

Abstract
Dopamine (DA) is a neurotransmitter involved in the control of locomotion, emotion, cognition, and reward. Administration of lithium salts is known to inhibit DA-associated behaviors in experimental animal models through unknown mechanisms. Here, we used a pharmacogenetic approach to show that DA can exert its behavioral effects by acting on a lithium-sensitive signaling cascade involving Akt/PKB and glycogen synthase kinase 3 (GSK-3). In the mouse striatum, increased DA neurotransmission arising either from administration of amphetamine or from the lack of the DA transporter results in inactivation of Akt and concomitant activation of GSK-3α and GSK-3β. These biochemical changes are not affected by activation of the cAMP pathway but are effectively reversed either by inhibition of DA synthesis, D2 receptor blockade, or administration of lithium salts. Furthermore, pharmacological or genetic inhibition of GSK-3 significantly reduces DA-dependent locomotor behaviors. These data support the involvement of GSK-3 as an important mediator of DA and lithium action in vivo and suggest that modulation of the Akt/GSK-3 pathway might be relevant to DA-related disorders, such as attention deficit hyperactivity disorder and schizophrenia.
Dopamine (DA) is a monoaminergic neurotransmitter that has been implicated in multiple brain disorders, such as Parkinson's disease, schizophrenia, attention deficit hyperactivity disorder, Tourette syndrome, addiction, and affective disorders (1-3). The major population of dopaminergic neurons in the brain arises from the substantia nigra pars compacta and projects to striatal neurons. The DA transporter (DAT) tightly controls the action of DA at the synaptic level by assuring its reuptake into presynaptic neurons, thus limiting extracellular DA concentration (3). Accordingly, mice lacking the DAT exhibit a persistent 5-fold elevation in extracellular striatal DA, leading to the appearance of locomotor hyperactivity and stereotypic movements when these mice are placed in a novel environment (4-6). At the cellular level the various physiological functions of DA are mediated by two classes of G protein-coupled receptors. The D1-like receptors (D1 and D5) are mostly coupled to Gsα and the D2-like receptors (D2, D3, and D4) are coupled to Gi/Goα (7). However, the signaling mechanisms mediating the action of DA on hyperactivity are still not fully understood. For instance, acute administration of lithium salts is known to antagonize the hyperactivity induced by various dopaminergic agonists (8-11). Nevertheless, the mechanism by which lithium interferes with DA-associated behavior remains uncharacterized. Here we show that one putative physiological target of lithium, glycogen synthase kinase 3 (GSK-3), is activated in response to sustained stimulation of DA receptors and that its inhibition interferes with the expression of DA-dependent behaviors.
Materials and Methods
Experimental Animals. C57BL/129SvJ DAT knockout (DAT-KO) mice (4) and their WT littermates, which were between 3 and 4 months of age and showed no signs of neurological motor symptoms (12), were used for all experiments. C57BL/6J GSK-3β heterozygote mice were described (13). WT C57BL/6J mice were obtained from The Jackson Laboratory. Before experiments, animals were housed four or five to a cage at 23°C on a 12 h light/12 h dark cycle with ad libitum access to food and water. Animal care was approved by the Institutional Animal Care and Use Committee and followed National Institutes of Health guidelines.
Antibodies. The anti-phospho-GSK-3α/β Ser-21/9, anti-phospho-Akt Thr-308, anti-phospho-Akt Ser-473, anti-total-Akt, and the anti-p-DA and cAMP-regulated phosphoprotein, apparent molecular weight of 32,000 (DARPP-32) Thr-34 antibodies were purchased from Cell Signaling Technology (Beverly, MA). The anti-DARPP-32 was obtained from BD Transduction Laboratories (Lexington, KY). The anti-GSK-3α/β clone 0011-A was from Santa Cruz Biotechnology, and the anti-actin clone c4 was from Chemicon. The specificity of anti-p-DARPP-32 antibody has been demonstrated (14). All other antibodies recognized only their target proteins on Western blot analysis (data not shown).
Western Blot Analysis. Mice were killed by decapitation, after which the heads of the animals were immediately cooled by immersion in liquid nitrogen for 6 sec. For each mouse, the right hemistriatum was rapidly dissected out (within 30 sec) on an ice-cold surface and frozen in liquid nitrogen before protein extraction. Tissue samples were homogenized in boiling 1% SDS solution supplemented with 2 μM okadaic acid and boiled for 10 min. Protein concentration was measured by using a DC-protein assay (Bio-Rad). Protein extracts (25 or 50 μg) were separated on 10% SDS/PAGE and transferred to nitrocellulose membranes. Blots were immunostained overnight at 4°C with the following primary antibodies: anti-phospho-GSK-3α/β Ser-21/9 (1:200 dilution); anti-phospho-Akt Thr-308 (1:100); anti-phospho-Akt Ser-473 (1:500); anti-GSK-3α/β clone 0011-A (1:5,000); anti-Akt (1:1,000); anti-DARPP-32 (1:1,000); anti-phospho-DARPP-32 Thr-34 (1:100); and anti-actin clone c4 (1:20,000). Immune complexes were revealed by using appropriate peroxidase-conjugated secondary antibodies (Jackson Immuno-Research) along with a chemiluminescent reagent (SuperSignal West-Pico, Pierce). Densitometric analysis was carried out within linear range by using imagequant V1.1 (Amersham Pharmacia Bioscience). For all experiments, representative Western blots displaying results obtained from two different mice for each group are shown.
