

Abstract
Our previous studies demonstrated that anti-CD40 mAb (anti-CD40) can synergize with CpG oligodeoxynucleotides (CpG) to mediate antitumor effects by activating myeloid cells, such as macrophages in tumor-bearing mice. Separate teams have shown that chemotherapy with gemcitabine (GEM) or 5-fluorouracil (5-FU) can reduce tumor-induced myeloid-derived suppressor cells (MDSC) in mice. In this study we asked if the same chemotherapy regimens with GEM or 5-FU will enhance the antitumor effect of anti-CD40 and CpG. Using the model of B16 melanoma growing intraperitoneally in syngeneic C57BL/6 mice, we show that these GEM or 5-FU treatment regimens reduced MDSC in the peritoneal cavity of tumor-bearing mice. Treatment of mice with GEM or 5-FU did not significantly affect the antitumor function of macrophages as assessed in vitro. In vivo, treatment with these GEM or 5-FU regimens followed by anti-CD40/CpG resulted in antitumor effects similar to those of anti-CD40/CpG in the absence of GEM or 5-FU. Likewise, reduction of MDSC by in vivo anti-Gr-1 mAb treatment did not significantly affect anti-CD40/CpG antitumor responses. Together, the results show that the GEM or 5-FU chemotherapy regimens did not substantially affect the antitumor effects induced by anti-CD40/CpG immunotherapy.
Keywords: anti-CD40, CpG, gemcitabine, 5-fluorouracil, immunotherapy
Introduction
Cytotoxic chemotherapy is effective treatment for some types of cancers; however, tumor cells often become drug-resistant, enabling many cancers to recur or progress. Combining chemotherapy with immunotherapy has been increasingly used in clinical practice to improve the clinical outcome [1]. Many experimental studies have shown that certain chemotherapeutic drugs such as cyclophosphamide (CY) given at low doses facilitate activation of antitumor T cells by depleting T suppressor (regulatory) cells [2,3]. Although many chemotherapeutics are immunosuppressive, certain cells of the innate immune system, particularly monocytes and macrophages (Mφ), are more resistant to chemotherapy than T cells in cancer patients [4]. Therefore, we hypothesize that chemotherapy will not prevent the efficacy of an antitumor immunotherapy designed to activate monocytes or Mφ and may even enhance it.
Agonistic anti-CD40 mAb (anti-CD40) interacts with the CD40 molecule expressed on the surface of dendritic cells and Mφ which leads to immune activation. Anti-CD40 has been shown to induce T cell-dependent [5, 6] and independent [7] antitumor responses in mice, which led to its clinical testing as a cancer treatment [8–11]. We have previously shown in mouse models that anti-CD40 induced antitumor effects via Mφ activation [12]. Combining anti-CD40 with CpG-oligodeoxynucleotides (CpG) in vivo resulted in synergistic activation of Mφ and induction of potent antitumor effects even in the absence of T- and NK-cells [13]. Combined treatment with CY and anti-CD40/CpG resulted in a greater reduction in tumor growth in B16 melanoma-bearing mice than was observed with either CY alone or anti-CD40/CpG [14]. Even multidrug chemotherapy consisting of vincristine, CY and doxorubicin, while suppressing the functions of T cells and NK cells, primed Mφ to secrete NO, IFN-γ and IL-12, and synergized with anti-CD40/CpG in inducing antitumor effects [15].
The antitumor effects of anti-CD40 with and without CpG involved Mφ and other myeloid cells [16, 17]. In our experiments [12–15], CY alone and in combination with vincristine and doxorubicin induced expansion of myeloid cells and synergized with anti-CD40/CpG [14, 15]. In contrast, other chemotherapeutic drugs, such as gemcitabine (GEM) and 5-fluorouracil (5-FU), with different mechanisms of action, were reported to substantially deplete tumor-induced myeloid cells, namely myeloid-derived suppressor cells (MDSC), in certain tumor models [18, 19]. As MDSC are present in large numbers in tumor-bearing mice (TBM) and inhibit aspects of immune function [20], in this study we asked whether the reduction of myeloid cells with the same GEM or 5-FU therapy regimens would enhance the antitumor effects of anti-CD40/CpG.
Material and Methods
Mice and cell lines
Female C57BL/6 mice 6 to 10 weeks old obtained from Taconic (Germantown, NY) were housed, cared for, and used in accordance with the Guide for Care and Use of Laboratory Animals (NIH publication 86-23, National Institutes of Health, Bethesda, MD, 1985). Mouse B16-F10 melanoma cell line was grown in RPMI 1640 complete medium supplemented with 10% FCS (Sigma Chemical, St Louis, MO), 2 mM L-glutamine, and 100U/ml of penicillin/streptomycin (all from Life Technologies, Inc. Grand Island, NY) at 37°C in a humidified 5% CO2 atmosphere.
