

Abstract
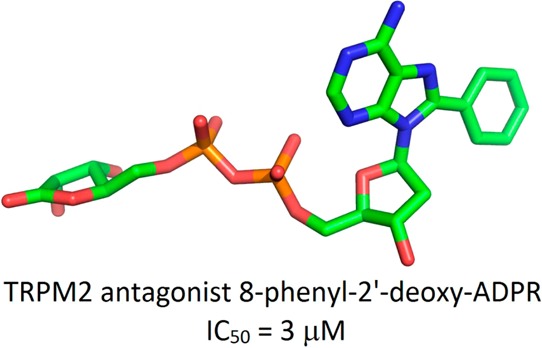
Adenosine 5′-diphosphoribose (ADPR) activates TRPM2, a Ca2+, Na+, and K+ permeable cation channel. Activation is induced by ADPR binding to the cytosolic C-terminal NudT9-homology domain. To generate the first structure–activity relationship, systematically modified ADPR analogues were designed, synthesized, and evaluated as antagonists using patch-clamp experiments in HEK293 cells overexpressing human TRPM2. Compounds with a purine C8 substituent show antagonist activity, and an 8-phenyl substitution (8-Ph-ADPR, 5) is very effective. Modification of the terminal ribose results in a weak antagonist, whereas its removal abolishes activity. An antagonist based upon a hybrid structure, 8-phenyl-2′-deoxy-ADPR (86, IC50 = 3 μM), is more potent than 8-Ph-ADPR (5). Initial bioisosteric replacement of the pyrophosphate linkage abolishes activity, but replacement of the pyrophosphate and the terminal ribose by a sulfamate-based group leads to a weak antagonist, a lead to more drug-like analogues. 8-Ph-ADPR (5) inhibits Ca2+ signalling and chemotaxis in human neutrophils, illustrating the potential for pharmacological intervention at TRPM2.
Introduction
Transient receptor potential (TRP) channels are six-transmembrane polypeptide subunits that assemble as tetramers to form cation-permeable pores.1 TRP subfamily melastatin, type 2 (TRPM2), is a Ca2+ permeant channel which is also permeant to Na+, K+, and Cs+ ions.2 TRPM2 is unique among the known ion channels as it contains a C-terminal domain which is homologous to NUDT9 ADPR-hydrolase, and this has led to considerable interest. The NUDT9-homology (NUDT9H) domain of human TRPM2 extends from residue 1236 to the C-terminus. NUDT9 was identified in an EST database screen for proteins with homology to the C-terminus of TRPM2. It is an enzyme of the Nudix family of pyrophosphatases, with adenosine 5′-diphosphoribose (ADPR, 1, Figure 1) as sole substrate. A NUDT9 crystal structure illustrated that this is a two-domain enzyme with a C-terminal ADPRase and N-terminal domain which enhances affinity for ADPR.3 Sequence alignment suggests that NUDT9H has the same features in TRPM2 channels and revealed that TRPM2 is gated by binding of ADPR,2,4 controlling cation entry through the plasma-membrane channel. Interestingly, TRPM2 is not a very effective hydrolase.5
Figure 1.

Formation of ADPR by CD38.
The TRPM2 channel is gated by micromolar cytosolic concentrations of ADPR (EC50 ∼ 100 μM). In contrast to other nicotinamide adenosine 5′-dinucleotide (NAD+) metabolites, such as cyclic adenosine 5′-diphosphoribose (cADPR, 3, Figure 1) and nicotinic acid adenine dinucleotide phosphate (NAADP), free ADPR has only recently been considered a second messenger.6 Cellular ADPR derives from NAD+ glycohydrolases such as CD38 that predominantly produce ADPR but also hydrolyze cADPR to ADPR (Figure 1) and other NAD+ metabolizing processes, including activation of poly-ADPribose polymerase (PARP) and poly-ADPribose glycohydrolase (PARG) pathways.7,8
TRPM2 is expressed in a variety of tissues, with highest transcript levels being detected in the brain, bone marrow, and cells of the immune system.9 Because the free cytosolic Ca2+ concentration ([Ca2+]i) sensitizes TRPM2 for activation by ADPR,10 resulting in a positive feedback loop and massive Ca2+ entry, it has been related to cell death from early on.11−13 Its activation downstream of reactive oxygen species suggested that it might be involved in the pathogenesis of neurodegenerative diseases,14 myocardial infarction,15 and type I and type II diabetes.16,17
More recent data point to an important physiological role for TRPM2 in cells of the innate immune system (reviewed in ref (18)). In macrophages/monocytes, TRPM2 is required for the production of the pro-inflammatory chemokine CXCL2 in response to reactive oxygen species19 and in the production of different cytokines in response to lipopolysaccharides.20 In neutrophils, TRPM2 is involved in Ca2+ signalling21,22 and the chemotactic response to fMLP.21 TRPM2 has also been shown to be involved in dendritic cell maturation and their chemotaxis to chemokines.23 Because TRPM2 is upregulated in T-lymphocytes after activation and appears to play a role in cytokine secretion and proliferation,24 it might also be important for the adaptive immune response.
In pancreatic β-cells, TRPM2 has been shown to be involved in modulation of insulin secretion.25,26 In neurons, it is much less clear whether TRPM2 also performs a physiological function besides its potential role in the pathophysiology of neurodegeneration. However, there have been initial reports of a contribution of TRPM2 in synaptic transmission.27,28
Research in these areas and identification of additional physiological processes involving ADPR/TRPM2 critically depend on the availability of specific small molecule tools. Over the years, some compounds that inhibit the TRPM2 channel have been described. The first TRPM2 inhibitor was flufenamic acid (FFA, IC50 70 μM),29 belonging to the fenamate group of nonsteroidal anti-inflammatory drugs. Other fenamates like mefenamic acid and niflumic acid also inhibit the channel although with lower potency (IC50 124 and 149 μM, respectively). A fenamate with reduced nonspecific effects, 2-(3-methylphenyl)aminobenzoic acid (3-MFA, IC50 76 μM), has recently been developed by Chen et al.30 Other TRPM2 inhibitors include the PLA2 inhibitor N-(p-amylcinnamoyl)anthranilic acid (ACA, IC50 1.7 μM)31 that shares the anthranilic acid motif with the fenamates, the antifungal imidazoles miconazole,25 econazole, and clotrimazole (IC50 < 3 μM for both),32 2-APB (IC50 ∼ 1 μM),25 and copper ions (IC50 2.6 μM).33 All of these pharmacological compounds apparently act as channel blockers and inhibit a variety of other ion channels (see ref (30) for citations). ACA affects other signal transduction pathways, thereby severely limiting its usefulness.
In 2007, some of us developed 8-Br-ADPR (4, Figure 1) as the first specific NUDT9H-TRPM2 antagonist.21 Both Ca2+ signalling and chemotaxis of murine neutrophils were very sensitive to 4.21 The introduction of an 8-bromo substituent turned the natural agonist ADPR into an antagonist, highlighting the sensitivity of ADPR to small structural modifications.
To better understand the ADPR/NUDT9H-TRPM2 system, it was necessary to develop further this early lead and begin to build a structure–activity relationship. Here, for the first time, we report a chemo-enzymatic approach involving chemical synthesis, coupled with use of Neurospora crassa NADase, to interrogate each major motif of the ADPR structure and subsequently evaluate the effect of these modifications on antagonist activity at the NUDT9 domain of the TRPM2 channel.
Results and Discussion
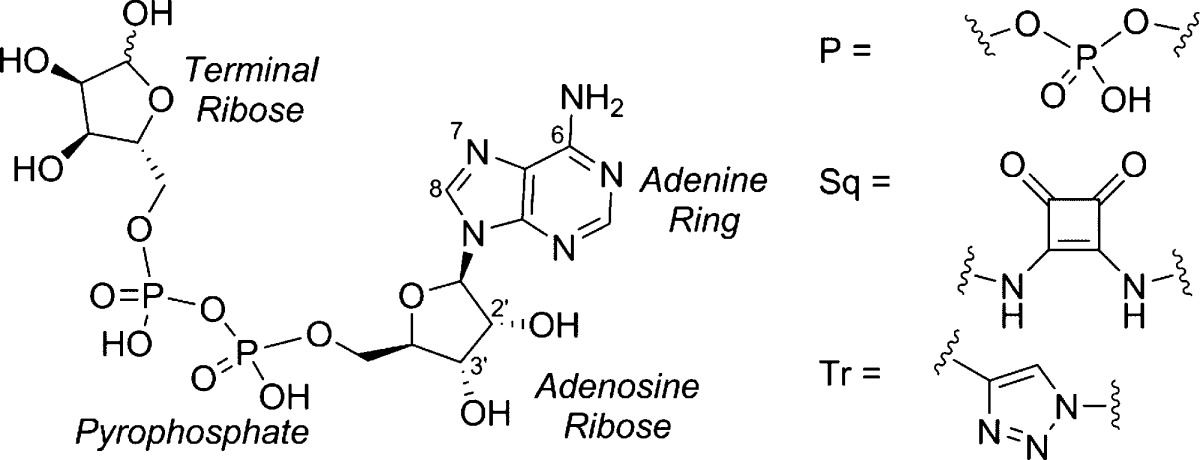
To explore the SAR of ADPR at TRPM2, we undertook a systematic approach, splitting ADPR into four major motifs: adenine, the adenosine ribose, the pyrophosphate, and the terminal ribose (Figure 1). We synthesized analogues with modifications in each of the four motifs to evaluate the resultant effect on antagonist activity and provide a comprehensive early SAR.
Adenine Modified Analogues: 8-Modified
The introduction of substituents at the 8-position of a nucleoside can directly affect the syn/anti orientation of the base residue. Following on from the discovery that 8-Br-ADPR (4) is an antagonist,21 we wanted to further explore the effect of other substituents in this area. We prepared three analogues with hydrophobic substitutions by Suzuki coupling (compounds 5–7, Scheme 1) and one more polar substitution in the form of 8-NH2-ADPR (12, Scheme 3).
Scheme 1. Synthesis of 8-Substituted ADPR Analogues.

Reagents and conditions: (i) NADase, Tris buffer (0.1 M, pH 7.2), rt, 92%; (ii) Br2, NaOAc buffer (0.5 M, pH 4), rt, 25%; (iii) Cs2CO3, Pd(OAc)2, TPPTS, X(BOH)2, MeCN–H2O, 125 °C, 5 min, 12–25%.
Scheme 3. Synthesis of 8-NH2–ADPR.

Reagents and conditions: (i) morpholine, dipyridyl disulfide, PPh3, DMSO, rt, 1 h then β-NMN+, MgSO4, 0.2 M MnCl2, 13%; (ii) NADase, Tris buffer (0.1M, pH 7.3), rt, 40%.
8-Br-ADPR (4) was prepared by treatment of commercially available NAD+ with NADase followed by bromination with Br2 in NaOAc buffer (pH 4). The Suzuki reaction of 4 with phenylboronic acid was not straightforward. The conditions that had been successfully applied to 8-Br-cIDPR34,35 did not result in any reaction with 4; possibly the less rigid phosphate conformation was able to bind with the palladium source, stalling the reaction. Re-examining the literature identified that Pd(OAc)2 as the palladium source, and the water-soluble triphenylphosphine TPPTS had been applied to similar compounds with success.36−38 These conditions were tested on a model system, the monophosphate 8-bromo-AMP 8, and the reaction with phenylboronic acid proceeded well. Unfortunately, this was not directly transferable to 4. As the reaction proceeded, a competing side reaction caused hydrolysis of the pyrophosphate, generating the 8-substituted AMP. Therefore, the reactions were carefully followed by HPLC, after which purification of the product by ion-exchange and reverse phase chromatography removed the monophosphate impurity and generated 8-phenyl-ADPR (5), 8-thiophenyl-ADPR (6), and 8-(3-acetylphenyl)-ADPR (7). Disappointingly, the reaction was not general for all boronic acids; only a fraction of those tried resulted in product, and those that failed just resulted in conversion of 4 to 8. To confirm that the lack of reactivity observed with the boronic acids was due to 4, the reaction with 4-phenylalanine (4-Ph-ala) boronic acid was repeated under the same conditions using 8 as reactant (Scheme 2). Unlike the reaction with 4 that generated no product, conversion was complete in 5 min and 8-(4-Ph-ala)-AMP 9 was isolated by combined ion-exchange and reverse phase chromatography.
Scheme 2. Synthesis of 8-(4-Ph-ala)-AMP.

Reagents and conditions: (i) TPPTS, Pd(OAc)2, Cs2CO3, MeCN–H2O, 18%.
8-NH2-ADPR (12) was prepared by the coupling of 8-NH2-AMP (10) with β-NMN+ to generate 8-NH2-NAD+ (11) that was then hydrolyzed by NADase to afford 12 (Scheme 3).
Adenine Modified Analogues: Base-Modified
To probe the other interactions between the adenine base and receptor, we also prepared analogues modified at C6 [IDPR (13) and 6-O-Me-IDPR (24)], N7 [7-deaza-8-Br-ADPR (15) and 7-deaza-IDPR (14)], and C2 [2-F-ADPR (16)]. Such modifications are challenging to introduce, as they usually require specialized preparation of a customized base that must then be coupled to the adenosine ribose and elaborated in a stepwise manner. Therefore, in the case of analogues previously known to the group, or in the literature, the NAD+ analogue was hydrolyzed enzymatically to generate the linear ADPR compound; NADase hydrolysis of NHD+ gave 13, 7-deaza-8-Br-NAD+39 gave 15, and 2-F-NAD+40 gave 16 (Figure 2). 7-Deaza-IDPR (14) was prepared by treatment of 7-deaza-NHD+ with ADPRC because in this instance the lack of N7 for cyclization results in the linear analogue on treatment with cyclase rather than a cyclic product.41
Figure 2.

Further purine modified ADPR analogues.
6-O-Methyl-IDPR (6-O-Me-IDPR, 24) was prepared from 6-chloropurine (17) and tetraacetyl-d-ribose (18) that were coupled together at N9 using Vorbrüggen conditions,42 followed by simultaneous deprotection of the three hydroxyls and conversion of the 6-chloro to a 6-methoxy substituent using sodium methoxide. Phosphorylation of the 5′-OH to generate 6-O-Me-AMP (21) was followed by activation of the phosphate, subsequent coupling to β-NMN+, and hydrolysis of the 6-O-Me-NHD+ (23) using NADase to generate the desired analogue 24 (Scheme 4).
Scheme 4. Synthesis of 6-O-Me-IDPR.

Reagents and conditions: (i) DBU, TMSOTf, MeCN, 60 °C, 1 h, 89%; (ii) 1 M NaOMe in MeOH, reflux, 1h, 95%; (iii) POCl3, TEP, H2O, 0 °C, 1 h; (iv) morpholine, dipyridyl disulfide, PPh3, DMSO, rt, 1 h; (v) 0.2 M MnCl2 in formamide, β-NMN+, rt, 16 h; (vi) NADase, Tris buffer (0.1M, pH 7.3), rt, 8% for steps iii–vi.
Adenosine Ribose Modified Analogues
Three modifications were prepared at the adenosine ribose: individual deletion of either the 2′- or 3′-hydroxyl group or complete removal of the ribose by replacing it with a more flexible and hydrophobic butyl chain. An acyclic analogue (acyclic-ADPR, 30) was prepared by introduction of an n-butyl chain at N9, via alkylation of 17 with chlorobutylacetate (25).43 Subsequent treatment with MeOH/NH3 simultaneously removed the acetate protecting group and substituted the 6-chloro substituent with a 6-amino group.44 Nucleoside analogue 27 was then phosphorylated under the standard POCl3/TEP conditions.41 Activation of the phosphate, followed by coupling with β-NMN+ and MnCl2 in formamide, delivered the desired linear acyclic-NAD+29. Treatment with NADase hydrolyzed the nicotinamide to afford 30 in very good yield (Scheme 5).
Scheme 5. Synthesis of Acyclic-ADPR.

Reagents and conditions: (i) DBU, DMF, 60 °C, 16 h, 48%; (ii) MeOH/NH3, 80 °C, 16 h, 88%; (iii) POCl3, TEP, 0 °C, 1 h, 72%; (iv) morpholine, dipyridyl disulfide, PPh3, DMSO, rt, 1 h then β-NMN+, MgSO4, 0.2 M MnCl2; (v) NADase, Tris buffer (0.1M, pH 7.3), rt, 81% for steps iv–v.
By removing an individual hydroxyl group, we hoped to uncover key interactions, i.e., the 2′-deoxy group in some cADPR analogues is critical for their antagonist activity.35 The preparation of 2′-deoxy-NAD+ (32) has previously been reported,45 and treating this with NADase generated 2′-deoxy-ADPR (33, Scheme 6).
Scheme 6. Synthesis of 2′-Deoxy-ADPR.

Reagents and conditions: (i) morpholine, dipyridyl disulfide, PPh3, DMSO, rt, 1 h then β-NMN+, MgSO4, 0.2 M MnCl2, 33%.; (ii) NADase, Tris buffer (0.1M, pH 7.3), rt, 85%.
The synthesis of 3′-deoxy-ADPR (43) was more challenging as 3′-deoxy-AMP (41) is not commercially available. Using a published synthesis,46 we prepared 3′-deoxyadenosine (37). However, in this instance, our usual phosphorylation conditions (POCl3, TEP) were not selective for the primary hydroxyl group, and a mixture of products was obtained. We therefore had to adopt a longer route, in which both hydroxyl groups were protected as the TBDMS ether, followed by selective cleavage of the primary ether and phosphitylation with a protected phosphoramidite, followed by oxidation under basic conditions using H2O2 and triethylamine. Simultaneous deprotection of the phosphate esters and 2′-hydroxyl group afforded 41 (Scheme 7).
Scheme 7. Synthesis of 3′-Deoxy-ADPR.

Reagents and conditions: (i) (a) triethylorthoacetate, pTsOH, MeCN, (b) AcBr, DCE, 29%; (ii) tBu3SnH, AIBN, toluene, 75%; (iii) NH3, MeOH, 90%; (iv) TBDMSCl, imidazole, DMF; (v) TFA-H2O, 0 °C; (vi) (a) iPr2N–P(OtBu)2, 5-Ph-1H-tetrazole, (b) H2O2, Et3N, 69%; (vii) TFA-H2O, 20 h, 90%; (viii) morpholine, dipyridyl disulphide, PPh3, DMSO, rt, 1 h then β-NMN+, MgSO4, 0.2 M MnCl2; (ix) NADase, Tris buffer (0.1M, pH 7.3), rt, 20% for steps viii–ix.
3′-Deoxy-AMP (41) was activated and coupled to β-NMN+ to generate 3′-deoxy-NAD+ (42), and the nicotinamide cleaved with NADase to generate 43.
Pyrophosphate-Modified Analogues
The diphosphate linkage is in most cases essential for biological activity. We prepared an analogue with increased negative charge, length, and flexibility in the form of three phosphate groups (ATPR, 47). ATPR (47) was prepared by degradation of cyclic adenosine triphosphoribose (cATPR,4746, Scheme 8). The cyclic precursor 46 was synthesized in two steps from the coupling of ADP (44) and β-NMN+ with EDC to afford the linear nicotinamide adenine trinucleotide (NAT, 45) that was cyclized by incubation with Aplysia cyclase as previously reported47 (Scheme 8).
Scheme 8. Synthesis of ATPR.

Reagents and conditions: (i) β-NMN+, MgCl2 (1M), HEPES (2M), EDC, 24 h, rt, 38%; (ii) Aplysia cyclase, HEPES (25 mM, pH 7.5), 3 days, rt, 39%; (iii) HEPES (25 mM, pH 7.5), reflux, 1 h, 54%.
In contrast, we developed analogues in which the pyrophosphate was replaced entirely by a phosphate or pyrophosphate bioisostere. The limited bioavailability of pyrophosphate-bearing compounds, their instability in a physiological environment, low membrane permeability, and difficulties in synthesis and scale up have led to considerable effort toward the development of isosteres. These include phosphate-containing, sulfur-containing, and carboxylate linkages as well as more unique cyclic mimics.48 However, no generally applicable bioisostere has so far been identified and with so many to choose from, our choices were based on ease of synthesis in the first instance. We therefore chose to utilize squarate, triazole, or sulfamate groups.
A small library of squarate compounds was prepared by coupling protected 5′-aminoadenosine 59 with a suitable squarate (Scheme 10). The analogue in which only the pyrophosphate is altered required 5′-aminoribofuranose (52) that was synthesized according to Scheme 9: One-step protection of the 1-, 2-, and 3-OH under acidic conditions generated 49.49 The 5-OH was then converted to the tosylate (50) that was displaced using sodium azide to generate 51 and the product reduced to the desired amine 52.
Scheme 10. Synthesis of the Squarate Series: ASqR, Cyclopentyl-ASq, Butyl-ASq, and Hexyl-ASq.

Reagents and conditions: (i) R-NH2, DIPEA, EtOH, rt, 1 h; (ii) diphenylphosphoryl azide, DBU, dioxane, rt, 3 h then TBAI, 15-crown-5, NaN3, reflux, 4 h, 88%; (iii) 10% Pd/C, EtOH, H2, rt, 16 h, 95%; (iv) DIPEA, EtOH, rt, 16 h; (v) 75% aq TFA, rt, 1 h.
Scheme 9. Preparation of 5′-Aminoribofuranose.