GSK-3 Activity Assay. For each determination of kinase activity, a complete mouse striatum was rapidly dissected out on an ice-cold surface and homogenized at 4°C in lysis buffer (20 mM Tris, pH 8.0/137 mM NaCl/10% glycerol/1% Nonidet P-40/0.5 mM sodium orthovanadate/2 μM okadaic acid and a mixture of protease inhibitors (Sigma-Aldrich). GSK-3α and -3β were immunoprecipitated at 4°C from 1 mg of protein extract with 2 μg of anti-GSK-3α/β monoclonal antibody and protein-A Sepharose (Amersham Pharmacia Bioscience). Immunoprecipitates were then incubated in assay buffer (20 mM Tris, pH 7.5/10 mM MgCl2/5mMDTT/0.2 mM ATP/0.5 μCi of [γ-32P]ATP) for 10 min at 30°C with 0.5 μg of recombinant phosphatase inhibitor 2 (New England Biolabs). Control assays were also carried out in the presence of the GSK-3 inhibitor Kenpaullone (2 μM, Sigma-Aldrich). Reactions were stopped by the addition of Laemmli loading buffer, boiled for 5 min, and resolved on SDS/10% PAGE. Gels were then stained with Coomassie blue and autoradiographed. Incorporation of 32P into recombinant phosphatase inhibitor 2 was measured by densitometry.
Drug Administration. LiCl, sodium valproate, raclopride, α-methyl-para-tyrosine (αMPT), and amphetamine were dissolved in saline and injected i.p.; SCH23390 was prepared in water and administered by s.c. injection. Alsterpaullone, SB 216763, indirubin-3′-monoxime, and 4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione (TDZD) were injected i.p. after suspension in a minimal amount of Tween and made up to volume with distilled water. 8-Br-cAMP was prepared in artificial cerebrospinal fluid and injected into the right lateral ventricle (intracerebroventricularly) in a volume of 4 μl at a rate of 1 μl/min as described (15). Cyclosporin A was prepared in 50% DMSO. Corresponding vehicle solutions were injected to respective control animals. All drugs were purchased from Sigma-Aldrich with the exception of SB 216763 (Tocris Cookson, Ellisville, MO) and TDZD (Calbiochem).
Measurement of Locomotor Activity. Locomotion was evaluated under illuminated conditions in an automated Omnitech Digiscan apparatus (AccuScan Instruments, Columbus, OH). Mice were initially placed into the activity monitor chamber for 30 min, then injected with vehicle (10 ml/kg of body weight i.p.) or the drug, returned to the chamber, and monitored for 2 h after injection. Locomotor activity was measured in terms of the total distance covered (horizontal activity), rearings were expressed as the number of vertical beam breaks (vertical activity), and the stereotypy time refers to the total time that stereotypic behaviors (repetitive beam breaks of a given beam or beams with intervals <1 sec) were observed (5).
Measurement of Extracellular DA Concentration. Extracellular striatal DA concentrations were measured by using in vivo microdialysis on freely moving animals followed by HPLC as described (5, 6).
Statistical Analysis. Data were analyzed by two-tailed t test, one-way ANOVA, or two-way ANOVA. Values in graphs were expressed as mean ± SEM.
Results
Lithium Antagonizes Behavioral Responses to DA in DAT-KO Mice. In DAT-KO mice the DA-dependent hyperactivity and stereotypy that develop after the exposure of the mice to a novel environment (5) can be significantly attenuated by LiCl. Administration of LiCl at doses known not to induce toxicity in mice (16-18) resulted in a rapid inhibition of horizontal activity that was maintained for at least 1 h after injection(Fig.1A). Quantification of behavioral data for a period of 60 min after drug administration revealed a dose-dependent inhibition of horizontal activity (Fig. 1B), vertical activity, and stereotypy (Fig. 7, which is published as supporting information on the PNAS web site) in DAT-KO mice treated with LiCl as compared with vehicle-treated DAT-KO mice. By comparison, LiCl (200 mg/kg of body weight i.p.) exerted only a minor effect on activity in habituated WT mice (data not shown).
Fig. 1.

Lithium antagonizes behavioral responses to DA in DAT-KO mice. (A) DAT-KO mice were placed in a locomotor activity monitor for an initial period of 30 min and were then injected (arrow) with vehicle (saline) or with LiCl (50, 100, or 200 mg/kg of body weight i.p.). Horizontal activity was recorded every 5 min for a 2-h period. (B) Horizontal activity was quantified for a period of 1 h after injection of either vehicle or different doses of LiCl. Note that LiCl-treated DAT-KO mice remained more active than vehicle-treated WT mice observed under the same conditions. (C-E) Densitometric Western blot analysis of phosphoprotein relative levels in extract prepared from the striatum of DAT-KO mice 30 min after injection of LiCl (200 mg/kg of body weight i.p.) or vehicle. Antibodies directed against p-Thr-34-DARPP-32 (C), p-Thr-308-Akt (open bar in D), and p-Ser-473-Akt (filled bar in D) and with an antibody recognizing both p-GSK-3α (Ser-21, filled bar in E) and β (Ser-9, open bars in E) were used. Phospho-independent antibodies directed against these different proteins were used as loading controls for densitometry. For all results, data are means ± SEM. *, P < 0.05; **, P < 0.005; ***, P < 0.001 as compared with vehicle-treated DAT-KO mice; #, P < 0.05 as compared with DAT-KO mice treated with 200 mg/kg LiCl. Numbers of animals per group (n) are indicated.