Antibodies and reagents
Anti-CD40 was prepared from the FGK 45.5 hybridoma cell line as described previously [12]. Endotoxin-free CpG1826 was purchased from Coley Pharmaceuticals Group (Wellesley, MA). 5-FU was dissolved in DMSO (both from Sigma Chemical, St Louis, MO) at 50 mg/ml. GEM-HCl (Eli Lilly and Company, Indianapolis, IN) was obtained from the UWHospital Pharmacy and prepared in phosphate-buffered saline (PBS). Bacterial LPS from Salmonella enteritidis was purchased from Sigma Chemical, St Louis, MO. Mouse recombinant IFN-γ was purchased from eBioscience, San Diego, CA.
In vivo tumor models and therapy
C57BL/6 mice were injected subcutaneously (s.c.) or intraperitoneally (i.p.) with 1x105 B16 melanoma cells in 0.1 or 0.5 ml PBS, respectively (day 0). TBM were injected i.p. with 0.5 mg anti-CD40 starting on day 7–9 after tumor implantation; 50μg CpG were injected i.p. three days after anti-CD40 injection (all i.p. injections were given in 0.5 PBS). Anti-Gr-1 (clone RB6-8C5) was injected intratumorally (i.t.) (0.2 mg in 0.1 PBS) on the same days as anti-CD40 (days 7 and 14) and CpG (days 10 and 17). 5-FU DMSO solution was diluted in PBS to achieve 50mg/kg and administered i.p. into mice. GEM (120 mg/kg) was injected i.p. in 0.5 PBS. Days of injection (following tumor implantation) are specified for each experiment. Antitumor effects were determined by measuring the perpendicular diameter of s.c tumors twice weekly, or extended survival of the mice in i.p. models. Tumor volumes were calculated according to the formula: (tumor length x tumor width2)/2.
Activation of Mφ
Peritoneal cells (PEC) were obtained via peritoneal cavity lavage with 5ml of ice-cold RPMI 1640 complete medium, supplemented with 1IU/ml of heparin (American Pharmaceutical Partners, Inc., Schaumburg, IL) when collected from TBM. Erythrocytes in TBM PEC were lysed by hypotonic shock. Collected PEC were placed into 96-well flat-bottom cell culture plates (Corning Inc., Corning, NY) at a concentration of 2x105 cells/well (or 1x105 cells/well for sorted cell populations). The peritoneal Mφ population was enriched by allowing PEC to adhere to plastic for 1.5–2 hrs followed by removal of non-adherent cells. For in vitro activation, total PEC or adherent Mφ were incubated in medium alone or in the presence of 10 U/ml of IFN-γ and 1 ng/ml of LPS.
Mφ mediated tumoristasis in vitro
Tumoristatic activity of Mφ was determined by the inhibition of DNA synthesis in tumor cells. Briefly, adherent PEC were stimulated in vitro as described above and simultaneously co-cultured with B16 tumor cells (1x104/well) for 48 h. To estimate DNA synthesis, cells were pulsed with 3H-TdR (1 μCi/well) during the last 6 h of incubation. 3H-TdR-incorporation was determined by β-scintillation of total cells harvested from the cell cultures onto glass fiber filters (Packard, Meriden, CT), using the Packard Matrix 9600 Direct β-counter (Packard, Meriden, CT). In this assay proliferation of B16 cells is >100 fold higher than that of PEC [12]. Results are presented as counts per 5 min for triplicate wells ± SD.
Nitric oxide production
Peritoneal Mφ were prepared and co-cultured with B16 cells for 48 h, as described above in the Mφ cytostatic assay. Supernatants were collected and nitrite accumulation was determined using Griess reagent (Sigma, St. Louis, MO). Equal volumes of supernatants and Griess reagent were mixed for 10 min, and the A570 was measured by a microplate reader and compared to a standard nitrite curve ranging from 0–125 μM.
Splenic T-cell proliferation assay
Splenocytes were prepared from the spleens pooled from two C57BL/6 mice by processing the spleens to a single-cell suspension, followed by lysis of erythrocytes by hypotonic shock. Two hundred thousand spleen cells were stimulated with 0.5μg/ml of monoclonal anti-CD3 and 5μg/ml of anti-CD28 (both from eBioscience, San Diego, CA) and cultured with sorted PEC in complete media in flat-bottomed 96-well plates for 72 hr. Cells were pulsed with 3H-TdR (1 μCi/well) during the last 6 h of incubation, and retained radioactivity was counted by β-scintillation of total cells harvested from the cell culture clusters onto glass fiber filters, using the Packard Matrix 9600 Direct β-counter. Results are presented as counts per 5 min for triplicate wells ± SD.
