

Abstract
Exposure of cells to inhibitors of DNA topoisomerase I (topo I) or topoisomerase II (topo II) leads to DNA damage that often involves formation of DNA double-strand breaks (DSBs). DNA damage, particularly induction of DSBs, manifests by phosphorylation of histone H2AX on Ser-139 which is mediated by one of the protein kinases of the phosphoinositide kinase family, namely ATM, ATR, and/ or DNA-PK. The presence of Ser-139 phosphorylated H2AX (γH2AX) is thus a reporter of DNA damage. This protocol describes quantitative assessment of γH2AX detected immunocytochemically in individual cells combined with quantification of cellular DNA content by cytometry. The bivariate analysis of γH2AX expression versus DNA content allows one to correlate DNA damage with the cell cycle phase or DNA ploidy. The protocol can also be used to assess activation (Ser-1981 phosphorylation) of ATM; this event also revealing DNA damage induced by topo I or topo II inhibitors. Examples where DNA damage was induced by topotecan (topo I) and etoposide (topo II) inhibitors are provided.
Keywords: Histone H2AX phosphorylation, ataxia telangiectasia mutated, ATM, DNA double-strand breaks, flow cytometry, apoptosis, cell cycle, topotecan, etoposide
1. Introduction
DNA topoisomerase I (topo I) and topoisomerase II (topo II) inhibitors are among the most widely used anticancer drugs. These drugs stabilize otherwise transient covalent complexes (cleavable complexes), consisting of DNA bound to topo I or topo II, in the process of untangling the DNA helix (1, 2). During DNA replication, the replication forks collide with these complexes converting them to DNA double-strand breaks (DSBs). DSBs can also be formed during transcription when the RNA polymerase molecules, progressing along the transcribed DNA strand, collide with the cleavable complexes (3). Binding of topo inhibitors to DNA in live cells also causes condensation of the DNA-inhibitor complexes (4). In addition to the mechanism involving formation of cleavable complexes, certain topo II inhibitors induce DNA damage, including formation of DSBs, by generating reactive oxygen intermediates (5, 6). Induction of DSBs may trigger apoptosis, and cells replicating their DNA were shown to be particularly susceptible to apoptosis when treated with topo I inhibitors of the camptothecin family or with the topo II inhibitors mitoxantrone or etoposide (6–8).
DNA damage, particularly involving formation of DSBs, triggers phosphorylation of histone H2AX (9), a variant of nucleosome core histone H2A (10). The phosphorylation, mediated by one of the members of the phosphoinositide kinase family, ATM, ATR, or DNA-PK, occurs on Ser-139 within the C terminus of H2AX. Nucleosomes containing this modified H2AX are located within a megabase domain of DNA flanking the DSBs (11). Phosphorylated H2AX, defined as γH2AX, can be detected immuno-cytochemically using γH2AX phospho-specific antibodies (Abs). Shortly after induction of DSBs, the appearance of γH2AX on chromatin manifests in the form of discrete immunofluorescent (IF) nuclear foci, each focus representing a single DSB (12). The intensity of γH2AX IF within an individual cell correlates with extent of DNA damage (frequency of DSBs) in its nucleus. Flow-or laser scanning-cytometry allows the rapid quantification of γH2AX IF in large cell populations, and multiparameter analysis of the cytometric data makes it possible to correlate DNA damage with other attributes of the cell, e.g., cell cycle phase or induction of apoptosis (13–16). Assessment of H2AX phosphorylation combined with the assessment of activation of ATM provides more specific and definitive evidence of DNA damage, particularly formation of DSBs, than analysis of H2AX phosphorylation alone, and the immunocytochemical probe to reveal ATM activation is available (5, 6, 8). This cytometric approach utilizing either γH2AX alone or both γH2AX and ATM-S1981P probes to measure DNA damage is rapid, more sensitive, and less cumbersome compared with the alternative, commonly used method, the comet assay (17).
