

The mitochondrial fission protein Drp1 binds to Mff on mitochondria, followed by entry into a complex with Fis1 at the ER–mitochondrial interface. Mutations in Fis1 disrupt disposal of defective mitochondria when fission is induced by stress. Fis1 thus acts in sequence with Mff to couple mitochondrial fission with downstream degradation processes.
Abstract
Mitochondrial fission is mediated by the dynamin-related protein Drp1 in metazoans. Drp1 is recruited from the cytosol to mitochondria by the mitochondrial outer membrane protein Mff. A second mitochondrial outer membrane protein, named Fis1, was previously proposed as recruitment factor, but Fis1−/− cells have mild or no mitochondrial fission defects. Here we show that Fis1 is nevertheless part of the mitochondrial fission complex in metazoan cells. During the fission cycle, Drp1 first binds to Mff on the surface of mitochondria, followed by entry into a complex that includes Fis1 and endoplasmic reticulum (ER) proteins at the ER–mitochondrial interface. Mutations in Fis1 do not normally affect fission, but they can disrupt downstream degradation events when specific mitochondrial toxins are used to induce fission. The disruptions caused by mutations in Fis1 lead to an accumulation of large LC3 aggregates. We conclude that Fis1 can act in sequence with Mff at the ER–mitochondrial interface to couple stress-induced mitochondrial fission with downstream degradation processes.
INTRODUCTION
Mitochondrial fission is mediated by dynamin-related proteins (Drp1 in metazoans and Dnm1 in yeast). These proteins are predominantly cytosolic, but a small fraction can assemble into spirals that wrap around the circumference of mitochondria and sever the membranes through constriction (Bleazard et al., 1999; Labrousse et al., 1999; Smirnova et al., 2001; Ingerman et al., 2005). Recruitment of Drp1 to mitochondria is mediated by proteins that are anchored in the mitochondrial outer membrane. Mammalian cells have three structurally distinct classes of recruitment factors on their mitochondrial outer membranes: Fis1, Mff, and the two related proteins MiD49 and MiD51 (MIEF1; James et al., 2003; Gandre-Babbe and van der Bliek, 2008; Otera et al., 2010; Palmer et al., 2011; Zhao et al., 2011).
Fis1 was first discovered in yeast, in which it is the sole recruitment factor on the outer membrane (Mozdy et al., 2000). This protein has two TPR motifs that bind to the yeast Drp1 homologue Dnm1 through adaptor proteins (Mdv1 and Caf4; Tieu and Nunnari, 2000; Griffin et al., 2005; Koch et al., 2005; Kobayashi et al., 2007; Koirala et al., 2010). Fis1 is present throughout the animal kingdom, but its functions in metazoans have been unclear. Fis1 can bind to human Drp1 in vitro, can promote fission when overexpressed, and has been implicated in a number of fission-dependent processes, such as apoptosis and autophagy (James et al., 2003; Yoon et al., 2003; Lee et al., 2004; Jofuku et al., 2005; Gomes and Scorrano, 2008; Twig et al., 2008). However, mammalian Fis1−/− cells have mild or no fission defect (Otera et al., 2010; Loson et al., 2013), suggesting that Fis1 plays an ancillary role in the fission process.
Mff is believed to be the principal recruitment factor for Drp1 on mitochondria in metazoans. Mff was shown to promote mitochondrial and peroxisome fission in mammalian cells (Gandre-Babbe and van der Bliek, 2008) and subsequently to be the main Drp1 receptor (Otera et al., 2010). MiD49 and MiD51 (MIEF1) proteins can act as alternative receptors because they also bind to Drp1 and affect fission when they are overexpressed or when their levels are altered in Mff-knockout cells (Loson et al., 2013; Palmer et al., 2013). Mff, MiD49, and MiD51 (MIEF1) proteins were shown to independently promote fission through Drp1 when they are expressed in a heterologous system (Koirala et al., 2013). These experiments show that Mff, Mid49/Mief, and Mid51 proteins are distinct Drp1 recruitment factors. However, MiD49 and MiD51 (MIEF1) proteins are found only in vertebrates, whereas Mff is present in all metazoans, so it would appear that MiD49 and MiD51 (MIEF1) proteins provide a vertebrate-specific function during fission.
While studying the Caenorhabditis elegans fission proteins, we observed essential roles for Mff and Drp1 in stress-induced fission. These observations are consistent with earlier results showing that mitochondrial fission is needed to separate healthy from defective parts of mitochondria (Twig et al., 2008). Moreover, genetic interactions between mutations in mitochondrial fission proteins and mutations in the Parkinson's proteins Pink1 and Parkin in Drosophila also suggest that mitochondrial fission is important for eliminating defective mitochondria (Deng et al., 2008; Poole et al., 2008; Yang et al., 2008). Pink1 and Parkin are key regulators of mitophagy, which is a specialized form of autophagy. Pink1 is a serine threonine kinase that is normally imported into mitochondria and rapidly degraded. Pink1 import and degradation are blocked when damaged mitochondria lose their membrane potential. De novo–synthesized Pink1 is then shunted to the mitochondrial outer membrane, where it recruits the E3 ubiquitin ligase Parkin from the cytosol (Chan et al., 2011; Youle and Narendra, 2011). Parkin-mediated ubiquitination leads to degradation of several key proteins, including mitofusins and Miro, which promote fusion between mitochondria and transport along microtubules (Youle and Narendra, 2011). By degrading these proteins, defective mitochondria are effectively isolated from the remaining population of mitochondria. It is not clear, however, to what extent and how directly mitochondrial fission proteins interact with mitophagy proteins.
Here we describe a new role for Fis1, acting in sequence with Mff. Fis1 mutants can still generate mitochondrial fragments upon treatment with stress-inducing chemicals, but the resulting autophagosomes form large LC3-positive aggregates that persist for many hours. Aggregate formation is inhibited by mutations in Drp1 and Mff and by mutations in Pink1, showing that Fis1 contributes to mitophagy. Chemicals that induce fission also promote interactions between Fis1, Drp1m and endoplasmic reticulum (ER) proteins, suggesting that Fis1 helps coordinate fission with changes in the ER–mitochondrial interface. We conclude that Fis1 acts after Drp1 and Mff initiate mitochondrial fission, guiding this process toward major cellular stress response pathways.
RESULTS
Effects of Fis1 and Mff on mitochondrial fission in C. elegans
To investigate the functions of Fis1 and Mff in C. elegans, we looked at strains with large deletions in each of the two C. elegans Fis1 and Mff genes. These were crossed, generating fis-1(tm1867); fis-2(gk414) and mff-1(tm2955); mff-2(tm3041) double mutants and a quadruple mutant with deletions in all four genes (hereafter called Fis1, Mff, and Fis1 Mff mutants). The effects on mitochondrial and peroxisome morphologies were compared with the strong fission defects in the drp-1(tm1108) deletion strain. Our results show that fis-1 and fis-2 single and double mutants have wild-type mitochondrial morphologies, as also shown by others (Breckenridge et al., 2008), that mff-1 and mff-2 single mutants have weak effects, and that the Mff double mutant has a mitochondrial fission defect similar to but not as strong as the drp-1 defect (Labrousse et al., 1999; Figure 1A and Supplemental Figure S1A). Analogous results were obtained with a peroxisome marker, showing punctate peroxisomes in wild-type and Fis1 mutants and tubular peroxisomes in drp-1 mutant and Mff double mutants (Supplemental Figure S1B). We conclude that C. elegans Mff homologues affect mitochondrial and peroxisome fission, whereas Fis1 homologues have no obvious effects.
FIGURE 1:
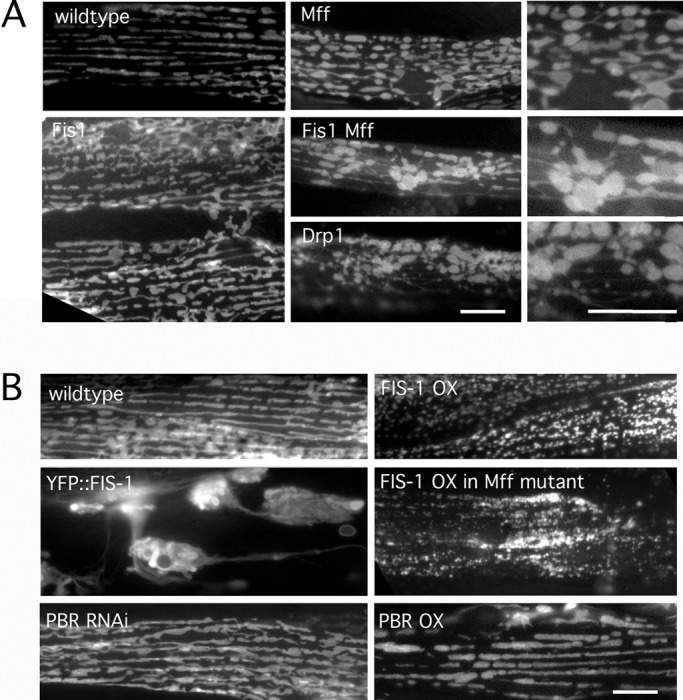
Effects of Mff and Fis1 on mitochondrial fission in C. elegans. (A) Mitochondria in C. elegans body wall muscles were labeled with mitochondrial outer membrane marker (YFP::Tom70, green) in strains as indicated. Right, enlargements of the Mff, Fis1 Mff, and Drp1 mutant images. (B) Effects of FIS-1 and YFP::FIS-1 overexpression with the muscle-specific myo-3 promoter. Mitochondria were labeled with YFP::TOM70. As a control for the effects of outer membrane proteins on mitochondrial morphology, we used feeding RNAi and an overexpression construct for another mitochondrial outer membrane protein (peripheral benzodiazepine receptor [PBR]). Bars, 10 μm.