Flow cytometric analysis and sorting
PEC from treated and control C57BL/6 mice were harvested and stained with FITC-conjugated anti-CD45, PE-conjugated anti-Gr-1, APC-conjugated anti-CD11b, or FITC-conjugated anti-B220 (all from eBioscience, San Diego, CA) for 40 min at 4 °C. Isotype-matched control rat IgG FITC, IgG APC and IgG PE, purchased from eBioscience or BD Pharmingen, were used as background controls. Cells were washed in ice-cold PBS supplemented with 0.5% FCS (flow buffer), subjected to flow cytometry using either a FACSCalibur flow cytometer or a MACSQuant Analyzer, and analyzed with FlowJo software (Ashland, OR). To calculate the absolute number of a PEC subset, the absolute number of total PEC (obtained via counting the viable cells on a haemocytometer) was multiplied by the percentage of that subset (%) obtained via flow cytometry analysis. Data were collected for 10,000 live events per sample.
Statistical Analysis
A two-tailed Student’s t-test was used to determine significant differences between groups within one experiment. Differences in the mean tumor growth rate of treatment groups were determined by fitting the tumor growth curve of each mouse to an exponential curve using the equation for exponential growth (Vt = Voert), where Vo is tumor volume, t is time and the rate constant r is the parameter taken to describe the overall tumor growth rate for each mouse. The parameter Vo was calculated to be the same for all mice within an experiment. The group mean r was compared between groups using a 2-tailed Student t-test. Survival data were analyzed with the Log-rank test. For all tests, P < 0.05 was considered statistically significant. Statistical analyses of nonlinear fit curves and Log-rank tests were performed using the GraphPad Prism 5.01 software. For all figures, * = P<0.05, ** = P < 0.01, *** = P<0.001, NS: =Non-significant.
Results
Effect of GEM and 5-FU on B16 melanoma-induced MDSC
It has been reported that GEM treatment (120 mg/kg) reduced the number of tumor-induced MDSC in several tumor models [18, 19, 21]. To determine whether GEM is effective in reducing the number of MDSC in the i.p. B16 melanoma model, C57BL/6 mice were injected i.p. with B16 cells, and treated with 120 mg/kg of GEM 11 days later. Peritoneal cells (PEC) were collected on day 14 and evaluated by flow cytometry for CD11b+ Gr-1+ cells to assess the number of MDSC. We recognize that different criteria and different markers have been used by different labs, and in different models, to characterize MDSC. As most analyses of MDSC in TBM show the MDSC are CD11b+ Gr-1+ cells [18–21], we are using this phenotypic definition of MDSC in this report; namely, we will refer to CD11b+ Gr-1+ cells as MDSC while recognizing that not all cells with this phenotype will necessarily function as MDSC. The results in Figs. 1A and 1B show that the percentage and number of CD11b+ Gr-1+ PEC were increased in TBM compared to naïve mice (p = 0.0005 and 0.0325, respectively). When the GEM treatment regimen was given to TBM mice, the percentage of CD11b+ Gr-1+ cells in the peritoneal cavity was significantly reduced compared to the TBM mice not receiving GEM (p=0.0037, Fig. 1A), whereas the reduction in absolute number of MDSC was noticeable, but did not reach statistical significance (p=0.089, Fig. 1B).
Figure 1.

Effect of GEM or 5-FU on i.p. B16 melanoma-induced MDSC. A, B: C57BL/6 mice were injected i.p. with 105 B16 cells on day 0. TBM (n= 6 per group) received either 120 mg/kg GEM (TBM GEM) or PBS (TBM) i.p. on day 11. On day 14, PEC were collected from TBM, TBM GEM and naïve mice (n=3–4 mice per group), stained with FITC-conjugated anti-CD45, PE-conjugated anti-Gr-1 and APC-conjugated anti-CD11b, and subjected to flow cytometry. Graphs A and B represent the results of one out of three similar experiments. C, D: B16 TBM (n=3 or 4 per group) received either 50 mg/kg of 5-FU (TBM 5-FU) or DMSO control (TBM) i.p. on day 7 and 14. PECs were collected on day 16 from TBM, TBM 5-FU and naïve mice (n=4), and subjected to flow cytometry using a similar protocol as A, B. Graphs C and D represent the results of one out of two similar experiments. The percentage (A, C) and absolute number (B, D) of CD45+ CD11b+Gr-1+ cells out of total PECs were calculated. The data are shown as Mean ± SD. * P<0.05, ** P < 0.01, *** P<0.001, NS: Non-significant.
In parallel to GEM we tested another chemotherapeutic drug, 5-FU, reported to be more effective than GEM in depleting MDSC [19]. The results in Figure 1C and 1D show that treatment of B16 TBM with 5-FU resulted in statistically significant reduction of the relative and absolute numbers of CD11b+ Gr-1+ PEC. Neither GEM nor 5-FU treatment affected number of CD11b+ Gr-1+ spleen cells in mice bearing i.p. B16 tumors (data not shown). In a separate experiment, MDSC were similarly reduced when 5-FU was given to TBM once, 2 days before collecting PEC, or twice, 9 days and 2 days before collecting PEC (data not shown).