The hyperactive behavior of DAT-KO mice depends on elevated DA tone in primary dopaminoceptive brain areas such as the striatum and can be blocked by inhibition of DA synthesis or DA receptor antagonists (5, 19). To assess if lithium antagonizes hyperactivity and stereotypy by affecting DA dynamics, extracellular DA levels were measured in the striatum of DAT-KO mice that received either vehicle or LiCl (200 mg/kg of body weight). In vivo microdialysis showed that LiCl at a dose that dramatically reduces activity of DAT-KO mice did not affect extracellular DA levels (Fig. 7), indicating that lithium affects the responsiveness to DA rather than DA dynamics.
Lithium Affects Akt and GSK-3 in DAT-KO Mice. Because lithium does not bind to DA receptors (20, 21), its potential action on cAMP-mediated DA signaling was assessed in the striatum. DARPP-32 is a known mediator of cAMP signaling whose phosphorylation on Thr-34 by PKA in response to cAMP is regulated by DA receptors (2, 14, 22). Western blots probed with an anti-phospho-DARPP-32 (Fig. 1C) antibody revealed no effect of lithium on Thr-34-DARPP-32 phosphorylation in the striatum of LiCl-treated (200 mg/kg of body weight i.p.) DAT-KO mice as compared with vehicle-treated mice, suggesting that under these conditions the cAMP/PKA/DARPP-32 signaling cascade plays little role in the action of lithium on DA-dependent behaviors.
We then examined whether inhibition of DA-associated behaviors by lithium could be related to changes in other signaling mechanisms. The Akt/GSK-3 signaling cascade represented an attractive possibility. Akt is mostly regulated through the phosphoinositol-dependent phosphorylation of both its Ser-473 and Thr-308 residues, resulting in its activation (23, 24). GSK-3α and GSK-3β are two substrates of Akt that are regulated through phosphorylation of serine residues on their N-terminal domain (Ser-21 for GSK-3α and Ser-9 for GSK-3β), which results in inactivation (25, 26). Previous reports from primary cell culture systems and from lithium-treated mice have shown that lithium can activate the serine/threonine kinase Akt by modulating the activity of phosphoinositol-regulated kinases or other mechanisms (27-30). In these systems, activation of Akt was associated with an increased phosphorylation of GSK-3, thus suggesting that lithium, a known direct inhibitor of GSK-3 in vitro (20, 31, 32), can inhibit GSK-3 activity in neurons both directly and indirectly (18, 27-32).
We used Western blot analysis to evaluate whether lithium can affect Akt and GSK-3 phosphorylation in DAT-KO mice. Phospho-specific Akt antibodies showed an ≈3-fold increase of phospho-Thr-308-Akt, whereas Ser-473 phosphorylation was unchanged in lithium-treated DAT-KO mice (Fig. 1D). The phosphorylation of GSK-3α and GSK-3β was then measured. Antibody selective for phosphorylated Ser-21/9 of GSK-3 revealed that lithium induced an ≈2-fold increase in site-specific phosphorylation of both forms of GSK-3 in the striatum of DAT-KO mice (Fig. 1E). These data indicate that regulation of the Akt/GSK-3 signaling cascade is affected by lithium in DAT-KO mice.
DA-Dependent Regulation of Akt and GSK-3. To explore whether a dysregulation of the Akt/GSK-3 pathway might contribute to DA-dependent hyperactivity, we then examined Akt and GSK-3 expression and regulation in the striatum of untreated DAT-KO mice versus WT littermates. Although no changes in the total levels of Akt, GSK-3α, and GSK-3β were observed in DAT-KO mice (Fig. 2A and Fig. 8, which is published as supporting information on the PNAS web site), phospho-dependent antibodies revealed that the phosphorylation of Thr-308-Akt and both GSK-3 isoforms were markedly reduced in the striatum of DAT-KO mice as compared with their WT littermates (Fig. 2 A and B). To confirm that the reduced phosphorylation of GSK-3 was associated with enhanced kinase activity, in vitro GSK-3 activity assays (Fig. 2C) were performed with a recombinant phosphatase inhibitor 2 as a substrate (33). These assays showed an ≈1.8-fold increase of GSK-3 activity in lysates prepared from DAT-KO mice (Fig. 2D). Moreover, kinase activity in the assays was reduced in the presence of the GSK-3 inhibitor kenpaullone (Fig. 2C Lower).
Fig. 2.

Inactivation of Akt and concomitant activation of GSK-3α and -3β in the striatum of DAT-KO mice. (A) Western blots showing the relative levels of total and phospho-Akt (Ser-473 and Thr-308) in the striatum of DAT-KO mice and WT littermates. (B) Densitometric analysis of the levels of p-Thr-308 (Upper, open bars), p-Ser-473-Akt (Upper, filled bars) and of phospho-GSK-3α and -3β (Lower) in mice from both genotypes. (C and D) GSK-3 activity assays performed after immunoprecipitation of striatal extracts by using a pan GSK-3α/β antibody. (C Upper) Autoradiogram showing the incorporation of 32P in recombinant phosphatase inhibitor 2 (I-2) by immunoprecipitates from WT and DAT-KO mice. (C Lower) Inhibition of kinase activity in the immunoprecipitates when the assay was performed in the presence of 2 μM GSK-3 inhibitor kenpaullone. Total inhibitor 2 was revealed by Coomassie blue staining. (D) Quantitative analysis revealed an ≈1.8-fold increase in GSK-3 activity in the striatum of DAT-KO mice. For densitometric analysis, optical densities obtained from specific phospho-independent antibodies were used as a loading control for the evaluation of phosphoprotein levels, and actin was used as a general loading control. Data are means ± SEM. *, P < 0.05 as compared with WT littermates.
