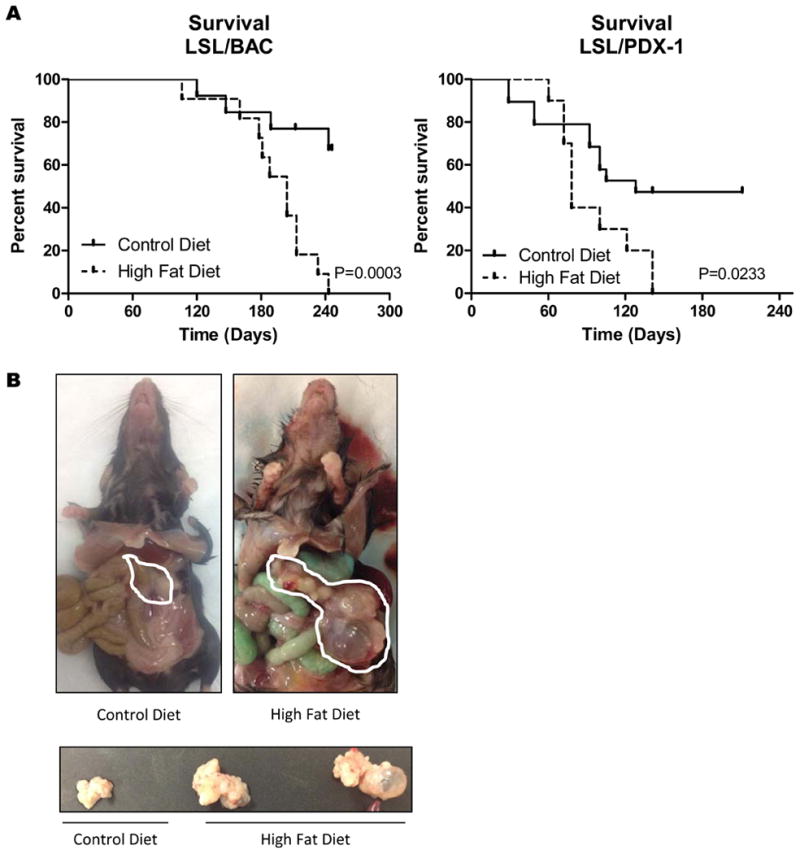
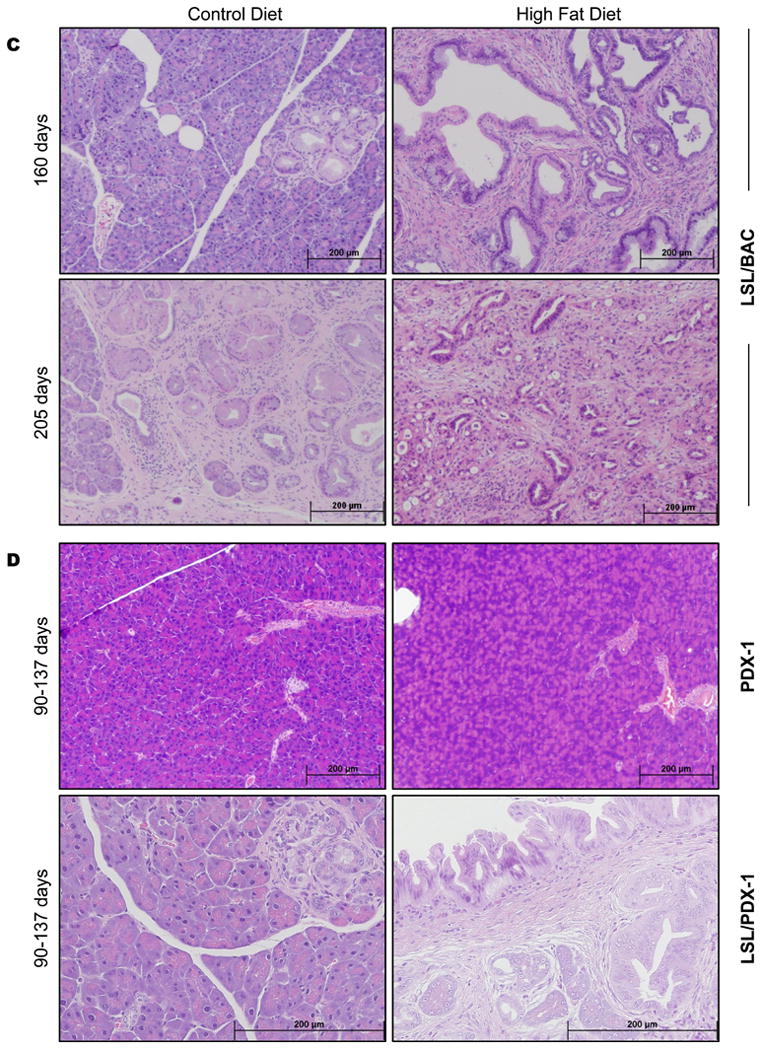
(A) The overall survival of LSL/BAC (CD; n=13 and HFD; n=11) and LSL/PDX-1 (CD; n=19 and HFD; n=10) animals on HFD was recorded. (B) Gross images of LSL/BAC animals on CD and HFD and the pancreases of these animals are shown. (C) Additionally shown are H&E stains of the pancreases of LSL/BAC animals on CD and HFD at 160 days and 205 days along with (D) histology of PDX-1 and Pdx-Cre-KrasG12D on CD and HFD from 90-137 days.
Along with overall survival, mice on HFD had an acceleration of PanIN development which progressed to cancer. Animals on the HFD had definite pancreatic tumors accompanied with large pancreatic cysts, which was not evident in mice on the CD (Figure 5B). Both LSL/BAC and LSL/Pdx-1 mice fed HFD had drastic pathological alterations in the pancreases (Figure 5C-D). PanIN progression to PanIN-2 and PanIN-3 lesions was greatly accelerated in the LSL/BAC mice on the HFD. At 205 days, LSL/BAC mice on the HFD had progressed to invasive cancer with metastasis to the lung, while the few early PanIN lesions in the pancreases of mice on the CD had not progressed to higher grade PanIN lesions (Figure 5C). Similarly, we examined the pancreas of Pdx-1 and LSL/Pdx-1 mice on CD or HFD. The level of inflammation, fibrosis, and the number of lobules containing PanIN lesions were increased HFD-fed mice (Supplementary Figure 3B). At 90-137 days when the mice were sacrificed, LSL/Pdx-1 mice on HFD had areas of PanIN-2 and PanIN-3 lesions in the pancreas which were not observed in CD fed mice (Figure 5D; Supplementary Figure 3B).
Discussion