The method presented in this chapter is designed to assess the intensity of γH2AX and/or ATM-S1981P immunofluorescence (IF) for evaluation of the extent of DNA damage. The detection of γH2AX or ATM-S1981P is combined with differential staining of DNA to measure cellular DNA contents and thus to define the cell cycle phase of the cells in which DNA damage is measured (see Note 1). The procedure is based on indirect IF detection of γH2AX using the primary Ab unlabeled and the secondary Ab conjugated with FITC or Alexa Fluor 488 (see Note 2). DNA is counterstained with propidium iodide (PI) whose emission spectrum (red) is separated from the green color emission of FITC or Alexa Fluor 488. The cells are briefly fixed in methanol-free formaldehyde and then transferred into 70% ethanol in which they can be kept briefly (≥2 h) or stored at −20°C for weeks or longer. Ethanol treatment makes the plasma membrane permeable to the γH2AX Ab; further permeabilization is achieved by including the detergent Triton X-100 in a solution used to incubate cells with the Ab. After incubation with the primary γH2AX Ab, the cells are incubated with FITC or Alexa Fluor 488-labeled secondary Ab and their DNA is counterstained PI in the presence of RNase A to remove RNA, which otherwise, similar to DNA, would be stained with PI. The intensity of cellular green (FITC or Alexa Fluor 488) and red (PI) fluorescence is measured by flow cytometry.
It should be noted that H2AX undergoes constitutive phosphorylation in healthy cells, not treated with radiation or genotoxic agents. This constitutive presence of γH2AX, which is more pronounced in S and G2M than in G1 cells, is considered to be in large part a reflection of oxidative DNA damage caused by metabolically generated oxidants (18, 19). Furthermore, extensive DNA fragmentation that takes place during apoptosis (20) leads to the formation of a large quantity of DSBs which triggers high level of H2AX phosphorylation (21). Strategies are presented in this chapter to differentiate between the DNA damage induced by topo inhibitors versus the constitutive damage occurring in untreated cells (see Note 3), or versus apoptosis-associated (AA) DSBs (see Note 4).
2. Materials
2.1. Reagents
Cells to be analyzed: 106–5 × 106 cells, untreated (control) and treated in culture with topo I or topo II inhibitors, are suspended in 1 ml of tissue culture medium. It is adequate to expose cells to 0.1–2 μM of camptothecin, topotecan, mitoxantrone, or etoposide for 1 h in culture to obtain extensive DNA damage that can be easily detected and measured according to this protocol.
70% ethanol.
Abs: There are several commercially available phospho-specific Abs monoclonal as well as polyclonal, unconjugated and FITC-or Alexa Fluor 488-conjugated, applicable to cytometry, that can be used to detect gH2AX or ATM-S1981P (e.g., from BioLegend, San Diego, CA; Cell Signaling/Santa Cruz Biotechnology, Danvers, MA; Molecular Probes/Invitrogen, Eugene, OR; Millipore/Upstate, Lake Placid, NY). The same companies also offer FITC- or Alexa 488-tagged secondary Abs to be used in conjunction with the unconjugated primary Abs.
12 × 75 mm polypropylene tubes.
Methanol-free formaldehyde fixative: Prepare 1% (v/v) solution of methanol-free formaldehyde (Polysciences, Warrington, PA) in phosphate-buffered saline (PBS). This solution may be stored at 40°C for up to 2 weeks.
BSA–T–PBS: Dissolve bovine serum albumin (BSA; Sigma) in PBS to obtain 1% (w/v) BSA solution. Add Triton X-100 (Sigma Chemical Co., St. Louis, MO) to obtain 0.2% (v/v) of its concentration. This solution may be stored at 40°C for up to 2 weeks.
PI stock solution: Dissolve propidium iodide (PI; Molecular Probes/Invitrogen) in distilled water to obtain a 1 mg/ml solution. This solution can be stored at 40°C in the dark (e.g., in the tube wrapped in aluminum foil) for several months.