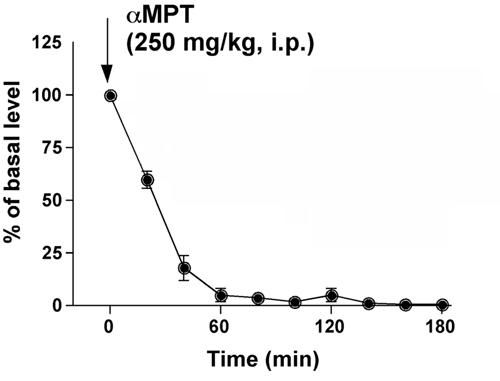
We then examined whether a causal relationship between elevated DA levels, inactivation of Akt and activation of GSK-3, existed. This was first done by depleting DA from the striatum of DAT-KO mice by using the inhibitor of DA synthesis αMPT. In DAT-KO mice, the absence of DAT-mediated DA recycling and storage renders DA neurons solely dependent on DA synthesis for the maintenance of extracellular DA levels (3). In agreement with previous data (5, 6), administration of αMPT (250 mg/kg of body weight i.p.) to DAT-KO mice resulted in a rapid decrease of extracellular striatal DA essentially below the limit of detection as measured by microdialysis for 3 h after injection (Fig. 9, which is published as supporting information on the PNAS web site). DA depletion resulted in an ≈3.5-fold increase in phospho-Thr-308-Akt and a concomitant ≈2-fold increase in the phosphorylation of GSK-3α and GSK-3β (Fig. 3A), indicating that DA regulates Akt and GSK-3 in DAT-KO mice.
Fig. 3.

Regulation of Akt and GSK-3 by DA in DAT-KO (A) Western blots showing increased levels of p-Thr-308-Akt, p-GSK-3α, and p-GSK-3β in the striatum of DAT-KO mice 2 h after inhibition of DA synthesis by injection ofαMPT (250 mg/kg of body weight i.p.). DA depletion in the striatum under these conditions was confirmed with microdialysis (Fig. 9). Data are means ± SEM. *, P < 0.05; ***, P < 0.001. (B) The D2-class receptor antagonist raclopride blocks the action of DA on the phosphorylation of Akt (Thr-308), GSK-3α, and GSK-3β at 1 h after injection. (C) No effect of the D1-class receptor antagonist SCH23390 (0.1 mg/kg of body weight s.c.) on Akt and GSK-3α/β phosphorylation as evaluated by Western blot analysis. (D and E) Intracerebroventricular injection of 8-Br-cAMP (50 nmol) (D) or of cyclosporin A (4 μl of a 10-μM solution) (E) increased phosphorylation of DARPP-32 (Thr-34) without affecting Akt and GSK-3α/β phosphorylation at 30 min after injection. Data are means ± SEM. *, P < 0.05; **, P ≤ 0.005; ***, P ≤ 0.001 as compared with vehicle-treated DAT-KO mice.
D1- and D2-class receptor antagonists were then used to further support these observations and to determine which DA receptors might be involved in the regulation of striatal Akt and GSK-3 by DA. Administration of the D2/D3 receptor antagonist raclopride (2 mg/kg of body weight i.p.) to DAT-KO mice led to a significant increase in the phosphorylation of Akt (Thr-308), GSK-3α, and GSK-3β as compared with vehicle-treated animals (Fig. 3B). On the other hand, the D1-class receptor antagonist SCH23390 (0.1 mg/kg of body weight s.c.) had no effect on Akt or GSK-3 phosphorylation (Fig. 3C). Thus, despite the fact that both these drugs antagonized hyperactivity of DAT-KO mice (data not shown), only D2-class receptors appear to contribute to the regulation of Akt and GSK-3 by DA.
According to known paradigms of DA receptor signaling, a blockade of D2-class receptor should result in an intracellular increase of cAMP levels, resulting in an activation of PKA (2, 7). Alternatively, D2 receptors can also exert their action by acting through cAMP-independent pathways. In this regard D2 receptor has been shown to activate the calcium/calmodulin-regulated phosphatase calcineurin, leading to the dephosphorylation of Thr-34 DARPP-32 (14). Thus, the possible involvement of PKA and calcineurin in the regulation of Akt and GSK-3 was investigated in DAT-KO mice. As might be expected, injection of a cell-permeable cAMP analog (8-Br-cAMP, 50 nmol) or of the calcineurin inhibitor cyclosporin A (4 μl of a 10-μM solution) in the lateral cerebral ventricle (intracerebroventricular injection) of DAT-KO mice induced a robust increase in Thr-34-DARPP-32 phosphorylation (Fig. 3 D and E). However, no effects of these treatments on Akt and GSK-3 phosphorylation were observed (Fig. 3 D and E). Taken together, these results provide evidence for a previously unappreciated cAMP-independent action of DA on the Akt/GSK-3 pathway in the mouse striatum. Moreover, lithium was able to antagonize both DA-associated behaviors and the action of DA on Akt and GSK-3 phosphorylation (Fig. 1), thus suggesting an important role of these kinases in the behavioral action of DA at least in this animal model.