Effect of GEM and 5-FU on Mφ function
We determined next whether the GEM and 5-FU treatments in vivo affected the functions of Mφ. Treatment of naïve mice with 5-FU did not significantly affect the ability of Mφ to suppress proliferation of B16 tumor cells (p=0.56, Fig. 2A) or produce NO (p=0.95, Fig. 2B) in vitro. Although it reduced the number of myeloid cells in TBM as shown in Fig. 1C, 5-FU did not significantly affect the ability of adherent PEC to produce NO (p=0.1, Fig. 2C). Similarly, in vivo administration of GEM to TBM did not cause any significant change in NO production by PEC (stimulated with IFNγ + LPS) (p=0.101, Fig. 2D).
Figure 2.

Effect of GEM or 5-FU on Mφ function. A, B: Two groups of naïve C57BL/6 mice (n=2 per group) were injected with either 50 mg/kg of 5-FU (5-FU) or DMSO (Control) i.p., and PEC were collected 5 days later. Total PEC (2x105/well) and B16 tumor cells (104/well) were placed in 96-well plates with medium or stimulated with IFN-γ (10 U/ml) and LPS (1 ng/ml). C: C57BL/6 mice were injected with B16 cells i.p. on day 0, and injected with either 50 mg/kg of 5-FU (TBM 5-FU) or DMSO (TBM, control) i.p. on days 5 and 10. PECs from naïve, TBM and TBM 5-FU (n=3–4 per group) were collected on day 14. Total PEC were stimulated with IFN-γ (10 U/ml) and LPS (1 ng/ml). The results of one out of two similar experiments are shown. D: B16 i.p. TBM were injected with 120 mg/kg of GEM (TBM GEM) or PBS (TBM) i.p. on day 11. Total PEC were collected on day 14 and placed in 96-well plates with medium or IFN-γ (10 U/ml) and LPS (1 ng/ml). All plates were incubated for 48 hours. D shows a combined graph of three similar experiments (8–9 mice per group). Counts of B16 cells were measured based on thymidine incorporation (A), and NO activity was determined by nitrite level in the supernatants (B, C, D). The data are shown as Mean ± SD. # Counts <150. NS: Non-significant.
To more precisely determine the effect of 5-FU on the function of myeloid cells, CD11b+ Gr-1+ PEC from 5-FU or PBS-treated B16 TBM were sorted by flow cytometry, and their ability to suppress tumor cells and secrete NO following activation in vitro, as well as to suppress proliferation of T cells, was compared. In two experiments, sorted myeloid cells from both untreated and 5-FU treated TBM substantially suppressed B16 proliferation (99.4 and 95.0% suppression, respectively) and induced similar moderate levels of NO production. As a control, sorted B220+ (B) cells from these same preparations were added to the tumor cells instead of the sorted myeloid cells, and, as expected, showed no significant inhibition of tumor cell proliferation (P= 0.071, Fig. 3A) and no production of NO (Fig. 3B). Purified CD11b+ Gr-1+ PEC from TBM caused substantial suppression of anti-CD3/CD28-induced proliferation of splenic T cells (Fig. 3C). The comparable MDSC from 5-FU treated TBM also showed substantial suppression of CD3/CD28 induced T cell proliferation (p<0.01), and were slightly more suppressive than those from the TBM not receiving 5-FU (97.4 vs. 94.9 % suppression, respectively, p < 0.05). These data would suggest that in vivo 5-FU treatment reduces the numbers of CD11b+ Gr-1+ cells (Fig. 1C–D), but does not keep the remaining CD11b+ Gr-1+ cells from suppressing the T cell proliferative response (Fig. 3C)
Figure 3.