PI staining solution: Dissolve DNase-free RNase A (e.g., from Sigma) in PBS to obtain 0.1% (w/v; 100 mg/ml) solution. Add an appropriate aliquot of PI stock solution (e.g., 5 μl per 1 ml) to obtain its 5 μg/ml final concentration. Store the PI staining solution in the dark. This solution may be stored at 40°C for up to 2 weeks.
2.2. Instrumentation
Flow cytometers of different types, offered by several manufacturers, can be used to measure cell fluorescence following staining according to the protocol given below. The most common flow cytometers are from Coulter Corporation (Miami, FL), Becton Dickinson Immunocytometry Systems (San Jose, CA), Cytomation/DAKO (Fort Collins, CO), and PARTEC (Zurich, Switzerland).
3. Methods
Centrifuge cells collected from tissue culture (suspended in culture medium) at 300g for 4 min at room temperature. Suspend cell pellet (1–2 × 106 cells) in 0.5 ml of PBS.
With a Pasteur pipette, transfer this cell suspension into a polypropylene tube (see Note 5) containing 4.5 ml of ice-cold 1% methanol-free formaldehyde solution in PBS. Keep on ice for 15 min.
Centrifuge at 300g for 4 min at room temperature and suspend the cell pellet in 4.5 ml of PBS. Centrifuge again as above and suspend the cell pellet in 0.5 ml of PBS. With a Pasteur pipette, transfer the suspension to a tube containing 4.5 ml of ice-cold 70% ethanol. The cells should be stored in the 70% ethanol at −20°C for at least 2 h, but may be stored under these conditions for up to 2 weeks.
Centrifuge at 200g for 4 min at room temperature, remove the ethanol, and suspend the cell pellet in 2 ml of BSA–T–PBS solution.
Centrifuge at 300g for 4 min at room temperature and suspend the cells again in 2 ml of BSA–T–PBS. Keep at room temperature for 5 min.
Centrifuge at 300g for 4 min at room temperature and suspend the cells in 100 μl of BSA–T–PBS containing 1 μg of the primary γH2AX Ab (see Notes 2 and 6).
Cap the tubes to prevent drying and incubate them overnight at 40°C (see Note 7).
Add 2 ml of BSA–T–PBS and centrifuge at 300g for 4 min at room temperature.
Centrifuge at 300g for 4 min at room temperature and suspend the cells in 2 ml of BSA–T–PBS.
Centrifuge at 300g for 4 min at room temperature and suspend the cell pellet in 100 μl of BSA–T–PBS containing the appropriate (anti-mouse or anti-rabbit, depending on the source of the primary Ab) FITC- or Alexa Fluor 488-tagged secondary Ab (see Note 6).
Incubate for 1 h at room temperature, occasionally gently shaking. Add 5 ml of BSA–T–PBS, and after 2 min, centrifuge at 300g for 4 min at room temperature.
Suspend the cells in 1 ml of the PI staining solution. Incubate at room temperature for 30 min in the dark.
Set up and adjust the flow cytometer for excitation within the blue wavelength range (488-nm laser line or BG-12 excitation filter).
Measure the intensity of green (530 ± 20 nm) and red (>600 nm) fluorescence of the cells by flow cytometry. Record the data.
Fig. 12.1. Bivariate flow cytometry.