Reagents and conditions: (i) H2SO4, acetone, MeOH, 48 h, 98%; (ii) pTsCl, DMAP, pyridine, 5 h, 81%; (iii) NaN3, DMF, 120 °C, 16 h, 91%; (iv) PPh3, THF, 16 h, 85%.
Four amines were coupled to diethylsquarate 53 in the presence of DIPEA and EtOH; 52 gave 54, cyclopentylamine gave 55, butylamine gave 56, and hexylamine gave 57. The squarate compounds 54–57 were then coupled to protected 5′-aminoadenosine 59 that was synthesized in two steps from 2′,3′-O-isopropylideneadenosine (58) via the 5′-azido intermediate to generate 60–63. The isopropylidene protecting group was then cleaved under standard acidic conditions, except in the case of the ribosyl compound 60 for which heating to 60 °C for 16 h was required to remove the 1-O-methyl protecting group, to furnish ADPR analogues 64–67 (Scheme 10).
An analogue was prepared in which the pyrophosphate was replaced by a 1,2,3-triazole. Suitable precursors were prepared by generating an azide at the 5′ of adenosine and introducing a propargyl ether on the terminal ribofuranose for copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition (“click” cyclization). 2′,3′-O-Isopropylidene adenosine (58) was treated with diphenylphosphoryl azide followed by sodium azide to generate 5′-azidoadenosine 68 that was then brominated in the 8-position. The propargyl counterpart was prepared by deprotonating 49 with sodium hydride, followed by addition of propargyl chloride to prepare the protected 5-O-propargyl ether 70. The two precursors 69 and 70 underwent a 1,3-dipolar cycloaddition promoted by Cu(I), generated in situ from copper sulfate and sodium ascorbate, to afford 71. Triazole 71 underwent Suzuki coupling with phenyl boronic acid at the preinstalled 8-bromo substituent to afford 72. This was deprotected under acidic conditions to generate 8-phenyladenosine-1,4-triazole ribose (8-Ph-ATrR, 73, Scheme 11). Despite multiple attempts, it was not possible to introduce the propargyl ether on the 5′-position of adenosine as the nucleoside was degraded during deprotonation with sodium hydride.
Scheme 11. Synthetic Route to the Triazole Analogue 8-Ph-ATrR.

Reagents and conditions: (i) DPPA, DBU, dioxane, then TBAI, 15-crown-5, NaN3, reflux, 88%; (ii) Br2, NaOAc buffer (1M, pH 3.9), rt, 24 h, 99%; (iii) propargyl chloride, NaH, TBAI, DMF, rt, 16 h, 72%; (iv) CuSO4·5H2O, sodium ascorbate, tBuOH-H2O, rt, 16 h, 71%; (v) Na2Cl4Pd, PhB(OH)2, TPPTS, Na2CO3, MeCN–H2O, 80 °C, 1 h, 21%; (vi) 0.1 M H2SO4, 80 °C, 24%.
The third pyrophosphate replacement explored was a sulfonamide. The sulfonamide was introduced to the 5′-hydroxyl group of a protected adenosine by treatment of 58 with sulfamoyl chloride. However, introduction of a terminal ribose proved difficult, both in the preparation of a suitable amide or carboxylic acid and in the condensation reaction. Condensation of 74 with salicylic acid afforded an opportunity to introduce a terminal ribose surrogate and generate a known phosphate bioisostere. The protected sulfonamide analogue 75 was then deprotected under acidic conditions to afford salicyl-adenosine monosulfamide (Sal-AMS, 76, Scheme 12).
Scheme 12. Synthesis of the Sulfonamide Analogue Sal-AMS.

Reagents and conditions: (i) H2NSO2Cl, Et3N, DMA, 0 °C → rt, 16 h, 85%, (ii) salicylic acid, CDI, DBU, MeCN–DMF, 60 °C, 3 h, 69%; (iii) 75% aq TFA, 36%.
Terminal Ribose-Modified Analogues
A cyclopentyl group was introduced in place of the terminal ribose to assess its importance in binding to TRPM2. This has none of the polar hydroxyl groups but a similar overall shape. Analogues modified at the terminal ribose have the advantage that they do not require coupling using expensive β-NMN+. Cyclopentanol (77) was phosphorylated and deprotected to give the corresponding free-phosphate 79 that was coupled to AMP-morpholidate to afford cyclopentyl-ADP (80, Scheme 13).
Scheme 13. Synthesis of Terminal Ribose Analogue Cyclopentyl-ADP.

Reagents and conditions: (i) (iPr)2N–P(OBn)2, 5-Ph-1H-tetrazole, DCM then mCPBA, 86%; (ii) cyclohexene, MeOH–H2O, Pd(OH)2/C, 80 °C, 2 h; (iii) AMP-morpholidate, MgSO4, MnCl2 in formamide (0.2M), rt, 16 h, 14%.
Combined Modifications
After initial positive results, we designed analogues combining substitutions that appeared to improve antagonist properties. Cyclopentyl-8-Ph-ADP (84, Scheme 14) was prepared by introduction of an 8-bromo substituent to 58, followed by Suzuki cross coupling with phenyl boronic acid to introduce an 8-phenyl substituent in 82. The free 5′-OH was phosphorylated to afford 8-Ph-AMP (83) that was activated and coupled to 79 to afford 84 (Scheme 14).
Scheme 14. Synthesis of Cyclopentyl-8-Ph-ADP.

Reagents and conditions: (i) Br2, NaOAc buffer, 89%; (ii) Na2Cl4Pd, PhB(OH)2, TPPTS, Na2CO3, MeCN–H2O, 80 °C, 1h, 81%; (iii) 75% aq TFA, rt; (iv) POCl3, TEP, 0 °C, 1 h; (v) morpholine, PPh3, dipyridyldisulfide, DMSO, rt, 1 h then cyclopentylphosphate 79, MnCl2 in formamide, MgSO4, rt, 16 h, 12%.
8-Phenyl-2′-deoxyadenosine-5′-diphosphoribose (8-Ph-2′-deoxy-ADPR, 86) was prepared by degradation of 8-phenyl-2′-deoxy-cADPR35 (85, Scheme 15).
Scheme 15. Synthesis of 8-Ph-2′-deoxy-ADPR.

Reagents and conditions: (i) KH2PO4 buffer, 70 °C, 2.5 h, 39%.
Pharmacology
To evaluate ADPR analogues for antagonist activity, HEK293 clones with stable expression of human TRPM2 were established. Clones derived from HEK293 cells transfected with an expression vector for human TRPM2 were selected according to their increase in [Ca2+]i in response to hydrogen peroxide (Figure 3A). Further, expression of TRPM2 at the protein level was confirmed in responsive clones by Western blot analysis (Figure 3B). The specificity of the antibody was checked by preincubation with a blocking peptide (Figure 3B, right). For different clones, maximum [Ca2+]i response to hydrogen peroxide increased with magnitude of TRPM2 expression (Figure 3).
Figure 3.
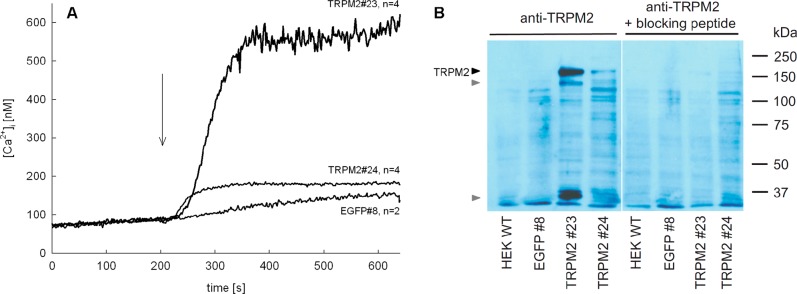
Characterization of HEK 293 cell clones stably expressing TRPM2. (A) Cell lines from limiting dilution cloning of transfected HEK293 cells were selected by measuring their [Ca2+]i response to H2O2 (300 μM, time of addition indicated by arrow). Characteristic tracings of different HEK293 clones (TRPM2#23, TRPM2#24) transfected with a vector for coupled expression of TRPM2 and EGFP and a HEK293 clone (EGFP#8) transfected with the empty vector (expressing EGFP) are shown (n = 2–4, mean). (B) TRMP2 protein expression of clones TRPM2#23, TRPM2#24, EGFP#8, and wild-type HEK293 cells was determined by Western blot analysis. Specificity of TRPM2 detection was tested by preincubation of the primary antibody with a corresponding blocking peptide (right part). Specific bands of full length TRPM2 (black arrow) and degradation products (gray arrows) are marked. None of these bands were seen in wild-type HEK293 cells, in EGFP#8 control cell line, or in the presence of blocking peptide.
Whole-cell patch-clamp experiments were performed to evaluate ADPR analogues using clone #24 showing intermediate expression of TRPM2. To avoid excessive inward currents, the sodium chloride within the bath solution for the patch clamp experiments was replaced by N-methyl-d-glucamine (NMDG) hydrochloride that does not pass through the TRPM2 channel. Under these conditions, activation of TRPM2, after break-in with 100 μM ADPR (1) in the pipet solution, resulted in a small but measurable inward current of Ca2+ and a more pronounced outward current mainly carried by K+ ions (Figure 4A, gray line) similar to previous observations.50 In contrast, no such currents were observed when a control cell line stably expressing EGFP (EGFP#8, Figure 4A, dashed gray line) was used instead of TRPM2#24 or the pipet was filled with a solution devoid of 1 (Figure 4A, dashed black line). As a read-out for TRPM2 activation the maximum net outward current at +15 mV obtained from repetitive voltage ramps between −85 and +20 mV was chosen.
Figure 4.

Stimulation of clone TRPM2#24 by infusion of 100 μM ADPR. Membrane currents of TRPM2#24 and EGFP#8 were measured in the whole-cell configuration using repetitive voltage ramps from −85 to +20 mV. Experiments were carried out with 1 mM Ca2+ and 140 mM NMDG instead of sodium in the extracellular solution. The free [Ca2+]i in the pipet solution was buffered to 200 nM by EGTA. (A) Characteristic tracings of membrane current at +15 mV and at −80 mV over time from representative experiments of TRPM2#24 (gray and black broken line) and EGFP#8 (gray broken line) stimulated with 100 μM ADPR (1) or vehicle (buffer) are shown. (B) Current–voltage relationship of the representative experiments shown in (A) at ramp 30 (corresponding to maximum ADPR induced current of TRPM2#24). (C) Maximum potassium outward current at +15 mV induced by infusion of buffer or 100 μM ADPR in TRPM2#24 or EGFP#8 indicated as mean ± SEM (n = 7–11), **, P < 0.01 (t test).
The first specific ADPR antagonist 8-Br-ADPR (4)21 significantly inhibited TRPM2 activation when applied at a 9-fold excess over ADPR (1). Thus, alongside the positive control (100 μM of 1) in most experiments, a combination of 100 μM 1 and 900 μM 4 was included as an inhibitor control (Figure 5). Most ADPR analogues were tested for antagonism to 100 μM of 1 at a concentration of 900 μM. Because some compounds were poorly soluble in water, they were resolubilized in DMSO. To avoid high concentrations of DMSO in the pipet solution, these compounds were tested for antagonist activity at a concentration of 100 μM in the presence of 100 μM of 1.
Figure 5.

Effect of purine-modified ADPR analogues on activation of TRPM2 by 1. Maximum outward currents were recorded as outlined in the Experimental Section. Pipet solutions contained either no nucleotide or 100 μM ADPR (1) alone or in combination with the indicated ADPR analogue. 8-Br-ADPR (4) was included as inhibitor control. Bars are grouped to indicate the position of purine modification (label on top). A horizontal dashed line marks the mean of the control (100 μM of 1). Data are shown as mean ± SEM (number of experiments denoted in or on top of the bar) and were analyzed by a nonparametric one-way ANOVA (Kruskal–Wallis test) followed by comparison against control (100 μM of 1) using Dunn’s correction for multiple testing. Results significantly different from control (p < 0.05) are indicated by asterisks.
Purine-Base Modifications
The ADPR analogues with modifications at C2 of the purine ring [2-F-ADPR (16)] or C6 [IDPR (13) and 6-O-Me-IDPR (24)] did not antagonize activation of TRPM2 by 1 (Figure 5). ADPR analogues with modifications at N7 of the purine ring [7-deaza-IDPR (14) and 7-deaza-8-Br-ADPR (15)] were also without significant effect. In contrast, introduction of a bulky, hydrophobic substitution at C8 as in 8-thiophenyl-ADPR (6), 8-(3-acetylphenyl)-ADPR (7), or 8-phenyl-ADPR (5) resulted in ADPR analogues that effectively antagonized TRPM2 activation at 900 μM (Figure 5), while a compound with a small polar substitution at C8, e.g., 8-NH2-ADPR 12, was inactive as an antagonist. This stark contrast in activity may be due to the alteration of the purine base conformation from anti to syn upon substitution with large groups in the 8-position.41 In contrast to an 8-bromo or 8-amino substituent, the 8-aryl group introduces a new conformational parameter, as not only will such groups tend to a predominantly syn configuration around the glycosidic bond, but they also introduce the possibility of torsional conformational effects of, as yet, unknown significance.
Adenosine Ribose Modifications
The adenosine ribose was replaced by an alkane chain, resulting in the weak antagonist acyclic-ADPR 30 that showed nearly complete inhibition at 900 μM, whereas at 100 μM, no inhibition of the ADPR induced current was observed, indicating a steep dose response relationship with an IC50 value in the upper micromolar range (Figure 6). It was not possible to measure the antagonist activity of 2′-deoxy- or 3′-deoxy-ADPR (33, 34). In contrast, 2′-deoxy-AMP (31) was active as a weak antagonist, whereas AMP 88 showed no antagonist activity in our hands (Figure 7A). There are conflicting reports on the antagonistic effect of 88; while it was previously described to inhibit TRPM2 activation completely with an IC50 value of 70 μM in whole cell patch clamp experiments,51 other studies have not shown any inhibition of ADPR-mediated currents in cell free patches from Xenopus oocytes expressing TRPM2, even at 60-fold excess of 88 over 1.52 Interestingly, the combination of this 2′-deoxy-modification and the 8-phenyl substitution at C8 in 8-Ph-2′-deoxy-ADPR 86 also completely inhibited activation of TRPM2 when applied at a concentration of 900 μM. Full concentration–response relationships for 4 and 86 are displayed in Figure 10. The adenosine ribose, particularly the C2′ position, seems to be important for binding of analogues to NUDT9H; possibly the absence of the hydroxyl group results in lower steric hindrance or lower repulsion between ligand and channel ultimately resulting in stronger binding.
Figure 6.

Effect of adenosine ribose-modified ADPR analogues on TRPM2 activation by ADPR (1). Maximum outward currents were recorded as outlined in the Experimental Section. Pipet solutions contained either no nucleotide or 100 μM of 1 alone or in combination with the indicated ADPR analogue. Data are shown as mean ± SEM (number of experiments denoted in or on top of the bar) and were analyzed by a one-way ANOVA followed by comparison against control (100 μM of 1) using Dunn’s correction for multiple testing. Results significantly different from control (p < 0.05) are indicated by asterisks.
Figure 7.

Effect of pyrophosphate-modified ADPR analogues on TRPM2 activation by ADPR (1) Maximum outward currents were recorded as outlined in the Experimental Section. Pipet solutions contained either no nucleotide or 100 μM of 1 alone or in combination with the indicated ADPR analogue. (A) Analogues that were soluble in buffer. (B) Analogues that were only soluble on addition of DMSO. Because squaryl and triazole compounds (64–67, 73) were resolubilized in DMSO, the pipet solution contained 0.1% DMSO. For the control conditions, DMSO was included in the pipet solution at the same final concentration. Data are shown as mean ± SEM (number of experiments denoted in or on top of the bar) and were analyzed by a nonparametric one-way ANOVA (Kruskal–Wallis test) followed by comparison against the respective control (100 μM of 1) using Dunn’s correction for multiple testing. Results significantly different from control (p < 0.05) are indicated by asterisks.
Figure 10.

Concentration–response relationship of ADPR antagonists. Because these data were recorded over a significant time frame, a certain variation of the mean currents obtained when using a pipet solution with 100 μM ADPR (1) occurred. In detail, the mean value ± SEM [in nA] of 100 μM ADPR (1) infusion was: for 5, 1.17 ± 0.13 (n = 16); 7, 1.25 ± 0.14 (n = 22); 6, 1.70 ± 0.25 (n = 20); 86, 1.23 ± 0.22 (n = 15); 4, 1.13 ± 0.14 (n = 16); and 84, 1.44 ± 0.20 (n = 8). To construct concentration–response curves and determine IC50 values, data were therefore normalized to the mean response of the control (100 μM of 1) for the corresponding set of experiments. Outward current as percentage of control is indicated as mean ± SEM. For estimation of IC50 values the level of 50% of control is marked by a dashed line. Except for 4, data were fitted to a three parameter logistic function constraining the top and bottom values to 100% and 0%, respectively. For 4, the course of the concentration–response curve was estimated by shifting a logistic function to the right.
Pyrophosphate Modifications
Introduction of an additional phosphate in ATPR (47) generated an analogue that appears to have some antagonist effect at 900 μM, although this was not statistically significant (Figure 7a). It remains unclear whether this is a result of increased length or the increased charge, the latter unattractive from a drug discovery point of view. Squarate analogues 64–67 and triazole analogue 73 could only be tested at 100 μM because the usual method of solubilizing the compounds in HEPES buffer was not successful, so DMSO was used. However, only as much as 0.1% (v/v) DMSO was tolerated in the pipet solution, allowing a maximum concentration of 100 μM to be reached. However, at this concentration, none of these compounds showed antagonist activity (Figure 7B). The acyl sulfamate analogue 76 was soluble in aqueous media and showed noticeable antagonist activity at 900 μM. Although this was not statistically significant, this analogue (in particular, because it combines both a neutral pyrophosphate bioisostere and simple substitution of the terminal ribose) represents a tractable lead toward future, more drug-like ADPR antagonists. Interestingly, Sal-AMS has previously been prepared as an antibiotic lead for the inhibition of siderophore biosynthesis in tuberculosis and plague.53,54 However, in ADPR, the pyrophosphate moiety is more than simply a “spacer” between the two riboses; to date, no analogues with a pyrophosphate substitution have shown a significant antagonist effect. These results indicate that the negative charge at the pyrophosphate is important for interaction with the NUDT9H domain. This finding is of great significance for all applications of such an antagonist in cell and animal models because the pyrophosphate moiety potentially may be attacked by pyrophosphatase enzyme activity. Thus, analogues containing the pyrophosphate moiety may suffer from a short half-life. Metabolically stable phosphonate-type analogues or alternative pyrophosphate replacement groups might be a future solution for this problem.55
Terminal Ribose Modifications
A number of analogues were prepared or are commercially available, including completely truncated “AMP-like” compounds. The latter are attractive analogues due to the presence of only one phosphate group; they are therefore likely to be more membrane permeant and are more easily synthetically accessible as they do not require the difficult coupling to expensive reagent β-NMN. In contrast to AMP (88) that was ineffective, 2′-deoxy-AMP (31) and 8-(4-phenylalanine)-AMP (9) suppressed activation of TRPM2 by 1 when applied at 900 μM (Figure 8). However, when equimolar to 1 (100 μM each), the antagonist effect was lost, indicating a steep concentration–response relationship with an IC50 value considerably higher than 100 μM. Interestingly, 8-Cl-AMP (87) showed no antagonist effect at 900 μM (Figure 8). The observed variation among AMP analogues suggests that 8-substitution plays a role in introducing antagonist properties, but these are much reduced in magnitude compared to the full-length analogues.
Figure 8.

Effect of AMP analogues on TRPM2 activation by ADPR (1). Maximum outward currents were recorded as outlined in the Experimental Section. Pipet solutions contained either no nucleotide and 100 μM of 1 alone or in combination with the indicated ADPR analogue. Data are shown as mean ± SEM (number of experiments denoted in or on top of the bar) and were analyzed by a nonparametric one-way ANOVA (Kruskal–Wallis test) followed by comparison against control (100 μM of 1) using Dunn’s correction for multiple testing. Results significantly different from control (p < 0.05) are indicated by asterisks.
Replacing only the terminal ribose, while maintaining the complete pyrophosphate, generated the antagonist cyclopentyl-ADP (80) that showed partial inhibition at 900 μM (Figure 9). This suggested that the hydroxyl groups on the terminal ribose may not be critical for antagonist activity. In contrast, the parent ADP (44) was inactive, suggesting that the five-membered ring does play a critical role in filling the binding site. Combining the terminal cyclopentyl substituent with the 8-phenyl substituent afforded cyclopentyl-8-phenyl-ADPR (84), which showed significant antagonist activity when applied in an equimolar concentration to 1 (100 μM each, Figure 9). Replacing the terminal ribose with a cyclopentyl group is attractive because, as well as removing any complication of both anomeric center stereochemistry and equilibrium with a ring-opened ribose form, it negates the possibility of intramolecular attack on the pyrophosphate by a ribose hydroxyl group, which may lead to instability. Thus, analogue 84 was analyzed in more detail (see Figure 10).
Figure 9.

