

Abstract
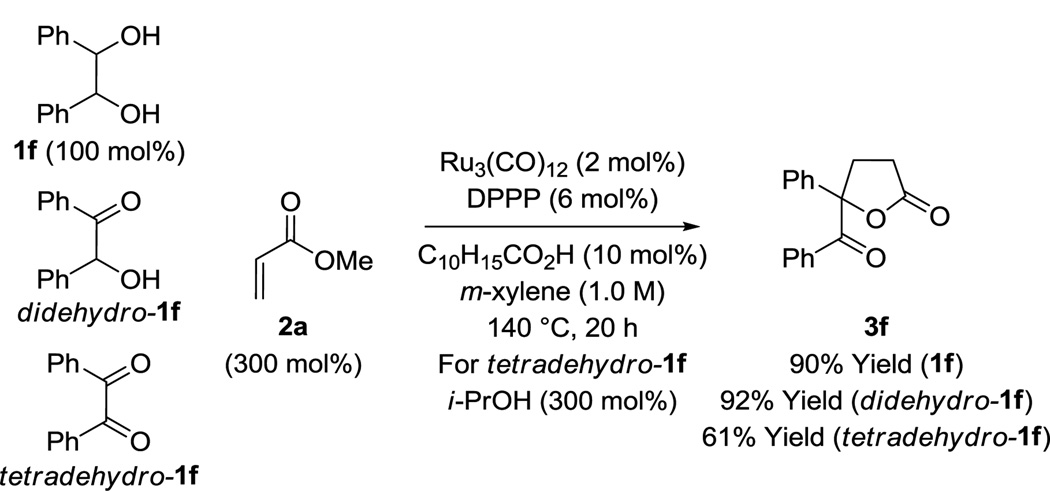
Under the conditions of ruthenium(0) catalyzed hydrohydroxyalkylation, vicinal diols 1a–1l and methyl acrylate 2a are converted to the corresponding lactones 3a–3l in good to excellent yield. The reaction of methyl acrylate 2a with hydrobenzoin 1f, benzoin didehydro-1f, and benzil tetradehydro-1f form the same lactone 3f product, demonstrating that this process may be deployed in a redox level-independent manner. A variety of substituted acrylic esters 2a–2h participate in spirolactone formation, as illustrated in the conversion of N-benzyl-3-hydroxyoxindole 1o to cycloadducts 4a–4h. Hydrohydroxyalkylation of hydroxyl-substituted methacrylate 2i with diols 1b, 1f, 1j and 1l forms α-exo-methylene-γ-butyrolactones 5b, 5f, 5j and 5l in moderate to good yield. A catalytic cycle involving 1,2-dicarbonyl-acrylate oxidative coupling to form oxaruthenacyclic intermediates is postulated. A catalytically competent mononuclear ruthenium(II) complex was characterized by single crystal X-ray diffraction. The influence of electronic effects on regioselectivity in reactions of nonsymmetric diols were probed using para-substituted 1-phenyl-1,2-propanediols 1g, 1m and 1n and density functional theory (DFT) calculations.
Introduction
Our laboratory has developed ruthenium and iridium “hydrohydroxyalkylations” wherein hydrogen transfer from primary alcohols to π-unsaturated reactants generates organometal-aldehyde pairs that combine to form products of carbonyl addition.1,2,3 Such C-C bond forming transfer hydrogenations may be viewed as alternatives to classical carbonyl additions, for which discrete alcohol-to-aldehyde oxidation and use of premetallated C-nucleophiles are often required. To expand the scope of this emerging family of C-C bond formations, the development of corresponding secondary alcohol mediated hydrohydroxyalkylations was undertaken. However, our initial efforts to promote hydrohydroxyalkylations with secondary alcohols using previously developed ruthenium2 and iridium3 catalysts resulted in conventional transfer hydrogenation to form ketone products. Products of C-C coupling were not observed.
It was postulated that secondary alcohols that form vicinal dicarbonyl compounds upon dehydrogenation, for example, α-hydroxy esters or vicinal cycloalkane diols, should engage more readily in carbonyl addition or oxidative coupling pathways en route to products of C-C coupling. However, this enhanced reactivity also renders vicinal dicarbonyl compounds more susceptible to reduction. Hence, if dehydrogenation is reversible, the short lifetime of the transient vicinal dicarbonyl might impede C-C coupling pathways. In view of this issue, we were inspired by recent reports of Ru3(CO)12 catalyzed aminations of 1,2-diols4a,b and α-hydroxy amides,4c which occur via reductive amination of transient vicinal dicarbonyl species. This result, along with Chatani’s observation of oxidative coupling pathways in Pauson-Khand reactions of 1,2-diones,5 suggested the feasibility of hydrohydroxyalkylations by way of oxidative coupling-secondary alcohol transfer hydrogenation pathways.
Ruthenium(0) catalysts derived from Ru3(CO)12 and phosphine ligands were found to promote the C-C coupling of α-hydroxy esters and amides to isoprene and myrcene to furnish products of prenylation and geranylation, respectively (Figure 1, top).6a,b More recently, a mechanistically related ruthenium(0) catalyzed [4+2] cycloaddition of vicinal diols via successive hydrohydroxyalkylation of dienes was developed (Figure 1, middle).6c Here, we report that ruthenium(0) catalyzed hydrohydroxyalkylation of acrylates with vicinal diols or their more highly oxidized congeners delivers spiro- and α-methylene-γ-butyrolactones, structural motifs that are ubiquitous in Nature (Figure 1, bottom).7
Figure 1.

Ruthenium(0) catalyzed hydrohydroxyalkylations.