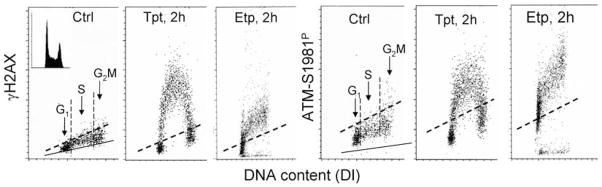
Detection of histone H2AX phosphorylation on Ser139 and ATM activation (phosphorylation on Ser-981) induced by topotecan (Tpt) or etoposide (Etp) in relation to the cell position in the cell cycle. Bivariate (cellular DNA content versus γH2AX IF or DNA content versus ATM-S1981P IF) distributions (scatterplots) of human lymphoblastoid TK6 cells are shown under the following conditions: untreated (Ctrl) or treated in cultures with 0.15 μM Tpt or 10 μM of Etp for 2 h (22). The cells were processed as described in the protocol; their fluorescence was measured by flow cytometry (FACScan; Becton Dickinson Immunochemistry, San Jose, CA). Note increased γH2AX and ATM-S1981P IF of S-phase cells from the Tpt-treated culture and the cell cycle phase-indiscriminative (overall) increase of γH2AX and ATM-S1981P IF of cells from the Etp-treated culture. Based on differences in DNA content, the subpopulations of cells in G1, versus S versus G2M phases of the cycle, may be distinguished and gated as shown in the Ctrl samples (vertical dashed lines). The mean γH2AX IF or ATP-S1981P IF may be then calculated for each cell cycle-phase subpopulation. To estimate the extent of the treatment-induced H2AX phosphorylation or ATM activation, the γH2AX or ATM-S10981P IF mean values of the untreated cells have to be subtracted from the respective G1, S, and G2M means of the treated samples to obtain the drug-induced increase (Δ) of the mean values. The dashed line represents the upper γH2AX or ATM-S1981P IF threshold for 95% of the cells from the untreated (Ctrl) cultures. The solid skewed lines represent the upper 95% threshold of the fluorescence intensity for the nonspecific isotype control. The cellular DNA content histogram of the cells subjected to Tpt or Etp treatment is shown in the left panel inset.
Acknowledgments
Supported by NCI grant RO1 28704.
Footnotes
On the bivariate distributions (scatterplots), subpopulations of cells in G1, versus S versus G2/M, are distinguished based on differences in their DNA content (intensity of PI fluorescence; see Fig. 12.1). To assess the mean extent of DNA damage for cells at a particular phase of the cycle, the mean values of γH2AX or ATM-S1981P IF are calculated separately for G1, S, and G2/M cells distinguished based on differences in DNA content, by the computer-interactive “gating” analysis. The gating analysis should be carried out to obtain mean values of γH2AX or ATM-S1981P IF for G1 (DNA index, DI =0.9–1.1), S (DI =1.2–1.8), and G2M (DI = 1.9–2.1) cell subpopulations.
If primary Ab conjugated with fluorochrome (e.g., with FITC or Alexa Fluor 488) is being used, replace the unlabelled Ab in step 5 with the conjugated one and move to step 10, omitting steps 6–9.
A low level of γH2AX or ATM-S1981P IF seen in the cells that have not been treated with inducers of DSBs represents the “background,” constitutive H2AX phosphorylation, and ATM activation, most of which reflects oxidative DNA damage induced by metabolically generated reactive oxygen species (ROS) (18, 19). The level of constitutive γH2AX phosphorylation and ATM activation is generally higher in S- and G2M- than in G1-phase cells and varies between different cell lines. To quantify the γH2AX or ATM-S1981P IF induced by external factors that damage DNA (e.g., topo I or topo II inhibitors), the constitutive component of γH2AX IF or ATM-S1981P has to be subtracted. Toward this end, the means of γH2AX or ATM-S1981P IF of G1, S, and G2/M-phase of the untreated cells are subtracted from the respective means of the G1, S, and G2/M subpopulations of the drug-treated cells, respectively (see Fig. 12.1). After the subtraction, the extent of increase in intensity of γH2AX (ΔγH2AX IF) or ATM-S1981P (ΔATM-S1981P IF) over the untreated sample represents the treatment-induced phosphorylation of this protein. The irrelevant isotype control can be used to estimate the nonspecific Ab binding component, although its use may be unnecessary when one is interested only in the assessment of the drug-induced increase (Δ) in IF.