Effect of terminal ribose-modified ADPR analogues on TRPM2 activation by ADPR (1). Maximum outward currents were recorded as outlined in the Experimental Section. Pipet solutions contained either no nucleotide or 100 μM of 1 alone or in combination with the indicated ADPR analogue. 8-Br-ADPR (4) was included as inhibitor control. Data are shown as mean ± SEM (number of experiments denoted in or on top of the bar) and were analyzed by a one-way ANOVA followed by comparison against control (100 μM of 1) using Dunn’s correction for multiple testing. Results significantly different from control (p < 0.05) are indicated by asterisks.
The compounds that showed enhanced antagonist activity were assessed in a concentration–response manner using patch-clamp experiments (Figure 10). The IC50 value of the previously reported 8-Br-ADPR (4) was around 300 μM. 8-Thiophenyl-ADPR (6) and 8-(3-Ac-Ph)-ADPR (7) showed a 6-fold increase in potency (IC50 values of 51 and 49 μM, respectively). This was further improved to 15 μM in cyclopentyl-8-phenyl-ADP (84) and to 11 μM in 8-phenyl-ADPR (5). Combining the 8-phenyl substitution on the adenine ring with removal of the hydroxyl group on C2′ of the adenosine ribose further enhanced the antagonist effect, resulting in an IC50 value of 3 μM for 8-Ph-2′-deoxy-ADPR (86), the best evaluated TRPM2 antagonist to date.
The five most potent inhibitors are all substituted with an aromatic ring at the 8-position of adenine. Comparison of the three 8-aromatic analogues 5–7 suggest that appended groups (e.g., 3-acetylphenyl-) or alternative ring sizes (e.g., thiophene) do not improve antagonist effects, although this has not been fully exploited due to the difficulties encountered with Suzuki chemistry to prepare these analogues. The small improvement in activity observed for cyclopentyl-8-phenyl-ADP (84) suggests that the terminal ribose is not critical.
Importantly, we also studied the biological activity of one of the best antagonist compounds with a low IC50 value, in neutrophil Ca2+ signalling and chemotaxis; a system where we have already established the physiologically important role of TRPM2.21 8-Phenyl-ADPR (5) was accessed via 4, as opposed to 8-phenyl-2′-deoxy-ADPR (86), which was prepared by degradation of its valuable cyclic counterpart. Thus, we assessed the effect of 5 on Ca2+ signalling and chemotaxis in human neutrophils stimulated with the chemotactic peptides A5 or fMLP (Figures 11 and 12).
Figure 11.

Partial antagonism of Ca2+ signalling in human neutrophils upon preincubation with 8-Ph-ADPR (5). Primary human PMN (polymorphonuclear neutrophils) were loaded with Fura-2/AM and analyzed by ratiometric Ca2+ imaging. Cells were stimulated by addition of 10 μM A5 peptide (A, gray line) or 1 μM fMLP (B, gray line). To determine the effect of 5 on fMLP or A5 peptide induced Ca2+ signalling, cells were preincubated with 100 μM of 5 for 15 min at rt (A,B black line). Data of single cells were synchronized to the first maximum after addition of A5 peptide or fMLP and represent mean values of Fura-2 ratio (primary y-axis). Corresponding [Ca2+]i in nM are shown on secondary y-axis. The number of cells for each condition (n) is indicated in the inset legend.
Figure 12.

Partial antagonism of chemotaxis in human neutrophils upon preincubation with 8-Ph-ADPR (5). Migration of freshly isolated primary human PMN was analyzed as detailed in the Experimental Section. (A) buffer, (B) buffer and 1.5 mM EGTA, (C) 50 nM fMLP gradient (indicated in green), (D) 50 nM fMLP gradient and 1.5 mM EGTA, (E) 50 nM fMLP gradient and 5. All cells were standardized to the center of the coordinate system as a start point. Black lines indicate trajectories of single cells during measurement. Blue dots represent the position of each cell at the end of measurement. The center of mass as mean of the position at the end of the measurement of all cells is marked by a red cross with ∥ showing mean movement in the y direction along fMLP gradient and ⊥ showing mean movement in the x-direction. Cells shifted in presence of fMLP (C) 49.4 μm, fMLP/EGTA (D) 32.6 μm, and fMLP/5 (E) 34.4 μm in the ∥ direction. In the absence of fMLP, cells showed less migration in the ∥ direction (buffer control (A), 9.8 μm, buffer/EGTA; (B), 5.6 μm).
To investigate the effect of NUDT9H-TRPM2 antagonism on Ca2+ signalling, freshly isolated primary human neutrophils from healthy donors and loaded with Fura2 were stimulated either by 1 μM fMLP (high affinity ligand for human FPR1) or 10 μM of the FPR2 specific A5 peptide.56,57 When the neutrophils were preincubated with 100 μM of 5 before application of the chemoattractant, the increase in [Ca2+]i upon stimulation was significantly reduced for both ligands (31.8% for fMLP and 31.4% for A5 peptide, Figure 11), indicating that like in mouse neutrophils,21 TRPM2 is activated by 1 and contributes to the Ca2+ signal by mediating Ca2+ entry downstream of chemoattractant receptors.
To analyze the effect of 8-phenyl-ADPR (5) on chemotaxis, we analyzed the migratory pattern of freshly isolated neutrophils from healthy donors. In the absence of a chemoattractant, the cells showed spontaneous migration that was largely nonvectorial as the center of gravity of all the cells changed only slightly (Figure 12A). This basal migration was nearly abolished when extracellular Ca2+ was removed by EGTA (Figure 12B), demonstrating the requirement for millimolar Ca2+ in the extracellular space. Upon a fMLP gradient, the cells moved toward the higher attractant concentration (Figure 12C). However, if extracellular Ca2+ was removed while the fMLP gradient was applied, directed migration was reduced but was still higher as compared to controls (compare parts D to C and A of Figure 12), indicating that fMLP induced chemotaxis partially depends on a high extracellular Ca2+ concentration. Importantly, when the neutrophils were preincubated with 100 μM of 5 and then placed within the fMLP gradient, the decrease in directed migration was similar to migration in the experiment with EGTA (compare parts E to D of Figure 12). Taken together, these data demonstrate the importance of ADPR mediated TRPM2 activation in directing the chemotactic response of neutrophils.
8-Phenyl-ADPR (5) partially reduced Ca2+ signalling by the FPR1 ligand fMLP and the FPR2 specific agonist A5 peptide in human neutrophils and also partially inhibited the directional migration in a gradient of fMLP. These results demonstrate that the novel ADPR analogue 5 not only inhibits activation of TRPM2 directly on target in the “cell free” whole-cell patch clamp system but also affects TRPM2-dependent processes in a cellular environment, making it a useful tool for the investigation of ADPR/TRPM2 dependent processes in other cell systems and physiological processes.
To date, to the best of our knowledge, 8-Br-ADPR (4) has been the only specific antagonist available for activation of TRPM2 by ADPR (1).21 Therefore, for the first time, our systematic study of ADPR antagonism allows us to build up an early SAR profile for the inhibition of TRPM2 by ADPR analogues (Figure 13 and Table1) and provides more potent agents for the study of TRPM2.
Figure 13.