We tested whether Mff is essential for mitochondrial fission in C. elegans, as suggested for mammalian cells (Otera et al., 2010), by inducing mitochondrial fission with the calcium ionophores ionomycin and A23187. These drugs convert mitochondria from their normal tubular shape to small, round, dispersed fragments in wild-type animals. As expected, calcium ionophore–induced fragmentation was not observed in drp-1(tm1108) mutants (Supplemental Figure S1C). However, fragmentation did occur in Fis1 and Mff double mutants and in the Fis1 Mff quadruple mutant, showing that Fis1 and Mff are not absolutely required for mitochondrial fission in C. elegans (Supplemental Figure S1C). To further test to what extent mutations in Fis1 or Mff inhibit mitochondrial fission, we conducted epistasis experiments with RNA interference (RNAi) for mitochondrial fusion genes. The drp-1 deletion completely reversed mitochondrial fragmentation caused by RNAi for the fusion proteins fzo-1 and eat-3 (Head et al., 2011), but deletions in Fis1 and Mff double mutants did not (Supplemental Figure S1, D and E). These results show that the effects of C. elegans Mff mutations are markedly less severe than the effects of a mutation in drp-1.
To test whether Mff mediates Drp1 recruitment in C. elegans, as it does is in mammals, we examined the localization of cyan fluorescent protein (CFP)::DRP-1 in muscle cells of mutant and wild-type animals. CFP::DRP-1 is observed in spots that mark impending fission events, as shown with time-lapse photography (Labrousse et al., 1999). This pattern was also observed in Fis1 and Mff double and Fis1 Mff quadruple mutants (Supplemental Figure S1F). Overexpression of CFP::DRP-1 changes mitochondrial morphologies in Mff double and Fis1 Mff quadruple mutants to a more normal tubular morphology, suggestive of Drp1-dependent fission without Mff (Supplemental Figure S1F). To verify that the different mutants have no effect on Drp1 localization, we conducted subcellular fractionation. Western blots show similar amounts of DRP-1 in the mitochondrial fractions of wild-type, Fis1 and Mff double mutants, and the Fis1 Mff quadruple mutant (Supplemental Figure S1G). C. elegans does not have MiD49 or MiD51 (MIEF1) homologues, which act as additional Drp1 recruitment factors in vertebrates (Palmer et al., 2011; Zhao et al., 2011), but other factors may still exist. Alternatively, Drp1 could bind directly to mitochondrial membranes, as suggested by in vitro binding to cardiolipin-containing liposomes (Montessuit et al., 2010). We conclude that Mff affects fission, but Mff and Fis1 are not essential for fission or for Drp1 recruitment to mitochondria in C. elegans.
Mutations in Fis1 lead to LC3/LGG-1 aggregates
To further investigate the role of Fis1, we tested whether Fis1 overexpression can induce mitochondrial fission in C. elegans. Our results show that FIS-1 overexpressed with the myo-3 promoter can cause fragmentation, similar to the effects of Fis1 overexpression in mammalian cells (James et al., 2003; Figure 1B). FIS-1 overexpression also causes fragmentation in the Mff double mutant, showing that it is able to overcome an Mff deficiency. However, overexpression of a yellow fluorescent protein (YFP)–tagged version of FIS-1 gives rise to grape-like clusters of mitochondria. These clusters are connected by thin tubules of mitochondrial outer membrane (Figure 1B). The few cells in which mitochondria are not clustered also have closed mitochondrial networks, suggesting that YFP::FIS-1 interferes with the fission process. Overexpression of an unrelated mitochondrial outer membrane protein does not have this effect (Figure 1B). These dominant effects show that overexpressed Fis1 can affect mitochondrial fission even though C. elegans Fis1 proteins are not required for fission.
The grape-like clusters of mitochondria in cells with YFP::FIS-1 are confined to small areas, unlike the dispersed mitochondrial distributions in wild-type animals and the drp-1(tm1108) mutant (Head et al., 2011). These clusters resemble autophagosome intermediates (Yoshii et al., 2011), which would be consistent with previous reports showing the induction of autophagy by overexpressed Fis1 and the inhibition of mitophagy by Fis1 small interfering RNA (siRNA) in mammalian cells (Gomes and Scorrano, 2008; Twig et al., 2008). To test whether loss of Fis1 function affects autophagy in C. elegans, we expressed the LC3 homologue LGG-1 fused to CFP in Fis1 and Mff mutants. Fis1 mutants showed a modest but consistent increase in the number and size of LGG-1::CFP clusters when compared with wild type or Mff mutants (Figure 2, A and B). This pattern was dramatically altered by treating worms for 1 h with the reactive oxygen species (ROS)–producing chemical Paraquat, followed by a 5-h recovery. The small LGG-1 clusters in untreated cells were replaced by much larger clusters (Figure 2, A and B). Similar results were obtained when worms were treated with the mitochondrion-specific inhibitor antimycin A (Figure 2, A and B).
FIGURE 2:

Fis1 genes trigger the formation of large autophagic aggregates. (A) CFP::LGG-1 (worm LC3, red) ectopically expressed in worm muscle cells along with YFP::TOM70 (green). Worms were untreated or incubated with Paraquat or antimycin A. Paraquat- and antimycin-treated worms were allowed to recover on plates without drugs (5 and 4 h, respectively). Bar, 10 μm. (B) Aggregate sizes, shown as percentages of cell surface area. Box plots represent 40 or more cells per condition (p values determined with unpaired Student's t test). (C) Surface renderings of LGG-1 aggregates (red) and mitochondria (green) in a Fis1 mutant treated with Paraquat as in A, but without recovery time. Two examples. These renderings were made from confocal microscope images (stacks of 27 images with steps of 230 nm). Bottom left, opacity of the red channel set at 50% to visualize embedded mitochondria (labeled as merged-50%). Otherwise, opacity was set at 100%. Bar, 1 μm.
To determine whether the LGG-1 clusters in Fis1 mutants contain remnants of mitochondria, we conducted triple labeling with the LGG-1 marker and mitochondrial outer membrane and matrix markers. Outer membrane and matrix markers were visible in inclusions in the LGG-1 aggregates, confirming that they contain portions of mitochondria (Supplemental Figure S2, A and B). Three-dimensional reconstructions of confocal images show that the aggregates consist of LGG-1 structures interspersed with mitochondria (Figure 2C). The aggregates disappear when Fis1 is reintroduced with transgenic DNA, confirming that they are caused by mutations in Fis1 (Supplemental Figure S2C). If the worms with aggregates were left to recover on plates without Paraquat, their aggregates persisted for many hours, but the mitochondrial marker (Tom70::YFP) disappeared over time (Supplemental Figure S2D), similar to the preferential degradation of mitochondrial outer membrane proteins in mammalian cells (Chan et al., 2011; Sarraf et al., 2013). CFP::DRP-1 was also initially present in and around the LGG-1 aggregates but then later disappeared (Supplemental Figure S2E). The aggregates disappear over the course of several days. It therefore seems likely that the aggregates are temporary structures formed by LGG-1–containing membranes that engulf portions of mitochondria. We conclude that mutations in Fis1 affect autophagy in C. elegans, causing the formation of large aggregates containing LGG-1, DRP-1, and remnants of mitochondria.
LC3/LGG-1 aggregates do not result from a compensatory response but instead are generated by aberrant mitochondrial fission
The large LGG-1 aggregates in Fis1 mutants may result from stalled mitophagy intermediates or from an alternative autophagy pathway induced to compensate for the loss of Fis1. A compensatory mechanism would be consistent with the surprising lack of brood-size defects in Fis1 mutants grown with increasing concentrations of Paraquat (Supplemental Figure S3A). We tested for compensatory mechanisms by growing Fis1 mutants on pha-4 and daf-16 RNAi bacteria. These two genes encode the FoxA and Foxo3 homologues, which are important transcriptional regulators of the major autophagic stress responses in C. elegans (Panowski et al., 2007), but Paraquat still induces large LGG-1 aggregates in Fis1 mutants grown with pha-4 or daf-16 RNAi (unpublished data), suggesting that these pathways are not required for aggregate formation. We also used quantitative PCR (qPCR) to determine the relative expression levels of autophagy genes controlled by pha-4 and daf-16, but the expression levels of these genes in the Fis1 double mutant was also similar to the expression levels in wild-type animals (Supplemental Figure S3B). These data show that genes encoding macroautophagy proteins are not induced in C. elegans Fis1 mutants.