This study investigated the mechanisms by which a HFD accelerates PDAC development. Specifically, our data linked the consumption of a HFD in the presence of physiologically relevant levels of oncogenic Kras in adult pancreatic acinar cells to elevated COX2 expression and Kras activity. The, likely mechanism involves a positive feedback inflammatory loop. High levels of Ras activity were associated with the development of PanINs leading to PDAC and reduced survival. These data support the concept of oncogenic Ras being largely inactive until externally stimulated, as we previously suggested. 14, 15 The current study indicates that a HFD can act as the external stimulus to induce inflammation that then promotes Ras activation. The data also supports the concept of a Ras-inflammatory positive feedback loop being required for chronic pancreas pathological responses. As was found in our earlier studies, inhibition of COX2 was able to break the inflammatory positive feedback mechanism. 14 These observations have important implications for cancer prevention.
To test the effects of a HFD on PDAC development this study primarily used an adult acinar specific Kras model. Expression of endogenous levels of oncogenic Kras in adult acinar cells is largely ineffective at initiating spontaneous pancreas pathologies, making this a good model to investigate the effects of potential environmental risk factors. 24,14,15 For comparison, we also examined the effects of the HFD on a PDX-1-Cre Kras model in which oncogenic Kras is activated during development. The developmental model gave similar results but with a higher background of spontaneous PanINs.