DNA undergoes extensive fragmentation during apoptosis (20), which leads to the appearance of a multitude of DSBs in apoptotic cells, triggering H2AX phosphorylation and ATM activation (6, 8, 21). It is often desirable, therefore, to distinguish between primary DSBs induced by DNA damaging agents (e.g., topo inhibitors) versus DSBs generated during apoptosis. The following attributes of γH2AF IF allow one to distinguish the cells with the radiation-, drug-or carcinogen-induced H2AX phosphorylation from the cells that have phosphorylation of this histone triggered by apoptosis-associated (AA) DNA fragmentation: (a) The γH2AX IF induced by external DNA damaging agents is seen rather early during the treatment (10 min to 2 h) whereas AA γH2AX IF is seen later (>3 h) (6, 8, 21); (b) The intensity of AA γH2AX IF is generally much higher than that of DNA damage-induced γH2AX IF, unless the cells are at a late stage of apoptosis (21); (c) The induction of AA γH2AX IF is prevented by cell treatment with the caspase inhibitor z-VAD-FMK, which precludes activation of endonucleases responsible for DNA fragmentation. In its presence, thus, the AA γH2AX IF is suppressed (16); and (d) the AA H2AX phosphorylation occurs concurrently with activation of caspase-3 in the same cells. Multiparameter analysis (active caspase-3 versus γH2AX IF), thus, provides a direct approach to distinguish cells in which DSBs were caused by inducers of DNA damage (active caspase-3 is undetectable) from the cells that have H2AX phosphorylation and ATM activation additionally triggered in response to apoptotic DNA fragmentation (active caspase-3 is present). These strategies are discussed in more detail elsewhere (16).
If the sample initially contains a small number of cells, they may be lost during repeated centrifugations. To minimize cell loss, polypropylene or siliconized glass tubes are recommended. Since transferring cells from one tube to another causes electrostatic attachment of a large fraction of cells to the surface of each new tube, all steps of the procedure (including fixation) preferentially should be done in the same tube. Addition of 1% BSA to rinsing solutions also decreases cell loss. When the sample contains very few cells, carrier cells (e.g., chick erythrocytes) may be included; they may be recognized during analysis based on differences in DNA content (intensity of PI fluorescence).
Quality of the primary and of the secondary antibody is of utmost importance. The ability to detect γH2AX or ATM-S1981P is often lost during improper transport or storage conditions of the Ab. We have occasionally observed that Abs provided by vendors are defective. Also of importance is the use of the Abs at optimal concentration. It is recommended that with the first use of every new batch, the primary and secondary Abs should be tested at serial dilutions (e.g., within the range between 0.2 and 2.0 μg/100 μl) to determine their optimal titer for detection of γH2AX or ATM-S1981P. The optimal titer is recognized as giving maximal signal-to-noise ratio, i.e., the maximal ratio of the mean IF intensity of the drug-treated cells to the untreated cells. The titer recommended by the vendor is not always the optimal one.
Alternatively, incubate for 1 h at 22–24°C. The overnight incubation at 4°C, however, appears to yield somewhat higher intensity of γH2AX IF or ATM-S1981P IF compared with 1-h incubation.
References
- 1.Hsiang YH, Lihou MG, Liu LF. Arrest of replication forks by drug stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- 2.D‘Arpa P, Beardmore C, Liu LF. Involvement of nucleic acid synthesis in cell killing mechanisms of topoisomerase poisons. Cancer Res. 1990;50:6916–6924. [PubMed] [Google Scholar]