We used genetic interactions with other fission mutants to test whether the LGG-1 aggregates in Fis1 mutants are by-products of faulty fission. The brood size of the drp-1(tm1108) deletion strain is reduced to zero when grown at 26°C instead of the normal temperature of 20°C (unpublished data). The brood size of the Mff double mutant is somewhat reduced at 26°C, and the brood size of the Fis1 double mutant is the least affected (Supplemental Figure S3C), consistent with the different degrees to which mitochondrial fission is affected in these strains. Importantly, the reduction in brood size is no worse in the Fis1 Mff quadruple mutants than in the Mff mutants, even though Fis1 mutations by themselves also reduce brood size. These data show that there is no additive or synergistic effect of Fis1 and Mff mutations on brood size, consistent with actions in the same pathway. This interpretation was confirmed by further analysis of LGG-1 aggregate formation at elevated temperatures. Wild-type and Mff mutants grown at 25 or 26°C have no aggregates, the Fis1 mutants have large aggregates, and the quadruple mutants have much smaller aggregates (Figure 3, A and B). Similar results were obtained with Paraquat and antimycin A (Figure 3C). Inhibition of aggregate formation by a block in Mff-dependent fission shows that Mff acts upstream of the Fis1-dependent step in this process.
FIGURE 3:

Mutations in Mff and Drp1 suppress aggregate formation. (A) The Fis1 double mutant has large aggregates when grown at 26°C. The Fis1 Mff quadruple mutant has much smaller aggregates under these conditions. Labeled as in Figure 2A. Bar, 10 μm. (B) Aggregate sizes, shown as percentages of cell surface area. Box plots represent 40 or more cells per condition (p values determined with unpaired Student's t test). Results are shown for growth at 25 and 26°C. (C) Mutations in Mff or Drp1 suppress Paraquat- and antimycin A–induced aggregate formation in Fis1 double mutants. Quantification of results is shown as in B.
The order of this pathway was additionally confirmed with Drp1 overexpression and Drp1 RNAi in Fis1-mutant animals. When Drp1 is overexpressed in Fis1-mutant animals, mitochondrial fission is induced, and more and larger aggregates are formed than in the Fis1 mutant alone (Supplemental Figure S3D). In contrast, Drp1 RNAi blocks the formation of aggregates. Moreover, a drp-1 fis-1 fis-2 triple mutant strain also had reduced number and size of aggregates when treated with Paraquat (Figure 3C). Together these results indicate that LGG-1 aggregates are formed during or after fission in Fis1 mutants.
LC3/LGG-1 aggregates are suppressed in a C. elegans pink-1 mutant
We used genetic crosses with a C. elegans Pink1 mutant to determine more directly whether Fis1 affects mitophagy. The C. elegans pink-1(tm1779) allele has a deletion encompassing the translation initiation site and the amino-terminal mitochondrial targeting sequence of Pink1. It therefore seems likely that this is a null allele with little or no mitophagy. One measure of mitophagy in mammalian cells is the formation of individual mitophagosomes, which appear as small spots with colocalizing LC3 and mitochondrial markers. Formation of these spots is blocked by dominant-negative mutations in Drp1 (Frank et al., 2012). We looked for similar spots of colocalization in C. elegans using our LGG-1 and mitochondrial markers. We observed colocalizing spots of ∼1-μm diameter in untreated wild-type and Fis1 mutant animals (Figure 4, A and B) and significantly reduced number of spots in Mff double and Fis1 Mff quadruple mutant strains (Figure 4, A and B). Spots that are still formed in Mff mutants could result from the low levels of fission that occurs in these animals, or they may represent stalled mitophagosome intermediates. In contrast, pink-1 single and pink-1 fis-1 fis-2 triple mutant animals had no colocalizing spots, as expected for a complete block of mitophagy (Figure 4, A and B). These results suggest that initial steps in the formation of mitophagosomes are not inhibited by Fis1 mutations, but their number is reduced by Mff mutations and eliminated by the Pink1 mutation.
FIGURE 4:

A mutation in Pink1 reduces the number of nascent mitophagosomes and suppresses aggregate formation in Fis1 mutants. (A) Mitophagy was induced by treatment for 1 h with Paraquat. Nascent mitophagosomes were detected by colocalization of CFP::LGG-1 (red) and YFP::TOM70 (green) in spots on the surface of mitochondria (arrows). As long as these spots were small (≤1 μm in diameter) they were classified as mitophagosomes. Larger spots were classified as aggregates. Strains were fis-1(tm1861); fis-2(gk414), pink-1(tm1799), and pink-1(tm1799) fis-1(tm1861); fis-2(gk414) (labeled as Fis1, Pink1, and Fis1 Pink1, respectively). Bar, 5 μm. (B) Boxplots showing the occurrence of CFP and YFP colocalizing spots per cell in the different strains (≥100 cells per condition, unpaired Student's t test). (C) Paraquat-, antimycin A–, and temperature-induced aggregate formation in Fis1, Pink1, and Fis1 Pink1 mutant strains. Aggregate sizes are shown as percentages of cell surface area. Box plots represent ≥40 cells per condition (p values determined with unpaired Student's t test). (D) C. elegans muscle cells labeled with (top) Parkin (PDR::CFP, red) and YFP::TOM70 (green) or (bottom) Parkin (PDR::CFP, red) and YFP::LGG-1 (green). Left, wild type, and right, Fis1 mutants treated with Paraquat (PQ). Bar, 10 μm.
The pink-1 fis-1 fis-2 triple mutant also had fewer and smaller LGG-1 aggregates than the Fis1 double mutant (Figure 4C). The suppression of LGG-1 aggregates by mutations in Pink1 could be due to changes in mitochondrial fission or fusion rates, since Pink1 and Parkin trigger the degradation of mitofusins in mammalian cells (Tanaka et al., 2010), and they may activate mitochondrial fission in Drosophila (Yang et al., 2008). Mitochondrial connectivity was somewhat increased in worms grown with Pink1 RNAi and in pink-1(tm1779) animals, but the effect was modest, and there were also other morphological abnormalities suggestive of mitochondrial defects (Supplemental Figure S4, A and B). Although this last experiment does not help decide between a role for Pink1 in fission or fusion, the foregoing experiments with aggregates and colocalizing spots do indicate that LGG-1 aggregates in Fis1 mutants are an aberrant product of Pink1-induced mitophagy. CFP-tagged Parkin also accumulates in LGG-1 aggregates in the Fis1 mutants, further suggesting that they are products of aberrant mitophagy (Figure 4D).
Mammalian Fis1−/− cells also form LC3 aggregates through stress-induced fission
Because of the importance of mitophagy for neurodegenerative diseases such as Parkinson's (Youle and Narendra, 2011) and the lack of an obvious fission defect in Fis1−/− HCT116 cells (Otera et al., 2010), we asked whether mammalian Fis1 could instead contribute to mitophagy as it does in worms. To complement the studies with the Fis1−/− cells, we generated an HCT116 cell line in which both alleles of the Mff gene were deleted (Figure 5A). Mff−/− cells have more elongated mitochondria than either wild-type or Fis1−/− cells, consistent with the role of Mff in recruiting Drp1 to mitochondria (Figure 5, B and C). It is worth noting that the phenotype of Mff−/− cells was not as strong as that of Mff siRNA–treated cells (Gandre-Babbe and van der Bliek, 2008), suggesting that there may be some adaptation through alternative recruitment proteins similar to C. elegans Mff mutants.
FIGURE 5:
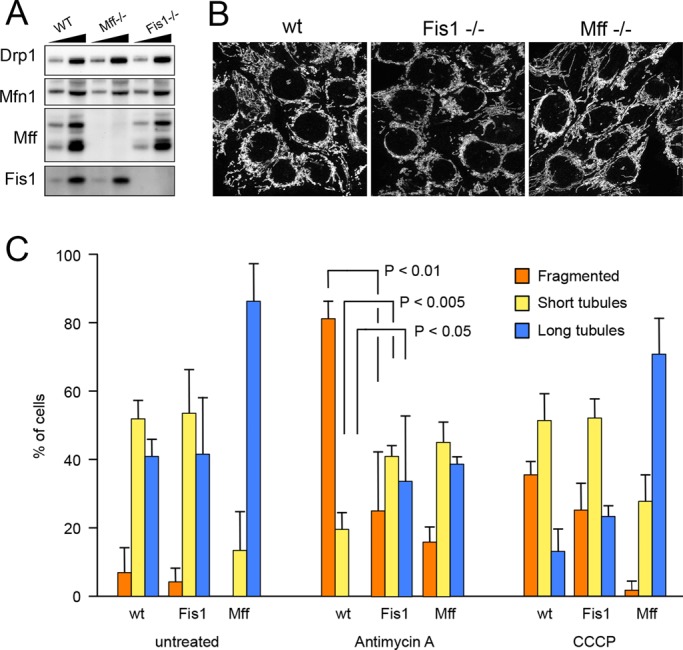
Characterization of mammalian Fis1−/− and Mff−/− cells. (A) Western blots showing that Fis1 is absent from HCT116 Fis1−/− cells and Mff is absent from HCT116 Mff−/− cells. Drp1 and Mfn1 expression levels were not changed. Low and high amounts of cell lysates were loaded on the gel for comparison. (B) Mitochondrial morphologies detected with anti-Tom20 antibody immunostaining in wild-type, Mff−/−, and Fis1−/− cells. (C) Quantification of mitochondrial morphologies in HCT116 wild-type, Fis1−/−, and Mff−/− cells that were left untreated or were treated for 10 min with antimycin A or CCCP. These cells were stained with MitoTracker Green and photographed. Thirty images for each condition were coded and pooled with the images for other conditions, allowing blind classification. Cells were classified as having predominantly fragmented, short tubular, or long tubular mitochondria. Mean and SD for three independent experiments. p values were determined with an unpaired Student's t test.