A point mutation in Kras is well recognized to be the earliest mutation that occurs in the transformation of normal pancreatic acinar cells to PanIN lesions. 8, 27-31 Interestingly, this mutation is present in a large number of healthy people who never develop PDAC. 8,32 Recently, we demonstrated that pancreatic inflammation associated with treatment of caerulein, lipopolysaccharide, or with elevated endogenous CCK levels induced a COX2-mediated positive feedback loop that drove sustained, high Kras activity. 14 In the present study, we found that the activity of Kras and its downstream pathways, including phospho-ERK, was greatly increased in Kras expressing mice fed a HFD. These data support the ability of a HFD to activate oncogenic Kras in the pancreas, creating a mechanistic link between obesity-induced acceleration of the transformation of normal acinar cells to pancreatic ductal lesions.
Another result of feeding LSL/BAC mice a HFD was increased PSC stimulation and stromal formation. PSC activation is universally present in the early stages of PDAC. 23 The role of PSCs and the stroma they produce in PDAC progression is currently unclear. Stellate cell activation is not simply a response to acinar cell damage and death, as acinar cell apoptosis does not activate stellate cells. 33 Apparently, inflammatory mediators released from acinar cells under the influence of high Ras activity are responsible for the initiation of pancreatic fibrosis. We recently found that stellate cells are directly stimulated by PGE2. 34 Therefore, COX2 activity induced by Ras is likely one of the mechanism of stromal formation. The present study demonstrated that a HFD can stimulate activation of oncogenic Kras, initiating the transformation of normal pancreatic cells to PanIN lesions, and further supports the importance of Kras as a driving mutation in the early development and progression to PDAC. Moreover, it highlights the increased risk of PDAC development and decreased survival in the presence of oncogenic Kras expression and with the consumption of a HFD.
We previously identified COX2 as an essential component of the positive feedback loop that enhances Kras activity in the presence of pancreatic inflammation. 14 The current study also found that COX2 activity was necessary for the effects of the HFD on Ras activity, inflammation, and fibrosis and PanIN development. Therefore, although oncogenic Kras is the primary driving force in promoting the early stages of PDAC, it requires COX2 to maintain the Ras inflammation feed-forward loop. This may provide a mechanistic explanation for previous studies indicating a protective effect of aspirin and COX2 inhibitors on the risk of pancreatic cancer. 35 Consumption of the HFD led to increased macrophage infiltration in pancreatic areas surrounding PanIN lesions, demonstrating the role of oncogenic Kras in the recruitment of inflammatory mediators to the pancreas, which may contribute to other factors that enhance PanIN formation. A model illustrating these interactions is provided (Figure 6).
Figure 6. Model of HFD induced pancreatic pathology via Kras activity.