Research Design and Methods
It was reasoned that ruthenium(0) catalyzed hydrohydroxyalkylation of acrylates with vicinal diols would provide transient oxaruthenacycles that would spontaneously cyclize to form lactone products (Figure 1, bottom). This method would complement alternate approaches to spirocyclic γ-butyrolactones,7a which include cationic rearrangements of epoxides8 and bromonium ions,9 Stetter type reactions,10 oxidative dearomatization,11 C-H hydroxylation of carboxylic acids,12 reductive cyclizations of α,β-unsaturated esters onto ketones,13 Pauson-Khand type reactions of olefins with vicinal diones,5 and the 2-(alkoxycarbonyl)allylation of carbonyl compounds.14
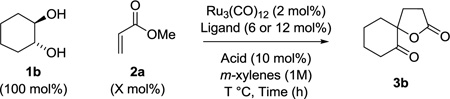
To probe the feasibility of the proposed transformation, racemic trans-1,2-cyclohexane diol 1b was exposed to methyl acrylate 2a (300 mol%) in the presence of Ru3(CO)12 (2 mol%) and various nitrogen or phosphorus containing ligands. It was found that the ruthenium catalyst modified by DPPP (6 mol%) was uniquely effective, providing the desired spirolactone 3b in 76% yield (Table 1, entry 4). Although increased loadings of methyl acrylate 2a were found to improve the isolated yield of spirolactone 3b (Table 1, entries 6 and 7), enhancing the intrinsic reaction efficiency so as to minimize the loading of methyl acrylate 2a was preferred. As further variation of the reaction parameters, including temperature (Table 1, entries 8 and 9), did not avail further improvement, carboxylic acid additives, which are known to co-catalyze hydrogenolysis of oxa- and azametallacycles, were evaluated.15 Using 1-adamantanecarboxylic acid (10 mol%) as a cocatalyst, the isolated yield of spirolactone 3b was increased from 76% to 96% (Table 1, entries 4 and 11).
Table 1.
Selected optimization experiments in the ruthenium catalyzed C-C coupling of diol 1b and methyl acrylate 2a.a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Additive | 2a (mol%) | Time (h) | T (°C) | Yield (%) |
| 1 | BIPY | – | 300 | 20 | 140 | trace |
| 2 | Phen | – | 300 | 20 | 140 | trace |
| 3 | PCy3 | – | 300 | 20 | 140 | trace |
| 4 | DPPP | – | 300 | 20 | 140 | 76 |
| 5 | DPPP | – | 200 | 20 | 140 | 56 |
| 6 | DPPP | – | 400 | 20 | 140 | 88 |
| 7 | DPPP | – | 500 | 20 | 140 | 79 |
| 8 | DPPP | – | 300 | 20 | 130 | 33 |
| 9 | DPPP | – | 300 | 20 | 150 | 69 |
| 10 | DPPP | Benzoic Acid | 300 | 20 | 140 | 93 |
| ⇨11 | DPPP | C10H15CO2H | 300 | 20 | 140 | 96 |
| 12 | DPPP | C10H15CO2H | 300 | 4 | 140 | 73 |
| 13 | DPPP | C10H15CO2H | 300 | 8 | 140 | 83 |
| 14 | DPPP | C10H15CO2H | 300 | 20 | 120 | 62 |
| 15 | DPPP | C10H15CO2H | 300 | 20 | 140 | 55b |
Cited yields are of material isolated by silica gel chromatography. C10H15CO2H refers to 1-adamantanecarboxylic acid.
0.5 mol% Ru3(CO)12. See Supporting Information for further experimental details.
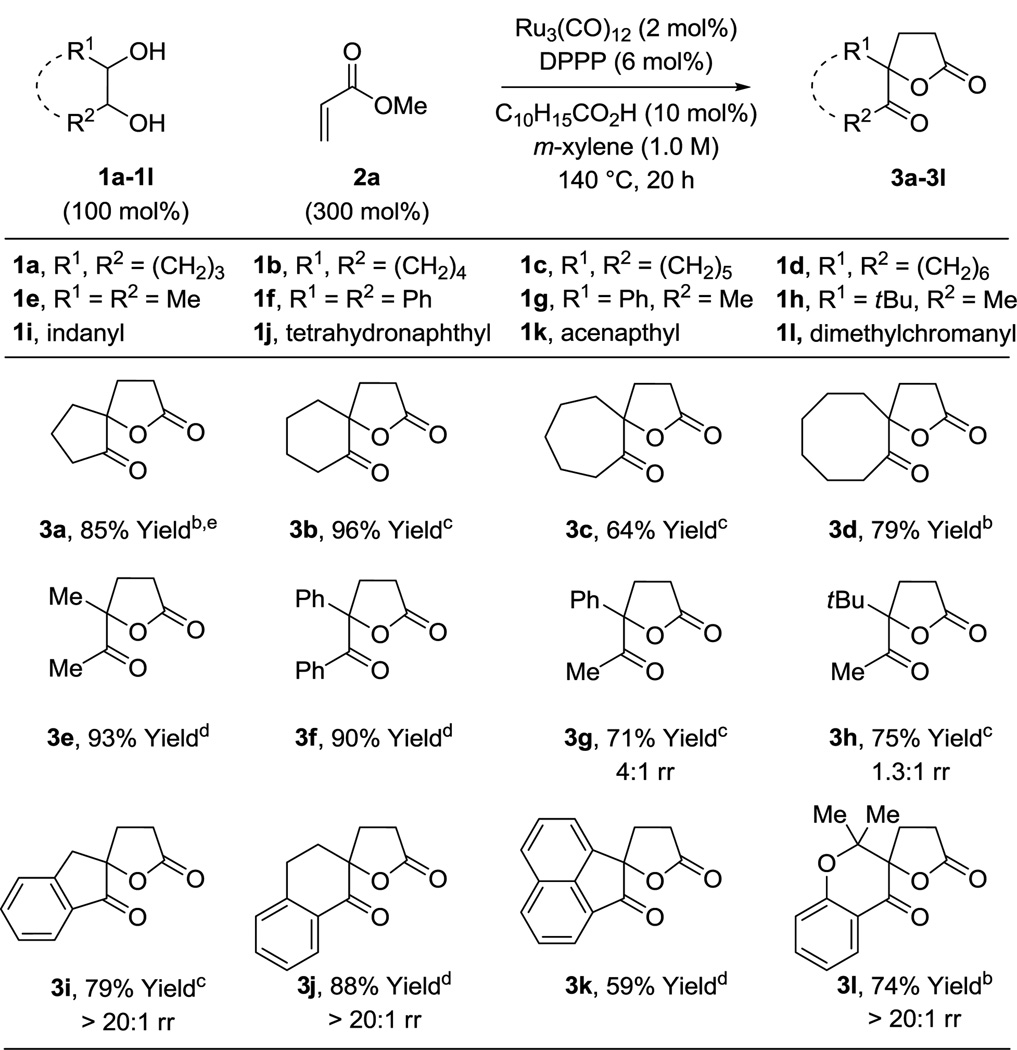