As previously reported, mutations in Fis1 do not normally affect the balance between mitochondrial fission and fusion (Figure 5, B and C), but it has also been suggested that Fis1 specifically affects stress-induced fission (Kim et al., 2011). To test this, we acutely induced fission by treating HCT116 cells for 10 min with antimycin A or carbonyl cyanide m-chlorophenyl hydrazone (CCCP) and monitored the effects on mitochondrial morphology by fluorescence microscopy. Almost all wild-type cells treated with antimycin A had fragmented mitochondria under these conditions, but fewer Fis1−/− cells had fully fragmented mitochondria (Figure 5C). Fragmentation was complete after prolonged incubations, so Fis1 is not required for fission, but mutations in Fis1 can slow stress-induced fission. This slowing was not observed with CCCP (Figure 5C), suggesting that CCCP and antimycin A trigger mitochondrial fission in different ways. Similar differences were previously observed with CCCP- and etoposide-treated Fis1−/− cells (Loson et al., 2013). We conclude that mutations in Fis1 have modest effects on fission induced by certain chemicals, such as antimycin A, but not by CCCP.
To test whether mammalian Fis1 mutants form LC3 aggregates, we treated the Fis1−/− cells with antimycin A. Because HCT116 cells have undetectably low Parkin expression levels (Ding et al., 2010), these cells were transfected with mCherry-Parkin, along with green fluorescent protein (GFP)–LC3 to follow aggregate formation. The diffuse and cytosolic pattern of GFP-LC3 in wild-type cells is converted to large aggregates in Fis1−/− cells that are treated with antimycin A. In contrast, parental HCT116 cells and Mff−/− cells have few or much smaller aggregates (Figure 6, A and C). Mitochondrial markers colocalize with GFP-LC3 aggregates in Fis1−/− cells like they do in C. elegans Fis1 mutants. The potassium ionophore valinomycin can similarly induce GFP-LC3 aggregates in HCT116 Fis1−/− cells (Supplemental Figure S5A). Immuno–electron microscopy shows that these aggregates contain clusters of mitochondria surrounded by LC3-decorated membranes (Figure 6D). In addition, dominant-negative mutant Drp1 (Drp1K38A; Smirnova et al., 2001) suppresses LC3 aggregate formation in mammalian Fis1−/− cells (Figure 6, B and C). We conclude that mammalian Fis1 mutants form LC3 aggregates and that these aggregates depend on Drp1.
FIGURE 6:

Large LC3 aggregates are formed in Fis1−/− mammalian cells. (A) LC3 aggregates induced by antimycin A in HCT116 Fis1−/− cells but not wild-type (WT) or Mff−/− cells. HCT116 WT, Fis1−/−, and Mff−/− cells were transfected with GFP-LC3 and mCherry-Parkin expression constructs, followed 18 h later by 3-h incubation with antimycin A to induce Parkin translocation onto mitochondria and immunostaining with anti-Tom20 antibody to label mitochondria. Bar, 10 μm. (B) GFP-LC3 aggregate formation is largely suppressed by transfection with a dominant-negative Drp1 mutant (Drp1K38A). Transfections and drugs as in A. (C) Quantification of results in A and B by classifying LC3 distributions as diffuse cytosolic, dispersed punctae, or large aggregates. Untreated cells (no antimycin A) had no aggregates (unpublished data). Mean and SD for three independent experiments (>100 cells/experiment). (D) Immunogold-labeled GFP-LC3 in an HCT116 WT cell and a Fis1−/− cell. Bar, 1 μm.
Longer incubations with antimycin A caused there to be more LC3 aggregates (Supplemental Figure S5B). Transfections with Fis1 shRNA followed by treatment with antimycin A also gave rise to a high percentage of cells with LC3 aggregates (Supplemental Figure S5, C–E), whereas these percentages were reduced with Fis1flox/− cells and with Fis1−/− cells that were transiently transfected with Fis1 (Supplemental Figure S5, C, F, and G). These results confirm that the effects are specific for Fis1. To determine whether the LC3 aggregates are caused by aberrant mitophagy, we manipulated Parkin expression in Fis1−/− cells. Similar to the effects of C. elegans Pink1 mutations, we find that Parkin expression is required for LC3 aggregate formation in mammalian cells (Supplemental Figure S5H). These results suggest that LC3 aggregates are caused by aberrant mitophagy.
A classic method for studying effects on mitophagy is assessment of mitochondrial clearance in HeLa cells that overexpress GFP-Parkin (Narendra et al., 2008). These cells were transfected with scrambled or Fis1 siRNA oligonucleotides, treated for 24 h with antimycin A, and monitored for clearance with fluorescence microscopy and Western blots. However, the results show no obvious differences in clearance between Fis1 siRNA and control cells (Figure 7, A and B). Moreover, clearance was suppressed to the same extent in both cell types when cells were additionally treated with the lysosome fusion inhibitor bafilomycin A1 (Yamamoto et al., 1998; Figure 7B). We conclude that mitophagic flux is unchanged, even though the dependence of LC3 aggregates on Pink1 and Parkin suggests that progression through the mitophagic pathway is abnormal.
FIGURE 7:

Fis1 acts before fusion to lysosomes in mammalian cells. (A) Clearance of mitochondria detected with fluorescence. HeLa cells that stably express GFP-Parkin were transfected with scrambled or Fis1 siRNA oligonucleotides and then treated for the indicated amounts of time with antimycin A. Cells were fixed and stained with Tom20 antibody (shown for 0- and 48-h treatments in the fluorescence images; bar, 10 μm). One hundred or more cells were assessed for the presence mitochondria and dispersal of Parkin as independent criteria at the time points shown in the histogram. (B) Clearance of mitochondria detected with Western blots. GFP-Parkin–expressing HeLa cells were transfected with siRNA and incubated for 48 h with solvent (dimethylsulfoxide), antimycin A, or antimycin A plus bafilomycin A1. Western blots were probed with antibodies for a mitochondrial matrix protein (ClpP), a cytosolic protein (tubulin), and three mitochondrial outer membrane proteins (Tom20, Tom40, and Fis1). The results show that mitochondrial outer membrane and matrix proteins are degraded to similar degrees in scrambled and Fis1 siRNA cells. (C) Western blots show accumulation of lipidated LC3 and inhibition of p62 degradation in Fis1−/− cells treated with antimycin A. HCT116 WT and Fis1−/− cell lines with stable EYFP-Parkin expression were treated with antimycin A for 3 or 6 h before cell lysis and immunoblotting. (D) Valinomycin induces increased LC3 lipidation in Fis1−/− cells, similar to antimycin A treatment, but this increase does not reflect an overall increase in autophagy, as shown with a block in further degradation by bafilomycin A1, nor does it reflect a block in mitochondrial degradation, as shown by the continued degradation of Mfn1 in valinomycin-treated Fis1−/− cells. HCT116 WT and Fis1−/− cell lines with stable enhanced YFP-Parkin expression were incubated for 1 h with or without valinomycin, followed by 5 h with or without bafilomycin A1. The asterisk indicates a nonspecific band. (E) Macroautophagy does not cause more LC3 lipidation in Fis1−/− than in WT cells. Macroautophagy was induced in EYFP-Parkin expressing HCT116 WT and Fis1−/− cells by 6-h amino acid starvation or with 4-h incubation with calcium phosphate precipitate (CPP). (F) Lack of LC3 clustering in cells with starvation-induced macroautophagy. GFP-LC3–transfected HCT116 WT and Fis1−/− cells were subjected to 6-h amino acid starvation and analyzed by fluorescence microscopy. Bar, 10 μm.
To determine how mitophagy might be affected, we probed Western blots for p62 and LC3. We found that the levels of lipidated LC3 are increased in Fis1−/− cells when compared with wild-type cells, especially after treatment with antimycin A (Figure 7C). Effects on LC3 lipidation were similarly observed with valinomycin (Figure 7D), which also induces mitophagy (Rakovic et al., 2010). However, the turnover of p62 was not increased in Fis1−/− cells treated with antimycin A or valinomycin, despite increased LC3 lipidation (Figure 7, C and D). In fact p62 degradation was reduced, suggesting that mutations in Fis1 slow the access of nascent mitophagosomes to downstream degradation processes. To further investigate which stages of autophagy are affected by Fis1, we also incubated valinomycin-treated cells with the downstream inhibitor bafilomycin A1 (Yamamoto et al., 1998). This treatment did not further increase the amount of LC3 lipidation or decrease the amount of p62 turnover, showing that inhibition by mutations in Fis1 occurs upstream of the actions of bafilomycin A1. Remarkably, Mfn1 is degraded to the same extent in wild-type and Fis1−/− cells treated with antimycin A (Figure 7D), showing that deletion of Fis1 does not inhibit the previously described Pink1- and Parkin-dependent proteosomal degradation of Mfn1 or entry onto the mitophagy pathway (Tanaka et al., 2010). Together these data suggest that the rates of entry into and exit from the mitophagy pathway are unchanged. Instead, an intermediate stage may be slowed down.