Early structure–activity relationship for ADPR antagonism.
Table 1. Summary of Analogues and Antagonist Activity at TRPM2f.
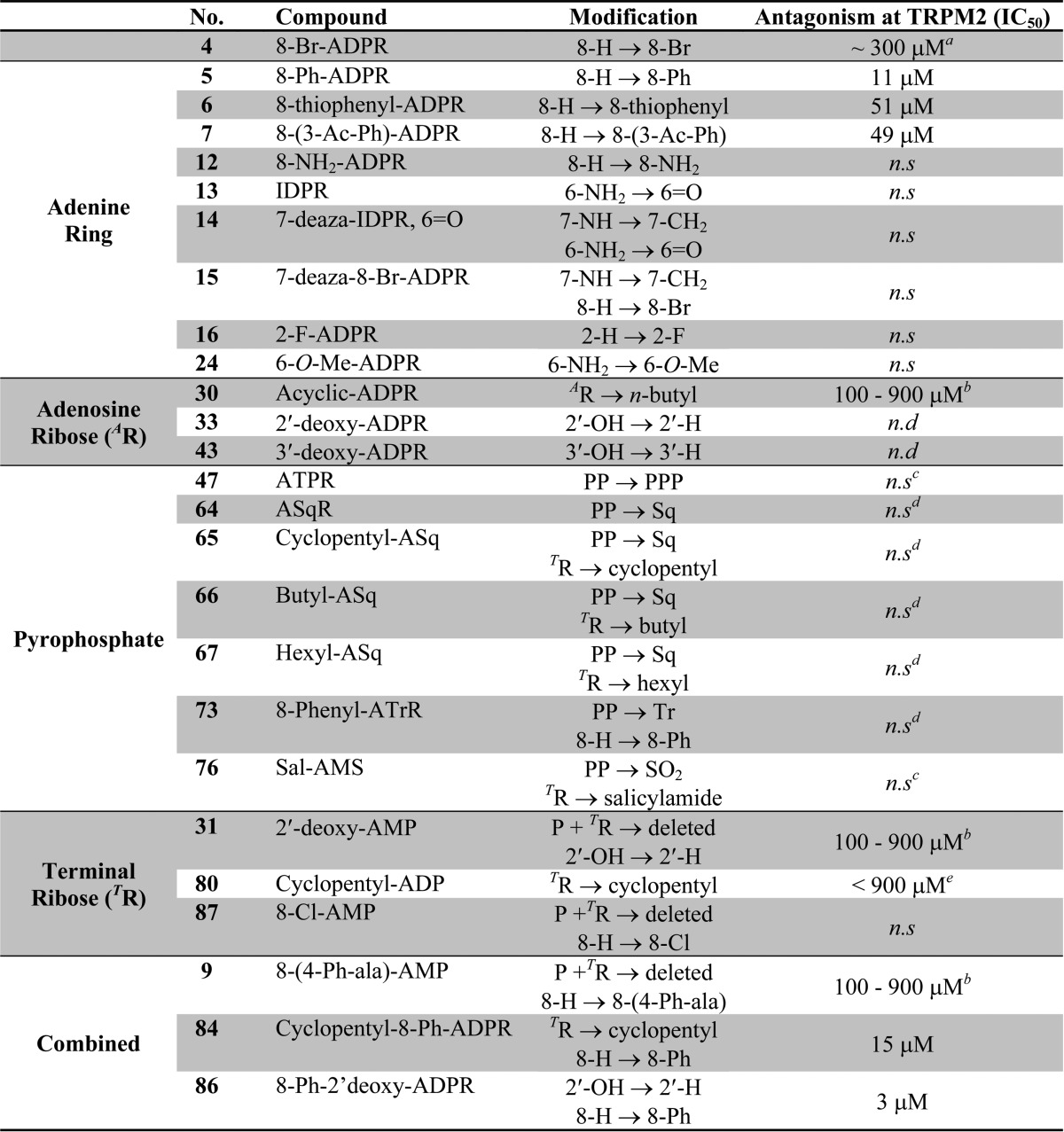
See ref (21).
No significant inhibition at 100 μM; compounds were not evaluated further.
Antagonistic effect observed but not statistically significant; see relevant figure.
Could only be tested at 100 μM due to solubility difficulties.
Partial inhibition was observed at 900 μM; not evaluated further.
n.d. = not determined. n.s. = no significant antagonist effect observed.
Conclusion
In this study, we systematically derivatized the new second messenger ADPR and monitored the antagonist activity of the derivatives at TRPM2 during activation by ADPR. The replacement of bromine by the bulky, flat, and torsionally mobile, hydrophobic phenyl substitutent at C8 of adenine together with a 2′-deoxy motif at the adenosine ribose converted the poor, but specific, antagonist 8-Br-ADPR (4) into the highly active specific NUDT9H-TRPM2 antagonist 8-Ph-2′-deoxy-ADPR (86). We have established that the amino/imino groups at C6 and the N7 nitrogen within the adenine base are important for antagonist activity. Likewise, the charges of the pyrophosphate bridge are critical for retaining antagonist activity because the bridge cannot be replaced by another noncharged linker with similar dimensions. However, substitution of the pyrophosphate by a sulfamate in Sal-AMS did lead to some antagonist activity and this compound is a promising lead for further development of neutral and more drug-like antagonists. Finally, we have also established that the terminal ribose is important but can be partially replaced by a cyclopentyl moiety. This SAR provides a key starting point for further development of potent modulators of TRPM2 and development of chemical biology tools to further probe this new signalling pathway.
Experimental Section
Chemistry
The purity of new tested compounds was determined to be ≥95% by analytical HPLC. HPLC analyses were carried out on a Waters 2695 Alliance module equipped with a Waters 2996 photodiode array detector (210–350 nm). The chromatographic system consisted of a Hichrom guard column for HPLC and a Phenomenex Synergi 4μ MAX-RP 80A column (150 mm × 4.60 mm), eluted at 1 mL/min with the following ion-pair buffer: 0.17% (m/v) cetrimide and 45% (v/v) phosphate buffer (pH 6.4) in MeOH.
General Procedure A: NADase Hydrolysis of NAD+ Analogues
NADase (from Neurospora crassa; Sigma; 0.9 U) in Tris-HCl buffer (2 mL, 0.1 M, pH 7.2–7.4) was added to a solution of the NAD+ analogue (30 μmol) in Tris-HCl buffer (1 mL, 0.1 M, pH 7.2–7.4). The reaction mixture was stirred at 37 °C until complete, followed by purification of the product by ion-exchange (Q-sepharose) chromatography eluting with a gradient (0–50%) of TEAB (1.0 M) in Milli-Q water. The appropriate fractions were collected and evaporated under reduced pressure. The residue was coevaporated several times with MeOH to remove excess TEAB to yield the desired ADPR analogue as a glassy solid in its triethylammonium (TEA) form.
General Procedure B: Suzuki Coupling
Cesium carbonate (0.24 mmol, 2.9 equiv) was added in one portion to a stirred solution of the corresponding boronic acid (0.103 mmol, 1.2 equiv), palladium acetate (0.004 mmol, 0.05 equiv), TPPTS (0.02 mmol, 0.24 equiv), and 8-Br-ADPR (TEA salt, 0.0823 mmol) in degassed MeCN–H2O (1:2 v/v; 2.4 mL) under an argon atmosphere. The reaction mixture was heated at 125 °C for 5 min; the reaction mixture turned black and HPLC analysis confirmed the reaction was complete. The reaction mixture was cooled to room temperature, QuadraPure TU (∼100 mg) added, and the mixture stirred for 16 h. The mixture was filtered and evaporated under reduced pressure to leave a crude product that was purified by ion-exchange (Q-Sepharose) chromatography eluted with a gradient (0–40%) of TEAB (1.0 M) in Milli-Q water followed by reverse phase (RP-18) column chromatography, eluted with 0–20% MeCN in TEAB (0.05 M) to isolate the desired 8-substituted ADPR product.
General Procedure C: Squarate Chemistry
To a solution of amine (0.443 mmol) and DIPEA (42 μL, 0.239 mmol) in EtOH (5 mL) was added diethylsquarate (72 μL, 0.487 mmol). The reaction was stirred at rt until TLC indicated completion of the reaction (ca. 1 h). The solvent was removed under reduced pressure, and the residue was purified on an Isco chromatographic system (DCM–acetone, 8:2 v/v) to yield the desired product.
General Procedure D: Isopropylidene Deprotection
The protected compound (0.1 mmol) was stirred in a 75% aq TFA solution (5 mL) at rt for 1 h. The solvents were evaporated under reduced pressure, and the residue was coevaporated with MeOH to remove any residual TFA. The remaining residue was purified on an Isco purification system (DCM–MeOH, 8:2 v/v) to yield the desired compound as a white solid.
Synthesis of 8-Modified ADPR Analogues
8-Phenyl Adenosine Diphosphoribose (8-Ph-ADPR, 5)
Phenylboronic acid (0.103 mmol, 21 mg) and 8-Br-ADPR 4 (TEA salt, 0.0823 mmol) were reacted under the general protocol B, yielding 8-Ph-ADPR (TEA salt, 6.0 by 1H NMR) (18 mg, 14.3 μmol, 19%) as a colorless solid. 1H (400 MHz, D2O) δ 8.14 (br s, 1H, H-2), 7.56–7.48 (br m, 5H, Ph), 5.78 (d, 1H, J = 5.9, H-1′), 5.16 (br, 0.4H, H-1″), 5.08 (br, 1H, H-2′), 5.05 (br, 0.6H, H-1″), 4.30 (br, 1H, H-2″), 3.82–4.18 (m, 8H, H-3′, H-4′, 2 × H-5′, H-3″, H-4″ and 2 × H-5″). 13C (100 MHz, D2O) δ 154.5, 153.4, 152.3, 150.3 (C-2), 131.3 (Ph, C–H), 129.7 (Ph, C–H), 129.2 (Ph, C–H), 127.9, 118.6, 101.3 (Cα/β-1″), 96.5 (Cα/β-1″), 89.0 (C-1′), 83.2 (C-4′ or Cα/β-4″), 81.9 (C-4′ or Cα/β-4″) 81.3 (C-4′ or Cα/β-4″), 75.3, 70.8, 70.6, 70.5 (C-2′), 70.2, 69.7 (C-2″), 66.5 (C-5′ or Cα/β-5″) 65.4 (C-5′ or Cα/β-5″) and 65.5 (C-5′ or Cα/β-5″); δP (162 MHz, D2O) −10.1 (very br). HRMS (ES–) calcd for C21H26N5O14P2, 634.0957 M–; found, 634.0970; and RT = 26.7 min.
8-(3-Acetylphenyl)adenosine Diphosphoribose [8-(3-Ac-Ph)-ADPR 7]
3-Acetylphenylboronic acid (0.1 mmol, 17 mg) and 8-Br-ADPR 4 (2 equiv TEA salt, 67 mg, 0.079 mmol) were reacted under the general protocol B to yield 8-(3-Ac-Ph)-ADPR (TEA salt, 3.0 equiv by 1H NMR) (11 mg, 9.3 μmol, 12%) as a colorless solid. 1H (400 MHz, D2O) δ 8.13–8.19 (m, 2H, Ar 2-H and H-2), 8.05 (d, 1H, J = 6.9, Ar 6-H), 7.86 (d, 1H, J = 6.9, Ar 4-H), 7.62 (t, 1H, J = 6.9, Ar 5-H), 5.78 (d, 1H, J = 5.9, H-1′), 5.19–5.23 (m, 1.4H, (1H) H-2′ and (0.4H) H-1″), 5.09 (d, 0.6H, J = 2.4, H-1″), 4.37–4.41 (m, 1H, H-2″), 3.87–4.21 (m, 8H, H-3′, H-4′, 2 × H-5′, H-3″, H-4″ and 2 × H-5″) and 2.62 (s, 3H,ArCOCH3). 13C (100 MHz, D2O) δ 202.4 (C=O), 155.1, 152.8 (C-2), 151.9, 150.1, 136.7, 134.4 (Ar 4-C), 130.6 (Ar 6-C), 129.6, 129.4 (Ar 5-C), 128.3 (Ar 2-C), 118.5, 101.1 (Cα/β-1″), 96.3 (Cα/β-1″), 88.8 (C-1′), 83.0 (C-4′ or Cα/β-4″, d, J 9.4), 81.7 (C-4′ or Cα/β-4″) 81.0 (C-4′ or Cα/β-4″, d, J = 9.4), 75.1, 70.6, 70.3, 70.3 (C-2′), 69.9, 69.5 (Cα/β-2″), 66.2 (C-5′ or Cα/β-5″, d, J = 7.1), 65.3 (C-5′ or Cα/β-5″, d, J = 7.1), 65.3 (C-5′ or Cα/β-5″, d, J = 7.1) and 26.3 (COCH3). 31P (162 MHz, D2O) δ −11.2 (br) and −11.4 (br). HRMS (ES–) calcd for C23H28N5O15P2, 676.1063 M–; found, 676.1076; and RT = 17.2 min.
8-(3-Thiophenyl)adenosine Diphosphoribose (8-Thiophenyl-ADPR, 6)
Thiophene-3-boronic acid (0.12 mmol, 16 mg) and 8-Br-ADPR 4 (TEA salt, 0.097 mmol) were reacted under the general protocol B to give 8-(3-thiophenyl)-ADPR (TEA salt, 2.3 equiv by 1H NMR) (25 mg, 24.7 μmol, 25%) as a colorless solid. 1H (400 MHz, D2O) δ 8.14 (s, 1H, H-2), 7.88 (br s, 1H, thiophenyl 2-H), 7.54 (dd, 1H, J = 4.7, 3.2, thiophenyl 4-H), 7.35 (d, 1H, J = 4.7, thiophenyl 5-H), 5.90 (d, 1H, J = 5.9, H-1′), 5.17–5.21 (m, 1.4H, (1H) H-2′ and (0.4H) H-1″), 5.09 (d, 0.6H, J = 1.9, H-1″), 4.37–4.40 (m, 1H, H-2″) and 3.88–4.18 (m, 8H, H-3′, H-4′, 2 × H-5′, H-3″, H-4″ and 2 × H-5″). 13C (100 MHz, D2O) δ 154.5, 152.1 (C-2), 149.9, 148.8, 129.3, 127.9, 127.8, 127.7, 118.2, 101.1 (Cα/β-1″), 96.3 (Cα/β-1″), 88.6 (C-1′), 82.8 (C-4′ or Cα/β-4″, d, = J 8.5), 81.7 (C-4′ or Cα/β-4″), 81.1 (C-4′ or Cα/β-4″, d, J = 8.5), 75.1, 70.6, 70.3, 70.1, 69.9, 69.4, 66.2 (C-5′ or Cα/β-5″), 65.3 (C-5′ or Cα/β-5″), 65.3 (C-5′ or Cα/β-5″). 31P (162 MHz, D2O) δ −11.2 (br) and −11.3 (br). HRMS (ES–) calcd for C19H24N5O14P2S, 640.0521 M–; found, 640.0527; and RT = 24.9 min.
8-(4-(2-Aminopropanoic acid)phenyl)adenosine-5′-monophosphate (8-(4-Ph-ala)-AMP, 9)
dl-4-Boronophenylalanine (0.1 mmol, 21 mg) and 8-Br-AMP 8 (0.75 equiv TEA salts, 40 mg, 0.08 mmol) were reacted under the general protocol B to give 8-(4-(2-aminopropanoic acid)phenyl)-AMP (TEA salt, 2.2 equiv by 1H NMR) (19 mg, 14.4 μmol, 18%) as a colorless solid. 1H (400 MHz, D2O) δ 8.17 (s, 1H, H-2), 7.61 (d, 2H, J = 8.2, Ar–H), 7.40 (d, 2H, J = 8.2, Ar–H), 5.77 (d, 1H, J = 5.8, H-1′), 5.16 (t, 1H, J = 6.3, H-2′), 4.35 (dd, 1H, J = 6.2, 5.1, H-3′), 3.88–4.09 (m, 4H, H-4′, H-5′ and NH2CHCH2), 3.26 (dd, 1H, J = 14.9, 5.3, NH2CHCHa/b) and 3.08 (1H, obscured by Et3N salt peak, NH2CHCHa/b). 13C (100 MHz, D2O) δ 192.2 (C=O), 155.1, 152.8, 152.6 (C-2), 150.1, 138.6, 129.9, 129.8, 126.9, 118.4, 88.6 (C-1′), 83.6 (C-4′), 70.0 (C-2′), 69.5 (C-3′), 63.4 (C-5′), 55.8 (NH2CHCH2) and 36.5 (ArCH2). 31P (162 MHz, D2O) δ −5.6 (s). HRMS (ES–) calcd for C19H22N6O9P, 509.1191 M–; found, 509.1174; and RT = 6.96 min.
8-Amino-nicotinamide Adenine-5′-dinucleotide (8-NH2-NAD+, 11)
Triphenylphosphine (130 mg, 0.5 mmol), morpholine (92 mL, 1.06 mmol), and 2,2′-dipyridyldisulfide (110 mg, 0.5 mmol) were added to a solution of 8-NH2-AMP 10 (55 mg, 0.15 mmol) in dry DMSO (600 μL). The mixture was stirred at rt for 4 h, and then a solution of sodium iodide in acetone (0.1 M) was added dropwise. The precipitate that formed was collected, washed with acetone, and redissolved in water and lyophilized to leave the crude morpholidate intermediate (39 mg) as a pale-yellow solid. The morpholidate was dissolved in a solution of MnCl2 in formamide (1 mL, 0.2 M), MgSO4 (48 mg, 0.4 mmol) and β-NMN+ (67 mmol, 0.2 mmol) were added, and the mixture was stirred for 2 days. The crude product was precipitated from the reaction mixture by the dropwise addition of MeCN, and the precipitate was collected, washed with MeCN, and dried. The crude product was purified by reverse phase column chromatography, eluting with 0–20% MeCN in TEAB (0.05 M). The sample was then treated with Chelex 100 (sodium form) to remove any paramagnetic material and lyophilized to yield the 8-amino-NAD (13 mg, 0.02 mmol, 13%) as a colorless solid. 1H (270 MHz, D2O) broad, possibly small amount of remaining Mn2+. HRMS (ES+) calcd for C21H29N8O14P2, 679.1273 M+; found, 679.1252; and RT = 3.03 min.
8-Aminoadenosine Diphosphoribose (8-NH2-ADPR, 12)
NADase (from Neurospora crassa; Sigma; 0.52 U) in Tris-HCl buffer (1 mL, 0.1 M, pH 7.2–7.4) was added to a solution of 8-NH2-NAD+11 (13 mg) in Tris-HCl buffer (4 mL, 0.1 M, pH 7.2–7.4). The reaction mixture was stirred at 35 °C and was monitored by HPLC. After 4 h, all of the starting material had been consumed, the reaction mixture was diluted with water until the conductivity <200 μS/cm, and the product purified by ion-exchange (Q-Sepaharose) chromatography eluting with a gradient (0–50%) of TEAB (1.0 M) in Milli-Q. Subsequent purification by reverse phase column chromatography, eluting with 0–30% MeCN in TEAB (0.05 M), left the desired 8-NH2-ADPR product (4.5 mg, 7.65 μmol, 40%) as a colorless solid. 1H (400 MHz, D2O) δ 7.98 (s, 1H, H-2), 5.99 (d, 1H, J = 7.6, H-1′), 5.24–5.31 (br, 0.4H, H-1′), 5.11–5.17 (br, 0.6H, H-1′), 4.68–4.64 (br m, 1H, H-2′), 4.38–4.44 (br m, 1H, H-2″), 3.91–4.31 (m, 8H, H-3′, H-4′, 2 × H-5′, H-3″, H-4″ and 2 × H-5″). HRMS (ES–) calcd for C15H23N6O14P2, 573.0753 M–; found, 573.0775; and RT = 12.2 min.
Synthesis of Purine Modified ADPR Analogues
Inosine-5′-diphosphoribose (IDPR, 13)
NHD+ (30 μmol) and NADase were reacted under the general protocol A to afford IDPR as a glassy solid (24.6 μmol, 82%). 1H (400 MHz, D2O) δ 8.44 (s, 1H, H-2), 8.19 (s, 1H, H-8), 6.11 (d, 1H, J1′,2′ = 6.1, H-1′), 5.31 (d, 1H, J1″,2″ = 4.1, H-1″β), 5.17 (d, 1H, J1″,2″ = 2.2, H-1″α), 4.76–4.72 (m, 1H, H-2′) and 4.53–3.96 (m, 9H, H-3′, H-4′, 2 × H-5′, H-2″, H-3″, H-4″ and 2 × H-5″). 31P (decoupled, 162 MHz, D2O) δ −10.2 (d, AB system, J = 18.8), −10.6 (d, AB system, J = 18.8). HRMS (ES–) calcd for C15H21N4O15P2, 559.0484 (M – H)−; found, 559.0480. UV (H2O, pH 7.2) λmax 248 nm (ε 14500).
7-Deaza-8-bromoadenosine Diphosphoribose (7-Deaza-8-bromo-ADPR, 15)
Nicotinamide-7-deaza-8-bromoadenine-5′-dinucletide39 (7-deaza-8-bromo-NAD+, 15 μmol) was treated with NADase under the general procedure A to afford 7-deaza-8-Br-ADPR as a glassy solid (12.7 μmol, 85%). 1H (270 MHz, D2O) δ 8.03 (s, 1H, H-2), 6.66 (s, 1H, H-7), 6.17 (d, 1H, J1′,2′ = 6.1, H-1′), 5.21–5.17 (m, 2H, H-2′ and H-1″), 4.54–4.50 (m, 1H, H-3′) and 4.18–3.94 (m, 8H, H-ribose). 31P (decoupled, 109 MHz, D2O) δ −10.5 (m) and −10.7 (m). HRMS (ES–) calcd for C12H22N4O14P279Br, 634.9797 (M – H)−; found 634.9787. HRMS (ES–) calcd for C12H22N4O14P281Br, 636.9776 (M – H)−; found, 636.9778. UV (H2O, pH 7.1) λmax 277 nm (ε 13250).
6-O-Methylinosine Diphosphoribose (6-O-Me IDPR, 24)
2′,3′,5′-Tri-O-acetyl-6-chloro adenosine 19
To a solution of 1,2,3,5-O-tetraacetate ribofuranose 18 (4.7 g, 14.7 mmol), 6-chloropurine 17 (2.5 g, 16.17 mmol), and DBU (6.5 mL, 44.1 mmol) in dry MeCN (100 mL) was added dropwise TMSOTf (10 mL, 58.8 mmol) at 0 °C. The resulting clear brown solution was stirred for 2 h at 60 °C, after which it was cooled to room temperature and aq satd NaHCO3 (400 mL) was added. The aqueous phase was extracted with DCM (3 × 300 mL), dried (Na2SO4), filtered, and evaporated under reduced pressure, giving a brown oil. The crude was purified by column chromatography on silica gel (DCM–acetone, 9:1 v/v) to afford the desired product as a white foam (4.9 g, 91%). 1H (270 MHz, CDCl3) δ 8.75 (s, 1H, H-8), 8.28 (s, 1H, H-2), 6.21 (d, 1H, J1′,2′ = 5.1, H-1′), 5.92 (app t, 1H, J2′,1′ = J2′,3′ = 5.1, H-2′), 5.62 (app t, 1H, J3′,2′ = J3′,4′ = 5.1, H-3′), 4.48–4.33 (m, 3H, H-4′, H-5′a and H-5′b), 2.13 (s, 3H, CH3), 2.10 (s, 3H, CH3) and 2.06 (s, 3H, CH3). 13C (68 MHz, CDCl3) δ 170.4, 169.7, 169.5 (all C=O), 152.4 (C-2), 151.7, 151.3 (2 × C), 143.7 (C-8), 86.9 (C-1′), 80.6 (C-4′), 73.2 (C-2′), 70.5 (C-3′), 62.9 (C-5′), 20.8, 20.6, and 20.5 (3 × CH3). Rf = 0.57 (DCM–acetone, 9:1 v/v).
6-O-Methylinosine 20
2′,3′,5′-Tri-O-acetyl-6-chloro adenosine 19 (1.45 g, 3.52 mmol) was added to a freshly prepared solution of NaOMe in MeOH (7.04 mmol in 10 mL). The solution was refluxed for one hour, after which it was cooled to rt and neutralized with AcOH. The solvent was evaporated, and the residue was purified by column chromatography on silica gel (DCM–acetone, 6:4 v/v) to yield the desired product as a white foam (943 mg, 95%). 1H (270 MHz, MeOH-d4) δ 8.49 (s, 1H, H-8), 8.42 (s, 1H, H-2), 6.04 (d, 1H, J1′,2′ = 5.9, H-1′), 4.77–4.73 (m, 1H, H-2′), 4.38 (dd, 1H, J3′,2′ = 5.1 and J3′,4′ = 3.1, H-3′), 4.18–4.15 (m, 1H, H-4′), 4.13 (s, 3H, CH3), 3.91 (dd, 1H, J5′a,5′b = 12.5 and J5′a,4′ = 2.6, H-5′a) and 3.77 (dd, 1H, J5′b,5′a = 12.5 and J5′b,4′ = 3.5, H-5′b). 13C (68 MHz, MeOH-d4) δ 160.5 (C-6), 151.7 (C-2), 150.8 (C), 142.6 (C-8), 121.3 (C), 89.9 (C-1′), 86.6 (C-4′), 74.3 (C-2′), 71.1 (C-3′), 61.9 (C-5′) and 53.7 (CH3); Rf = 0.09 (DCM–acetone, 6:4 v/v). MS (APCI+) m/z 283.4 [(MH)+, 100%]. HRMS (ES+) calcd for C11H15N4O5, 283.1037 (MH)+; found, 283.1038.
6-O-Methylinosine-5′-monophosphate (6-O-Me-IMP, 21)
6-O-Methylinosine 20 (80 mg, 0.264 mmol) was dissolved in triethylphosphate (1 mL) by heating with a heatgun. The resulting colorless solution was cooled to 0 °C, and water (2 μL) was added followed by POCl3 (0.1 mL, 1.056 mmol). It was stirred at 0 °C until disappearance of starting material and formation of a single peak as shown by HPLC. After 1 h, the reaction mixture was quenched by addition of ice/water (15 mL) and stirred for 15 min at 0 °C, after which it was warmed up to rt. Triethylphosphate was extracted with EtOAc (6 × 6 mL), and the aqueous phase was neutralized with 2 M NaOH. It was then applied to a reverse phase gradifrac column eluted with a gradient of 5–65% MeCN in 0.05 M TEAB. The appropriate fractions were collected and lyophilized overnight to afford the desired monophosphate as its triethylammonium salt. 1H (270 MHz, D2O) δ 9.01 (s, 1H, H-8), 8.51 (s, 1H, H-2), 6.14 (d, 1H, J1′,2′ = 3.7, H-1′), 4.63 (app t, 1H, J2′,1′ = J2′,3′ = 4.2, H-2′), 4.41–4.37 (m, 1H, H-3′), 4.31 (br s, 1H, H-3′), 4.22–4.15 (m, 1H, H-5′a) and 4.11 (m, 4H, OCH3 and H-5′b). 13C (68 MHz, CDCl3) δ 159.5 (C-6), 153.6 (C-2), 149.4 (C-4), 129.6 (C-8), 115.7 (C-5), 89.6 (C-1′), 83.7 (C-4′, J = 8.7), 74.7 (C-2′), 69.5 (C-3′), 64.2 (C-5′) and 55.9 (OCH3). 31P (109 MHz, CDCl3) δ 0.6. MS: (ES–) m/z 361.5 [(M – H)−, 100%]. HRMS (ES–) calcd for C11H14N4O8P, 361.0555 [(M – H)−]; found, 361.0558.