Optimal conditions identified for formation of spirolactone 3b were applied to the C-C coupling of cyclic and acyclic diols 1a–1l and methyl acrylate 2a. The corresponding lactones 3a–3l were generated in good to excellent yield (Table 2). Both cis- and trans-diols react with equal efficiency. As illustrated in the conversion of diols 1a–1d to 3a–3d, five-, six-, seven- and eight-membered ring cycloalkanes participate in spirolactone formation. Acyclic vicinal diols 1e–1h form lactone products 3e–3h. Whereas nonsymmetric diols 1g and 1h are converted to lactones 3g and 3h with incomplete control of regioselectivity, the reactions of cyclic diols 1i, 1j, and 1l are completely regioselective, providing spirolactones 3i, 3j, and 3l as single constitutional isomers.
Table 2.
Ruthenium(0) catalyzed hydrohydroxyalkylation of methyl acrylate 2a with diols 1a–1l to form lactones 3a–3l.a.
 |
Cited yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
The cis-1,2-diol was employed.
The trans-1,2-diol was employed.
A mixture of cis- and trans-1,2-diols was employed.
2a (400 mol%).
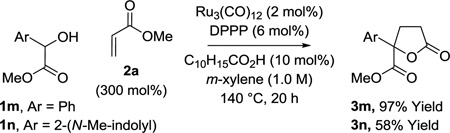
As illustrated in the conversion of hydrobenzoin 1f, benzoin didehydro-1f, and benzil tetradehydro-1f to lactone 3f, catalytic C-C coupling may be accomplished in oxidative, redox-neutral, and reductive modes, respectively (Table 3). For the latter reaction involving benzil tetradehydro-1f, isopropanol (300 mol%) is employed as terminal reductant. Additionally, it was found that other vicinally deoxygenated compounds participate in lactone formation. For example, exposure of α-hydroxy esters 1m and 1n to methyl acrylate 2a under standard reaction conditions provided the corresponding spirolactones 3m and 3n in 97% and 58% yields, respectively (eq. 1).
 |
(eq. 1) |
Table 3.
Redox level-independent formation of lactone 3fa.
 |
Yields are of material isolated by silica gel chromatography. C10H15CO2H refers to 1-adamantanecarboxylic acid. See Supporting Information for further details.
Having explored the scope of the diol and hydroxyester partners 1a–1n, substituted α,β-unsaturated esters 2a–2h were investigated. Attempted reactions of esters 2b–2h with diols 1a–1l under standard conditions did not provide products of C-C coupling. In contrast, the reactions of N-benzyl-3-hydroxyoxindole 1o with esters 2a–2h proceed in good to excellent yield to furnish spirooxindole products 4a–4h (Table 4). As illustrated, β-substituted acrylic esters 2b, 2c, 2f, 2g and 2h provide the corresponding spirolactones 4b, 4c, 4f, 4g and 4h, respectively, in good to excellent isolated yields as single diastereomers. Relative stereochemistry was assigned by single crystal X-ray diffraction analysis of 4b. The relative stereochemistry of cycloadducts 4c, 4f, 4g and 4h is assigned in analogy to 4b. As will be discussed in greater detail, for cycloadditions of acrylic esters 2a–2h with N-benzyl-3-hydroxyoxindole 1o, catalytic amounts of potassium tert-butoxide are required to enforce complete levels of diastereoselectivity. Finally, it is notable that even β,β-substituted acrylic ester 2e participates in spirolactone formation, albeit in moderate yield.
Table 4.
Ruthenium(0) catalyzed hydrohydroxyalkylation of acrylic esters 2a–2h with N-benzyl-3-hydroxyoxindole 1o to form spirolactones 4a–4h.a.