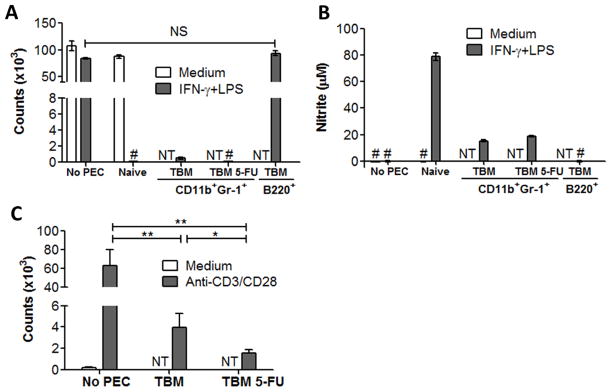
Sorting CD11b+Gr-1+ Cells from 5-FU treated mice. C57BL/6 mice were injected i.p. with 105 B16 cells on day 0. TBM received either 50 mg/kg of 5-FU (n=9 per group, TBM 5-FU) or DMSO control (n=5 per group, TBM) i.p. on days 5 and 11. PEC were collected on day 14 from all TBM and two naïve mice. A, B: PEC were stained with FITC-conjugated anti-B220, PE-conjugated anti-Gr-1 and APC-conjugated anti-CD11b. CD11b+Gr-1+ cells from TBM 5-FU and TBM, as well as B220+ cells from TBM were sorted. In 96-well plates, adherent naïve PEC or sorted PEC (105/well) were incubated with B16 cells (104/well) in medium or IFN-γ (10 U/ml) and LPS (1 ng/ml) for 48 hours. Counts of B16 cells (A) inhibited by PEC were measured based on thymidine incorporation in tumor cells, and NO activity was determined by nitrite level (B) in the supernatants. Results in panel A and B are representative of two independent experiments. C: Spleen cells pooled from two naïve mice were placed 2x105/well with sorted PEC (1x105/well) in medium or anti-CD3 (0.5μg/ml)/anti-CD28 (5μg/ml). PEC were obtained from TBM treated with 5-FU (TBM-5-FU) or without treatment (TBM). Counts of spleen cells were measured by thymidine incorporation assay 48 hours later. Panel C represents the combined results from two similar experiments. The data are shown as Mean ± SD. * P < 0.05, ** P < 0.01. # counts < 150 or nitrite level < 1μM. NS: Non-significant. NT: Not tested.
Antitumor effects of GEM and 5-FU in combination with CD40/CpG
To determine if GEM or 5-FU affects the immune mediated antitumor responses of anti-CD40/CpG in vivo, we first determined the sensitivity of B16 melanoma cells to GEM and 5-FU in vitro. Each of these chemotherapeutic drugs mediated dose-dependent inhibition of B16 cell proliferation in vitro (data not shown).
Then we tested the effects of chemotherapy with GEM and 5-FU in vivo, separately and in combination with anti-CD40/CpG immunotherapy. In the subcutaneous B16 tumor model, GEM alone was not effective, whereas anti-CD40/CpG treatment reduced tumor growth (Fig. 4A). Combined treatment with GEM followed by anti-CD40/CpG resulted in an antitumor effect similar to that of anti-CD40/CpG alone (Fig. 4A). Similarly, combining 5-FU with anti-CD40/CpG did not significantly modify the antitumor effect of anti-CD40/CpG alone against s.c. B16 tumors (Fig. 4B); treatment with 5-FU alone showed a trend toward tumor growth inhibition, but this was not statistically significant (p=0.11, Fig. 4B). In addition, combined treatment with 5-FU and anti-CD40/CpG was not significantly different from the immunotherapy alone in the i.p. B16 melanoma model (Fig. 4C).
Figure 4.

Antitumor effect of GEM, 5-FU or Gr-1+ cell depletion in combination with anti-CD40/CpG in vivo. C57BL/6 mice were injected s.c. (A, B, D) or i.p. (C) with 105 B16 cells on day 0. A: TBM ( n=4 or 5 per group) had no treatment (control) or were treated with GEM i.p. on days 7 and 14, anti-CD40/CpG i.p. on days 8/11 and 15/18, or a combination of GEM and anti-CD40/CpG. B: TBM (n=6 or 7 per group) had no treatment or were treated with 5-FU i.p. on day 6, anti-CD40/CpG i.p. on days 7/10 and 14/17, or a combination of 5-FU and anti-CD40/CpG. C: TBM (n=11 per group) had no treatment or were treated with 5-FU i.p. on days 5 and 15, anti-CD40/CpG i.p. on day 9/12, or a combination of 5-FU and anti-CD40/CpG. The graph represents the combination of two similar experiments. D: C57BL/6 mice were injected s.c. with 105 B16 cells on day 0. TBM (n=6 or 7 per group) had no treatment or were treated with 0.2mg of anti-Gr-1 i.t. on days 7, 10, 14 and 17, anti-CD40/CpG i.p. on days 7/10 and 14/17, or a combination of anti-Gr-1 and anti-CD40/CpG. Panels A, C and D represent single experiments, while B represents two similar experiments. Means ± SE of tumors volumes (A, B, D) are presented. Control mice received DMSO, PBS, or Rat IgG. * P < 0.05, ** P < 0.01 and *** P < 0.001 for control group versus treatment groups. There was no statistically significant difference between the anti-CD40/CpG and combined treatment groups. Arrows indicate treatment schedule.