Nicotinamide 6-O-Methyl-hypoxanthine-5′-dinucleotide (6-O-Me-NHD+, 23)
6-O-Me-IMP 21 (120 mg, 0.331 mmol) was dissolved in dry DMSO (2 mL) and coevaporated with dry DMF (5 × 3 mL). The residue was dissolved in DMSO (1 mL) to which was added morpholine (150 μL, 1.724 mmol), dipyridyldisulfide (182 mg, 0.827 mmol), and triphenylphosphine (217 mg, 0.827 mmol), at which point the solution became bright yellow. It was stirred for 1 h at rt, after which HPLC analysis showed formation of a new peak. Precipitation of the product occurred by dropwise addition of a solution of NaI in acetone (0.1 M). The resulting precipitate was filtered and washed with acetone to yield the desired product as a pale-yellow solid, which was used in the next step without further purification. 6-O-Me-IMP morpholidate (100 mg, 0.232 mmol), β-NMN+ (85 mg, 0.253 mmol), and MgSO4 (54 mg, 0.464 mmol) were dissolved in a 0.2 M solution of MnCl2 in formamide (1.7 mL) and stirred at rt for 16 h, after which HPLC analysis showed completion of the reaction (RT (β-NMN) = 2.1 min and RT (6-O-Me-NHD) = 3.8 min). MeCN was added to precipitate the product, which was filtered, dissolved in Milli-Q, and applied to a reverse phase gradifrac column eluted with a gradient of 5–65% MeCN in 0.05 M TEAB. Further treatment with Chelex 100 to remove any paramagnetic particles afforded the desired product as the sodium salt (18 mg, 8%). 1H (400 MHz, D2O) δ 9.21 (s, 1H, HN2), 9.07 (d, 1H, J6,5 = 6.3, HN6), 8.67 (d, 1H, J4,5 = 8.2, HN4), 8.39 (s, 1H, H-8), 8.27 (s, 1H, H-2), 8.09–8.06 (m, 1H, HN5), 5.96 (d, 1H, J1″,2″ = 5.9, H-1″), 5.94 (d, 1H, J1′,2′ = 5.5, H-1′), 4.62 (app t, 1H, J2′,1′ = J2′,3′ = 5.5, H-2′) and 4.38–4.06 (m, 9H, Hsugar). 13C (100 MHz, D2O) δ 165.1 (C=O), 160.7 (C-6), 151.1 (C-8), 151.0 (C-4), 145.7 (CN4), 142.4 (CN6), 141.5 (C-2), 139.8 (CN2), 133.6 (CN3), 128.6 (CN5), 120.4 (C-5), 99.9 (C-1″), 87.0 (C-1′), 86.8 (C-4″, d, J = 9.2), 83.7 (C-4′, d, J = 9.2), 77.4 (C-2″), 74.0 (C-2′), 70.4 (C-3′), 70.2 (C-3″), 64.8, 63.3 (2 × CH2) and 54.9 (CH3). 31P (109 MHz, D2O) δ −11.4 (d, J = 20.7) and −11.7 (d, J = 20.7). MS (ES–) m/z 678.2 [(M – H)−, 100%]. HRMS (ES–) calcd for C22H28N6O15P2, 678.1093 [(M – H)−]; found, 678.1088. UV (H2O) λmax 251 nm (ε 19200).
6-O-Methyl-inosine-5′-diphosphate Ribose (6-O-Me-IDPR, 24)
6-O-Me-NHD+ sodium salt 23 (17.3 mg, 25.5 μmol) was incubated with Aplysia cyclase (40 μL) in a 25 mM HEPES buffer (35 mL, pH 7.4) at rt. After 4 h at rt, HPLC analysis showed completion of the reaction (Rt (nicotinamide) = 1.7 min and Rt (product) = 15.9 min). The mixture was then applied to a Q-sepharose ion exchange column eluted with 1 M TEAB buffer. The appropriate fractions were collected and evaporated under vacuum, and the residue was coevaporated with MeOH to afford the hydrolyzed product 6-O-Me-IDPR as a triethylammonium salt. 1H (270 MHz, D2O) δ 8.58 (s, 1H, H-2), 8.45 (s, 1H, H-8), 6.15 (d, 1H, J1′,2′ = 5.6, H-1′), 5.26 (d, 0.5 H, J1″,2″ = 4.2, Hβ-1″), 5.16 (d, 0.5 H, J1″,2″ = 2.2, Hα-1″), 4.78 (1H, hidden under HDO peak), 4.48–4.47 (m, 1H), 4.34 (br s, 1H), 4.27–4.17 (m, 3H), 4.12 (s, 3H, OMe), 4.08–3.92 (m, 3H) and 3.84–3.82 (m, 1H). 31P (109 MHz, D2O) δ −10.2 (d, J = 21.1) and −10.6 (d, J = 21.1). MS: (ES–) m/z 573.4 [(M – H)−, 80%]. HRMS (ES–) calcd for C16H23N4O15P2, 573.0641 [(M – H)−]; found, 573.0646. UV (H2O) λmax 260 nm (ε 18800).
Synthesis of Adenosine Ribose Modified ADPR Analogues
2′-Deoxyadenosine Diphosphoribose (2′-Deoxy-ADPR, 33)
2′-Deoxy-NAD+32 (22 μmol) was reacted with NADase under general protocol B to yield the desired hydrolyzed product (18.7 μmol, 85%). 1H (270 MHz, D2O) δ 8.41 (s, 1H, H-2), 8.17 (s, 1H, H-8), 6.48–6.43 (m, 1H, H-1′), 5.26 (d, 1H, J1″,2″ = 4.1, H-1″α), 5.15 (d, 1H, J1″,2″ = 2.2, H-1″β), 4.71 (m, 1H, partially hidden under HDO peak, H-2′), 4.27–3.87 (m, 8H, H-3′, H-4′, H-5′, H-3″, H-4″ and H-5″), 2.83–2.78 (m, 1H, H-2′a) and 2.55 (ddd, 1H, J2′b,2′a = 14.0, J2′b,1′ = 6.3 and J2′b,3′ = 3.3, H-2′b). 31P (decoupled, 109 MHz, D2O) δ −10.4 (br s), −10.5 (br s). HRMS (ES–) calcd for C15H22N5O13P2, 542.0695 (M – H)−; found, 542.0681. UV (H2O, pH 7.4) λmax 259 nm (ε 15400).
3′-Deoxyadenosine-5′-diphosphoribose (3′-Deoxy-ADPR, 43)
To a solution of 3′-deoxy-NAD+42 (16 μmol) in Tris buffer (100 mM, pH 7.3, 5 mL) was added NADase (200 μL). The reaction was left for 2 h at 37 °C, after which HPLC analysis showed no remaining starting material. The volatiles were evaporated under reduced pressure, and the residue was applied to a semipreparative C18 column developed with a linear gradient of 0.1 M TEAB against MeCN. The appropriate fractions were evaporated, and excess TEA salt was removed by coevaporation with MeOH to leave the desired ADPR analogue (2.6 μmol, 20%) as a glassy solid TEA salt. 1H (400 MHz, D2O) δ 8.37 (s, 1H, H-8), 8.16 (s, 1H, H-2), 6.03 (d, 1H, J1′,2′ = 5.0, H-1′), 5.20 (d, 1H, J1″,2″ = 4.1, H-1″α), 5.10 (d, 1H, J1″,2″ = 2.2, H-1″β), 4.71–4.63 (m, 2H, H-sugar), 4.23–3.85 (m, 7H, H-sugar), 2.35 (dd, 1H, J3′a,3′b = 10.0 and J3′a,4′ = 5.8, H-3′a) and 2.17–2.12 (m, 1H, H-3′b). 31P (decoupled, 162 MHz, D2O) δ −11.1 (br m). HRMS (ES–) calcd for C15H22N5O13P2, 542.3090 (M – H)−; found, 542.3098. UV (H2O, pH 7.3) λmax 260 nm (ε 15400).
Synthesis of Acyclic-ADPR 30
9-(4-Hydroxybutyl)adenine-5′-monophosphate 28
9-(4-Hydroxybutyl)adenine 27 (80 mg, 0.386 mmol) was dissolved in trimethylphosphate (1.3 mL) by heating with a heatgun. Phosphorus oxychloride (144 μL, 1.545 mmol) and water (2 μL) were added at 0 °C, and the resulting solution was stirred at rt for 3 h. Ice/water (15 mL) was then added, and the mixture was stirred for further 15 min, after which it was extracted with EtOAc (×6). The aqueous layer was neutralized with 5 N NaOH and applied to a reverse phase column and the product eluted with a gradient of 0.05 M TEAB against MeCN. The appropriate fractions were combined and evaporated. The residue obtained was coevaporated with MeOH to remove excess TEA salt, leaving the desired monophosphate as its triethylammonium salt (92 mg, 72%). 1H (270 MHz, DMSO-d6) δ 8.13 (s, 1H, H-2 or H-8), 7.92 (s, 1H, H-8 or H-2), 7.88 (br s, 2H, NH2), 4.03 (t, 2H, J = 7.2, CH2-N), 3.85 (q, 2H, J = 7.2, CH2-O), 1.80–1.75 (m, 2H, O-CH2-CH2) and 1.56–1.49 (m, 2H, O-CH2-CH2). 31P (109 MHz, D2O) 1.4 (s).
Nicotinamide-9-(4-hydroxybutyl)adenine-5′-dinucleotide 29
9-(4-Acetoxybutyl)adenine-5′-monophosphate·1TEA 28 (92 mg, 0.277 mmol) was dissolved in dry DMSO (1 mL) and coevaporated with dry DMF (5 × 3 mL). The residue was dissolved in DMSO (400 μL) to which was added morpholine (106 μL, 1.233 mmol), dipyridyldisulfide (130 mg, 0.592 mmol), and triphenylphosphine (155 mg, 0.592 mmol), at which point the solution became bright yellow. It was stirred for 2 h at rt, after which HPLC analysis showed completion of the reaction. Precipitation of the product occurred by dropwise addition of a solution of NaI in acetone (0.1 M, 20 mL). The resulting precipitate was filtered, washed with acetone, and dried (31P: δ = 6.7 ppm). It was then reacted with β-NMN+ (84 mg, 0.250 mmol) and MgSO4 (53 mg, 0.454 mmol) in a 0.2 M solution of MnCl2 in formamide (1.5 mL) at rt overnigh,t after which HPLC analysis showed completion of the reaction (Rt = 2.9 min). Precipitation occurred by dropwise addition of MeCN. The precipitate was filtered, dissolved in Milli-Q, and applied to a reverse phase column eluted with a 5–65% gradient of MeCN in 0.05 M TEAB. Further treatment with Chelex 100 to remove any paramagnetic particles afforded the desired dinucleotide as its sodium salt.
9-(4-Hydroxybutyl)adenine-5′-diphosphoribose (Acyclic-ADPR, 30)
Nicotinamide-9-(4-acetoxybutyl)adenine-5′-dinucleotide 29 (10 μmol) was treated with NADase under general procedure B to leave the desired acyclic-ADPR (8.1 μmol, 81%). 1H (270 MHz, D2O) δ 8.09 (s, 2H, H-2 and H-8), 5.25 (d, 0.4H, J1″,2″ = 3.8, H-1″β), 5.17 (d, 0.6H, J1″,2″ = 1.9, H-1″α), 4.20–4.02 (m, 3H, H-2′ and CH2-N), 3.97–3.90 (m, 5H, H-3′, H-4′, H-5′ and CH2-O), 1.90–1.83 (m, 2H, O-CH2CH2) and 1.62–1.55 (m, 2H, O-CH2CH2). 31P (decoupled, 109 MHz, D2O) δ −10.2 (m). HRMS (ES–) calcd for C14H22N5O11P2, 498.0795 (M – H)−; found, 498.0786. UV (H2O, pH 7.2) λmax 261 nm (ε 16000).
Synthesis of Pyrophosphate Modified ADPR Analogues
Adenosine Triphosphoribose (ATPR, 47)
A solution of cATPR 46 (5 μmol) in Tris HCl (100 mM, pH 7) was heated to 100 °C for 1 h, after which HPLC analysis showed conversion to a new product. The solution was applied to a reverse phase column eluted with a 5–65% gradient of MeCN in 0.05 M TEAB. The appropriate fractions were collected and evaporated to afford the desired nucleotide as its triethylammonium salt (2.7 μmol, 54%). 1H (270 MHz, D2O) δ 8.54 (s, 1H, H-2), 8.26 (s, 1H, H-8), 6.11 (d, 1H, J1′,2′ = 5.8, H-1′), 5.31 (d, 0.4H, J1″,2″ = 4.1, H-1″β), 5.15 (d, 0.6H, J1″,2″ = 2.3, H-1″α), 4.55–4.52 (m, 1H, H-2′) and 4.37–3.96 (m, 9H, H-3′, H-4′, H-5′, H-2″, H-3″, H-4″ and H-5″). 31P (decoupled, 109 MHz, D2O) δ −11.6 (br s), −23.4 (br s, O-P-O). HRMS (ES–) calcd for C15H23N5O17P2, 638.0307 (M – H)−; found, 638.0331. UV (H2O, pH 7.2) λmax 259 nm (ε 17180).
Synthesis of Sulfonamide Analogue: Salicylic Adenosine Monosulfamide (Sal-AMS)
Synthesis was carried out without protection of the 6-amino group to generate Sal-AMS. For details, see Supporting Information.
Synthesis of Squarate Analogues: Adenosine Squaryl (ASq)
2′,3′-O-Isopropylidene-5′-amino-5′-deoxy adenosine 59
10% Pd/C (110 mg) was added to a solution of 2′,3′-O-isopropylidene-5′-azido-5′-deoxyadenosine (1.0 g, 3.01 mmol) in EtOH. The mixture was stirred for 16 h under a hydrogen atmosphere, after which the palladium was filtered and the solvent was removed under vacuum, yielding the desired compound as a white solid (0.9 g, 95%). 1H (400 MHz, DMSO-d6) δ 8.34 (s, 1H, H-8), 8.14 (s, 1H, H-2), 7.29 (br s, 2H, NH2), 6.11 (d, 1H, J1′,2′ = 3.0, H-1′), 5.42 (dd, 1H, J2′,3′ = 6.2 and J2′,1′ = 3.0, H-2′), 5.01 (dd, 1H, J3′,2′ = 6.2 and J3′,4′ = 2.9, H-3′), 4.20–4.16 (m, 1H, H-4′), 2.91–2.81 (m, 2H, 2 × H-5′), 1.52 (s, 3H, CH3) and 1.30 (s, 3H, CH3). 13C (100 MHz, DMSO-d6) δ 156.1 (C-6), 152.7 (C-8), 148.8 (C-4), 140.0 (C-2), 119.2 (C-5), 113.3 (C), 89.2 (C-1′), 878.0 (C-4′), 82.7 (C-2′), 81.6 (C-3′), 43.7 (C-5′), 27.0 and 25.2 (2 × CH3). HRMS (ES+) calcd for C13H19N6O3, 307.1513 (MH)+; found, 307.1511.
1-O-Methyl-2,3-O-isopropylidene-5-O-p-toluenesulfonyl-β-d-ribofuranose 50
To a solution of 1-O-methyl-2,3-O-isopropylidene-β-d-ribofuranose 49 (0.61 g, 2.989 mmol) in dry pyridine (1 mL), externally cooled with ice, was added p-toluenesulfonyl chloride (0.7 g, 3.668 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred at rt under nitrogen for 5 h. Water (0.3 mL) was added and stirring continued for 30 min. This mixture was extracted with chloroform (3 × 10 mL) and the combined organic phases washed sequentially with CuSO4 (10% w/v, aq satd), NaHCO3 (aq satd) and water and then dried over anhydrous sodium sulfate. The solvent was evaporated, and the residue was purified on an Isco chromatographic system (petrol–EtOAc, 7:3 v/v) to yield the desired compound as a white solid (0.92 g, 81%). 1H (400 MHz, CDCl3) δ 7.71 (d, 2H, J = 8.7 2 × Ar–H), 7.26 (d, 2H, J = 8.0, Ar–H), 4.83 (s, 1H, H-1′), 4.51 (dd, 1H, J3′,2′ = 6.0 and J3′,4′ = 0.6, H-3′), 4.44 (d, 1H, J2′,3′ = 6.0, H-2′), 4.21 (dt, 1H, J4′,5′ = 7.1 and J4′,3′ = 0.6, H-4′), 3.93–3.91 (m, 2H, H-5′), 3.14 (s, 3H, OMe), 2.36 (s, 3H, CH3), 1.35 (s, 3H, CH3) and 1.19 (s, 3H, CH3). 13C (100 MHz, CDCl3) δ 144.9 (C-SO2), 132.8 (C-Me), 129.8 (2C), 127.9 (2C) (all CH), 112.6 (C), 109.4 (C-1′), 84.8 (C-4′), 83.5 (C-2′), 81.3 (C-3′), 69.1 (C-5′), 54.9 (OMe), 26.2, 24.8 (2 × CH3) and 21.5 (CH3-Ph). HRMS (ES+) calcd for C16H22NaO7S, 381.0978 (MH)+; found, 381.0969.
1-O-Methyl-2,3-O-isopropylidene-5-azido-5-deoxy-β-d-ribofuranose 51
To a solution of 1-O-methyl-2,3-O-isopropylidene-5-O-p-toluenesulfonyl-β-d-ribofuranose 50 (2.4 g, 6.7 mmol) in DMF (20 mL) was added NaN3 (5.2 g, 80.4 mmol), and the reaction mixture was stirred at 120 °C for 16 h. After cooling to rt, acetone (20 mL) was added and the solid was removed by filtration. The solvents were evaporated under reduced pressure, and the residue was dissolved in DCM (50 mL) and washed successively with water (50 mL), satd aq NaHCO3 (50 mL), and water (50 mL). The organic layer was dried (Na2SO4), filtered, and evaporated to leave an oil which was purified on an Isco chromatographic system (petrol–EtOAc, 1:1 v/v), yielding the title compound as a colorless oil (1.4 g, 91%). 1H (400 MHz, CDCl3) δ 4.90 (s, 1H, H-1′), 4.50 (s, 2H, H-2′ and H-3′), 4.19 (ddd, 1H, J4′,5′a = 7.6, J4′,5′b = 6.8 and J4′,3′ = 0.6, H-4′), 3.35 (dd, 1H, J5′a,5′b = 12.5 and J5′a,4′ = 7.6, H-5′a), 3.28 (s, 3H, OMe), 3.17 (dd, 1H, J5′b,5′a = 12.5 and J5′b,4′ = 6.8, H-5′b), 1.39 (s, 3H, CH3) and 1.22 (s, 3H, CH3). 13C (100 MHz, CDCl3) δ 112.6 (C), 109.8 (C-1′), 85.3 (C-4′), 85.1 (C-2′), 82.0 (C-3′), 55.1 (OMe), 53.7 (C-5′), 26.4 and 24.9 (2 × CH3). HRMS (ES+) calcd for C9H15N3NaO4, 252.0955 (MH)+; found, 252.0949.
1-O-Methyl-2,3-O-isopropylidene-5-amino-5-deoxy-β-d-ribofuranose 52
PPh3 (1.95 g, 7.45 mmol) was added to a solution of 1-O-methyl-2,3-O-isopropylidene-5-azido-5-deoxy-β-d-ribofuranose 51 (1.4 g, 6.11 mmol) in THF (7 mL). The reaction mixture was stirred at rt for 16 h, after which water (7 mL) was added and it was stirred for further 7 h. Evaporation of the solvents followed by purification on an Isco chromatographic system (petrol–EtOAc, 1:1 v/v) gave the title compound as a colorless oil (1.04 g, 85%). 1H (400 MHz, CDCl3) δ 4.84 (s, 1H, H-1′), 4.49–4.46 (s, 2H, H-2′ and H-3′), 4.05–4.01 (m, 1H, H-4′), 3.24 (s, 3H, OMe), 2.71–2.62 (m, 2H, 2 × H-5′), 1.36 (s, 3H, CH3) and 1.19 (s, 3H, CH3). 13C (100 MHz, CDCl3) δ 112.2 (C), 109.5 (C-1′), 88.8 (C-4′), 85.4 (C-2′), 82.1 (C-3′), 54.9 (OMe), 45.4 (C-5′), 26.4 and 24.8 (2 × CH3). HRMS (ES+) calcd for C9H18NO4, 204.1230 (MH)+; found, 204.1226.
3-(1′-O-Methyl-2′,3′-O-isopropylidene-5′-amino-5′-deoxy-β-d-ribofuranose)-4-ethoxycyclobut-3-ene-1,2-dione 54
1-O-Methyl-2,3-O-isopropylidene-5-amino-5-deoxy-β-d-ribofuranose 52 (90 mg, 0.443 mmol), DIPEA (42 μL, 0.239 mmol), and diethylsquarate (72 μL, 0.487 mmol) were reacted under the general protocol C to yield the desired product as a white foam (137 mg, 95%). 1H (400 MHz, CDCl3) δ 4.91 (s, 1H, H-1′), 4.66 (q, 4H, J = 6.9, CH2), 4.53–4.49 (m, 2H, H-2′ and H-3′), 4.29 (app t, 1H, J = 5.6, H-4′), 3.73–3.71 (br m, 1H, H-5′a), 3.51–3.49 (br m, 1H, H-5′b, 3.31 (s, 3H, OMe), 1.38 (s, 3H, CH3), 1.37 (t, 3H, J = 6.9, CH3) and 1.22 (s, 3H, CH3). 13C (100 MHz, CDCl3) δ 189.4 (C=O), 184.3 (C=O), 172.6 (2 × C=C), 112.8 (C), 109.9 (C-1′), 85.8 (C-4′), 85.2 (C-2′), 81.5 (C-3′), 69.7 (CH2), 55.5 (OMe), 46.4 (C-5′), 26.3, 24.8 (2 × CH3 isopropyl) and 15.7 (CH3). HRMS (ES+) calcd for C15H22NO7, 328.1391 (MH)+; found, 328.1408.
3-(2′,3′-Isopropylidene-5′-amino-5′-deoxyadenosine)-4-(1″-O-methyl-2″,3″-O-isopropylidene-5″-amino-5″-deoxy-β-d-ribofuranose)cyclobut-3-ene-1,2-dione 60
To a solution of 3-(1′-O-methyl-2′,3′-O-isopropylidene-5′-amino-5′-deoxy-β-d-ribofuranose)-4-ethoxycyclobut-3-ene-1,2-dione (91 mg, 0.305 mmol) and DIPEA (26 μL, 0.152 mmol) in EtOH (2 mL) was added 2′,3′-isopropylidene-5′-amino-5′-deoxyadenosine (98 mg, 0.320 mmol). The reaction was stirred at rt for 1 h. The solvent was removed under reduced pressure, and the residue was purified on an Isco chromatographic system (DCM–MeOH, 8:2 v/v) to yield the desired product as a white foam (106 mg, 60%). 1H (400 MHz, DMSO-d6) δ 8.30 (s, 1H, H-2), 8.16 (s, 1H, H-8), 7.29 (br s, 2H, NH2), 6.18 (d, 1H, J1′,2′ = 2.5, H-1′), 5.42 (dd, 1H, J2′,3′ = 6.3 and J2′,1′ = 2.5, H-2′), 5.0 (dd, 1H, J3′,2′ = 6.3 and J3′,4′ = 3.5, H-3′), 4.91 (s, 1H, H-1″), 4.63 (d, 1H, J3″,2″ = 6.0, H-3″), 4.55 (d, 1H, J2″,3″ = 6.0, H-2″), 4.26–4.22 (m, 1H, H-4′), 4.12 (app t, 1H, J4″,5″ = 7.0, H-4″), 3.91 (br s, 1H, H-5′a), 3.75–3.68 (m, 1H, H-5′b), 3.64 (br s, 1H, H-5″a), 3.49–3.47 (m, 1H, H-5″b), 3.27 (s, 3H, OMe), 1.52 (s, 3H, CH3), 1.34 (s, 3H, CH3), 1.30 (s, 3H, CH3) and 1.22 (s, 3H, CH3). 13C (100 MHz, DMSO-d6) δ 182.7, 182.6 (2 × C=O), 167.6 (2 × C=C), 156.1 (C-6), 152.8 (C-2), 148.8 (C-4), 139.9 (C-8), 119.2(C-5), 113.7, 111.6 (2 × C), 108.8 (C-1″), 88.7 (C-1′), 85.5 (C-4″), 85.1 (C-4′), 84.5 (C-2″), 83.2 (C-2′), 81.2 (C-3″), 81.1 (C-3′), 54.4 (OCH3), 46.4 (C-5″), 45.1 (C-5′), 27.0, 26.2, 25.3, and 24.7 (4 × CH3). HRMS (ES+) calcd for C26H34N7O9, 588.2413 (MH)+; found, 588.2429.