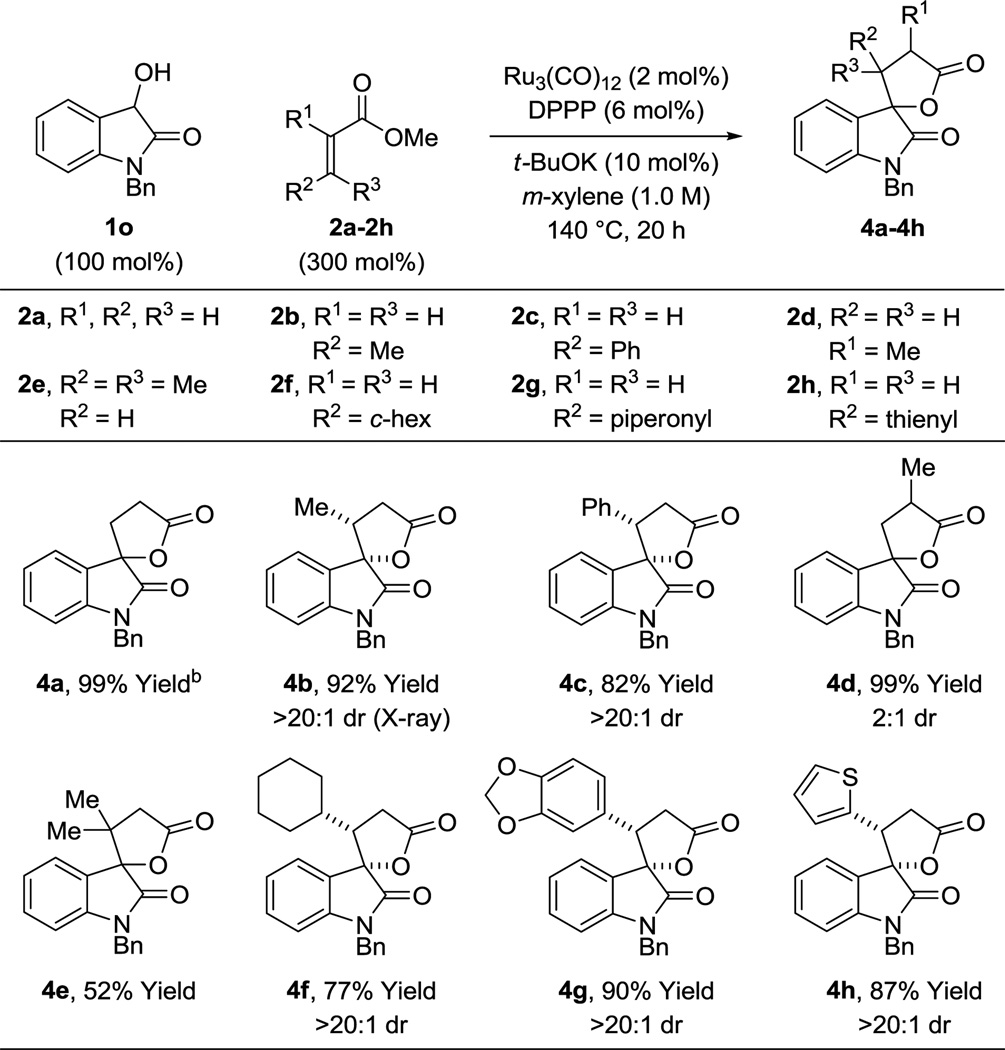 |
Cited yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
The fact that diols 1a–1l did not react with substituted α,β-unsaturated esters 2b–2h may be due to reversible oxaruthenacycle formation. If so, one can envision decorating the enoate reactant such that the transient metallacyclic intermediate is captured and driven to product. As the oxaruthenacycle intermediate may be viewed as a ruthenium enolate (Figure 1, bottom), it was reasoned that the hydroxyl-substituted methacrylate 2i might engage in E1cB elimination to furnish α-methylene-γ-butyrolactones. In the event, upon exposure of diols 1b, 1f, 1j and 1l to acrylic ester 2i under standard conditions the α-exo-methylene γ-butyrolactones 5b, 5f, 5j and 5l, respectively, were formed in moderate yield.14 The modest yields in the formation of 5b, 5f, 5j and 5l are, in part, attributed to reduction of the exocyclic double bond (Table 5).
Table 5.
Hydrohydroxyalkylation of hydroxyl-substituted methacrylate 2i with diols 1b, 1f, 1j and 1l to form α-exomethylene-γ-butyrolactones 5b, 5f, 5j and 5l.a
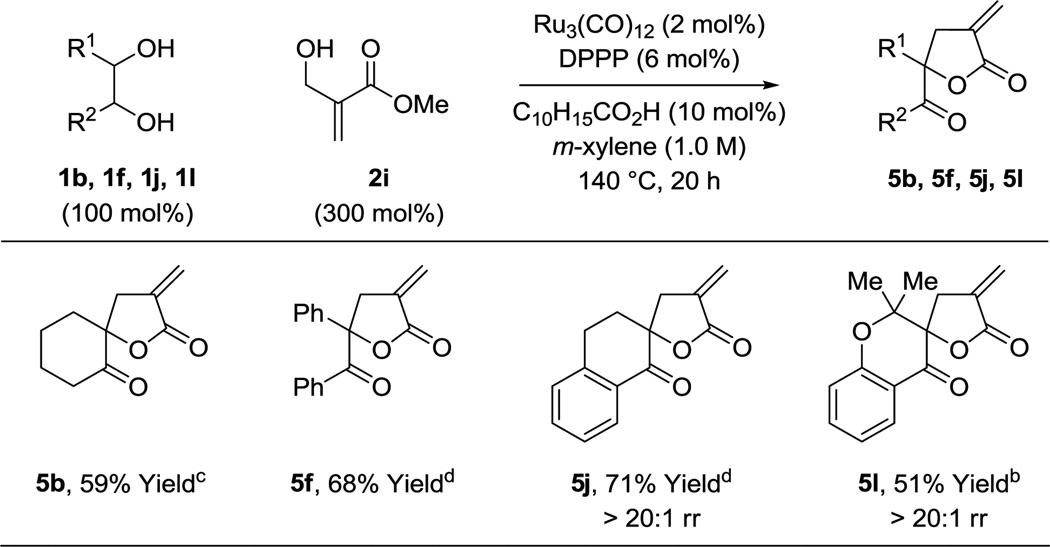 |
Yields are of material isolated by silica gel chromatography. C10H15CO2H refers to 1-adamantanecarboxylic acid. See Supporting Information for further details.