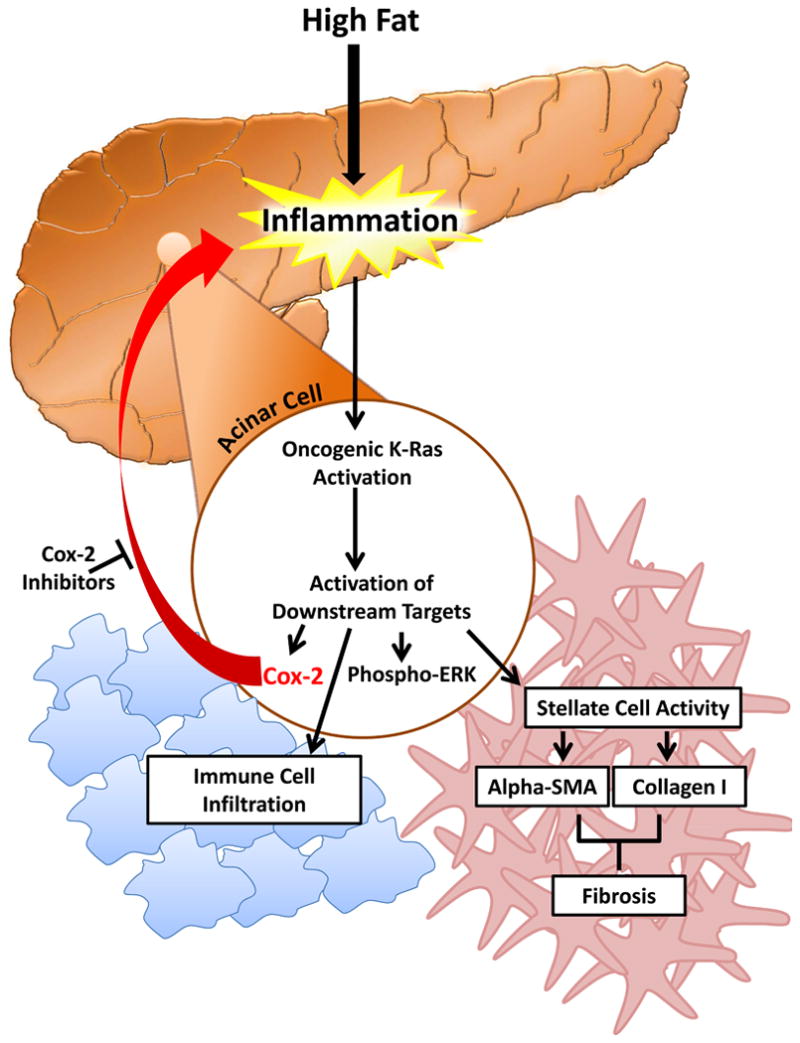
A HFD causes increased Kras activity in animals bearing oncogenic Kras. Activation of oncogenic Kras leads to downstream activation of COX2, Phospho-ERK, and infiltration of macrophages into the stroma, as well as activation of quiescent PSCs producing αSMA and collagen I. COX2 forms a critical part of a positive feed-forward loop maintaining Kras activity and further increasing inflammation, fibrosis, and recruitment of inflammatory mediators to the pancreas. COX2 inhibitors or genetic deletion of COX2 can interfere with the feed-forward mechanism and prevent HFD induced pathology.
A previous study using a p48 developmental promoter observed accelerated PanIN progression induced by a HFD. These researchers suggested that tumor promotion was associated with inflammation and specifically showed that TNFR1 signaling and TNF-α played a role in enhancing PanIN formation.18 Data from this study are entirely consistent with increased Ras activity and supports the importance of an inflammatory mechanism. Based on these data and the current study, it seems likely that several inflammatory mechanisms may be involved in the postive feed-back loop that maintains Ras activity. Therefore, there may be several potential means of blocking this loop and preventing cancer formation.
Previous studies have also suggested an association between obesity and the growth of xenograft tumors, including PDAC. 2, 4, 25 For example, mice with orthotopic implantation of Mia PaCa2 human pancreatic tumor cells fed a HFD had much greater tumor burdens compared to mice fed a CD. 26 Although these studies showed a strong connection between obesity and pancreatic tumor burden, the mechanisms involved are unclear. Since it was observed that Ras activity can be elevated by external stimuli in PDAC cells, 15 it is possible that Ras activity and inflammatory positive feed-back mechanisms are involved in the growth of fully developed cancer. However, it is currently unknown whether the mechanisms involved in the growth of fully developed PDAC are different from those involved in cancer initiation that we have identified.
In conclusion, the findings of the present study revealed for the first time a mechanism linking a HFD with the development of PDAC involving inflammation-induced Kras activity. Because oncogenic Kras expression is present in many healthy individuals, the consumption of a HFD by individuals possessing oncogenic Ras mutations in pancreatic cells likely increases their risk of developing PDAC. Therefore, limiting consumption of fat and increasing intake of COX2 inhibitors may prevent pancreatic inflammation and fibrosis and the initiation of the PanIN-PDAC sequence.