- 3.Wu J, Liu LF. Processing of topoisomerase I cleavable complexes into DNA by transcription. Nucleic Acids Res. 1997;25:4181–4186. doi: 10.1093/nar/25.21.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kapuscinski J, Darzynkiewicz Z. Relationship between the pharmacological activity of antitumor drugs Ametantrone and Mitoxantrone (Novantrone) and their ability to condense nucleic acids. Proc Natl Acad Sci USA. 1986;83:6302–6306. doi: 10.1073/pnas.83.17.6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang X, Kurose A, Tanaka T, Traganos F, Dai W, Darzynkiewicz Z. Activation of ATM and histone H2AX phosphorylation induced by mitoxantrone but not by topotecan is prevented by the antioxidant N-acetyl-L-cysteine. Cancer Biol Ther. 2006;5:959–964. doi: 10.4161/cbt.5.8.2878. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka T, Halicka HD, Traganos F, Seiter K, Darzynkiewicz Z. Induction of ATM activation, histone H2AX phosphorylation and apoptosis by etoposide: relation to the cell cycle phase. Cell Cycle. 2007;6:371–376. doi: 10.4161/cc.6.3.3835. [DOI] [PubMed] [Google Scholar]
- 7.Del Bino G, Bruno S, Yi PN, Darzynkiewicz Z. Apoptotic cell death triggered by camptothecin or teniposide: the cell cycle specificity and effects of ionizing radiation. Cell Prolif. 1992;25:537–548. doi: 10.1111/j.1365-2184.1992.tb01458.x. [DOI] [PubMed] [Google Scholar]
- 8.Kurose A, Tanaka T, Huang X, Halicka HD, Traganos F, Dai W, Darzynkiewicz Z. Assessment of ATM phosphorylation on Ser-1981 induced by DNA topoisomerase I and II inhibitors in relation to Ser-139-histone H2AX phosphorylation, cell cycle phase and apoptosis. Cytometry A. 2005;68A:1–9. doi: 10.1002/cyto.a.20186. [DOI] [PubMed] [Google Scholar]
- 9.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 10.West MH, Bonner WM. Histone 2A, a heteromorphous family of eight protein species. Biochemistry. 1980;19:3238–3245. doi: 10.1021/bi00555a022. [DOI] [PubMed] [Google Scholar]
- 11.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sedelnikova OA, Rogakou EP, Panuytin IG, Bonner W. Quantitative detection of 125IUdr-induced DNA double-strand breaks with γ-H2AX antibody. Radiat Res. 2002;158:486–492. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 13.MacPhail SH, Banath JP, Yu TY, Chu EH, Lambur H, Olive PL. Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. Int J Radiat Biol. 2003;79:351–358. doi: 10.1080/0955300032000093128. [DOI] [PubMed] [Google Scholar]
- 14.Huang X, Okafuji M, Traganos F, Luther E, Holden E, Darzynkiewicz Z. Assessment of histone H2AX phosphorylation induced by DNA topoisomerase I and II inhibitors topotecan and mitoxantrone and by DNA cross-linking agent cisplatin. Cytometry. 2004;58A:99–110. doi: 10.1002/cyto.a.20018. [DOI] [PubMed] [Google Scholar]
- 15.Huang X, Halicka HD, Traganos F, Tanaka T, Kurose A, Darzynkiewicz Z. Cytometric assessment of DNA damage in relation to cell cycle phase and apoptosis. Cell Prolif. 2005;38:223–243. doi: 10.1111/j.1365-2184.2005.00344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka T, Huang X, Halicka HD, Zhao H, Traganos F, Albino AP, Dai W, Darzynkiewicz Z. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A. 2007;71A:648–661. doi: 10.1002/cyto.a.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Speit G, Hartmann A. The comet assay (single cell gel test) Methods Mol Biol. 1999;113:203–212. doi: 10.1385/1-59259-675-4:203. [DOI] [PubMed] [Google Scholar]
- 18.Zhao H, Tanaka T, Halicka HD, Traganos F, Zarebski M, Dobrucki J, Darzynkiewicz Z. Cytometric assessment of DNA damage by exogenous and endogenous oxidants reports the aging-related processes. Cytometry A. 2007;71A:905–914. doi: 10.1002/cyto.a.20469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle. 2006;5:1940–1945. doi: 10.4161/cc.5.17.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kajstura M, Halicka HD, Pryjma J, Darzynkiewicz Z. Discontinuous fragmentation of nuclear DNA during apoptosis revealed by discrete “sub-G1” peaks on DNA content histograms. Cytometry A. 2007;71A:125–131. doi: 10.1002/cyto.a.20357. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka T, Kurose A, Huang X, Dai W, Darzynkiewicz Z. ATM kinase activation and histone H2AX phosphorylation as indicators of DNA damage by DNA topoisomerase I inhibitor topotecan and during apoptosis. Cell Prolif. 2006;39:49–60. doi: 10.1111/j.1365-2184.2006.00364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]