The cis-1,2-diol was employed.
The trans-1,2-diol was employed.
A mixture of cis- and trans-1,2-diols was employed.
Mechanism and Discussion
A plausible general catalytic mechanism for the ruthenium catalyzed C-C coupling of cyclohexanediol 1b and methyl acrylate 2a to form spirolactone 3b is as follows (Scheme 1). Based on literature precedent, intervention of a discrete, mononuclear ruthenium(0) complex is anticipated.16 Consistent with this expectation, upon heating a solution of Ru3(CO)12, DPPP, and 1-adamantanecarboxylic acid, the mononuclear ruthenium (II) species, Ru(CO)(dppp)(C10H15CO2)2 is formed, as established by single crystal X-ray diffraction analysis (Figure 2). It should be noted that Ru(CO)(dppp)(C10H15CO2)2 is a competent precatalyst for catalytic C-C coupling (eq. 2). Oxidative coupling of tetradehydro-1b and methyl acrylate 2a forms oxaruthenacycle I,5,6 which is anticipated to reside as
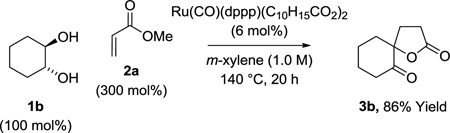 |
(eq. 2) |
the O-bound haptomer.17 The requisite dione tetradehydro-1b likely arises through Ru3(CO)12 catalyzed oxidation of cyclohexanediol 1b employing methyl acrylate 2a as the hydrogen acceptor.18–20 Protonation of oxaruthenacycle I15 by cyclohexanediol 1b or didehydro-1b may be slow compared to protonation of oxaruthenacycle I by 1-adamantanecarboxylic acid to form ruthenium carboxylate II, which lactonizes to form the spirocycle 3b. The resulting ruthenium(II) complex III may engage in substitution with cyclohexanediol 1b or α-hydroxy ketone dihydro-1b. Upon β-hydride elimination, dihydro-1b or tetradehydro-1b would be generated, respectively, along with a ruthenium hydride, which upon O-H reductive elimination would regenerate ruthenium(0).
Scheme 1.

A plausible general mechanism for ruthenium(0) catalyzed spirolactone formation as illustrated in the coupling of diol 1b and methyl acrylate 2a.
Figure 2.

Single crystal X-ray diffraction data of Ru(CO)(dppp)(C10H15CO2)2.a
aDisplacement ellipsoids are scaled to the 50% probability level. Hydrogen atoms have been omitted for clarity. C10H15CO2H refers to 1-adamantanecarboxylic acid.
Whereas couplings of methyl acrylate 2a with diols 1a–1l require an acidic cocatalyst (Table 2), cycloadditions of acrylic esters 2a–2h with N-benzyl-3-hydroxyoxindole 1o require catalytic amounts of potassium tert-butoxide to enforce complete levels of diastereoselectivity. Based on the postulated mechanism (Scheme 1), one possible interpretation is as follows. If oxidative coupling is reversible via retro-Michael addition, complete kinetic stereoselectivity will be eroded if transfer hydrogenolysis of the metallacycle is not fast. Deprotonation of N-benzyl-3-hydroxyoxindole 1o may accelerate transfer hydrogenolysis with respect to retro-Michael addition through alkoxide exchange as indicated (Scheme 2).
Scheme 2.

Diastereoselection in the formation of spirolactones 4b, 4c, 4f, 4g and 4h and potential effect of tert-butoxide.
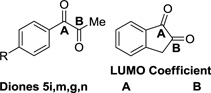
The inversion in regioselectivity observed in the reaction of diol 1g versus diols 1i and 1j merits discussion. As observed across numerous carbonyl additions, 1,2-indanedione reacts at the carbonyl moiety distal to the aromatic ring,21 whereas 1-phenyl-2,3-propanedione reacts predominantly at the carbonyl moiety proximal to the aromatic ring.22 Such trends in regioselectivity are evident in metal catalyzed transformations, for example, hydrogenations of 1,2-indanedione and 1-phenyl-1,2-propanedione.23 Naturally, regioselectivities observed in the aforementioned carbonyl additions and the present ruthenium catalyzed C-C couplings are governed by the interaction of frontier molecular orbitals. Thus, notwith-standing steric effects, C-C coupling will occur predominantly at the dione carbonyl bearing the largest LUMO coefficient. Indeed, as posited by Hoffmann, the conversion of polarized bis(olefin) complexes to metallacyclopentanes should occur such that C-C bond formation occurs at the atom bearing the largest LUMO coefficient.24
To challenge this hypothesis, a series of para-substituted 1-phenyl-1,2-propanediols 1g, 1m and 1n were prepared and subjected to standard conditions for spirolactone formation (eq. 3). For the para-methoxy-substituted diol 3m, the electrophilicity of the resulting dione at the carbonyl moiety proximal to the arene is attenuated and the proportion of regioisomer derived from C-C coupling to this position decreases. Conversely, for the 1,2-dione derived from the para-carbomethoxy-substituted diol 3n, the electrophilicity of the carbonyl moiety proximal to the arene is now augmented and the proportion of regioisomer derived from C-C coupling to this position increases. To more quantitatively correlate regioselectivity with the magnitude of the respective dione LUMO coefficients, density functional theory (DFT) calculations were used to evaluate the dione LUMO coefficients. Although these data correspond to the diones in the ground state, and not the ruthenium bound diones that would be evident in the transition state, the observed trends are in alignment with the experimental results. That is, while the LUMO coefficients are always larger at the carbonyl moiety proximal to the arene, the proportion of regioisomers derived from coupling to the carbonyl moiety distal to the arene increases as the difference between the LUMO coefficients become smaller. Notably, indane diol 1i, engages in completely regioselective coupling, and the corresponding dione 5i is predicted to have the smallest difference between the LUMO coefficients (Table 6).