Supplementary Material
Acknowledgments
Grant support: NIH-DK052067 and The Lockton Endowment (to C.D.L.), NIDDK Diversity Supplement DK052067-08 (to C.D.L. for Z.C.-M.), and NIH T32 CA009599 Ruth L. Kirschstein National Research Service Award (to C.L.R.). This research is supported in part by the MD Anderson Cancer Center Support Grant CA016672.
Abbreviations List
- α-SMA
α-smooth muscle actin
- BAC
bacterial-artificial-chromosome
- BW
body weight
- CD
control diet
- Cox
cyclooxygenase
- Ela
elastase
- ERK
extracellular signal-regulated kinase
- GEMMs
genetically engineered mouse models
- GEFs
guanine exchange factor
- GAPs
GTPase activating protein
- GDP
guanosine diphosphate
- GTP
guanosine triphosphate
- H&E
hematoxylin and eosin
- HFD
high-fat diet
- MAPK
mitogen-activated protein kinase
- IHC
immunohistochemistry
- PanIN
pancreatic intraepithelial neoplasia
- PBS
phosphate-buffered saline
- PDAC
pancreatic ductal adenocarcinoma
- PI3K
phosphoinositide 3-kinase
- Pdx
pancreatic and duodenal homeobox 1
- PSC
pancreatic stellate cell
- RBD
Raf-Ras binding domain
- RT
room temperature
Footnotes
Disclosures The authors declare no conflicts of interest.
Author contributions: B.P.: study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content, and technical support; C.L.R., J.D., Y.L., D.C., S,B.G., B.J., H.H., H.W., and J.B.F.: technical and material support; Z.C.-M. and C.D.L.: study concept and design, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content, obtainment of funding, technical support, study supervision, and study concept and design.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Arslan AA, Helzlsouer KJ, Kooperberg C, et al. Anthropometric measures, body mass index, and pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan) Arch Intern Med. 2010;170:791–802. doi: 10.1001/archinternmed.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li D, Morris JS, Liu J, et al. Body mass index and risk, age of onset, and survival in patients with pancreatic cancer. JAMA. 2009;301:2553–62. doi: 10.1001/jama.2009.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nature Reviews Cancer. 2004;4:579–91. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 5.Rapp K, Schroeder J, Klenk J, et al. Obesity and incidence of cancer: a large cohort study of over 145,000 adults in Austria. Br J Cancer. 2005;93:1062–7. doi: 10.1038/sj.bjc.6602819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larsson SC, Permert J, Hakansson N, et al. Overall obesity, abdominal adiposity, diabetes and cigarette smoking in relation to the risk of pancreatic cancer in two Swedish population-based cohorts. British Journal of Cancer. 2005;93:1310–1315. doi: 10.1038/sj.bjc.6602868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.WHO fact sheet 311 Obesity and Overweight; 2012. [assessed 2012 Nov 8];2012 Available from http://www.who.int/mediacentre/factsheets/fs311/en/
- 8.Iovanna J, Mallmann MC, Goncalves A, et al. Current knowledge on pancreatic cancer. Front Oncol. 2012;2:6. doi: 10.3389/fonc.2012.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herreros-Villanueva M, Hijona E, Cosme A, et al. Mouse models of pancreatic cancer. World J Gastroenterol. 2012;18:1286–94. doi: 10.3748/wjg.v18.i12.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Remmers N, Bailey JM, Mohr AM, et al. Molecular pathology of early pancreatic cancer. Cancer Biomark. 2010;9:421–40. doi: 10.3233/CBM-2011-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jimeno A, Hidalgo M. Molecular biomarkers: their increasing role in the diagnosis, characterization, and therapy guidance in pancreatic cancer. Mol Cancer Ther. 2006;5:787–96. doi: 10.1158/1535-7163.MCT-06-0005. [DOI] [PubMed] [Google Scholar]