Inhibition of GSK-3 Antagonizes DA-Mediated Behaviors. To further examine the involvement of the Akt/GSK-3 pathway in the behavioral effects of DA, we then tested whether the DA-dependent hyperactivity of DAT-KO mice could be reversed by inhibition of GSK-3. DAT-KO mice were treated with different compounds that share the ability to inhibit the catalytic activity of GSK-3. Five structurally unrelated GSK-3 inhibitors, SB 216763 (34), alsterpaullone (35), indirubin-3-monoxime (36), sodium valproate (37), and TDZD (38), were used. At all doses tested, SB 216763 (3, 5, and 10 mg/kg of body weight i.p.) significantly inhibited horizontal activity (Fig. 4), stereotypy, and vertical activity (Fig. 10, which is published as supporting information on the PNAS web site) of DAT-KO mice as measured for 30 min after injection. Likewise, administration of indirubin-3-monoxime (5, 10, and 20 mg/kg of body weight i.p.) or alsterpaullone (3, 5, and 10 mg/kg of body weight i.p.) also resulted in significant reductions of horizontal activity (Fig. 4), vertical activity, and stereotypy (Fig. 10). In agreement with a previous study of mice with reduced levels of DAT expression (39), significant reductions in activity (Fig. 4) and stereotypy (Fig. 10) were also obtained, albeit with a 45-min delay, after the administration of sodium valproate. Finally, injection of the catalytic binding site GSK-3 inhibitor TDZD (30 mg/kg of body weight i.p.) to DAT-KO mice resulted in a marked inhibition of hyperactivity (Fig. 4) and stereotypy (Fig. 10), thus providing a compelling indication that GSK-3 activity is important for the expression of DA-associated behaviors and that its inhibition can reproduce the behavioral effects of lithium in DAT-KO mice.
Fig. 4.

Inhibition of horizontal activity in DAT-KO mice by different GSK-3 inhibitors. DAT-KO mice were placed in a locomotor activity monitor for an initial period of 30 min and were then injected with vehicle or with SB 216763 (3, 5, or 10 mg/kg of body weight i.p.), Indirubin-3′-monoxime (5, 10, or 20 mg/kg of body weight i.p.), Alsterpaullone (3, 5, and 10 mg/kg of body weight i.p.), Sodium valproate (100, 200, or 300 mg/kg of body weight i.p.) or TDZD (30 mg/kg of body weight i.p.). All effects were monitored for a period of 30 min starting 5 min after injection, except for sodium valproate for which measurement started 45 min after injection, and compared with a separate set of vehicle-treated mice observed under the same conditions. Data are means ± SEM. *, P < 0.05; ***, P < 0.001 as compared with vehicle-treated mice. Numbers of animals per group (n) are indicated.
GSK-3β Is Implicated in the Actions of Amphetamine. To validate our observations in normal animals, we then examined whether DA could inhibit Akt and activate GSK-3 in WT mice. Amphetamine is an indirect DA-receptor agonist as it elevates extracellular DA levels in the mouse striatum (3). Administration of an effective dose of amphetamine (2 mg/kg of body weight i.p.) (3, 5) resulted in a reduction of Akt, GSK-3α, and GSK-3β phosphorylation in the striatum of WT mice (Fig. 5A), as measured 90 min after injection. However, no apparent change in the phosphorylation of these proteins was detected at 30 min after amphetamine injection (Fig. 5A), whereas Akt phosphorylation was significantly reduced at 60 min after injection (data not shown). Furthermore, a similar time-dependent reduction of Akt phosphorylation was also observed after administration of the D1/D2 DA agonist apomorphine (data not shown), thus suggesting that Akt inactivation and GSK-3 activation are late biochemical responses to DA receptor stimulation.
Fig. 5.

Involvement of GSK-3 in the action of amphetamine in vivo. (A) Quantification of Akt (Thr-308), GSK-3α, and GSK-3β phosphorylation as measured by Western blot analysis at 0, 30, and 90 min after amphetamine injection. *, P < 0.05; P < 0.005 as compared with saline-treated animals. (B and C) Reduced response to amphetamine in GSK-3β+/- mice. (B) Changes in horizontal activity induced by different doses of amphetamine (0, 1, or 2 mg/kg of body weight i.p., n = 6 mice per group) in GSK-3β+/- mice and WT littermates as measured for a period of 90 min after injection. (C) Locomotor activity curve of WT and GSK-3β+/- mice after injection of 2 mg of amphetamine/kg of body weight (i.p.). Data are means ± SEM. *, P < 0.05; **, P < 0.005 as compared with WT mice receiving the same dose of amphetamine; and ##, P < 0.005; ###, P < 0.001 as compared with vehicle-treated mice from the same genotype.