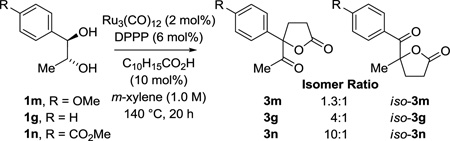 |
(eq. 3) |
Table 6.
Magnitude of dione LUMO coefficients determined by DFT calculations correlate to regioselectivity.a
 |
Δ (A – B) | Experimental Isomeric Ratio (A:B) |
||
|---|---|---|---|---|
| LUMO Coefficient | ||||
| Diones 5i,m,g,n | A | B | ||
| 5i, indane dione | −0.12189 | −0.10896 | 0.013 | 1:>20 |
| 5m, R = OMe | −0.13750 | −0.11802 | 0.019 | 1.3:1 |
| 5g, R = H | −0.13943 | −0.11213 | 0.027 | 4:1 |
| 5n, R = CO2Me | −0.13353 | −0.09587 | 0.038 | 10:1 |
Density functional theory (DFT) calculations were carried out with QChem 4.0 using the B3LYP hybrid functional and 6-311G(d,p) basis set.
To further challenge the veracity of the proposed oxidative coupling mechanism, several control experiments were performed. To evaluate the possibility of an mechanistic pathway involving conventional Michael addition, benzoin didehydro-1f was converted to the methyl ether O-Me-didehydro-1f and subjected to standard conditions for ruthenium(0) catalyzed lactone formation, however, no reaction was observed and the starting materials were recovered unchanged (eq. 4). Additionally, hydroxyketone didehydro-1j was subjected to methyl acrylate 2a in the presence of various Lewis acids (RuCl3, B(OMe)3, InCl3, ZnI2, MgCl2). Here, only small quantities of the spirolactone were obtained along with recovered starting materials (eq. 5).
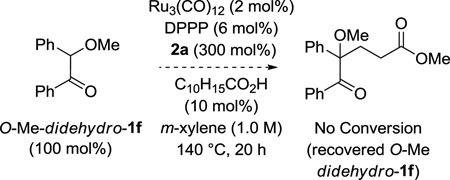 |
(eq. 4) |
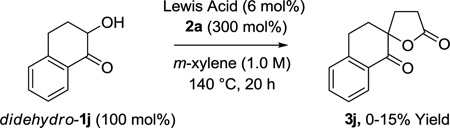 |
(eq. 5) |
Conclusions
In summary, we report a convergent synthesis of γ-butyrolactones, including spiro- and α-methylene-γ-butyrolactones, through the ruthenium(0) catalyzed C-C coupling of vicinal diols and acrylic esters. As demonstrated in the reactions of methyl acrylate 2a with hydrobenzoin 1f, benzoin didehydro-1f, and benzil tetradehydro-1f, such transformations can be conducted in a redox level-independent manner. As shown in the conversion of α-hydroxy esters 1m and 1n to lactones 3m and 3n, respectively, the reaction is applicable to other vicinally dioxygenated systems. Additionally, diverse α,β-unsaturated esters 2a–2h participate in spirolactone formation to form cycloadducts 4a–4h. A catalytically competent ruthenium(II) complex Ru(CO)(dppp)(C10H15CO2)2 was characterized by single crystal X-ray diffraction, and the influence of electronic effects on regioselectivity in reactions of nonsymmetric diols were probed experimentally and computationally. Future studies will focus on the development of related atom efficient C-C couplings that result in formal alcohol C-H functionalization.
Experimental Section
General Experimental Procedure for Hydrohydroxyalkylation of Methyl Acrylate with Diol 1b
To a resealable pressure tube (13 × 100 mm) equipped with a magnetic stir bar was added trans-1,2-cyclohexanediol 1b (35 mg, 0.30 mmol), Ru3(CO)12 (3.8 mg, 0.006 mmol, 2 mol%), 1,3-bis(diphenylphosphino)propane (7.4 mg, 0.018 mmol, 6 mol%), and 1-adamantanecarboxylic acid (5.4 mg, 0.03 mmol, 10 mol%). The tube was sealed with a rubber septum and purged with argon. Methyl acrylate 2a (81 µL, 0.90 mmol, 300 mol%) and m-xylenes (0.22 mL) were added. The rubber septum was replaced with a screw cap. The mixture was heated at 140 °C (oil bath temperature) for 20 h, at which point the reaction mixture was allowed to cool to ambient temperature. The reaction mixture was concentrated and the residue was subjected to flash column chromatography (SiO2; hexanes:ethyl acetate = 1:1) to furnish the title compound (48.4 mg, 0.29 mmol, 96%) as a clear yellow oil.