To further test the potential influence of MDSC depletion on anti-CD40/CpG therapy, mice bearing s.c. B16 melanoma were treated i.p. with anti-Gr-1 mAb, RB6, an approach shown previously to deplete Gr-1+ cells in vivo [22–24]. Flow cytometric analysis confirmed a significant depletion of CD11b+Gr-1+ PEC in RB6-injected TBM compared with Rat IgG-treated TBM (3.14 ± 0.7% vs. 40.13 ± 4.08%, respectively; P<0.005). Reduction of MDSC by anti-Gr-1 mAb treatment did not significantly inhibit B16 tumor growth, and did not significantly affect anti-CD40/CpG antitumor responses (Fig. 4D); this was comparable to the results of our studies in mice receiving 5-FU (Figs. 4B–C) or GEM treatments (Fig. 4A). Similar to i.p injections, intra-tumoral injections of anti-Gr-1 mAb did not influence the antitumor effect of anti-CD40/CpG against s.c. B16 melanoma (data not shown).
Discussion
Some combinatory approaches using chemotherapy and immunotherapy have showed antitumor synergy in experimental studies and have been increasingly implemented in the clinic [25, 26]. These immunotherapy approaches primarily focus on the facilitation of T cell-mediated antitumor responses [25]. We have recently shown that chemotherapy and immunotherapy can also synergize in activating innate immunity to combat cancer in TBM. Thus, chemotherapy with CY [14] or CY in combination with doxorubicin and vincristine [15] can synergize with anti-CD40/CpG in inducing antitumor effects by activated Mφ.
In a separate study we showed that not only Mφ but other myeloid cells expressing CD11b and Gr-1 markers can be activated in TBM to mediate antitumor effects [17]. As these CD11b and Gr-1 markers also characterize MDSC, which substantially increase with tumor progression in tumor-bearing hosts [20], we thought to determine the role of these CD11b+Gr-1+ cells in the antitumor effects induced by anti-CD40/CpG. We hypothesized two possible and opposing outcomes of depleting CD11b+ Gr-1+ cells. First, these immature myeloid cells might secrete NO, one of the effector molecules induced by CD40 ligation [27], and potentially be activated with anti-CD40/CpG to mediate antitumor effects. In this case, reducing the numbers of CD11b+ Gr-1+ cells could inhibit the antitumor effects of anti-CD40/CpG. Alternatively, CD11b+ Gr-1+ MDSC can polarize Mφ to the M2 phenotype, inhibiting M1 effector responses [28], and thus could inhibit Mφ activation with anti-CD40/CpG. If the second condition were dominant, reducing MDSC in TBM would likely promote the M1 Mφ phenotype, resulting in better antitumor effects in response to anti-CD40/CpG. Although MDSC are known to inhibit T cell function [20], their suppression of tumor-specific T cell immunity should not play a significant role in our findings as the antitumor effect of anti-CD40/CpG against B16 melanoma was shown to be T cell-independent [7, 13–15].
To reduce MDSC in vivo we used three separate published approaches, namely treating with GEM, 5-FU or anti-Gr-1 mAb. GEM was found to selectively reduce the number of MDSC in several mouse tumor models [18, 21, 28]. Another chemotherapeutic agent, 5-FU, was compared with GEM and found to be more effective in depleting MDSC [19]. Anti-Gr-1 mAb has been shown to reduce the number of two MDSC populations: Gr-1+ neutrophils and monocytes [24].
In our experiments, treatment with the GEM regimen used in other studies (120 mg/kg) significantly reduced the percentage, and to a lesser degree the number, of MDSC in the i.p. B16 tumor model. As published data on GEM-induced MDSC reduction in TBM have not evaluated the GEM-induced MDSC reduction in the B16 melanoma model, it remains to be determined whether GEM is more effective in reducing MDSC in some murine tumor models than in others. GEM treatment did not significantly enhance or suppress the antitumor effect of anti-CD40/CpG in the s.c. B16 tumor model (Fig. 4A). This result is in keeping with the findings from Beatty et al., who used a model of spontaneous pancreatic cancer in mice to show that GEM did not enhance the antitumor effect of anti-CD40 [11]. Similar to GEM, 5-FU (Fig. 1C–D) reduced MDSC in the i.p. B16 tumor model. Our results indicate that reducing the number of MDSC by GEM or 5-FU neither enhances nor inhibits the ability of Mφ from these mice to be activated in vitro by co-culture with IFNγ and LPS (Fig. 2A–D). Furthermore, the residual CD11b+ Gr-1+ cells from 5-FU treated mice retain their ability to suppress T cell proliferation (Fig. 3C). Finally, reducing the number of MDSC by 5-FU or anti-Gr-1 mAb does not augment or inhibit the in vivo antitumor effects of anti-CD40/CpG (Fig. 4A–D).