To further establish the involvement of GSK-3 in the expression of DA-induced behaviors, we assessed the behavioral effects of amphetamine in GSK-3β mutant mice and in their WT littermates. Homozygote GSK-3β knockout mice die during embryogenesis (13). However, heterozygote mice (GSK-3β+/- mice) develop normally without any overt phenotypes (13) and show a reduction of ≈50% (n = 6, P ≤ 0.005) in striatal GSK-3β as revealed by Western blot analysis (Fig. 11, which is published as supporting information on the PNAS web site). In locomotor activity tests, unchallenged GSK-3β+/- mice were not different from WT littermates either in horizontal activity (Fig. 5B), vertical activity, or stereotypy (data not shown). However, quantification of horizontal activity for 90 min after drug administration (Fig. 5B) revealed that GSK-3β+/- showed a markedly blunted response to acute amphetamine (1 or 2 mg/kg of body weight i.p.). In contrast to WT littermates, GSK-3β+/- mice were not significantly activated after injection of 1 mg/kg amphetamine (Fig. 5B), whereas, at a dose of 2 mg/kg of body weight, GSK-3β+/- mice exhibited reduced (≈50% less) locomotor activation (Fig. 5B). Detailed analysis of the locomotor response to amphetamine over time revealed that differences between WT and GSK-3β+/- mice only became significant after the first 30 min after injection, thus correlating with the time course of activation of GSK-3 by amphetamine (Fig. 5D). Taken together, these results suggest that despite the apparent temporal delay in the response of Akt and GSK-3 to dopaminergic stimulation, modulation of this pathway contributes significantly to the overall psychomotor response of animals.
Discussion
Our data reveal that lithium can exert its acute effect on DA-dependent behaviors by interfering with the action of DA on an Akt/GSK-3 signaling cascade in the brain of living mice. In the striatum of DAT-KO mice, the elevated DA tone leads to activation of GSK-3α and -3β through a signaling cascade involving D2-class receptors and reduced Akt activity. Similar changes in Akt and GSK-3 activity were also observed after acute administration of the indirect DA agonist amphetamine to WT mice. Moreover, pharmacological or genetic inhibition of GSK-3 reproduced the effect of lithium and reduced behavioral responses to pharmacologically or genetically elevated dopaminergic tone, thus establishing this cascade as an important mediator of DA action in vivo.
The results presented here suggest that cAMP-independent DA receptor signaling can play a significant role in the expression of DA-associated behaviors (Fig. 6). However, it is important to keep in mind that inhibition of GSK-3 either by lithium, GSK-3 inhibitors, or a gene-targeting approach did not completely abolish DA-mediated behavior in mice. Moreover, D1-class receptor blockade did not affect the Akt/GSK-3 pathway while reducing hyperactivity in DAT-KO animals (19). These findings suggest that DA functions are mediated by an intricate signaling network (Fig. 6) in which both cAMP-dependent and -independent events play important and perhaps cooperative roles in the expression of a full behavioral response.
Fig. 6.

Working model of potential mechanisms of DA receptor signaling and sites of action of the different pharmacological agents used in this study. In red, regulation of the Akt/GSK-3 pathway by DA. Elevated DA tones lead to activation of GSK-3 through a signaling cascade involving D2-class receptors and the inactivation of Akt caused by a reduction of its phosphorylation on Thr-308. This reduction in Akt activity results in a reduced phosphorylation/increased activation of GSK-3α and GSK-3β that, in turn, regulate DA-associated behaviors. Note that, in contrast to DARPP-32, the phosphorylation of Akt and GSK-3 was not affected by cAMP. Dashed lines indicate the effect of different drugs on this signaling cascade. D1R, DA D1 receptor; D2R, DA D2 receptor; AC, adenylyl cyclase.
Our results in WT animals also indicate that the inactivation of Akt/activation of GSK-3 by DA only occurs after a sustained (>30 min) stimulation of DA receptors. In fact, analysis of the behavioral response of GSK-3β+/- mice to amphetamine suggests, on the one hand, that GSK-3β does not play a central role in the initial response to amphetamine (first 30 min). On the other hand, even minor (≈25%) changes in total GSK-3 activity are sufficient to significantly blunt DA-dependent locomotor behavior over the full time course of the response. This observation is also consistent with data obtained from DAT-KO mice in which DA receptors are continuously stimulated. In this regard, our results differ from previous reports of a D2 receptor activation of Akt and inactivation of GSK-3β after short-term exposure of cell cultures to DA agonists (40-42) or within 15 min after treatment of mice with cocaine (40) or amphetamine (43). Further detailed studies of DA receptor signaling at different time points after stimulation will be necessary to explain these discrepancies.
Apart from the regulation of the Akt/GSK-3 pathway demonstrated here, D2-class receptors can also act independently from cAMP to regulate ion-channel permeability, inositol signaling, and phosphatase activity (7). It will be interesting in the future to examine whether any of these responses to D2 receptor stimulation are responsible for the regulation of Akt and GSK-3 by DA or if they correspond to different modalities of DA-receptor signaling. In this regard, modulation of phosphoinositol signaling and activity of related kinases by DA represent an attractive possibility because the phosphoinositol pathway has been associated with both the regulation of Akt and the action of lithium. Moreover, the time course of Akt dephosphorylation after DA receptor activation suggests that processes involved in receptor desensitization may also contribute to the regulation of Akt. However, the choice of a proper model system will undoubtedly be important for examining these possibilities. Indications are that DA receptors may not have the same signaling characteristics in different model systems. For instance, stimulation of D2 receptor has been shown to enhance inositol signaling in PC12 cells and to reduce it in rat lactotroph cells (41, 42, 44). Such differences in signaling may be explained by the coupling of DA receptors to different signaling machinery in different model systems or in response to different artificial DA agonists (41). These differences underscore the importance of studying functional DA signaling in native tissues in vivo.