Supplementary Material
ACKNOWLEDGMENT
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) are acknowledged for partial support of this research. Professor Eric Anslyn and Brette Chapin are acknowledged for kindly conducting density functional theory (DFT) calculations.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures and spectroscopic data for all new compounds (1H NMR, 13C NMR, IR, HRMS), including images of NMR spectra. Single crystal X-ray diffraction data for spirolactone 4b and the ruthenium complex Ru(CO)(dppp)(C10H15CO2)2. This material is available free of charge via the internet at http://pubs.acs.org
REFERENCES
- 1.For selected reviews on the direct redox-triggered C-C coupling of alcohols, see: Bower JF, Krische MJ. Top. Organomet. Chem. 2011;43:107. doi: 10.1007/978-3-642-15334-1_5. Hassan A, Krische MJ. Org. Proc. Res. Devel. 2011;15:1236. doi: 10.1021/op200195m. Moran J, Krische MJ. Pure Appl. Chem. 2012;84:1729. doi: 10.1351/PAC-CON-11-10-18.
- 2.For selected examples of ruthenium(II) catalyzed C-C couplings of primary alcohols, see: Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x. Patman RL, Williams VM, Bower JF, Krische MJ. Angew. Chem. Int. Ed. 2008;47:5220. doi: 10.1002/anie.200801359. Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:14120. doi: 10.1021/ja805356j. Zbieg JR, McInturff EL, Krische MJ. Org. Lett. 2010;12:2514. doi: 10.1021/ol1007235. Zbieg JR, McInturff EL, Leung JC, Krische MJ. J. Chem. Soc. 2011;133:1141. doi: 10.1021/ja1104156. Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ. Science. 2012;336:324. doi: 10.1126/science.1219274. McInturff EL, Yamaguchi E, Krische MJ. J. Am. Chem. Soc. 2012;134:20628. doi: 10.1021/ja311208a.
- 3.For selected examples of iridium catalyzed C-C couplings of primary alcohols, see: Bower JF, Skucas E, Patman RL, Krische MJ. J. Am. Chem. Soc. 2007;129:15134. doi: 10.1021/ja077389b. Bower JF, Patman RL, Krische MJ. Org. Lett. 2008;10:1033. doi: 10.1021/ol800159w. Han SB, Kim IS, Han H, Krische MJ. J. Am. Chem. Soc. 2009;131:6916. doi: 10.1021/ja902437k. Zbieg JR, Fukuzumi T, Krische MJ. Adv. Synth. Catal. 2010;352:2416. doi: 10.1002/adsc.201000599. Moran J, Preetz A, Mesch RA, Krische MJ. Nature Chem. 2011;3:287. doi: 10.1038/nchem.1001. Geary LM, Woo SK, Leung JC, Krische MJ. Angew. Chem. Int. Ed. 2012;51:2972. doi: 10.1002/anie.201200239.
- 4.For Ru3(CO)12 catalyzed secondary alcohol amination via alcohol mediated hydrogen transfer, see: Bahn S, Tillack A, Imm S, Mevius K, Michalik D, Hollmann D, Neubert L, Beller M. ChemSusChem. 2009;2:551. doi: 10.1002/cssc.200900034. Pingen D, Müller C, Vogt D. Angew. Chem. Int. Ed. 2010;49:8130. doi: 10.1002/anie.201002583. Zhang M, Imm S, Bahn S, Neumann H, Beller M. Angew. Chem. Int. Ed. 2011;50:11197. doi: 10.1002/anie.201104309.
- 5.(a) Chatani N, Tobisu M, Asaumi T, Fukumoto Y, Murai SJ. Am. Chem. Soc. 1999;121:7160. [Google Scholar]; (b) Tobisu M, Chatani N, Asaumi T, Amako K, Ie Y, Fukumoto Y, Murai S. J. Am. Chem. Soc. 2000;122:12663. [Google Scholar]
- 6.For ruthenium(0) catalyzed C-C couplings of secondary alcohols, see: Leung JC, Geary LM, Chen T-Y, Zbieg JR, Krische MJ. J. Am. Chem. Soc. 2012;134:15700. doi: 10.1021/ja3075049. Chen T-Y, Krische MJ. Org. Lett. 2013;15:2994. doi: 10.1021/ol401184k. Geary LM, Glasspoole BW, Kim MM, Krische MJ. J. Am. Chem. Soc. 2013;135:3796. doi: 10.1021/ja400691t.
- 7.For reviews of γ-butyrolactone and α-methylene-γ-butyrolactones, respectively, see: Bartoli A, Rodier F, Commeiras L, Parrain J-L, Chouraqui G. Nat. Prod. Rep. 2011;28:763. doi: 10.1039/c0np00053a. Kitson RRA, Millemaggi A, Taylor RJK. Angew. Chem. Int. Ed. 2009;48:9426. doi: 10.1002/anie.200903108.
- 8.For selected examples of spirolactone formation via epoxide ring opening, see: Yokoyama T, Izui N. Bull. Chem. Soc. Japan. 1965;38:1501. Faraj H, Claire M, Rondot A, Aumelas A, Auzou G. J. Chem. Soc., Perkin Trans. 1990;1:3045. Eipert M, Maichle-Mossmer C, Maier ME. Tetrahedron. 2003;59:7949. Fujioka H, Matsuda S, Horai M, Fujii E, Morishita M, Nishiguchi N, Hata K, Kita Y. Chem. Eur. J. 2007;13:5238. doi: 10.1002/chem.200601341.
- 9.For selected examples of spirolactone formation via bromonium ion intermediates, see: Mandal AK, Jawalkar DG. Tetrahedron. Lett. 1986;27:99. Mandal AK, Jawalkar DG. J. Org. Chem. 1989;54:2364.
- 10.For selected examples of spirolactone formation via N-heterocyclic carbene catalyzed Stetter type reactions, see: Burstein C, Glorius F. Angew. Chem. Int. Ed. 2004;43:6205. doi: 10.1002/anie.200461572. Ye W, Cai G, Zhuang Z, Jia X, Zhai H. Org. Lett. 2005;7:3769. doi: 10.1021/ol051422m.