- 12.Koorstra JB, Hustinx SR, Offerhaus GJ, et al. Pancreatic carcinogenesis. Pancreatology : official journal of the International Association of Pancreatology. 2008;8:110–25. doi: 10.1159/000123838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maitra A, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20:211–26. doi: 10.1016/j.bpg.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Daniluk J, Liu Y, Deng DF, et al. An NF-kappa B pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. Journal of Clinical Investigation. 2012;122:1519–1528. doi: 10.1172/JCI59743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang H, Daniluk J, Liu Y, et al. Oncogenic K-Ras requires activation for enhanced activity. Oncogene. 2013 doi: 10.1038/onc.2012.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maitra A, Adsay NV, Argani P, et al. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Modern Pathology. 2003;16:902–912. doi: 10.1097/01.MP.0000086072.56290.FB. [DOI] [PubMed] [Google Scholar]
- 17.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–7. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khasawneh J, Schulz MD, Walch A, et al. Inflammation and mitochondrial fatty acid beta-oxidation link obesity to early tumor promotion. Proc Natl Acad Sci U S A. 2009;106:3354–9. doi: 10.1073/pnas.0802864106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji B, Song J, Tsou L, et al. Robust acinar cell transgene expression of CreErT via BAC recombineering. Genesis. 2008;46:390–5. doi: 10.1002/dvg.20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 22.Ishikawa TO, Herschman HR. Conditional knockout mouse for tissue-specific disruption of the cyclooxygenase-2 (Cox-2) gene. Genesis. 2006;44:143–9. doi: 10.1002/gene.20192. [DOI] [PubMed] [Google Scholar]
- 23.Omary MB, Lugea A, Lowe AW, et al. The pancreatic stellate cell: a star on the rise in pancreatic diseases. Journal of Clinical Investigation. 2007;117:50–59. doi: 10.1172/JCI30082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Calle EE, Rodriguez C, Walker-Thurmond K, et al. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. New England Journal of Medicine. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 26.Wang F, Kumagai-Braesch M, Herrington MK, et al. Increased lipid metabolism and cell turnover of MiaPaCa2 cells induced by high-fat diet in an orthotopic system. Metabolism. 2009;58:1131–6. doi: 10.1016/j.metabol.2009.03.027. [DOI] [PubMed] [Google Scholar]
- 27.Deramaudt T, Rustgi AK. Mutant KRAS in the initiation of pancreatic cancer. Biochim Biophys Acta. 2005;1756:97–101. doi: 10.1016/j.bbcan.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Lohr M, Kloppel G, Maisonneuve P, et al. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17–23. doi: 10.1593/neo.04445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hruban RH, van Mansfeld AD, Offerhaus GJ, et al. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele- specific oligonucleotide hybridization. Am J Pathol. 1993;143:545–54. [PMC free article] [PubMed] [Google Scholar]
- 30.Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57:2140–3. [PubMed] [Google Scholar]
- 31.DiGiuseppe JA, Hruban RH, Offerhaus GJ, et al. Detection of K-ras mutations in mucinous pancreatic duct hyperplasia from a patient with a family history of pancreatic carcinoma. Am J Pathol. 1994;144:889–95. [PMC free article] [PubMed] [Google Scholar]
- 32.Tada M, Ohashi M, Shiratori Y, et al. Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology. 1996;110:227–31. doi: 10.1053/gast.1996.v110.pm8536861. [DOI] [PubMed] [Google Scholar]
- 33.Gaiser S, Daniluk J, Liu Y, et al. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut. 2011;60:1379–88. doi: 10.1136/gut.2010.226175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charo C, Holla V, Arumugam T, et al. Prostaglandin E2 Regulates Pancreatic Stellate Cell Activity Via the EP4 Receptor. Pancreas. 2012 doi: 10.1097/MPA.0b013e318264d0f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan XL, Reid Lombardo KM, Bamlet WR, et al. Aspirin, nonsteroidal anti-inflammatory drugs, acetaminophen, and pancreatic cancer risk: a clinic-based case-control study. Cancer Prev Res (Phila) 2011;4:1835–41. doi: 10.1158/1940-6207.CAPR-11-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.