The molecular mechanism by which lithium acts on behavior is still the subject of debate. Two major hypotheses are that lithium may either perturb inositol signaling or directly inhibit GSK-3 (20, 21, 30). In our system, lithium can inhibit GSK-3 activity either directly (18) or through an indirect mechanism involving the activation of Akt (27-29), thus opposing the effect of DA on this pathway. Future studies involving mice lacking components of the Akt/GSK-3 signaling cascade in specific brain regions will be useful to further validate these observations. Whether the acute effect of lithium on DA-receptor signaling is pertinent to its chronic action in the treatment of mood disorders will also be important to examine. However, it is interesting that similar changes in Akt and GSK-3 phosphorylation have been reported to occur in the brain of mice that were chronically treated with lithium or valproic acid (28).
DA is associated with many neurological and psychiatric conditions. Thus, the elucidation of a connection between sustained DA-receptor signaling, Akt, GSK-3, and aberrant behavior raises the possibility that a dysregulation of the Akt/GSK-3 pathway may contribute to the etiology of DA-associated neuropsychiatric conditions. During the review process of this article, modifications of Akt/GSK-3 signaling, similar to those observed in our study in response to excess of DA, were described in individuals with schizophrenia (45). Finally, the ability of GSK-3 inhibitors to normalize behavior in DAT-KO mice raises the possibility that targeting of the Akt/GSK-3 signaling pathway may provide a previously unappreciated approach to manage conditions associated with altered DA neurotransmission (1-3).
Supplementary Material
Acknowledgments
We thank Susan Suter for excellent assistance in the maintenance of the DAT-KO mouse colony. J.-M.B. thanks Rossio Motta for her inspiration in the conduct of this research. This work was supported in part by National Institutes of Health Grants DA-13511 and MH-40159 (to M.G.C.). J.-M.B. is the recipient of a long-term fellowship from the Human Frontier Research Program, J.R.W. is an International Scholar, and M.G.C. is an Investigator of the Howard Hughes Medical Institute.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: GSK-3, glycogen synthase kinase 3; DA, dopamine; DAT, DA transporter; DAT-KO, DAT knockout; αMPT, α-methyl-para-tyrosine; DARPP-32, dopamine- and cAMP- regulated phosphoprotein, apparent molecular weight of 32,000; TDZD, 4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione.
References
- 1.Carlsson, A. (1987) Annu. Rev. Neurosci. 10, 19-40. [DOI] [PubMed] [Google Scholar]
- 2.Greengard, P. (2001) Science 294, 1024-1030. [DOI] [PubMed] [Google Scholar]
- 3.Gainetdinov, R. R. & Caron, M. G. (2003) Annu. Rev. Pharmacol. Toxicol. 43, 261-284. [DOI] [PubMed] [Google Scholar]
- 4.Giros, B., Jaber, M., Jones, S. R., Wightman, R. M. & Caron, M. G. (1996) Nature 379, 606-612. [DOI] [PubMed] [Google Scholar]
- 5.Gainetdinov, R. R., Wetsel, W. C., Jones, S. R., Levin, E. D., Jaber, M. & Caron, M. G. (1999) Science 283, 397-401. [DOI] [PubMed] [Google Scholar]
- 6.Jones, S. R., Gainetdinov, R. R., Jaber, M., Giros, B., Wightman, R. M. & Caron, M. G. (1998) Proc. Natl. Acad. Sci. USA 95, 4029-4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Missale, C., Nash, S. R., Robinson, S. W., Jaber, M. & Caron, M. G. (1998) Physiol. Rev. 78, 189-225. [DOI] [PubMed] [Google Scholar]
- 8.Cox, C., Harrison-Read, P. E., Steinberg, H. & Tomkiewicz, M. (1971) Nature 232, 336-338. [DOI] [PubMed] [Google Scholar]
- 9.Flemenbaum, A. (1977) Biol. Psychiatry 12, 563-572. [PubMed] [Google Scholar]
- 10.Aylmer, C. G., Steinberg H. & Webster, R. A. (1987) Psychopharmacology 91, 198-206. [DOI] [PubMed] [Google Scholar]
- 11.Barnes, J. C., Costall, B., Domeney, A. M. & Naylor, R. J. (1986) Psychopharmacology 89, 311-316. [DOI] [PubMed] [Google Scholar]
- 12.Cyr, M., Beaulieu, J. M., Laakso, A., Sotnikova, T. D., Yao, W. D., Bohn, L. M., Gainetdinov, R. R. & Caron, M. G. (2003) Proc. Natl. Acad. Sci. USA 100, 11035-11040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O. & Woodgett, J. R. (2000) Nature 406, 86-90. [DOI] [PubMed] [Google Scholar]
- 14.Nishi, A., Snyder, G. L. & Greengard, P. (1997) J. Neurosci. 17, 8147-8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mantella, R. C., Vollmer, R. R., Li, X. & Amico, J. A. (2003) Endocrinology 144, 2291-2296. [DOI] [PubMed] [Google Scholar]