To test whether the increased lipidation of LC3 is caused by macroautophagy, we grew wild-type and Fis1−/− cells under two conditions that induce macroautophagy (starvation and calcium phosphate precipitation; Gao et al., 2008). Neither treatment caused changes in LC3 lipidation in Fis1−/− cells when compared with wild-type cells, nor are aggregates formed in starved Fis1−/− cells, suggesting that Fis1 plays no role in starvation-induced macroautophagy (Figure 7, E and F). We conclude that human Fis1 acts during the mitophagy process before defective mitochondria are fully eliminated by core autophagy proteins.
Drp1 enters into a complex with Fis1 and ER proteins at the interface between mitochondria and ER
To further investigate processes affected by Fis1, we conducted coimmunoprecipitation experiments with Drp1. It was previously shown that overexpressed Mff and to a lesser extent overexpressed Fis1 will coimmunoprecipitate with Drp1 when a membrane-permeable cross-linker is added before cell lysis (Otera et al., 2010). We were unable to coimmunoprecipitate endogenous Fis1 under these conditions but were able to coimmunoprecipitate endogenous Mff (Figure 8A). This situation changed dramatically upon treatment with CCCP. After 20 min, endogenous Fis1 was readily detectable in coimmunoprecipitates, while at the same time the signal for Mff appeared to decrease. This decrease may reflect transfer of Drp1 from Mff to Fis1 or decrease in the total amount of Drp1 on mitochondria because of completing the fission cycle.
FIGURE 8:
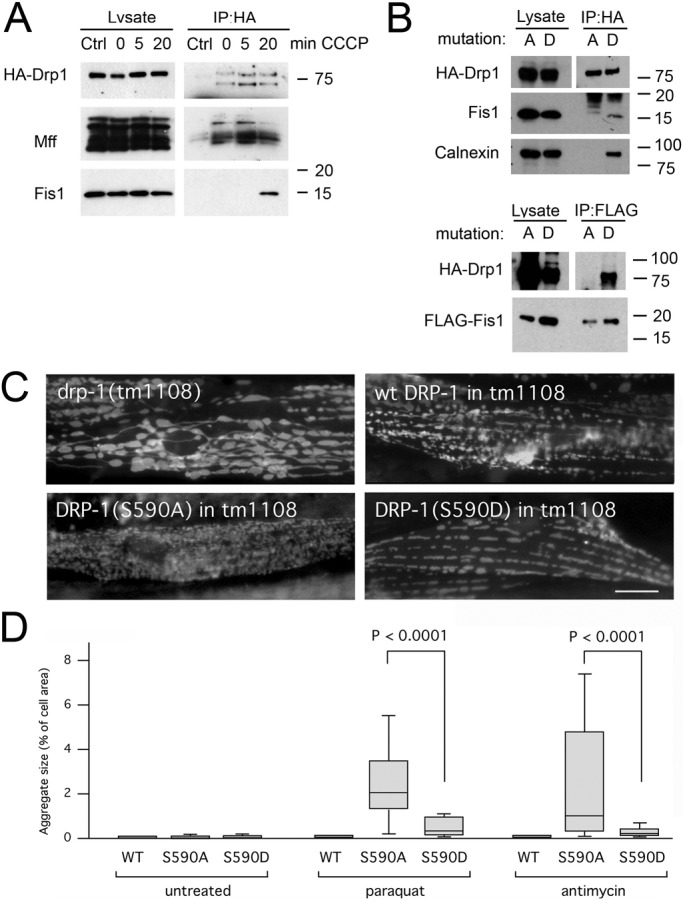
Drp1 enters into a complex with Fis1 when fission is induced. (A) HeLa cells were transfected with HA-Drp1 and treated for the indicated amounts of time with CCCP to induce mitochondrial fission. The cells were then treated with DSP and lysed with 1% SDS before coimmunoprecipitation with mock (Ctrl) or mouse HA antibody. Western blots were probed with rabbit anti-HA, Mff, and Fis1 antibodies. (B) Top, results of HeLa cell transfections with HA-Drp1(S600A) or HA-Drp1(S600D), treatment with DSP, coimmunoprecipitation with HA antibody, and probing for endogenous Fis1. Calnexin also coimmunoprecipitates with HA-Drp1(S600D) but not with HA-Drp1(S600A). Bottom, results obtained after cotransfection with FLAG-tagged Fis1 and coimmunoprecipitation with FLAG antibody. (C) Mitochondrial morphologies in C. elegans muscle cells detected with the mitochondrial outer membrane marker (Pmyo-3::Tom70::YFP). Mitochondria in drp-1(tm1108) worms are highly connected. Mitochondrial fission was restored with transgenic DRP-1(wt), transgenic DRP-1(S590A), or transgenic DRP-1(S590D). Bar, 10 μm. (D) LGG-1 aggregate sizes in muscle cells of drp-1(tm1108) worms treated with chemicals as indicated. Mitochondrial fission was restored with transgenic DRP-1(wt), transgenic DRP-1(S590A), or transgenic DRP-1(S590D). Aggregate sizes are shown as percentages of cell surface area as in Figure 2B (n > 40 cells/condition, unpaired Student's t test).
It was previously shown that Drp1 can bind to Fis1 when it is phosphorylated at Ser-600 by Ca/Cam kinase Iα (Han et al., 2008). Independently, it was shown that this residue is also phosphorylated by Cdk1, Rock1, and protein kinase C δ when fission is induced (Taguchi et al., 2007; Qi et al., 2011; Wang et al., 2012). We tested whether Drp1 mutations that mimic the dephosphorylated or phosphorylated states of Ser-600 promote coimmunoprecipitation with endogenous Fis1 and conversely whether transfected FLAG-tagged Fis1 coimmunoprecipitates either one or both of the Drp1 mutants. We observed robust coimmunoprecipitation with Drp1(S600D) but not Drp1(S600A), suggesting that phosphorylation at Ser-600 is enough to drive Drp1 into a complex with Fis1 (Figure 8B).
We tested whether the interactions promoted by the phosphorylation of Drp1 contribute to LC3/LGG-1 aggregate formation, using mutations in the corresponding phosphorylation site of worm Drp1. We expressed DRP-1(S590D) and DRP-1(S590A) in drp-1(tm1108) animals, which have no endogenous Drp1. Both mutant proteins restored mitochondrial fission, as shown by tubular and fragmented mitochondria, in contrast to the drp-1(tm1108) strain by itself, which has highly connected mitochondria (Figure 8C). Paraquat and antimycin A induced more and larger LGG-1 aggregates in worms with DRP-1(S590A) than in worms with DRP-1(S590D) or wild-type DRP-1 transgene (Figure 8D). DRP-1(S590A) thereby mimics the effects of Fis1 deletions in vivo. We conclude that Drp1 phosphorylation defects cause LC3/LGG-1 aggregate formation similar to Fis1 defects, suggesting that critical interactions with phosphorylated Drp1 prevent aggregate formation.
To test which fission-inducing conditions promote Fis1 coimmunoprecipitation, we treated cells with antimycin A (mitophagic fission), the phorbol-ester phorbol-12-myristate-13-acetate (PMA; fission through kinase activation), and staurosporine (apoptotic fission). We found that each of these chemicals strongly increased coimmunoprecipitation of Fis1 with Drp1 (Figure 9A). The effect was specific, because other mitochondrial outer membrane proteins (Tom20, Tom40, and VDAC) did not coimmunoprecipitate (Figure 9A). We conclude that Drp1 does not normally bind to Fis1 but can coimmunoprecipitate Fis1 when fission is induced under a range of conditions, including apoptosis and mitophagy.
FIGURE 9:

The Drp1-Fis1 complex also contains ER proteins. (A) HeLa cells were transfected with HA-Drp1 and treated as in Figure 8A, but instead of CCCP they were incubated for 3 h with antimycin A (Ant), phorbol myristic acid (PMA), or staurosporine (STS), followed by cross-linking and lysis with RIPA. Blots were probed with HA, Fis1, calnexin, Bap31, Tom40, porin, and Tom20 antibodies. Bap31 is partially degraded by caspase 8–dependent cleavage when cells are treated with staurosporine (Iwasawa et al., 2011), but this treatment clearly also induces coimmunoprecipitation with Drp1. (B) HeLa cells were transfected with scrambled or Fis1 siRNA oligonucleotides, followed after 2 d by transfection with HA-Drp1, and then treated as in A. Surprisingly, the overall amount of calnexin was reduced by Fis1 siRNA, but this did not prevent coimmunoprecipitation with HA-Drp1. (C) Triple labeling with mls::mCherry (red), the ER marker ss:YFP::KDEL (green), and CFP::LGG-1 (blue). The images show that LGG-1 clusters contain ER. Bar, 10 μm. (D) Aggregate sizes in muscle cells of wild-type worms and Fis1 mutants grown with RNAi for C. elegans PACS-2 (T18H9.7) or Bap31 (Y54G2A.18) homologues, followed by treatment with Paraquat. Aggregate sizes are shown as percentages of cell surface area. Boxplots represent ≥40 cells/condition (p values determined with unpaired Student's t test). (E) Diagram showing where Drp1, Mff, and Fis1 act during mitochondrial fission and subsequent degradation through mitophagy.