Our previous findings suggest that the increased number of myeloid cells following certain chemotherapy regimens may be associated with the increased antitumor effects of the combination of these chemotherapy regimens and anti-CD40/CpG immunotherapy. Thus, since CY and/or doxorubicin increase levels of myeloid cells [29], they could enhance the efficacy of anti-CD40/CpG treatment. In our studies, chemotherapy regimens with CY alone or in combination with doxorubicin and vincristine did indeed enhance the antitumor effects of anti-CD40/CpG immunotherapy against B16 melanoma [14, 15]. It is possible, therefore, that some myeloid cells induced by the tumor, chemotherapy or both are activated by anti-CD40/CpG to induce antitumor activity. This antitumor effect by myeloid cells might potentially balance the inhibitory effects demonstrated by MDSC. In other words, we hypothesized that the depletion of MDSC might blunt some of their immunosuppressive effects, thereby enabling immunotherapy to be more effective. However, we did not observe this hypothesized result. The fact that we did not observe any detectible change in the antitumor effect of anti-CD40/CpG by reducing the number of myeloid cells in the current study might be explained by the existence of two or more concurrent independent myeloid mechanisms. That is to say, it is possible that 5-FU or anti-Gr-1 mAb depletes myeloid cells that have opposite functions: antitumor effectors as well as immunosuppressive MDSC, with the net effect being that both functions are cancelled out. Tumor-induced myeloid cells might be activated to become antitumor effectors [7], but they can also suppress M1 Mφ [28]. Therefore, reduction of both these cell populations might not modify the resulting antitumor effect of anti-CD40/CpG immunotherapy. Another possibility may be that influencing the magnitude of the in vivo antitumor effect requires a more complete in vivo depletion of MDSC than we were able to achieve in this model with GEM, 5-FU or anti Gr-1 mAb. Alternatively, activated MDSC may kill tumor cells in parallel with other effector populations that must also be simultaneously depleted in order to inhibit the detected antitumor effect. This would be analogous to studies in a model of spontaneous tumor regression where NK cells, Mφ and neutrophils were each independently involved in the resistance: depletion of one or two of these cell subsets did not reduce resistance to tumor growth, while depletion of all 3 subsets interfered with the protective antitumor effect [30].
In summary, the findings presented here suggest that GEM or 5-FU does not enhance the antitumor effects of anti-CD40/CpG, and that partial MDSC depletion neither enhances nor interferes with the in vivo antitumor effects induced against B16 tumors in C57BL/6 mice by anti-CD40/CpG.
Highlights.
5-fluorouracil (5-FU) or gemcitabine (GEM) reduced the number of tumor-associated myeloid cells.
5-FU or GEM did not affect macrophage functions.
Chemotherapy with 5-FU or GEM did not affect the antitumor effect of anti-CD40 and CpG-ODN in mice.
Acknowledgments
This study was supported by NIH-NCI grants CA87025 and CA32685, a grant from the Midwest Athletes Against Childhood Cancer (MACC) Fund, support from The Crawdaddy Foundation, and a UW-Madison Hilldale Undergraduate Research Fellowship (to X.Q.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mitchell MS. Combinations of anticancer drugs and immunotherapy. Cancer Immunol Immunother. 2003;52:686–92. doi: 10.1007/s00262-003-0427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med. 1982;155:1063–74. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lutsiak ME, Semnani RT, De PR, Kashmiri SV, Scholom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. 2005;105:2862–8. doi: 10.1182/blood-2004-06-2410. [DOI] [PubMed] [Google Scholar]
- 4.Solomayer EF, Feuerer M, Bai L, Umansky V, Beckhove P, Meyberg GC, Bastert G, Schirrmacher V, Diel IJ. Influence of adjuvant hormone therapy and chemotherapy on the immune system analysed in the bone marrow of patients with breast cancer. Clin Cancer Res. 2003;9:174–80. [PubMed] [Google Scholar]
- 5.Fonsatti E, Maio M, Altomonte M, Hersey P. Biology and clinical application of CD40 in cancer treatment. Semin Oncol. 2010;37:517–23. doi: 10.1053/j.seminoncol.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Khong A, Nelson DJ, Nowak AK, Lake RA, Robinson BW. The use of agonistic anti-CD40 therapy in treatments for cancer. Int Rev Immunol. 2012;31:246–66. doi: 10.3109/08830185.2012.698338. [DOI] [PubMed] [Google Scholar]
- 7.Rakhmilevich AL, Alderson KL, Sondel PM. T cell-independent antitumor effects of CD40 ligation. Int Rev Immunol. 2012;31:267–78. doi: 10.3109/08830185.2012.698337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, Sullivan P, Mahany JJ, Gallagher M, Kramer A, Green SJ, O’Dwyer PJ, Running KL, Huhn RD, Antonia SJ. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25:876–83. doi: 10.1200/JCO.2006.08.3311. [DOI] [PubMed] [Google Scholar]