3-(5′-Amino-5′-deoxyadenosine)-4-(5″-amino-5″-deoxy-β-d-ribofuranose)cyclobut-3-ene-1,2-dione 64
3-(2′,3′-Isopropylidene-5′-amino-5′-deoxyadenosine)-4-(1″-O-methyl-2″,3″-O-isopropylidene-5″-amino-5″-deoxy-β-d-ribofuranose) cyclobut-3-ene-1,2-dione 60 (40 mg, 0.07 mmol) was deprotected by stirring in 0.1 M H2SO4 for 16 h at 80 °C to yield the desired compound as a white solid (15 mg, 45%). 1H (400 MHz, DMSO-d6) δ 8.31 (s, 1H, H-2), 8.16 (s, 1H, H-8), 7.27 (br s, 2H, NH2), 6.17 (d, 1H, J1′,2′ = 2.5, H-1′), 4.91 (s, 1H, H-1″), 4.35–4.24 (m, 4H, H-2′, H-2″, H-3′, H-3″), 4.22–4.18 (m, 1H, H-4′), 4.12 (app t, 1H, J4″,5″ = 7.0, H-4″), 3.92 (br s, 1H, H-5′a), 3.75–3.68 (m, 1H, H-5′b), 3.65 (br s, 1H, H-5″a), 3.49–3.47 (m, 1H, H-5″b). 13C (100 MHz, DMSO-d6) δ 182.8, 182.6 (2 × C=O), 167.7 (2 × C=C), 156.2 (C-6), 152.8 (C-2), 148.9 (C-4), 139.9 (C-8), 119.2(C-5), 108.8 (C-1″), 88.7 (C-1′), 85.5 (C-4″), 85.1 (C-4′), 84.5 (C-2″), 83.2 (C-2′), 81.2 (C-3″), 81.1 (C-3′), 46.4 (C-5″), 45.1 (C-5′). HRMS (ES+) calcd for C19H23N7O9, 493.1557 (MH)+; found, 493.1564.
3-Cyclopentylamino-4-ethoxycyclobut-3-ene-1,2-dione 55
Cyclopentylamine (104 μL, 0.863 mmol), DIPEA (99 μL, 0.570 mmol), and diethylsquarate (172 μL, 1.162 mmol) were reacted under the general protocol C to yield the desired product as a white foam (137 mg, 95%). 1H (400 MHz, CDCl3) δ 7.04 (br s, 1H, NH), 4.75–4.73 (m, 2H, OCH2), 4.08–4.03 (m, 1H, CH), 1.99–1.96 (m, 2H), 1.74–1.72 (m, 2H), 1.59–1.56 (m, 4H) (4 × CH2) and 1.41 (t, 3H, J = 6.6, CH3). 13C (100 MHz, CDCl3) δ 189.6 (C-2), 182.6 (C-1), 177.3 (C-3), 171.8 (C-4), 69.4 (CH2), 56.4 (CH), 33.8 (CH2), 23.5 (CH2) and 15.7 (CH3). HRMS (ES+) calcd for C11H15NO3, 210.1130 (MH)+; found, 211.1127. Rf = 0.3 (petrol–EtOAc, 6:4 v/v).
3-(2′,3′-O-Isopropylidene-5′-amino-5′-deoxyadenosine)-4-(cyclopentylamino)-cyclobut-3-ene-1,2-dione 61
To a solution of 3-cyclopentylamino-4-ethoxycyclobut-3-ene-1,2-dione 55 (110 mg, 0.526 mmol) and DIPEA (50 μL, 0.284 mmol) in EtOH (3 mL) was added 2′,3′-O-isopropylidene-5′-amino-5′-deoxyadenosine 59 (177 mg, 0.578 mmol). The reaction mixture was stirred at rt for 18 h. The solvent was removed under reduced pressure, and the residue was purified on an Isco chromatographic system (DCM–MeOH, 8:2 v/v) to yield the desired product as a white solid (106 mg, 43%). 1H (400 MHz, MeOH-d4) δ 8.32 (s, 1H, H-8), 8.29 (s, 1H, H-2), 6.25 (d, 1H, J1′,2′ = 2.6, H-1′), 5.55 (dd, 1H, J2′,3′ = 6.4 and J2′,1′ = 2.6, H-2′), 5.16 (dd, 1H, J3′,2′ = 6.4 and J3′,4′ = 3.8, H-3′), 4.44–4.40 (m, 1H, H-4′), 4.07 (dd, 1H, J5′a,5′b = 14.1 and J5′a,4′ = 4.4, H-5′a), 3.93 (dd, 1H, J5ba,5′a = 14.1 and J5′b,4′ = 6.7, H-5′b), 3.78 (sept, 1H, J = 6.6, CH), 1.72–2.07 (m, 8H, 4 × CH2), 1.64 (s, 3H, CH3) and 1.43 (s, 3H, CH3). 13C (100 MHz, MeOH-d4) δ 183.9 (C-2), 183.5 (C-1), 169.4 (C-3), 169.0 (C-4), 157.4 (C-6), 154.2 (C-2), 150.3 (C-4), 141.9 (C-8), 120.7 (C-5), 115.9 (C), 91.4 (C-1′), 87.0 (C-4′), 85.2 (C-2′), 82.9 (C-3′), 55.9 (CH), 46.6 (C-5′), 43.8, 35.1 (4 × CH2), 27.5 and 25.6 (2 × CH3). HRMS (ES+) calcd for C22H28N7O5, 470.2146 (MH)+; found, 470.2158.
3-(5′-Amino-5′-deoxyadenosine)-4-(cyclopentylamino)-cyclobut-3-ene-1,2-dione 65
3-(2′,3′-O-Isopropylidene-5′-amino-5′-deoxyadenosine)-4-(cyclopentylamino)-cyclobut-3-ene-1,2–dione 61 (50 mg, 0.11 mmol) was deprotected under general protocol D to yield the desired compound as a white solid (40 mg, 85%). 1H (400 MHz, DMSO-d6) δ 9.32, 9.17 (2 × NH), 8.39 (s, 1H, H-8), 8.30 (s, 1H, H-2), 7.74 (br s, 2H, NH2), 5.94 (d, 1H, J = 5.9, H-1′), 4.74–4.63 (m, 4H, 2 × CH2), 4.25–4.14 (m, 2H, H-2′, H-3′), 3.99–3.96 (m, 1H, H-4′), 3.84–3.80 (m, 1H, H-5′a), 3.75–3.67 (m, 2H, CH, H-5′b), 1.42 (t, 2H, J = 6.8) and 1.33 (t, 2H, J = 6.8)(2 × CH2). 13C (100, DMSO-d6) δ 189.2 (C=O), 182.3 (C=O), 176.8 (C=C), 172.8 (C=C), 155.3 (C-6), 151.3 (C-2), 148.8 (C-4), 140.7 (C-2), 119.5 (C-5), 88.3 (C-1′), 83.5 (C-2′), 72.5 (C-3′), 70.8 (C-4′), 68.8 (2C, 2 × CH2), 45.8 (C-5′), 15.5 (2C, 2 × CH2). HRMS (ES+) calcd for C19H24N7O5, 430.1833 (MH)+; found, 418.1838.
3-Butylamino-4-ethoxycyclobut-3-ene-1,2-dione 56
Butylamine (135 μL, 1.367 mmol), DIPEA (141 μL, 0.811 mmol), and diethylsquarate 53 (222 μL, 1.503 mmol) were reacted under general protocol C to leave the desired product as a white foam (230 mg, 91%). 1H (400 MHz, CDCl3) δ 4.77 (q, 2H, J = 7.2, OCH2-Me), 3.66 (t, 1H, J = 7.0, CHH), 3.48 (t, 1H, J = 7.0, CHH), 1.69–1.62 (m, 2H, CH2), 1.53–1.40 (br m, 5H, CH2 and CH3) and 1.01 (t, 3H, J = 7.2, CH3). 13C (100 MHz, CDCl3) δ 189.9 (C-2), 184.5 (C-1), 177.6 (C-3), 174.7 (C-4), 70.7, 45.3, 33.7, 20.6 (4 × CH2), 16.2 (Et:CH3) and 14.0 (Bu:CH3). HRMS (ES+) calcd for C10H16NO3, 198.1125 (MH)+; found, 198.1124. Rf = 0.5 (petrol–EtOAc, 6:4 v/v).
3-(2′,3′-O-Isopropylidene-5′-amino-5′-deoxyadenosine)-4-butylamino-cyclobut-3-ene-1,2-dione 62
To a solution of 3-butylamino-4-ethoxycyclobut-3-ene-1,2-dione 56 (200 mg, 1.081 mmol) and DIPEA (92 μL, 0.531 mmol) in EtOH (5 mL) was added 2′,3′-O-isopropylidene-5′-amino-5′-deoxyadenosine 59 (300 mg, 0.983 mmol). The reaction mixture was stirred at rt for 24 h. The solvent was removed under reduced pressure, and the residue was purified on an Isco chromatographic system (DCM–MeOH, 8:2 v/v) to yield the desired product as a white foam (364 mg, 81%). 1H (400 MHz, DMSO-d6) δ 8.38 (s, 1H, H-8), 8.23 (s, 1H, H-2), 7.39 (br s, 2H, NH2), 6.25 (d, 1H, J1′,2′ = 2.4, H-1′), 5.50 (dd, 1H, J2′,3′ = 6.3 and J2′,1′ = 2.4, H-2′), 5.04 (dd, 1H, J3′,2′ = 6.3 and J3′,4′ = 3.5, H-3′), 4.33–4..29 (m, 1H, H-4′), 3.95 (br, 1H, H-5′a), 3.81–3.75 (br, 1H, H-5′b), 3.51 (br, 2H, CH2), 3.38–3.34 (m, 1H, CHH), 1.60 (s, 3H, CH3), 1.51 (br, 1H, CHH), 1.32 (s, 3H, CH3), 1.32 (q, 2H, J = 7.3, CH2-Me) and 0.91 (t, 3H, J = 7.3, CH3). 13C (100 MHz, DMSO-d6) δ 182.7 (C=O), 182.2 (C=O), 167.7 (C=C), 167.1 (C=C), 156.1 (C-6), 152.8 (C-2), 148.8 (C-4), 139.9 (C-8), 119.1 (C-5), 113.7 (C), 88.7 (C-1′), 85.1 (C-4′), 83.6 (C-2′), 81.2 (C-3′), 45.0 (CH2), 42.9 (C-5′), 32.7 (CH2), 27.0, 25.2 (2 × CH3), 18.9 (CH2) and 13.4 (CH3). HRMS (ES+) calcd for C21H28N7O5, 458.2146 (MH)+; found, 458.2142.
3-(5′-Amino-5′-deoxyadenosine)-4-butylamino-cyclobut-3-ene-1,2-dione 66
3-(2′,3′-O-Isopropylidene-5′-amino-5′-deoxyadenosine)-4-butylamino-cyclobut-3-ene-1,2-dione 62 (50 mg, 0.109 mmol) was deprotected under general protocol D to give the desired compound as a white solid (41 mg, 91%). 1H (400 MHz, DMSO-d6) δ 8.40 (s, 1H, H-8), 8.26 (s, 1H, H-2), 7.56–7.45 (br m, 4H, NH2 and 2 × NH), 5.98 (d, 1H, J1′,2′ = 5.8, H-1′), 4.73 (app t, 1H, J2′,3′ = J2′,1′ = 5.8, H-2′), 4.23 (app t, 1H, J3′,2′ = J3′,4′ = 5.8, H-3′), 4.10–4.06 (m, 1H, H-4′), 3.86–3.79 (m, 2H, 2 × H-5′), 3.53–5.52 (m, 2H, CH2), 1.51–1.49 (m, 2H, CH2), 1.34 (q, 2H, J = 7.3) and 0.92 (t, 3H, J = 7.3). 13C (100 MHz, DMSO-d6) δ 182.2 (2 × C=O), 167.6 (C-3), 167.2 (C-4), 154.9 (C-6), 151.2 (C-2), 149.2 (C-4), 140.4 (C-8), 119.2 (C-5), 87.8 (C-1′), 83.7 (C-4′), 72.9 (C-2′), 70.8 (C-3′), 45.2 (C-5′), 33.4, 24.8, 23.8 (3 × CH2) and 15.8 (CH3). HRMS (ES+) calcd for C18H24N7O5, 418.1833 (MH)+; found, 418.1834.
3-Hexylamino-4-ethoxycyclobut-3-ene-1,2-dione 57
Hexylamine (130 μL, 0.988 mmol), DIPEA (92 μL, 0.533 mmol), and diethylsquarate 53 (161 μL, 1.087 mmol) were reacted under general protocol C to yield the desired product as a white foam (200 mg, 95%). 1H (400 MHz, CDCl3) δ 4.80–4.75 (m, 2H, CH2-Me), 3.65 (t, 1H, J = 7.0, CHH-NH), 3.48 (t, 1H, J = 7.0, CHH-NH), 1.69–1.64 (m, 2H, CH2), 1.53–1.44 (m, 9H, 3 × CH2 and CH3) and 0.99–0.96 (m, 3H, CH3). 13C (100 MHz, CDCl3) δ 189.9 (C-2), 184.5 (C-1), 177.5 (C-3), 174.8 (C-4), 70.7 (Et:CH2), 45.6, 32.5, 31.6, 27.1, 23.6 (5 × CH2), 16.2 (Et:CH3) and 14.5 (hex:CH3). HRMS (ES+) calcd for C12H20NO3, 226.1438 (MH)+; found, 226.1443. Rf = 0.62 (petrol–EtOAc, 6:4 v/v).
3-(2′,3′-O-Isopropylidene-5′-amino-5′-deoxyadenosine)-4-(hexylamino)cyclobut-3-ene-1,2-dione 63
To a solution of 3-hexylamino-4-ethoxycyclobut-3-ene-1,2-dione 57 (190 mg, 0.892 mmol) and DIPEA (76 μL, 0.438 mmol) in EtOH (5 mL) was added 2′,3′-O-isopropylidene-5′-amino-5′-deoxyadenosine 59 (248 mg, 0.811 mmol). The reaction was stirred at rt for 20 h. The solvent was removed under reduced pressure, and the residue was purified on an Isco chromatographic system (DCM–MeOH, 8:2 v/v) to yield the desired product as a white foam (291 mg, 74%). 1H (400 MHz, DMSO-d6) δ 8.37 (s, 1H, H-8), 8.23 (s, 1H, H-2), 7.38 (br s, 4H, NH2 and 2 × NH), 6.26 (d, 1H, J1′,2′ = 2.4, H-1′), 5.49 (dd, 1H, J2′,3′ = 6.2 and J2′,1′ = 2.4, H-2′), 5.07 (dd, 1H, J3′,2′ = 6.2 and J3′,4′ = 3.5, H-3′), 4.33–4.29 (m, 1H, H-4′), 3.96 (br, 1H, H-5′a), 3.81–3.74 (br, 1H, H-5′b), 3.50 (br, 2H, CH2), 1.60 (s, 3H, CH3), 1.51 (br, 1H, CHH), 1.38 (s, 3H, CH3), 1.30 (br, 7H, 3 × CH2 and CHH) and 0.91–0.88 (m, 3H, CH3). 13C (100 MHz, DMSO-d6) δ 182.7 (C-2), 182.2 (C-1), 167.9 (C-3), 168.5 (C-4), 156.1 (C-6), 152.8 (C-2), 148.8 (C-4), 139.9 (C-8), 119.2 (C-5), 113.7 (C), 88.7 (C-1′), 85.2 (C-4′), 83.1 (C-2′), 81.2 (C-3′), 45.1 (CH2), 43.2 (C-5′), 30.7, 30.6 (2 × CH2), 27.0 (CH3), 25.4 (CH2), 25.3 (CH3) 21.9 (CH2) and 13.8 (CH3). HRMS (ES+) calcd for C23H32N7O5, 486.2459 (MH)+; found, 486.2475.
3-(5′-Amino-5′-deoxyadenosine)-4-(hexylamino)cyclobut-3-ene-1,2-dione 67
3-(2′,3′-O-Isopropylidene-5′-amino-5′-deoxyadenosine)-4-(hexylamino)cyclobut-3-ene-1,2-dione 63 (50 mg, 0.102 mmol) was deprotected under general protocol D to yield the desired compound as a white solid (41 mg, 91%). 1H (400 MHz, DMSO-d6) δ 8.42 (s, 1H, H-8), 8.27 (s, 1H, H-2), 7.65 (br s, 2H, NH2), 7.44 (br s, 2H, 2 × NH), 5.98 (d, 1H, J1′,2′ = 5.7, H-1′), 4.72 (app t, 1H, J = 5.0, H-2′), 4.22 (app t, 1H, J = 4.3, H-3′), 4.10–4.06 (m, 2H, H-4′ and H-5′a), 3.85–3.79 (m, 1H, H-5′b), 3.51 (br s, 2H, CH2-NH), 1.51–1.29 (m, 8H, 4 × CH2) and 0.91–0.88 (m, 3H, CH3). 13C (100 MHz, DMSO-d6) δ 182.6 (C-2), 182.3 (C-1), 167.9 (2 × C=C), 155.2 (C-6), 151.5 (C-2), 149.2 (C-4), 140.2 (C-8), 119.2 (C-5), 87.6 (C-1′), 83.7 (C-4′), 72.9 (C-2′), 70.8 (C-3′), 45.5 (CH2), 43.2 (C-5′), 30.7, 30.6, 25.4, 21.9 (4 × CH2) and 13.8 (CH3). HRMS (ES+) calcd for C20H28N7O5, 446.2146 (MH)+; found, 446.2157. Rf = 0.55 (DCM–MeOH, 8:2 v/v).
Synthesis of Click Analogue: 8-Phenyladenosine-1,4-Triazole Ribose (8-Ph-ATrR)
1-O-Methyl-2,3-O-isopropylidene-5-O-propargyl-β-d-ribofuranose 70
A solution of 1-O-methyl-2,3-O-isopropylidene-β-d-ribofuranose 49 (600 mg, 2.94 mmol) in DMF (40 mL) was cooled to 0 °C, and NaH (156 mg, 3.91 mmol, 60% in mineral oil) was added. The mixture was stirred at 0 °C for 30 min, after which TBAI (65 mg, 0.176 mmol) and propargyl chloride (0.25 mL, 3.528 mmol) were added. The reaction mixture was stirred at rt for 16 h, the solvent was removed under reduced pressure, and the residue was purified by column chromatography using an Isco chromatographic system (petrol–EtOAc, 1:0 → 0:1 v/v). The product was obtained as a colorless liquid (512 mg, 72%). 1H (400 MHz, CDCl3) δ 4.95 (s, 1H, H-1′), 4.65 (d, 1H, J2′,3′ = 6.0, H-2′), 4.55 (d, 1H, J3′,2′ = 6.0, H-3′), 4.33–4.30 (m, 1H, H-4′), 4.17 (d, 2H, J = 2.4, CH2), 3.58 (dd, 1H, J5′a,5′b = 9.5 and J5′a,4′ = 6.5, H-5′a), 3.52–3.47 (m, 1H, H-5′b), 3.32 (s, 3H, OMe), 2.42 (t, 1H, J = 6.3 Hz, CH), 1.46 (s, 3H, CH3) and 1.30 (s, 3H, CH3). 13C (100 MHz, CDCl3) δ 112.3 (C), 109.2 (C-1′), 85.0 (C-4′), 84.8 (C-2′), 81.9 (C-3′), 79.3 (C), 74.6 (HC≡), 70.5 (C-5′), 58.3 (CH2), 54.8 (OCH3), 26.4 and 24.9 (2 × CH3). HRMS (ES+) calcd for C12H18NaO5, 265.1046 (MH)+; found, 265.1042. Rf = 0.9 (petrol–EtOAc, 1:1 v/v).
2′,3′-O-Isopropylidene-5′-azido-5′-deoxy-8-bromoadenosine 69
2′,3′-O-Isopropylidene-5′-azido-5′-deoxyadenosine 68 (100 mg, 0.30 mmol) was taken up in NaOAc buffer (pH 4, 1 M, 15 mL) and Br2 (12 μL, 0.45 mmol) added. The resulting solution was stirred in the dark for 24 h and then a solution of NaHSO3 (4 M, aq) added until the solution was colorless. All solvents were evaporated and the residue purified by column chromatography using an Isco chromatographic system (DCM–acetone, 6:4 v/v). The title compound was obtained as an off-white solid (123 mg, 99%). 1H (400 MHz, CDCl3) δ 8.27 (s, 1H, H-2), 6.20 (d, 1H, J1′,2′ = 1.8 Hz, H-1′), 5.99 (br s, 2H, NH2), 5.68 (dd, 1H, J2′,3′ = 6.3 and J2′,1′ = 1.8, H-2′), 5.15 (dd, 1H, J3′,2′ = 6.3 and J3′,4′ = 3.6, H-4′), 4.36–4.31 (m, 1H, H-4′), 3.54–3.43 (m, 2H, 2 × H-5′), 1.61 (s, 3H, CH3) and 1.39 (s, 3H, CH3). 13C (100 MHz, CDCl3) δ 154.4 (C-6), 152.8 (C-2), 150.3 (C-4), 127.5 (C-8), 120.1 (C-5), 114.5 (C), 91.2 (C-1′), 86.4 (C-4′), 83.4 (C-2′), 82.4 (C-3′), 52.1 (C-5′), 27.0 and 25.3 (2 × CH3). HRMS (ES+) calcd for C13H16N8O379Br, 411.0523 (MH)+; found, 411.0532; and calcd for C13H16N8O381Br, 413.0503 (MH)+; found, 413.0522. Rf = 0.58 (DCM–acetone, 3:2 v/v).
1-(2′,3′-O-Isopropylidene-5′-deoxy-8-bromoadenosine)-4-(2″,3″-O-isopropylidene-5″-O-methylribosyl)-1,2,3-triazole 71
To a solution of 2′,3′-O-isopropylidene-5′-deoxy-5′-azido-8-bromoadenosine 69 (123 mg, 0.30 mmol) in a mixture of tBuOH-H2O (1:1 v/v, 6 mL) was added CuSO4·5H2O (2 mg, 0.015 mmol), sodium ascorbate (6 mg, 0.03 mmol), and 1-O-methyl-2,3-O-isopropylidene-5-O-propargyl-β-d-ribofuranose 70 (73 mg, 0.30 mmol). The reaction mixture was stirred at rt for 16 h, the solvents were removed under vacuum, and the residue was purified on an Isco chromatographic system (DCM–acetone, 6:4 v/v) to yield the product as a pale-yellow solid (140 mg, 71%). 1H (400 MHz, CDCl3) δ 8.22 (s, 1H, H-2), 7.32 (s, 1H, CH-triazole), 6.29 (br s, 2H, NH2), 6.17 (d, 1H, J1′,2′ = 1.7 Hz, H-1′), 5.55 (dd, 1H, J2′,3′ = 6.3 and J2′,1′ = 1.7, H-2′), 5.22 (dd, 1H, J3′,2′ = 6.3 and J3′,4′ = 3.9, H-3′), 4.88 (s, 1H, H-1″), 4.74 (dd, 1H, J5′a,5′b = 13.9 and J5′a,4′ = 4.1, H-5′a), 4.62–4.48 (m, 6H, H-4′, H-2″, H-3″, H-5′b and CH2-triazole), 4.25–4.21 (m, 1H, H-4″), 3.50–3.39 (m, 2H, 2 × H-5″), 3.20 (s, 3H, OMe), 1.55 (s, 3H, CH3), 1.40 (s, 3H, CH3), 1.33 (s, 3H, CH3) and 1.21 (s, 3H, CH3). 13C (100 MHz, CDCl3) δ 154.6 (C-6), 153.1 (C-2), 150.1 (C-4), 144.9 (C-triazole), 127.0 (C-8), 123.5 (CH-triazole), 120.1 (C-5), 114.7, 112.3 (2 × C), 109.2 (C-1″), 90.9 (C-1′), 85.9 (C-4′), 85.0 (C-4″), 84.9 (C-2″), 83.9 (C-2′), 81.9 (2C, C-3″ and C-3′), 71.3 (C-5″), 64.6 (OCH2-Tr), 54.6 (OCH3), 51.5 (C-5′), 27.0, 26.3, 25.3, and 24.9 (4 × CH3). HRMS (ES+) calcd for C25H33N8NaO879Br, 675.1497 (MH)+; found, 675.1469; calcd for C25H33N8NaO881Br, 677.1476 (MH)+; found, 677.1451. Rf = 0.58 (DCM–acetone, 3:2 v/v).
1-(2′,3′-O-Isopropylidene-5′-deoxy-8-phenyladenosine)-4-(2″,3″-O-isopropylidene-5″-O-methylribosyl)-1,2,3-triazole 72
To 1-(2′,3′-O-isopropylidene-5′-deoxy-8-bromoadenosine)-4-(2″,3″-O-isopropylidene-5″-O-methylribosyl)-1,2,3-triazole 71 (140 mg, 0.21 mmol), Na2Cl4Pd (3 mg, 5 mol %), PhB(OH2) (32 mg, 0.27 mmol), TPPTS (30 mg, 25 mol %), and Na2CO3 (68 mg, 0.64 mmol) was added degassed MeCN–H2O (3 mL, 1:2 v/v) and the resulting solution stirred at 80 °C for 1 h. All solvents were evaporated and the residue purified by column chromatography using an Isco chromatography system (DCM–acetone, 6:4 v/v) to yield the product (30 mg, 21%). 1H (400 MHz, CDCl3) δ 8.27 (s, 1H, H-2), 7.70–7.48 (m, 5H, Ph), 7.35 (s, 1H, CH-triazole), 6.90 (br s, 2H, NH2), 6.04 (d, 1H, J1′,2′ = 1.6, H-1′), 5.57 (dd, 1H, J2′,3′ = 6.2 and J2′,1′ = 1.6, H-2′), 5.28 (dd, 1H, J3′,2′ = 6.2 and J3′,4′ = 3.5, H-3′), 4.87 (s, 1H, H-1″), 4.82 (dd, 1H, J5′a,5′b = 14.2 and J5′a,4′ = 4.7, H-5′a), 4.71 (dd, 1H, J5′b,5′a = 14.2 and J5′b,4′ = 8.0, H-5′a), 4.59–4.46 (m, 5H, H-4′, H-2″, H-3″ and CH2-triazole), 4.23–4.20 (m, 1H, H-4″), 3.47 (dd, 1H, J5″a,5″b = 9.7 and J5″a,4″ = 6.5, H-5″a), 3.43–3.40 (m, 1H, H-5″b), 3.18 (s, 3H, OMe), 1.45 (s, 3H, CH3), 1.38 (s, 3H, CH3), 1.29 (s, 3H, CH3) and 1.21 (s, 3H, CH3). 13C (100 MHz, CDCl3) δ 155.6 (C-6), 152.6 (C-2), 151.5 (C-8), 150.0 (C-4), 144.8 (C-triazole), 130.5 (2C), 129.6 (2C), 128.9 (5 × CH-phenyl), 128.6 (C-phenyl), 123.5 (CH-triazole), 119.4 (C-5), 114.3, 112.2 (2 × C), 109.1 (C-1″), 90.4 (C-1′), 86.1 (C-4′), 85.0 (C-4″), 84.9 (C-2″), 83.8 (C-2′), 82.5 (C-3′), 81.9 (C-3″), 71.3 (C-5″), 64.6 (OCH2-Tr), 54.6 (OCH3), 51.8 (C-5′), 26.9, 26.3, 25.2, and 24.9 (4 × CH3). HRMS (ES+) calcd for C31H38N8NaO8, 673.2705 (MH)+; found, 673.2678. Rf = 0.41 (DCM–acetone, 3:2 v/v).
1-(5′-Deoxy-8-phenyladenosine)-4-(2″,3″-O-isopropylidene-5″-O-methylribosyl)-1,2,3-triazole 73