We also tested whether Drp1 becomes part of a larger complex at the ER–mitochondrial interface (mitochondrion-associated membrane [MAM]), because Fis1 was previously shown to coimmunoprecipitate with Bap31 (an ER protein in the MAM; Iwasawa et al., 2011). Our results show that hemagglutinin (HA)-tagged Drp1 coimmunoprecipitates endogenous Bap31 and calnexin (another MAM protein; Myhill et al., 2008) when cells are treated with antimycin A or PMA (Figure 9A). These treatments induce mitochondrial fragmentation (Supplemental Figure S5I), but the downstream effects are different. Antimycin A induces mitophagy, whereas PMA induces mitochondrial fission, and over a longer period it leads to apoptosis. A third treatment, in which we used staurosporine to more quickly induce apoptosis, does promote coimmunoprecipitation of Fis1 with Drp1, but there was less coprecipitation of calnexin and little or none of Bap31, most likely because Bap31 is cleaved by a caspase during apoptosis (Iwasawa et al., 2011). We conclude that different fission-inducing chemicals cause Drp1 to enter into a fission complex with Fis1 and other MAM proteins.
Similarly, calnexin was found to coimmunoprecipitate with HA-Drp1(S600D) but not HA-Drp1(S600A) (Figure 8B). These results suggest that the S600D mutation promotes entry of Drp1 into the fission complex in the MAM. To determine whether Fis1 is required for this step, we cotransfected Fis1 siRNA oligonucleotides. We noted that Fis1 siRNA caused a reduction in the overall amounts of calnexin but did not prevent coimmunoprecipitation of Bap31 and calnexin with Drp1, suggesting that Fis1 is not required for complex formation (Figure 9B). Instead, Fis1 may contribute to other aspects of fission—for example, helping to coordinate changes at the ER–mitochondrial interface with downstream effects.
ER has been connected with mitochondrial fission (Friedman et al., 2011) and is a source of autophagosomal membrane (Hamasaki et al., 2013), raising the possibility that ER contributes to the aggregates in Fis1 mutants. In support, we can detect an ER marker in the LGG-1 aggregates in C. elegans Fis1 mutants (Figure 9C). Relevance of the MAM for aggregate formation was tested by treating worms with RNAi for C. elegans PACS-2 (an adaptor for calnexin) and Bap31 homologues (Figure 9D). We found that PACS-2 RNAi reduces the size and prevalence of LGG-1 aggregates in Paraquat-treated Fis1-mutant animals (Figure 9D). Because PACS-2 is required for maintaining the ER–mitochondrial interface (Myhill et al., 2008), our result suggests that the MAM contributes to the formation of LGG-1 aggregates in the Fis1 mutant. Taken together, our data show that Drp1 enters into a complex at the MAM when mitochondrial fission is induced. This complex promotes the orderly and efficient removal of defective mitochondria. Fis1 is part of the MAM complex and contributes to a late stage of the removal process but is not required for entry of Drp1 into the MAM or fission.
DISCUSSION
Although Fis1 plays a well-established role in yeast mitochondrial fission (Mozdy et al., 2000), the functions of Fis1 in metazoans have remained a matter of debate. Earlier experiments suggested that Fis1 contributes to mitochondrial fission in mammalian cells (James et al., 2003), and it was suggested that Fis1 can promote stress-induced fission (Kim et al., 2011), but these functions were challenged by the apparent absence fission defects in Fis1−/− cells (Otera et al., 2010). Modest effects on fission and partial redundancy with Mff have, however, also been observed (Loson et al., 2013). Our data confirm this modest effect with specific fission-inducing conditions, but they also reveal much more pronounced effects on downstream stages of removal through mitophagy. Mutations in Fis1 disrupt mitophagy by generating LC3/LGG-1 aggregates upon treatment with ROS-producing toxins such as Paraquat, antimycin A, and valinomycin. These aggregates are suppressed by mutations in Drp1 and Mff, showing that Fis1 acts in sequence with Mff during or after the mitochondrial fission process. We conclude that Fis1 can affect fission and is part of the larger fission complex but is not required for fission and has much more dramatic effects on an intermediate stage of the mitophagy pathway.
A range of different approaches for inactivating Fis1 in mammalian cells and worms give rise to LGG-1/LC3 aggregates, showing that these aggregates do not result from secondary mutations in other genes. Specificity is further supported by our ability to suppress aggregates through Fis1 overexpression in C. elegans. Our data also show that the aggregates were not a secondary response to starvation- or heat shock–induced macroautophagy. Several earlier reports suggested that Fis1 might affect autophagy. It was shown, for example, that Fis1 siRNA reduces the number of mitophagy intermediates (Twig et al., 2008), whereas Fis1 overexpression can induce autophagy in mammalian cells (Gomes and Scorrano, 2008). It was also suggested that C. elegans Fis1 might indirectly affect fission and autophagy (Breckenridge et al., 2008; Kim et al., 2011), and Drosophila studies showed genetic interactions with Pink1 and Parkin mutants (Yang et al., 2008). Our results confirm and extend these observations by showing that LGG-1/LC3 aggregates form when Fis1 mutants are treated with chemicals that disrupt mitochondrial function. These aggregates can contain mitochondrial and ER markers, along with tagged versions of Pink1 and Parkin proteins. More important, they are suppressed by mutations in Pink1 and Parkin genes. Together these results show that the LGG-1/LC3 aggregates in Fis1 mutants are products of aberrant mitophagy.
Mitochondrial clearance, resulting from prolonged incubation with chemicals such as CCCP, is inhibited by mutations in Pink1 and Parkin (Narendra et al., 2008). We find, however, that mutations in Fis1 have little or no effect on clearance. Mfn1 is still degraded at early time points, and the remaining mitochondrial proteins are removed at later time points after incubating in Fis1 mutants with mitochondrial toxins. If clearance is not blocked, then why is there a buildup of LGG-1/LC3 aggregates in Fis1 mutants? Our Western blots show that mutations in Fis1 increase the amount of LC3 but not of p62. There is no further increase when bafilomycin A is added, showing that mutations in Fis1 do not increase the total amount of mitophagy or affect later stages of mitophagy. It seems likely that the total number of mitochondria that enter the mitophagy pathway remains the same but mutations in Fis1 slow the disposal process at a stage before fusion to lysosomes, causing a temporary buildup of intermediates in the disposal pathway and resulting in LC3/LGG-1 aggregates. On the basis of these results, we propose that Fis1 contributes to mitophagy, after Mff and Drp1 initiate mitochondrial fission but before defective mitochondrial fragments are fully engaged in the degradation process (Figure 9E).
Does Fis1 couple fission with mitophagy? Our data show that Drp1 can coimmunoprecipitate Fis1 when mitochondrial fission is chemically induced. Drp1 then also coimmunoprecipitates ER proteins, such as calnexin and Bap31, suggesting that Fis1 is part of a larger protein complex at the ER–mitochondrial interface. The inclusion of ER proteins in the fission complex is consistent with previously observed mitochondrial fission events at the MAM (Friedman et al., 2011) and with active recruitment of Drp1 to the MAM by an ER protein (Korobova et al., 2013). Proteins in the MAM also promote apoptosis (Alirol et al., 2006), as shown by the contributions of Bap31 and Fis1 to this process (Iwasawa et al., 2011). It therefore seems likely that Fis1 acts at a critical junction in the fission process when the products of fission are directed toward normal mitochondrial homeostasis, mitophagy, or apoptosis.
Little is known about the organization of the fission complex or how it might affect the different outcomes of the fission process. Drp1 can bind to Fis1 in vitro (Yoon et al., 2003; Han et al., 2008), but our data do not show a requirement for direct interactions in vivo. Drp1 can still coimmunoprecipitate Bap31 and calnexin when Fis1 is knocked down, and fission is at best mildly impaired in Fis1 mutants. Moreover, binding between Fis1 and a Rab-GAP (Onoue et al., 2013) and between Fis1 and Bap31 (Iwasawa et al., 2011) raises the possibility that Fis1 has other functions during or after fission. Further studies of these interactions may help clarify the roles of Fis1 in mitophagy and apoptosis. Our results do, however, already show that Fis1 acts at a critical junction in major stress-response pathways. Depending on the stress conditions, Fis1 can contribute to apoptosis or to the orderly disposal of defective mitochondria through mitophagy.