- 16.Wood, A. J., Goodwin, G. M., De Souza, R. & Green, A. R. (1986) Neuropharmacology 25, 1285-1288. [DOI] [PubMed] [Google Scholar]
- 17.Wissocq, J. C., Heurteaux, C. & Thellier, M. (1983) Neuropharmacology 22, 227-232. [DOI] [PubMed] [Google Scholar]
- 18.Phiel, C. J., Wilson, C. A., Lee, V. M. & Klein, P. S. (2003) Nature 423, 435-439. [DOI] [PubMed] [Google Scholar]
- 19.Ralph, R. J., Paulus, M. P., Fumagalli, F., Caron, M. G. & Geyer, M. A. (2001) J Neurosci. 21, 305-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phiel, C. J. & Klein, P. S. (2001) Annu. Rev. Pharmacol. Toxicol. 41, 789-813. [DOI] [PubMed] [Google Scholar]
- 21.Berridge, M. J., Downes, C. P. & Hanley, M. R. (1989) Cell 59, 411-419. [DOI] [PubMed] [Google Scholar]
- 22.Fienberg, A. A., Hiroi, N., Mermelstein, P. G., Song, W., Snyder, G. L., Nishi, A., Cheramy, A., O'Callaghan, J. P., Miller, D. B., Cole, D. G., et al. (1998) Science 281, 838-842. [DOI] [PubMed] [Google Scholar]
- 23.Alessi, D. R., Andjelkovic, M., Caudwell, B., Cron, P., Morrice, N., Cohen, P. & Hemmings, B. A. (1996) EMBO J. 15, 6541-6551. [PMC free article] [PubMed] [Google Scholar]
- 24.Scheid, M. P. & Woodgett, J. R. (2001) Nat. Rev. Mol. Cell. Biol. 2, 760-768. [DOI] [PubMed] [Google Scholar]
- 25.Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M. & Hemmings, B. A. (1995) Nature 378, 785-789. [DOI] [PubMed] [Google Scholar]
- 26.Frame, S. & Cohen, P. (2001) Biochem. J. 359, 1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chalecka-Franaszek, E. & Chuang, D. M. (1999) Proc. Natl. Acad. Sci. USA. 96, 8745-8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Sarno, P., Li, X. & Jope, R. S. (2002) Neuropharmacology 43, 1158-1164. [DOI] [PubMed] [Google Scholar]
- 29.Zhang, F., Phiel, C. J., Spece, L., Gurvich, N. & Klein, P. S. (2003) J. Biol. Chem. 278, 33067-33077. [DOI] [PubMed] [Google Scholar]
- 30.Jope, R. S. (2003) Trends Pharmacol. Sci. 24, 441-443. [DOI] [PubMed] [Google Scholar]
- 31.Klein, P. S. & Melton, D. A. (1996) Proc. Natl. Acad. Sci. USA 93, 8455-8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stambolic, V., Ruel, L. & Woodgett, J. R. (1996) Curr. Biol. 6, 1664-1668. [DOI] [PubMed] [Google Scholar]
- 33.Aitken, A., Holmes, C. F., Campbell, D. G., Resink, T. J., Cohen, P., Leung, C. T. & Williams, D. H. (1984) Biochim. Biophys. Acta 790, 288-291. [DOI] [PubMed] [Google Scholar]
- 34.Coghlan, M. P., Culbert, A. A., Cross, D. A., Corcoran, S. L., Yates, J. W., Pearce, N. J., Rausch, O. L., Murphy, G. J., Carter, P. S., Roxbee Cox, L., et al. (2000) Chem. Biol. 7, 793-803. [DOI] [PubMed] [Google Scholar]
- 35.Leost, M., Schultz, C., Link, A., Wu, Y. Z., Biernat, J., Mandelkow, E. M., Bibb, J. A., Snyder, G. L., Greengard, P., Zaharevitz, D. W., et al. (2000) Eur. J. Biochem. 267, 5983-5994. [DOI] [PubMed] [Google Scholar]
- 36.Leclerc, S., Garnier, M., Hoessel, R., Marko, D., Bibb, J. A., Snyder, G. L., Greengard, P., Biernat, J., Wu, Y. Z., Mandelkow, E. M., et al. (2001) J. Biol. Chem. 276, 251-260. [DOI] [PubMed] [Google Scholar]
- 37.Chen, G., Huang, L. D., Jiang, Y. M. & Manji, H. K. (1999) J. Neurochem. 72, 1327-1330. [DOI] [PubMed] [Google Scholar]
- 38.Martinez, A., Alonso, M., Castro, A., Perez, C. & Moreno, F. J. (2002) J. Med. Chem. 45, 1292-1299. [DOI] [PubMed] [Google Scholar]
- 39.Ralph-Williams, R. J., Paulus, M. P., Zhuang, X., Hen, R. & Geyer, M. A. (2003) Biol. Psychiatry 53, 352-359. [DOI] [PubMed] [Google Scholar]
- 40.Brami-Cherrier, K., Valjent, E., Garcia, M., Pages, C., Hipskind, R. A. & Caboche, J. (2002) J. Neurosci. 22, 8911-8921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nair, V. D. & Sealfon, S. C. (2003) J. Biol. Chem. 278, 47053-47061. [DOI] [PubMed] [Google Scholar]
- 42.Kihara, T., Shimohama, S., Sawada, H., Honda, K., Nakamizo, T., Kanki, R., Yamashita, H. & Akaike, A. (2002) J. Neurosci. Res. 70, 274-282. [DOI] [PubMed] [Google Scholar]
- 43.Svenningsson, P., Tzavara, E. T., Carruthers, R., Rachleff, I., Wattler, S., Nehls, M., McKinzie, D. L., Fienberg, A. A., Nomikos, G. G. & Greengard, P. (2003) Science 302, 1412-1415. [DOI] [PubMed] [Google Scholar]
- 44.Vallar, L., Vicentini, L. M. & Meldolesi, J. (1988) J. Biol. Chem. 263, 10127-10134. [PubMed] [Google Scholar]
- 45.Emamian, E. S., Hall, D., Birnbaum, M. J., Karayiorgou, M. & Gogos, J. A. (2004) Nat. Genet. 36, 131-137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}