1-(2′,3′-O-Isopropylidene-5′-deoxy-8-phenyladenosine)-4-(2″,3″-O-isopropylidene-5″-O-methylribosyl)-1,2,3-triazole 72 (30 mg, 0.046 mmol) was deprotected by stirring in 0.1 M H2SO4 for 16 h at 80 °C to yield the desired compound (6 mg, 24%) as a white solid. 1H (400 MHz, CDCl3) δ 8.27 (s, 1H, H-2), 7.70–7.48 (m, 5H, Ph), 7.35 (s, 1H, CH-triazole), 6.90 (br s, 2H, NH2), 6.04 (d, 1H, J1′,2′ = 1.6, H-1′), 5.57 (dd, 1H, J2′,3′ = 6.2 and J2′,1′ = 1.6, H-2′), 5.28 (dd, 1H, J3′,2′ = 6.2 and J3′,4′ = 3.5, H-3′), 4.87 (s, 1H, H-1″), 4.82 (dd, 1H, J5′a,5′b = 14.2 and J5′a,4′ = 4.7, H-5′a), 4.71 (dd, 1H, J5′b,5′a = 14.2 and J5′b,4′ = 8.0, H-5′a), 4.59–4.46 (m, 5H, H-4′, H-2″, H-3″ and CH2-triazole), 4.23–4.20 (m, 1H, H-4″), 3.47 (dd, 1H, J5″a,5″b = 9.7 and J5″a,4″ = 6.5, H-5″a) and 3.43–3.40 (m, 1H, H-5″b). 13C (100 MHz, CDCl3) δ 155.6 (C-6), 152.6 (C-2), 151.5 (C-8), 150.0 (C-4), 144.8 (C-triazole), 130.5 (2C), 129.6 (2C), 128.9 (5 × CH-phenyl), 128.6 (C-phenyl), 123.5 (CH-triazole), 119.4 (C-5), 109.1 (C-1″), 90.4 (C-1′), 86.1 (C-4′), 85.0 (C-4″), 84.9 (C-2″), 83.8 (C-2′), 82.5 (C-3′), 81.9 (C-3″), 71.3 (C-5″), 64.6 (OCH2-Tr) and 51.8 (C-5′). HRMS (ES+) calcd for C24H29N8O8, 557.2103 (MH)+; found, 557.2097.
Synthesis of Terminal Ribose Modification: Synthesis of Cyclopentyl-ADP
Cyclopentyl-dibenzylphosphoramidite 78
To a solution of tetrazole (81 mg, 1.16 mmol) and diisopropyl-dibenzylphosphoramidite (300 mg, 0.871 mmol) in DCM (10 mL) was added cyclopentanol 77 (50 mg, 0.58 mmol). The reaction mixture was stirred at rt for 20 min, after which TLC analysis showed total conversion of starting material to a single phosphite (petrol–EtOAc, 6:4 v/v, Rf = 0.32). The solution was cooled to 0 °C, and mCPBA (200 mg, 1.16 mmol) was added in one portion. The mixture was warmed up to rt, diluted with EtOAc (20 mL), and washed with 10% Na2SO3 (20 mL), satd aq NaHCO3 (20 mL), and brine (20 mL). The organic phase was collected, dried (Na2SO4), filtered, and evaporated to dryness. The residue was purified with an Isco chromatographic system (petrol–EtOAc, 7:3 v/v) to yield the title compound as a colorless oil (173 mg, 86%). 1H (400 MHz, CDCl3) δ 7.36–7.20 (m, 10H, H-benzyl), 5.06–4.97 (m, 4H, 2 × CH2), 4.90–4.85 (m, 1H, CH–O) and 1.86–1.50 (m, 8H, 4 × CH). 31P (161 MHz, CDCl3, decoupled) δ −1.6 (s).
Cyclopentyl-monophosphate 79
The above material (78, 173 mg, 0.5 mmol) was dissolved in a mixture of MeOH–H2O–cyclohexane (10:1:5 v/v/v, 16 mL) to which was added Pd(OH)2/C (20%). The solution was heated to 80 °C for 2 h, after which the palladium was removed by filtration through Celite and the filtrate was evaporated under reduced pressure, leaving a residue which was used directly in the next step.
β-Cyclopentyl-ADP 80
AMP·Na+ salt (190 mg, 0.547 mmol) was passed through a small Dowex column (TEA form) and eluted with Milli-Q water. The solvent was evaporated to leave a residue, which was dissolved in DMSO and coevaporated with DMF (3 × 3 mL). The residue obtained was dissolved in DMSO (3 mL) and morpholine (0.25 mL, 2.845 mmol), dipyridyldisulfide (301 mg, 1.367 mmol), and triphenylphosphine (358 mg, 1.367 mmol) were added in this order. The resulting yellow solution was stirred for 90 min, after which a 0.1 M solution of NaI in acetone was added. The precipitate obtained was collected by filtration and used directly in the next step. To a solution of AMP-morpholidate (154 mg, 0.350 mmol) and cyclopentane monophosphate 79 (64 mg, 0.380 mmol) in 0.2 N MnCl2 in formamide (2 mL) was added MgSO4 (82 mg, 0.70 mmol), and it was stirred for 16 h at rt, after which HPLC analysis showed product formation. Precipitation of the product occurred on addition of MeCN and purification on RP-18 afforded (after treatment with Chelex 100) the desired dinucleotide as a glassy solid (55 μmol, 14% over 2 steps). 1H (400 MHz, D2O) δ 8.44 (s, 1H, H-2), 8.19 (s, 1H, H-8), 6.07 (s, 1H, H-1′), 4.73–4.71 (m, 1H, CH–O), 4.66 (br s, 1H, H-2′), 4.48 (br s, 1H, H-3′), 4.32 (br s, 1H, H-4′), 4.15 (br s, 2H, 2 × H-5′), 1.62–1.60 (m, 4H), 1.52–1.50 (m, 2H) and 1.36–1.34 (m, 2H) (4 × CH2). 31P (161 MHz, D2O) δ 11.5 (m). 13C (100 MHz, D2O) δ 158.2 (C-6), 153.0 (C-8), 149.3 (C-4), 140.0 (C-2), 113.3 (C-5), 87.0 (C-1′), 84.0 (C-4′), 79.9 (CH), 74.3 (C-2′), 70.5 (C-3′), 65.2 (C-5′), 33.4 and 22.7 (2 × CH2). HRMS (ES–) calcd for C15H22N5O10P2, 494.0847(M – H)−; found, 494.0839. UV (H2O, pH 7.4) λmax 259 nm (ε 17700).
Synthesis of Cyclopentyl-8-Phenyl-ADP
8-Phenyl-2′,3′-O-isopropylidene-adenosine 82
A flask containing 8-bromo-2′,3′-O-isopropylidene-adenosine 81 (200 mg, 0.519 mmol), Na2Cl4Pd (5 mol %), TPPTS (25 mol %), PhB(OH)2 (190 mg, 1.562 mmol), and Na2CO3 (165 mg, 1.557 mmol) was purged with argon, and a degassed mixture of MeCN–H2O (1:1 v/v, 6 mL) was added. The resulting mixture was refluxed for 1 h, then water (6 mL) was added and the solution neutralized with 1 M HCl. The white precipitate obtained was collected by filtration and dried under vacuum (161 mg, 81%). 1H (400 MHz, DMSO-d6) δ 8.16 (s, 1H, H-2), 7.73–7.71 (m, 2H, Ar–H), 7.60–7.58 (m, 3H, Ar–H), 7.44 (s, 2H, NH2), 5.84 (d, 1H, J1′,2′ = 3.4, H-1′), 5.56 (dd, 1H, J2′,3′ = 6.1 and J2′,1′ = 3.4, H-2′), 5.38–5.36 (m, 1H, 5′–OH), 5.03 (dd, 1H, J3′,2′ = 6.1 and J3′,4′ = 2.5, H-3′), 4.17 (dd, 1H, J4′,5′ = 5.1 and J4′,3′ = 2.5, H-4′), 3.63 (dd, 1H, J5′a,5′b = 11.5 and J5′a,4′ = 5.1, H-5′a), 3.56–3.51 (m, 1H, H-5′b), 1.41 (s, 3H, CH3) and 1.28 (s, 3H, CH3). 13C (100 MHz, DMSO-d6) δ 156.1 (C-6), 152.4 (C-2), 150.1 (C-4), 149.6 (C-Ph), 130.3, 129.6 (2 × CH), 129.1 (C-8), 128.8 (CH), 118.8 (C-5), 113.0 (C), 90.4 (C-1′), 86.5 (C-4′), 81.9 (C-2′), 81.8 (C-3′), 61.9 (C-5′), 27.0 and 25.2 (2 × CH3). HRMS (ES+) calcd for C19H21N5O4, 384.1672 (MH)+; found, 384.1686.
8-Phenyladenosine-monophosphate 83
8-Phenyl-2′,3′-O-isopropylidene-adenosine (82, 150 mg, 0.39 mmol) was deprotected under general protocol D to yield the desired compound as a white solid which was used directly in the next step. 8-Phenyl-adenosine (0.39 mmol) was dissolved in triethylphosphate (1 mL) by heating with a heatgun. The resulting colorless solution was cooled to 0 °C, and water (2 μL) was added followed by POCl3 (0.15 mL, 1.56 mmol), then stirred at 0 °C until disappearance of the starting material and formation of a single peak was observed as shown by HPLC. After 1 h, the reaction was quenched by addition of ice–water (15 mL) and the mixture was stirred for 15 min at 0 °C, after which it was warmed to rt. Triethylphosphate was extracted with EtOAc (6 × 6 mL), and the aqueous phase was neutralized with 2 N NaOH. It was then applied to a reverse phase gradifrac column eluted with a 5–65% gradient of MeCN in 0.05 M TEAB. The appropriate fractions were collected and lyophilized to afford the desired monophosphate as its triethylammonium salt. 1H (400 MHz, DMSO-d6) δ 8.16 (s, 1H, H-2), 7.73–7.71 (m, 2H, Ar–H), 7.60–7.58 (m, 3H, Ar–H), 7.44 (s, 2H, NH2), 5.84 (d, 1H, J1′,2′ = 3.4, H-1′), 5.56 (dd, 1H, J2′,3′ = 6.1 and J2′,1′ = 3.4, H-2′), 5.03 (dd, 1H, J3′,2′ = 6.1 and J3′,4′ = 2.5, H-3′), 4.17 (dd, 1H, J4′,5′ = 5.1 and J4′,3′ = 2.5, H-4′), 3.63 (dd, 1H, J5′a,5′b = 11.5 and J5′a,4′ = 5.1, H-5′a) and 3.56–3.51 (m, 1H, H-5′b). 13C (100 MHz, DMSO-d6) δ 156.1 (C-6), 152.4 (C-2), 150.1 (C-4), 149.6 (C-Ph), 130.3, 129.6 (2 × CH), 129.1 (C-8), 128.8 (CH), 118.8 (C-5), 113.0 (C), 90.4 (C-1′), 86.5 (C-4′, d, J = 8.3 Hz), 81.9 (C-2′), 81.8 (C-3′), 61.9 (C-5′, d, J = 8.8 Hz). 31P (109 MHz, D2O) δ 1.6 (s). HRMS (ES–) calcd for C16H17N5O7, 422.0871 (M – H)−; found, 422.0868.
Cyclopentyl-8-phenyladenosine-diphosphate (Cyclopentyl-8-Ph-ADP 84)
8-Ph-AMP·Na+ salt (83, 53 mg, 0.092 mmol) was passed through a small Dowex column (TEA form) and eluted with Milli-Q water. The solvent was evaporated to leave a residue, which was dissolved in DMSO and coevaporated with DMF (3 × 3 mL). The residue obtained was dissolved in DMSO (90 μL) and morpholine (42 μL, 0.478 mmol), dipyridyldisulfide (51 mg, 0.23 mmol), and triphenylphosphine (60 mg, 0.23 mmol) were added in this order. The resulting yellow solution was stirred for 90 min, after which a 0.1 M solution of NaI in acetone was added. The precipitate obtained was collected by filtration and used directly in the next step. To a solution of 8-Ph-AMP-morpholidate (31 mg, 0.060 mmol) and cyclopentane monophosphate 79 (11 mg, 0.066 mmol) in 0.2 N MnCl2 in formamide (0.5 mL) was added MgSO4 (14 mg, 0.12 mmol), and it was stirred for 16 h at rt, after which HPLC analysis showed product formation. Precipitation of the product occurred on addition of MeCN and purification on RP-18 afforded (after treatment with Chelex 100) the desired dinucleotide as a glassy solid (11 μmol, 12% over 2 steps). 1H (400 MHz, D2O) δ 8.21 (s, 1H, H-2), 7.67 (d, 1H, J = 6.2), 7.60–7.56 (m, 2H), 7.28 (d, 2H, J = 8.1) (5 × ArH), 5.82 (d, 1H, J = 6.4, H-1′), 5.15 (app t, 1H, H-2′), 4.31 (dd, 1H, J = 6.4, 4.5, H-3′), 4.08–3.98 (m, 4H, H-4′, 2 × H-5′, CH–O), 1.62–1.60 (m, 4H), 1.52–1.50 (m, 2H) and 1.36–1.34 (m, 2H) (4 × CH2). 31P (161 MHz, D2O) δ 11.5 (m). 13C (100 MHz, D2O) δ 158.3 (C-6), 152.1 (C-8), 149.3 (C-4), 140.1 (C-2), 132.4 (2C), 129.7 (2C), 128.6 (5 × ArCH), 113.2 (C-5), 87.2 (C-1′), 83.8 (C-4′), 79.7 (CH), 73.9 (C-2′), 70.4 (C-3′), 65.3 (C-5′), 33.6 and 22.9 (2 × CH2). HRMS (ES–) calcd for C21H27N5O10P2, 570.1155 (M – H)−; found, 570.1149. UV (H2O, pH 7.4) λmax 276 nm (ε 17600).
Synthesis of 8-Phenyl-2′-Deoxy-ADPR 86
8-Phenyl-2′-deoxy-cADPR 85 (13 μmol) was incubated in KH2PO4 buffer (100 mM, pH 7.4) at 70 °C for 2.5 h, after which HPLC analysis showed a new peak at Rt = 28 min. The volatiles were removed under reduced pressure, and the residue was applied to a C18 semipreparative column eluted with a gradient of 0.1 M TEAB in MeCN. The appropriate fractions were combined and evaporated to leave the desired product as a glassy solid in its triethylammonium form (5.06 μmol, 39%). 1H (500 MHz, D2O) δ 8.21 (s, 1H, H-2), 7.65–7.55 (m, 5H, Ar-H), 6.29–6.27 (m, 1H, H-1′), 5.21 (d, 1H, J1″,2‴ = 4.5, H-1″α), 5.10 (d, 1H, J1″,2‴ = 2.2, H-1″β), 4.54–3.81 (m, 9H, H-ribose), 3.22–3.16 (m, 1H, H-2′a) and 2.20–2.13 (m, 1H, H-2′b). 31P (decoupled, 109 MHz, D2O) δ −11.1 (br s). 13C (100 M, D2O) 153.0 (C-6), 152.6 (C-8), 150.3 (C-2), 148.4 (C-4), 131.1, 129.7, 129.0, 128.5 (5 × CH phenyl), 101.2 (C-1″), 91.1 (C-1′), 86.6 (C-4″/C-4′), 75.2 (C-2″), 74.8 (C-2′), 70.9 (C-3″), 70.4 (C-3′), 65.4 (C-5″) and 62.8 (C-5′). HRMS (ES–) calcd for C21H26N5O13P2, 618.1008 (M – H)−; found, 618.1013. UV (H2O, pH 7.4) λmax 277 nm (ε 15600).
Pharmacology
Materials
BSA (bovine serum albumin), Fura-2/AM, NaCl, EGTA, NMDG, Tris base, and Histopaque-1119 were purchased from Sigma Aldrich (München, Germany). KCl, MgSO4, MgCl2, CaCl2, NaH2PO4, d-glucose, l-ascorbic acid, Tween, ionomycin, and EDTA were obtained from Merck (Darmstadt, Germany). HEPES was procured from Biomol (Hamburg, Germany). Fibronectin, DMEM, and penicillin/streptomycin were supplied by Invitrogen (Darmstadt, Germany). FBS (fetal bovine serum) and G418 sulfate were purchased from Biochrom (Berlin, Germany). 8-Cl-AMP was obtained from Biolog (Bremen, Germany). The anti-TRPM2 antibody was procured from Novus Biologicals (Littleton, USA). The goat antirabbit antiserum was purchased from Dianova (Hamburg, Germany). PVDF membrane was acquired from Millipore (Darmstadt, Germany). Percoll and ECL western blotting detection reagents were supplied by GE Healthcare (Uppsala, Sweden).
Cell Culture
HEK293 cells were maintained in DMEM medium containing Glutamax I complemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin at 37 °C in the presence of 5% CO2. HEK293 clones expressing TRPM2/EGFP (or EGFP for control) were cultured under the same conditions, while the medium was supplemented with 400 μg/mL G418 sulfate.
Transfection of HEK293 Cells and Generation of Cell Lines
HEK293 wild-type cells were transfected with two different expression vectors coding either for human TRPM2 and EGFP (pIRES2-EGFP-TRPM2) or for EGFP alone (pIRES2-EGFP) as described previously.58,59 Cells carrying the expression constructs were selected by addition of 400 μg/mL G418 sulfate (Biochrom) to the culture medium. Clonal cell lines with stable expression were generated using the limiting dilution technique.
Patch Clamp Measurements
Cells were seeded at low density the day before use. During the experiments, cells were kept at room temperature in bath solution (1 mM CaCl2, 140 mM NMDG, 5 mM KCl, 3.3 mM MgCl2, 1 mM CaCl2, 5 mM d-glucose, 10 mM HEPES, pH 7.4). Patch pipets with resistances of 1.7–3.5 MΩ were pulled from 1.5 mm diameter borosilicate glass capillaries and filled with pipet solution (120 mM KCl, 8 mM NaCl, 1 mM MgCl2, 10 mM HEPES, 10 mM EGTA, 5.6 mM CaCl2), resulting in 200 nM free [Ca2+] as calculated by CaBuf software (G. Droogmans, formerly available from ftp://ftp.cc.kuleuven.ac.be/pub/droogmans/cabuf.zip). Patch clamp experiments were carried out in the whole-cell configuration.60 Data were acquired with an EPC10 amplifier and PatchMaster software (HEKA Elektronik, Germany) and were compensated for fast and slow capacity transients. The cells were held at −50 mV and current was measured during 140 ms voltage ramps from −85 to −20 mV every 5 s over a period of 450 s. Series resistance was compensated by 70%. For activation of TRPM2, ADPR was added to the pipet solution at a concentration of 100 μM. Antagonist activity of the ADPR analogues was tested by adding them at different concentrations to a pipet solution with 100 μM ADPR. During some experiments, the pipet solution contained 0.1% DMSO because stock solutions of the more lipophilic ADPR analogues were prepared in DMSO.
Purification of Human Neutrophils
Fresh blood with EDTA as anticoagulant was obtained from indiscriminately selected volunteers. Neutrophils were isolated as described elsewhere.61 In brief, the blood was fractionated with a Histopaque-1119 density gradient and subsequently neutrophils were further purified by the use of Percoll layers ranging from 65 to 85% in density. After a final washing step, the cells were resuspended in Ca2+ measurement buffer (140 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1 mM CaCl2, 1 mM NaH2PO4, 4 mM glucose, 20 mM HEPES, pH 7.4) and kept on ice until use. To avoid premature activation of the neutrophils, all buffers used during the isolation were supplemented with 2 mM EDTA, cell concentrations exceeding 5 × 106 cells/mL were avoided, and only endotoxin free materials and solutions were used. All experiments were performed within 6 h of blood donation.
Ratiometric Ca2+ Imaging of Human Neutrophils
Neutrophils were incubated with 4 μM Fura2/AM for 30 min at 37 °C in the dark, washed twice, and resuspended in Ca2+ measurement buffer (see above) at a concentration of 1 × 106 cells/mL. For each measurement, 5 × 104 cells were transferred to a small chamber consisting of a rubber O-ring fixed with silicon grease on a glass coverslip coated subsequently with 25 ng of BSA and 250 ng of fibronectin. The cells were incubated for 15 min at ambient temperature in the presence of 10 mM l-ascorbic acid (pH 7.4) and, if applicable, varying concentrations of 8-phenyl-ADPR. The loaded coverslip was mounted on the stage of a PerkinElmer/Imrovision imaging system built around a Leica DM IRE 2 fluorescence microscope. Approximately 70 s after the beginning of the measurement, the cells were stimulated by addition of fMLP (final concentration 1 μM) or A5 peptide (final concentration 10 μM). Ratiometric Ca2+ imaging was performed as described previously.62
Chemotaxis Measurement of Human Neutrophils
The migration of neutrophils was observed microscopically in microfluidic devices (μ-Slide Chemotaxis, Ibidi, Martinsried, Germany). First the μ-Slides were coated with 50 μg/mL fibronectin for 30 min at ambient temperature before washing three times and drying. Isolated neutrophils were resuspended to a concentration of 3 × 106 cells/mL in Ca2+ measurement buffer supplemented with 10% (v/v) plasma obtained from the same donor. If applicable, 8-phenyl-ADPR or EGTA was added and the whole slide was loaded according to the manufacturer’s instructions and incubated at room temperature for 15 min. Adding 18 μL of fMLP (125 nM) to the upper reservoir resulted in a chemotactical gradient from 0 → 50 nM fMLP across the observation chamber. Plasma supplemented buffer without chemoattractant was used in control experiments. The slide was mounted on the stage of the imaging system, and the main chamber observed at 10 times magnification in bright-field mode. After a 5 min resting period, greyscale images with a resolution of 672 × 510 pixels were recorded every 30 s for 1 h using Openlab Software 4.0.4. Cell migration was tracked manually with a 5 × 5 pixel maximum intensity centering correction using the manual-tracking plugin for ImageJ (1.45e). Migrational parameters were calculated from the movement paths using the Chemotaxis and Migration Tool software (v2.0, Ibidi GmbH).