MATERIALS AND METHODS
Materials
Paraquat (MP Biomedicals, Santa Ana, CA), A23187, and ionomycin (Calbiochem/Merck, Darmstadt, Germany) were used at the indicated concentrations. Phorbol myristic acid (Calbiochem) was used at 1 μM. CCCP, antimycin A, valinomycin, and bafilomycin A1 were from Sigma-Aldrich (St. Louis, MO) and used at final concentrations of 20 μM, 40 μg/ml, 10 μM, and 100 nM, respectively. Staurosporine (Tocris Biosciences, Bristol, UK) was used at 2 μM. Mff and Mfn1 antibodies were described previously (Gandre-Babbe and van der Bliek, 2008; Tanaka et al., 2010). The following antibodies were purchased: FLAG (Genescript, Piscataway, NJ), HA (Roche, Basel, Switzerland), Tom20, Tom40, Parkin, and normal mouse immunoglobulin G (IgG; Santa Cruz Biotechnology, Santa Cruz, CA), calnexin and Drp1 (BD Transduction Laboratories, Franklin Lakes, NJ), p62 (Abnova, Taipei City, Taiwan), porin (MitoSciences, Eugene, OR), LC3B (Sigma-Aldrich), Fis1 (Alexis Biochemicals and Proteintech, Farmingdale, NY), α-tubulin (Invitrogen, Carlsbad, CA), and actin (Sigma-Aldrich). Rabbit polyclonal antibodies were generated against polypeptide fragments of C. elegans DRP-1 protein and used along with other antibodies to probe blots of C. elegans extracts as described previously (Head et al., 2011).
C. elegans strains, plasmids, and imaging
C. elegans fis-1(tm1867), mff-1(tm2955), mff-2(tm3041), drp-1(tm1108), and pink-1(tm1779) strains were provided by S. Mitani (National Bioresource Project of Japan, Tokyo Women's Medical University School of Medicine, Tokyo, Japan). The fis-2(gk414) strain was provided by the Caenorhabditis Genetics Center (University of Minnesota, St. Paul, MN). These strains were backcrossed four to six times with wild-type N2 animals to remove adventitious mutations. Transgenic strains were made by microinjection, using rol-6 as a transformation marker. Feeding RNAi was as described (Timmons et al., 2001). Full-length fis-1, fis-2, mff-1, and mff-2 cDNAs were cloned in the pPD96.52 expression vector with the myo-3 promoter for muscle cell expression as described previously (Head et al., 2011). YFP coding sequences (Labrousse et al., 1999) were added into the NheI site of the pPD96.52 expression before FIS-1 coding sequences to create the Pmyo-3::YFP::FIS-1 construct. Mitochondrial outer membrane and matrix markers with GFP derivatives under control of the myo-3 promoter were described previously (Labrousse et al., 1999). An mCherry marker was made by replacing GFP with mCherry coding sequences. The CFP::LGG-1 marker, modeled after a previously reported GFP::LGG-1 construct (Melendez et al., 2003), was made by cloning CFP and LGG-1 coding sequences behind the myo-3 promoter in the pPD96.52 vector. The ER marker Pmyo-3::SS::YFP::KDEL was described previously (Labrousse et al., 1999). DRP-1(S590A) and DRP-1(S590D) mutants were made using QuikChange site-directed mutagenesis and Pmyo-3::DRP-1 (Labrousse et al., 1999) as template. The C. elegans Parkin marker Pmyo-3::PDR-1::CFP was made by PCR cloning in the pPD96.52 vector. Feeding RNAi for the peripheral benzodiazepine receptor was achieved with a bacterial construct for the corresponding gene (C41G7.3). Overexpression was achieved with the myo-3 promoter fused to the coding region of C41G7.3. This construct was injected into an rde-1(ne219) (RNAi defective) to prevent antisense RNA.
To determine expression levels with qPCR, C. elegans cultures were synchronized with bleach, cultured with OP50 bacteria, and harvested as young adults. RNA was extracted by repeated freezing and thawing of worms suspended in TRIzol (Invitrogen). RNA preparations were treated with DNaseI, followed by cDNA synthesis using Thermoscript with protocols provided by the manufacturers (Invitrogen and Ambion/Life Technologies, Carlsbad, CA). Primers for qPCR were designed as in Haynes et al. (2007) with the primers TCGGTATGGGACAGAAGGAC and CATCCCAGTTGGTGACGATA for pan-actin (act-1,3,4), TCAATGGATTTCGGTTGGGA and ACGCTCCAACAGGATCTCTA for bec-1 (T19E7.3), ACTTCCTCATCAGAAAACGC and TTCCTCGTGATGGTCCTGGT for lgg-1 (C32D5.9), AGCCCAGGAAGCTCAAGAAG and GGAGACGATCGAATGTCTTT for p62 (T12G3.1), GGTTGAAGAGTCCGAGTTCT and AAGTTGGATGGATGTCTGTC for let-363 (B0261.2), GGGAAGAAACGTGATCATCGA and CAGCCTCCTCATTAGCCTTG for hsp-60 (Y22D7AL.5), and CAAACTCCTGTGTCAGTATCATGGAAGG and GCTGGCTTTGACAATCTTGTATGGAACG for hsp-6 (C37H5.8.3). Each qPCR contained cDNA made from 1 µg of total RNA. qPCRs were performed in triplicate with SYBR Green reagent (Invitrogen) with annealing and extension at 63°C in an Mx3000P Thermal Cycler, followed by analysis with MxPro Software (Agilent, Santa Clara, CA). Dissociation curves confirmed individual PCR products. The Ct values were converted to percentages, and expression ratios were calculated for three independent RNA preparations.
Where indicated, worms were incubated for 1 h at 20°C in M9 media with 80 mM Paraquat and bacteria or for 2 h with 1 mM antimycin A, after which they were allowed to recover on nematode growth media plates with bacteria but no drugs for 5 and 4 h, respectively, at 20°C. Mitochondrial fragmentation was induced by incubating worms for 1 h at 20°C in M9 medium with 50 μM A23187 or ionomycin. For temperature shift experiments, worms were grown for 48 h at 25 or 26°C before imaging. Brood sizes were determined by placing single L1 larvae on individual plates and counting progeny that survive to the L4 larval stage. For subcellular fractionation, synchronized cultures of C. elegans were harvested, homogenized with a glass tissue homogenizer, and fractionated by differential centrifugation (Head et al., 2011). The P2 fraction (14,000 × g pellet) was washed twice with STEG (250 mM sucrose, 5 mM Tris-HCl, 1 mM EGTA, pH 7.4) and protease inhibitors.
Fluorescence images of live worms were made with a Zeiss Axiovert 200M microscope equipped with a 100×/1.45 numerical aperture (NA) α-Plan-Fluar objective (Carl Zeiss, Jena, Germany) and an ORCA ER-CCD camera (Hamamatsu, Shizuoka, Japan). Images in the figures were processed with Photoshop (Adobe, San Jose, CA). Because the muscle cells that we used for this analysis are flat, we could approximate aggregate sizes by measuring their surface areas. These areas were measured by tracing cell and aggregate outlines with ImageJ software (National Institutes of Health, Bethesda, MD; 40 or more cells for each condition). Aggregate sizes are shown as percentages of cell area in boxplots. The horizontal line inside the box is the median. The bottom of the box shows where the first quartile ends and the second begins, and the top shows where the third quartile ends and the fourth begins. The whiskers show minimum and maximum values of the data set, as long as these values do not differ from the median by >1½ times the interquartile range.
Surface renderings of mitochondria and LGG-1 aggregates were made with stacks of 20 fluorescence images collected over a distance of 1 μm with a spinning disk confocal microscope (Marianas System; Intelligent Imaging Innovations, Santa Monica, CA). The images were processed with the three-dimensional software package provided by the manufacturer (Slidebook 5.5). Surface rendering was set with an outline width of 1 pixel and a threshold of 50% for the red channel (LGG-1 fluorescence) and 25% for the green channel (mitochondrial outer membrane marker). Where indicated, opacity of the red channel was set at 50% to visualize embedded mitochondria. Otherwise, opacity was set at 100%.
Mammalian cell culture and transfections
Fis1−/−, Fis1flox/−, and HCT116 with lentivirus-infected control shRNA and Fis1 shRNA were reported previously (Otera et al., 2010). Transfections were done at 18–24 h after transfer using FuGENE 6 (Roche) or Lipofectamine LTX (Invitrogen) as recommended by the manufacturer. Amino acid starvation was induced by washing cells twice with starvation buffer (140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM glucose, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4), followed by a 6-h incubation with starvation buffer containing 1% (wt/vol) bovine serum albumin (BSA). Calcium phosphate precipitates were made by mixing 50 mM HEPES, pH 7.1, and 3 mM Na2HPO4 with an equal volume of 256 mM CaCl2 just before adding this mixture to cultured cells at a final calcium concentration of 12.8 mM. Cells were then incubated for 4 h to induce macroautophagy. The FLAG-tagged Fis1 construct was purchased from Genecopeia (Rockville, MD). Fis1 siRNA oligonucleotides for coimmunoprecipitation experiments were from Sigma-Aldrich (ID SAS_Hs01_00171949_AS). These were transfected into HeLa cells with Oligofectamine as described (Head et al., 2009). HA-tagged Drp1 was described previously (Smirnova et al., 2001). Here we used an N- and C-terminal–tagged version. S600A and S600D mutants were made with a QuikChange protocol using DpnI restriction enzyme and Phusion DNA polymerase from New England BioLabs (Ipswich, MA).