- 9.Advani R, Forero-Torres A, Furman RR, Rosenblatt JD, Younes A, Ren H, Harrop K, Whiting N, Drachman JG. Phase I study of the humanized anti-CD40 monoclonal antibody dacetuzumab in refractory or recurrent non-Hodgkin’s lymphoma. J Clin Oncol. 2009;27:4371–7. doi: 10.1200/JCO.2008.21.3017. [DOI] [PubMed] [Google Scholar]
- 10.Hussein M, Berenson JR, Niesvizky R, Munshi N, Matous J, Sobecks R, Harrop K, Drachman JG, Whiting N. A phase I multidose study of dacetuzumab (SGN-40; humanized anti-CD40 monoclonal antibody) in patients with multiple myeloma. Haematologica. 2010;95:845–8. doi: 10.3324/haematol.2009.008003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buhtoiarov IN, Lum H, Berke G, Paulnock D, Sondel PM, Rakhmilevich AL. CD40 ligation induces antitumor reactivity of murine macrophages via an IFN gamma-dependent mechanism. J Immunol. 2005;174:6013–22. doi: 10.4049/jimmunol.174.10.6013. [DOI] [PubMed] [Google Scholar]
- 13.Buhtoiarov IN, Lum HD, Berke G, Sondel PM, Rakhmilevich AL. Synergistic Activation of Macrophages via CD40 and TLR9 results in T cell independent antitumor effects. J Immunol. 2006;176:309–18. doi: 10.4049/jimmunol.176.1.309. [DOI] [PubMed] [Google Scholar]
- 14.Johnson EE, Buhtoiarov IN, Baldeshwiler MJ, Felder MA, Van Rooijen N, Sondel PM, Rakhmilevich AL. Enhanced T-cell-independent antitumor effect of cyclophosphamide combined with anti-CD40 mAb and CpG in mice. J Immunother. 2011;34:76–84. doi: 10.1097/CJI.0b013e318200b28a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buhtoiarov IN, Sondel PM, Wigginton JM, Buhtoiarova TN, Yanke EM, Mahvi DA, Rakhmilevich AL. Anti-tumour synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumour-associated macrophages. Immunol. 2011;132:226–39. doi: 10.1111/j.1365-2567.2010.03357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lum HD, Buhtoiarov IN, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL. In vivo CD40 ligation can induce T cell-independent antitumor effects that involve macrophages. J Leuk Biol. 2006;79:1181–92. doi: 10.1189/jlb.0405191. [DOI] [PubMed] [Google Scholar]
- 17.Rakhmilevich AL, Baldeshwiler MJ, Van De Voort TJ, Felder MA, Yang RK, Kalogriopoulos NA, Koslov DS, Van Rooijen N, Sondel PM. Tumor-associated myeloid cells can be activated in vitro and in vivo to mediate antitumor effects. Cancer Immunol Immunother. 2012;61:1683–97. doi: 10.1007/s00262-012-1236-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. 2009;9:900–9. doi: 10.1016/j.intimp.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 19.Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rebe C, Ghiringhelli F. 5-fluoruracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–61. doi: 10.1158/0008-5472.CAN-09-3690. [DOI] [PubMed] [Google Scholar]
- 20.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature Reviews. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–21. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 22.Rakhmilevich AL. Neutrophils are essential for resolution of primary and secondary infection with Listeria monocytogenes. J Leuk Biol. 1995;57:827–31. doi: 10.1002/jlb.57.6.827. [DOI] [PubMed] [Google Scholar]
- 23.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leuk Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 24.Fortin C, Huang X, Yang Y. NK cell response to vaccinia virus is regulated by myeloid-derived suppressor cells. J Immunol. 2012;189:1843–9. doi: 10.4049/jimmunol.1200584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang T, Herlyn D. Combination of active specific immunotherapy or adoptive antibody or lymphocyte immunotherapy with chemotherapy in the treatment of cancer. Cancer Immunol Immunother. 2009;58:475–92. doi: 10.1007/s00262-008-0598-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moschella F, Proietti E, Capone I, Belardelli F. Combination strategies for enhancing the efficacy of immunotherapy in cancer patients. Ann N Y Acad Sci. 2010;1194:169–78. doi: 10.1111/j.1749-6632.2010.05464.x. [DOI] [PubMed] [Google Scholar]
- 27.Lum HD, Buhtoiarov IN, Schmidt BE, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL. Tumoristatic effects of anti-CD40 mAb-activated macrophages involve nitric oxide and tumor-necrosis factor-α. Immunol. 2006;118:261–70. doi: 10.1111/j.1365-2567.2006.02366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–83. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 29.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hicks AM, Riedlinger G, Willingham MC, Alexander-Miller MA, Von Kap-Herr C, Pettenati MJ, Sanders AM, Weir HM, Du W, Kim J, Simpson AJG, Old LJ, Cui Z. Transferable anticancer innate immunity in spontaneous regression/complete resistance mice. Proc Natl Acad Sci U S A. 2006;103:7753–8. doi: 10.1073/pnas.0602382103. [DOI] [PMC free article] [PubMed] [Google Scholar]