Acknowledgments
We thank the Wellcome Trust for Project Grant 084068 (to B.V.L.P. and A.H.G.) and Programme Grant 082837 (to B.V.L.P.). We are also grateful to the Deutsche Forschungsgemeinschaft for continuous support (grant no. GU360/13-1 to A.H.G. and R.F. and GU360/15-1 to A.H.G.).
Glossary
Abbreviations Used
- ACA
N-(p-amylcinnamoyl)anthranilic acid
- ADP
adenosine 5′-diphosphate
- ADPR
adenosine 5′-diphosphoribose
- ADPRC
ADPR cyclase
- AIBN
azobisisobutyronitrile
- AMP
adenosine 5′-monophosphate
- ASqR
adenosine squaryl ribose
- ATPR
adenosine 5′-triphosphate ribose
- ATrR
adenosine triazole ribose
- [Ca2+]i
free cytosolic Ca2+ concentration
- cADPR
cyclic adenosine 5′-diphosphoribose
- cATPR
cyclic adenosine 5′-triphosphate ribose
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCC
N,N′-dicyclohexylcarbodiimide
- DIPEA
N,N-diisopropylethylamine
- FFA
flufenamic acid
- fMLP
formyl-methionyl-leucyl-phenylalanine
- IDPR
inosine-5′-diphosphoribose
- NAADP
nicotinic acid adenine dinucleotide phosphate
- NAD+
nicotinamide adenosine 5′-dinucleotide
- NHD+
nicotinamide hypoxanthine 5′-dinucleotide
- NMDG
N-methyl-d-glucamine
- β-NMN+
β-nicotinamide 5′-mononucleotide
- NUDT9H
NudT9-homology domain
- PARG
poly-ADPribose-glycohydrolase
- PARP
poly-ADP ribose-polymerase
- 4-Ph-ala
4-phenylalanine
- SAR
structure–activity relationship
- Sal-AMS
salicyl-adenosine monosulfamide
- TEAB
triethylammonium bicarbonate
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- TEP
triethylphosphate
- TPPTS
trisodium tris(m-sulfonatophenyl)phosphine)
- TRP
transient receptor potential channels
- TRPM2
transient receptor potential channel, subfamily melastatin, member 2
Supporting Information Available
General experimental information, experimental procedures for known compounds, and 1H NMR spectra for compounds 5, 6, 7, 84, and 86. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written by contributions from all authors and all authors have approved the final version. C.M and T.K. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Clapham D. E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [DOI] [PubMed] [Google Scholar]
- Perraud A. L.; Fleig A.; Dunn C. A.; Bagley L. A.; Launay P.; Schmitz C.; Stokes A. J.; Zhu Q. Q.; Bessman M. J.; Penner R.; Kinet J. P.; Scharenberg A. M. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599. [DOI] [PubMed] [Google Scholar]
- Shen B. W.; Perraud A. L.; Scharenberg A.; Stoddard B. L. The crystal structure and mutational analysis of human NUDT9. J. Mol. Biol. 2003, 332, 385–398. [DOI] [PubMed] [Google Scholar]
- Sano Y.; Inamura K.; Miyake A.; Mochizuki S.; Yokoi H.; Matsushime H.; Furuichi K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science 2001, 293, 1327–1330. [DOI] [PubMed] [Google Scholar]
- Perraud A. L.; Schmitz C.; Scharenberg A. M. TRPM2 Ca2+ permeable cation channels: from gene to biological function. Cell Calcium 2003, 33, 519–531. [DOI] [PubMed] [Google Scholar]
- Koch-Nolte F.; Haag F.; Guse A. H.; Lund F.; Ziegler M. Emerging roles of NAD+ and its metabolites in cell signaling. Sci. Signaling 2009, 2, mr1. [DOI] [PubMed] [Google Scholar]
- Fonfria E.; Marshall I. C.; Benham C. D.; Boyfield I.; Brown J. D.; Hill K.; Hughes J. P.; Skaper S. D.; McNulty S. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br. J. Pharmacol. 2004, 143, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buelow B.; Song Y.; Scharenberg A. M. The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J. Biol. Chem. 2008, 283, 24571–24583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfria E.; Murdock P. R.; Cusdin F. S.; Benham C. D.; Kelsell R. E.; McNulty S. Tissue Distribution Profiles of the Human TRPM Cation Channel Family. J. Recept. Signal Transduction 2006, 26, 159–178. [DOI] [PubMed] [Google Scholar]
- McHugh D.; Flemming R.; Xu S.-Z.; Perraud A.-L.; Beech D. J. Critical Intracellular Ca2+ Dependence of Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel Activation. J. Biol. Chem. 2003, 278, 11002–11006. [DOI] [PubMed] [Google Scholar]
- Hara Y.; Wakamori M.; Ishii M.; Maeno E.; Nishida M.; Yoshida T.; Yamada H.; Shimizu S.; Mori E.; Kudoh J.; Shimizu N.; Kurose H.; Okada Y.; Imoto K.; Mori Y. LTRPC2 Ca2+-Permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol. Cell 2002, 9, 163–173. [DOI] [PubMed] [Google Scholar]
- Miller B. A. Inhibition of TRPM2 function by PARP inhibitors protects cells from oxidative stress-induced death. Br. J. Pharmacol. 2004, 143, 515–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Hirschler-Laszkiewicz I.; Tong Q.; Conrad K.; Sun S. C.; Penn L.; Barber D. L.; Stahl R.; Carey D. J.; Cheung J. Y.; Miller B. A. TRPM2 is an ion channel that modulates hematopoietic cell death through activation of caspases and PARP cleavage. Am. J. Physiol.: Cell Physiol. 2006, 290, C1146–C1159. [DOI] [PubMed] [Google Scholar]
- Aarts M.; Tymianski M. TRPMs and neuronal cell death. Pfluger’s Arch.—Eur. J. Physiol. 2005, 451, 243–249. [DOI] [PubMed] [Google Scholar]
- Yang K. T.; Chang W. L.; Yang P. C.; Chien C. L.; Lai M. S.; Su M. J.; Wu M. L. Activation of the transient receptor potential M2 channel and poly(ADP-ribose) polymerase is involved in oxidative stress-induced cardiomyocyte death. Cell Death Differ. 2006, 13, 1815–1826. [DOI] [PubMed] [Google Scholar]
- Bari M. R.; Akbar S.; Eweida M.; Kühn F. J. P.; Gustafsson A. J.; Lückhoff A.; Islam M. S. H2O2-induced Ca2+ influx and its inhibition by N-(p-amylcinnamoyl) anthranilic acid in the β-cells: involvement of TRPM2 channels. J. Cell. Mol. Med. 2009, 13, 3260–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharenberg A. M. TRPM2 and pancreatic beta-cell responses to oxidative stress. Islets 2009, 1, 165–166. [DOI] [PubMed] [Google Scholar]
- Knowles H.; Li Y.; Perraud A.-L. The TRPM2 ion channel, an oxidative stress and metabolic sensor regulating innate immunity and inflammation. Immunol. Res. 2013, 55, 241–248. [DOI] [PubMed] [Google Scholar]
- Yamamoto S.; Shimizu S.; Kiyonaka S.; Takahashi N.; Wajima T.; Hara Y.; Negoro T.; Hiroi T.; Kiuchi Y.; Okada T.; Kaneko S.; Lange I.; Fleig A.; Penner R.; Nishi M.; Takeshima H.; Mori Y. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nature Med. 2008, 14, 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrhahn J.; Kraft R.; Harteneck C.; Hauschildt S. Transient receptor potential melastatin 2 is required for lipopolysaccharide-induced cytokine production in human monocytes. J. Immunol. 2010, 184, 2386–2393. [DOI] [PubMed] [Google Scholar]
- Partida-Sanchez S.; Gasser A.; Fliegert R.; Siebrands C. C.; Dammermann W.; Shi G.; Mousseau B. J.; Sumoza-Toledo A.; Bhagat H.; Walseth T. F.; Guse A. H.; Lund F. E. Chemotaxis of Mouse Bone Marrow Neutrophils and Dendritic Cells Is Controlled by ADP-Ribose, the Major Product Generated by the CD38 Enzyme Reaction. J. Immunol. 2007, 179, 7827–7839. [DOI] [PubMed] [Google Scholar]
- Pantaler E.; Lückhoff A. Inhibitors of TRP channels reveal stimulus-dependent differential activation of Ca2+ influx pathways in human neutrophil granulocytes. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 380, 497–507. [DOI] [PubMed] [Google Scholar]
- Sumoza-Toledo A.; Lange I.; Cortado H.; Bhagat H.; Mori Y.; Fleig A.; Penner R.; Partida-Sanchez S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011, 25, 3529–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer N.; Hicking G.; Göbel K.; Wiendl H. TRPM2 Cation Channels Modulate T Cell Effector Functions and Contribute to Autoimmune CNS Inflammation. PLoS One 2012, 7, e47617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi K.; Inada H.; Tominaga M. Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br. J. Pharmacol. 2008, 153, 1324–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K.; Tominaga M. TRPM2 modulates insulin secretion in pancreatic beta-cells. Islets 2011, 3, 209–211. [DOI] [PubMed] [Google Scholar]
- Xie Y. F.; Belrose J. C.; Lei G.; Tymianski M.; Mori Y.; Macdonald J. F.; Jackson M. F. Dependence of NMDA/GSK-3beta mediated metaplasticity on TRPM2 channels at hippocampal CA3-CA1 synapses. Mol. Brain 2011, 4, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. R.; Machold R. P.; Witkovsky P.; Rice M. E. TRPM2 Channels Are Required for NMDA-Induced Burst Firing and Contribute to H2O2-Dependent Modulation in Substantia Nigra Pars Reticulata GABAergic Neurons. J. Neurosci. 2013, 33, 1157–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill K.; Benham C. D.; McNulty S.; Randall A. D. Flufenamic acid is a pH-dependent antagonist of TRPM2 channels. Neuropharmacology 2004, 47, 450–460. [DOI] [PubMed] [Google Scholar]
- Chen G.-L.; Zeng B.; Eastmond S.; Elsenussi S. E.; Boa A. N.; Xu S.-Z. Pharmacological comparison of novel synthetic fenamate analogues with econazole and 2-APB on the inhibition of TRPM2 channels. Br. J. Pharmacol. 2012, 167, 1232–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft R.; Grimm C.; Frenzel H.; Harteneck C. Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br. J. Pharmacol. 2006, 148, 264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill K.; McNulty S.; Randall A. D. Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004, 370, 227–237. [DOI] [PubMed] [Google Scholar]
- Zeng B.; Chen G. L.; Xu S. Z. Divalent copper is a potent extracellular blocker for TRPM2 channel. Biochem. Biophys. Res. Commun. 2012, 424, 279–284. [DOI] [PubMed] [Google Scholar]
- Wagner G. K.; Guse A. H.; Potter B. V. L. Rapid synthetic route toward structurally modified derivatives of cyclic adenosine 5′-diphosphate ribose. J. Org. Chem. 2005, 70, 4810–4819. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Wagner G. K.; Weber K.; Garnham C.; Morgan A. J.; Galione A.; Guse A. H.; Potter B. V. L. 2′-Deoxy cyclic adenosine 5′-diphosphate ribose derivatives:importance of the 2′-hydroxyl motif for the antagonistic activity of 8-substituted cADPR derivatives. J. Med. Chem. 2008, 51, 1623–1636. [DOI] [PubMed] [Google Scholar]
- Čapek P.; Pohl R.; Hocek M. Cross-coupling reactions of unprotected halopurine bases, nucleosides, nucleotides and nucleoside triphosphates with 4-boronophenylalanine in water. Synthesis of (purin-8-yl)- and (purin-6-yl)phenylalanines. Org. Biomol. Chem 2006, 4, 2278–2284. [DOI] [PubMed] [Google Scholar]
- Čapek P.; Cahová H.; Pohl R.; Hocek M.; Gloeckner C.; Marx A. An Efficient Method for the Construction of Functionalized DNA Bearing Amino Acid Groups through Cross-Coupling Reactions of Nucleoside Triphosphates Followed by Primer Extension or PCR. Chem.—Eur. J. 2007, 13, 6196–6203. [DOI] [PubMed] [Google Scholar]
- Černa I.; Pohl R.; Klepetářová B.; Hocek M. Synthesis of 6,8,9-Tri- and 2,6,8,9-Tetrasubstituted Purines by a Combination of the Suzuki Cross-coupling, N-Arylation, and Direct C–H Arylation Reactions. J. Org. Chem. 2008, 73, 9048–9054. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Bailey V. C.; Potter B. V. L. Chemoenzymatic synthesis of 7-deaza cyclic adenosine 5′-diphosphate ribose analogues, membrane permeant modulators of intracellular calcium release. J. Org. Chem. 2007, 73, 1693–1703. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Muller-Steffner H.; Schuber F.; Potter B. V. L. Nicotinamide 2-fluoroadenine dinucleotide unmasks the NAD(+) glycohydrolase activity of Aplysia californica adenosine 5′-diphosphate ribosyl cyclase. Biochemistry 2007, 46, 4100–4109. [DOI] [PubMed] [Google Scholar]
- Moreau C.; Wagner G. K.; Weber K.; Guse A. H.; Potter B. V. L. Structural determinants for N1/ N7 cyclization of nicotinamide hypoxanthine dinucleotide derivatives by ADP-ribosyl cyclase from Aplysia californica: Ca2+-mobilizing activity of 8-substituted cyclic inosine 5′-diphosphoribose analogs in T-lymphocytes. J. Med. Chem. 2006, 49, 5162–5176. [DOI] [PubMed] [Google Scholar]
- Kristinsson H.; Nebel K.; O’Sullivan A. C.; Struber F.; Winkler T.; Yamaguchi Y. A novel synthesis of sulfamoyl nucleosides. Tetrahedron 1994, 50, 6825–6838. [Google Scholar]
- Galeone A.; Mayol L.; Oliviero G.; Piccialli G.; Varra M. Synthesis of a novel N-carbocyclic, N-9 butyl analogue of cyclic ADP-ribose (cADPR). Tetrahedron 2002, 58, 363–368. [Google Scholar]
- Volpini R.; Mishra R. C.; Kachare D. D.; Ben D. D.; Lambertucci C.; Antonini I.; Vittori S.; Marucci G.; Sokolova E.; Nistri A.; Cristalli G. Adenine-Based Acyclic Nucleotides as Novel P2X3 Receptor Ligands. J. Med. Chem. 2009, 52, 4596–4603. [DOI] [PubMed] [Google Scholar]
- Ashamu G. A.; Sethi J. K.; Galione A.; Potter B. V. L. Roles for adenosine ribose hydroxyl groups in cyclic adenosine 5′-diphosphate ribose-mediated Ca2+-release. Biochemistry 1997, 36, 9509–9517. [DOI] [PubMed] [Google Scholar]
- Norman D. G.; Reese C. B. A Convenient Preparation of 3′-Deoxyadenosine. Synthesis 1983, 1983, 304–306. [Google Scholar]
- Zhang F. J.; Yamada S.; Gu Q. M.; Sih C. J. Synthesis and characterization of cyclic ATP-ribose: a potent mediator of calcium release. Bioorg. Med. Chem. Lett. 1996, 6, 1203–1208. [Google Scholar]
- Elliott T. S.; Slowey A.; Ye Y.; Conway S. J. The use of phosphate bioisosteres in medicinal chemistry and chemical biology. MedChemComm 2012, 3, 735–751. [Google Scholar]
- Leonard N. J.; Carraway K. L. 5-Amino-5-deoxyribose derivatives. Synthesis and use in the preparation of “reversed” nucleosides. J. Heterocycl. Chem. 1966, 3, 485–489. [Google Scholar]
- Starkus J.; Beck A.; Fleig A.; Penner R. Regulation of TRPM2 by extra- and intracellular calcium. J. Gen. Physiol. 2007, 130, 427–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolisek M.; Beck A.; Fleig A.; Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell 2005, 18, 61–69. [DOI] [PubMed] [Google Scholar]
- Tóth B.; Csanády L. Identification of Direct and Indirect Effectors of the Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel. J. Biol. Chem. 2010, 285, 30091–30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreras J. A.; Ryu J.-S.; Di Lello F.; Tan D. S.; Quadri L. E. N. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nature Chem. Biol. 2005, 1, 29–32. [DOI] [PubMed] [Google Scholar]
- Somu R. V.; Boshoff H.; Qiao C.; Bennett E. M.; Barry C. E.; Aldrich C. C. Rationally Designed Nucleoside Antibiotics That Inhibit Siderophore Biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 2005, 49, 31–34. [DOI] [PubMed] [Google Scholar]
- Zha M.; Guo Q.; Zhang Y.; Yu B.; Ou Y.; Zhong C.; Ding J. Molecular Mechanism of ADP-Ribose Hydrolysis By Human NUDT5 From Structural and Kinetic Studies. J. Mol. Biol. 2008, 379, 568–578. [DOI] [PubMed] [Google Scholar]
- Klein C.; Paul J. I.; Sauvé K.; Schmidt M. M.; Arcangeli L.; Ransom J.; Trueheart J.; Manfredi J. P.; Broach J. R.; Murphy A. J. Identification of surrogate agonists for the human FPRL-1 receptor by autocrine selection in yeast. Nature Biotechnol. 1998, 16, 1334–1337. [DOI] [PubMed] [Google Scholar]
- Partida-Sánchez S.; Goodrich S.; Kusser K.; Oppenheimer N.; Randall T. D.; Lund F. E. Regulation of Dendritic Cell Trafficking by the ADP-Ribosyl Cyclase CD38: Impact on the Development of Humoral Immunity. Immunity 2004, 20, 279–291. [DOI] [PubMed] [Google Scholar]
- Fliegert R.; Glassmeier G.; Schmid F.; Cornils K.; Genisyuerek S.; Harneit A.; Schwarz J. R.; Guse A. H. Modulation of Ca2+ entry and plasma membrane potential by human TRPM4b. FEBS J. 2007, 274, 704–713. [DOI] [PubMed] [Google Scholar]
- Kirchberger T.; Moreau C.; Wagner G. K.; Fliegert R.; Siebrands C. C.; Nebel M.; Schmid F.; Harneit A.; Odoardi F.; Flugel A.; Potter B. V. L.; Guse A. H. 8-Bromo-cyclic inosine diphosphoribose: towards a selective cyclic ADP-ribose agonist. Biochem. J. 2009, 422, 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill O. P.; Marty A.; Neher E.; Sakmann B.; Sigworth F. J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pfluger’s Arch.—Eur. J. Physiol. 1981, 391, 85–100. [DOI] [PubMed] [Google Scholar]
- Brinkmann V.; Laube B.; Abu Abed U.; Goosemann C.; Zychlinsky A. Neutrophil extracellular traps: how to generate and visualize them. J. Vis. Exp. 2010, 36, e1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunerth S.; Mayr G. W.; Koch-Nolte F.; Guse A. H. Analysis of subcellular calcium signals in T-lymphocytes. Cell. Signalling 2003, 15, 783–792. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.