- 11.For selected examples of spirolactone formation via oxidative dearomatization, see: Tamura Y, Yakura T, Haruta J, Kita Y. J. Org. Chem. 1987;52:3927. Cox C, Danishefsky SJ. Org. Lett. 2000;2:3493. doi: 10.1021/ol006531+. Uyanik M, Yasui Takeshi, Ishihara K. Tetrahedron. 2010;66:5841. Dohi T, Takenaga N, Nakae T, Toyoda Y, Yamasaki M, Shiro M, Fujioka H, Maruyama A, Kita Y. J. Am. Chem. Soc. 2013;135:4558. doi: 10.1021/ja401074u.
- 12.For selected examples of spirolactone formation via C-H hydroxylation, see: Uyanik M, Suzuki D, Yasui T, Ishihara K. Angew. Chem. Int. Ed. 2011;50:5331. doi: 10.1002/anie.201101522. Bigi MA, Reed SA, White MC. Nat. Chem. 2011;3:216. doi: 10.1038/nchem.967.
- 13.For a recent review on spirolactone formation via reductive cyclization of α,β-unsaturated esters onto ketones, see: Streuff J. Synthesis. 2013:281.
- 14.For synthesis of α-methylene-γ-butyrolactones via carbonyl 2-(alkoxycarbonyl)allylation, see: Montgomery TP, Hassan A, Park BY, Krische MJ. J. Am. Chem. Soc. 2012;134:11100. doi: 10.1021/ja303839h. and references cited therein.
- 15. Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:280. doi: 10.1021/ja0670815. and references cited therein.
- 16.Ru3(CO)12 reacts with dppe in benzene solvent to provide Ru(CO)3(dppe): Sanchez-Delgado RA, Bradley JS, Wilkinson GJ. Chem. Soc. Dalton Trans. 1976:399.
- 17.For an O-bound nickel enolate derived via intramolecular enalalkyne oxidative coupling, see: Amarasinghe KKD, Chowdhury SK, Heeg MJ, Montgomery J. Organometallics. 2001;20:370.
- 18.For Ru3(CO)12 catalyzed oxidation of alcohols employing olefins and alkynes as hydrogen acceptors, see: Blum Y, Reshef D, Shvo Y. Tetrahedron Lett. 1981;22:1541. Shvo Y, Blum Y, Reshef D, Menzin M. J. Organomet. Chem. 1982;226:C21. Meijer RH, Ligthart GBWL, Meuldijk J, Vekemans JAJM, Hulshof LA, Mills AM, Kooijman H, Spek AL. Tetrahedron. 2004;60:1065.
- 19.For mechanistically related Ru3(CO)12 catalyzed transfer hydrogenation of ketones mediated by isopropanol, see: Johnson TC, Totty WG, Wills M. Org. Lett. 2012;14:5230. doi: 10.1021/ol302354z.
- 20.For mechanistically related Ru3(CO)12 catalyzed secondary alcohol amination via alcohol mediated hydrogen transfer, see: Baehn S, Tillack A, Imm S, Mevius K, Michalik D, Hollmann D, Neubert L, Beller M. ChemSusChem. 2009;2:551. doi: 10.1002/cssc.200900034. Pingen D, Müller C, Vogt D. Angew. Chem. Int. Ed. 2010;49:8130. doi: 10.1002/anie.201002583. Zhang M, Imm S, Bahn S, Neumann H, Beller M. Angew. Chem. Int. Ed. 2011;50:11197. doi: 10.1002/anie.201104309.
- 21.1,2-Indanedione reacts with nucleophiles exclusively at the carbonyl moiety distal to the aromatic ring. For selected examples, see: Fatiadi AJ. Synthesis. 1978:165. Petrovskaia O, Taylor BM, Hauze DB, Carroll PJ, Joullie MM. J. Org. Chem. 2001;66:7666. doi: 10.1021/jo0105179. and references therein. Vanden Eynden MJ, Kunchithapatham K, Stambuli JP. J. Org. Chem. 2010;75:8542. doi: 10.1021/jo1019283.
- 22.1-Phenyl-2,3-propanedione reacts with nucleophiles predominantly at the carbonyl moiety proximal to the aromatic ring suggesting the LUMO coefficient is largest at this position. For selected examples, see: Inaba S-i, Rieke RD. Synthesis. 1984;844 Nishigaichi Y, Orimi T, Takuwa A. J. Organomet. Chem. 2009;694:3837. Dhondi PK, Carberry P, Choi LB, Chisholm JD. J. Org. Chem. 2007;72:9590. doi: 10.1021/jo701643h. Ramachandran PV, Rudd MT, Burghardt TE, Reddy MVR. J. Org. Chem. 2003;68:9310. doi: 10.1021/jo034954u. Gewald R, Kira M, Sakurai H. Synthesis. 1996;111 Kang S-K, Baik T-G, Jiao Z-H. Synth. Comm. 2002;32:75.
- 23.(a) Busygin I, Rosenholm M, Toukoniitty E, Murzin DY, Leino R. Catal. Lett. 2007;117:91. [Google Scholar]; (b) Langvik O, Maki-Arvela P, Aho A, Saloranta T, Murzin DY, Leino R. Catal. Lett. 2013;143:142. [Google Scholar]; (c) Nieminen V, Taskinen A, Hotokka M, Murzin DY. J. Catal. 2007;245:228. [Google Scholar]
- 24.Stockis A, Hoffmann R. J. Am. Chem. Soc. 1980;102:2952. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.