Mff−/− cells were made from HCT116 cells. The first allele was knocked out with rAAV-based homologous recombination (Topaloglu et al., 2005) by targeting exon 4 (present in all Mff isoforms). Because targeting efficiency of rAAV was low (1.4%), transcription activator–like effector nucleases (TALENs) were used for the second allele of Mff (Miller et al., 2011). A synthetic gene coding for Tale (+63) with N- and C-terminal regions of Tale, a half T repeat, and a FokI domain were cloned into the pcDNA3.1/Zeo (+) vector (Invitrogen) to make pcDNA-Talen (+63). An NheI site near the multiple cloning site of pcDNA3.1-Talen (+63) was removed by site-directed mutagenesis. The remaining NheI site was then used to clone the assembled left and right Tale repeats (Huang et al., 2011), generating Mff-Talen-L and Mff-Talen-R constructs. The targeted sequence of Mff was GCTGTTCGCCAAAATGGACAGCTGGTCAGAAATGATTCTCTGTGAGT. A donor DNA similar to that used for rAAV-based gene-targeting vector was constructed into pSEPT-puro vector (pSEPT vector with the neomycin marker replaced by puromycin). Lipofectamine LTX (Invitrogen) was used to transfect HCT116 cells with 0.1 μg of Mff-Talen-L, 0.1 μg of Mff-Talen-R, and 0.8 μg of donor DNA in a 12-well plate. One day after transfection, cells were transferred to 96-well plates with 0.4 μg/ml puromycin and grown for 10 d. Resistant clones were expanded and genotyped by PCR to ensure correct targeting. Cells were grown with McCoy's 5A medium, 10% fetal bovine serum, 1 mM glutamine, and nonessential amino acids.
Immunoblotting, immunostaining, and coimmunoprecipitation of mammalian cells
Total cell lysates were harvested for Western blot analysis by rinsing twice with phosphate buffered saline (PBS), followed by lysis with 2% (vol/vol) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate in PBS with protease inhibitor cocktail (Boehringer). The lysates were incubated for 30 min on ice and clarified by centrifugation (14,000 × g for 15 min at 4°C), and Laemmli sample buffer was added to the supernatants. From 30 to 60 µg of proteins was subjected to 4–12% Bis-Tris SDS–PAGE and transferred to polyvinylidene fluoride membranes. After blocking with 5% milk in PBS-Tween 20 (PBS-T) buffer, membranes were incubated with primary antibodies at room temperature. Membranes were washed three times with PBS-T and incubated with horseradish peroxidase–conjugated secondary antibodies (GE Healthcare, Little Chalfont, UK). Immunoreactive protein bands were detected by ECL Plus reagents (GE Healthcare) and analyzed with a ChemiDoc (BioRad, Hercules, CA). For immunostaining, cells were grown in dishes with borosilicate slides, fixed for 25 min with 4% paraformaldehyde in PBS, and permeabilized for 15 min with 0.15% (vol/vol) Triton X-100 in PBS. Cells were then blocked for 45 min with 10% BSA in PBS and incubated with primary antibodies. Secondary antibodies were Alexa Fluor 594– or 647–conjugated goat anti-mouse or rabbit IgG (Invitrogen). Mammalian cells were imaged with an LSM510 confocal microscope equipped with a 63×/1.4 NA Plan-Apochromat lens (Carl Zeiss). Images were processed with Photoshop.
For immuno–electron microscopy, HCT116 WT and Fis1−/− cells stably expressing YFP-LC3 and mCherry-Parkin were treated with valinomycin for 3 h and fixed for 30 min with 4% paraformaldehyde and 0.05% glutaraldehyde in PBS. The fixed cells were washed four times with PBS, followed by permeabilization for 40 min with 0.1% saponin and 5% goat serum in PBS. The cells were then incubated for 1 h with mouse anti-GFP antibody (clone 3E6 from Invitrogen), followed by 1 h with nanogold-conjugated anti-mouse IgG antibody (Nanoprobes, Yaphank, NY) and further processing as described (Tanner et al., 1996). Thin sections (∼80 nm) were counterstained with uranyl acetate and lead citrate. The sections were examined with a JEOL 200 CX transmission electron microscope. Images were collected with a digital charge-coupled device camera (XR-100 CCD; AMT, Danvers, MA).
For coimmunoprecipitation, HeLa cells were grown in 10-cm dishes, transfected with FuGENE HD (Promega, Madison, WI), and harvested by scraping cells and washing them with PBS, followed by 30 min at 25°C with 1 mM DSP cross-linker (Pierce/Thermo, Rockford, IL) and quenching for 15 min on ice with 10 mM Tris-HCl, pH 7.5. Lysis was done with 1% SDS as described (Otera et al., 2010) or with RIPA buffer. Cell lysates were sonicated for 10 s, followed by centrifugation for 15 min at 21,000 × g. The supernatant was incubated with immune-precipitating antibody and Protein G Dynabeads (Invitrogen). The control antibody was mouse normal IgG.
Supplementary Material
Acknowledgments
We thank other members of the van der Bliek and Youle labs for helpful discussions. Strains were provided by the Caenorhabditis Genetics Center and S. Mitani. Technical support for electron microscopy was provided by the National Institute of Neurological Disorders and Stroke EM Facility. This work was supported by grants from the National Institutes of Health (GM051866) and the National Science Foundation (0552271) to A.M.V.D.B. B.P.H. received a National Institutes of Health Training Grant (T32-GM07104). S.K. was supported by a fellowship from the Naito Foundation and the Nakatomi Foundation. N.H. was supported by the Japan Society for the Promotion of Science, and K.Y. was supported by a Japan Society for the Promotion of Science Fellowship for Research Abroad.
Abbreviations used:
- CCCP
carbonyl cyanide m-chlorophenyl hydrazone
- CFP
cyan fluorescent protein
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- MAM
mitochondrion-associated membrane
- PMA
phorbol-12-myristate-13-acetate
- YFP
yellow fluorescent protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-09-0525) on November 6, 2013.
Present addresses: *Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC 27710.
†Division of Biology Education, College of Education, Chosun University, Gwangju 501-759, Republic of Korea.
REFERENCES
- Alirol E, James D, Huber D, Marchetto A, Vergani L, Martinou JC, Scorrano L. The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis. Mol Biol Cell. 2006;17:4593–4605. doi: 10.1091/mbc.E06-05-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. 1999;1:298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge DG, Kang BH, Kokel D, Mitani S, Staehelin LA, Xue D. Caenorhabditis elegans drp-1 and fis-2 regulate distinct cell-death execution pathways downstream of ced-3 and independent of ced-9. Mol Cell. 2008;31:586–597. doi: 10.1016/j.molcel.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci USA. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285:27879–27890. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta. 2012;1823:2297–2310. doi: 10.1016/j.bbamcr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Ding WX, Stolz DB, Yin XM. Induction of macroautophagy by exogenously introduced calcium. Autophagy. 2008;4:754–761. doi: 10.4161/auto.6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta. 2008;1777:860–866. doi: 10.1016/j.bbabio.2008.05.442. [DOI] [PubMed] [Google Scholar]
- Griffin EE, Graumann J, Chan DC. The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol. 2005;170:237–248. doi: 10.1083/jcb.200503148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head BP, Zulaika M, Ryazantsev S, van der Bliek AM. A novel mitochondrial outer membrane protein, MOMA-1, that affects cristae morphology in Caenorhabditis elegans. Mol Biol Cell. 2011;22:831–841. doi: 10.1091/mbc.E10-07-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol. 2011;29:699–700. doi: 10.1038/nbt.1939. [DOI] [PubMed] [Google Scholar]
- Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, Nunnari J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–1027. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasawa R, Mahul-Mellier AL, Datler C, Pazarentzos E, Grimm S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011;30:556–568. doi: 10.1038/emboj.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- Jofuku A, Ishihara N, Mihara K. Analysis of functional domains of rat mitochondrial Fis1, the mitochondrial fission-stimulating protein. Biochem Biophys Res Commun. 2005;333:650–659. doi: 10.1016/j.bbrc.2005.05.154. [DOI] [PubMed] [Google Scholar]
- Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell. 2011;44:532–544. doi: 10.1016/j.molcel.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Tanaka A, Fujiki Y. Fis1, DLP1, and Pex11p coordinately regulate peroxisome morphogenesis. Exp Cell Res. 2007;313:1675–1686. doi: 10.1016/j.yexcr.2007.02.028. [DOI] [PubMed] [Google Scholar]
- Koch A, Yoon Y, Bonekamp NA, McNiven MA, Schrader M. A role for Fis1 in both mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2005;16:5077–5086. doi: 10.1091/mbc.E05-02-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala S, Bui HT, Schubert HL, Eckert DM, Hill CP, Kay MS, Shaw JM. Molecular architecture of a dynamin adaptor: implications for assembly of mitochondrial fission complexes. J Cell Biol. 2010;191:1127–1139. doi: 10.1083/jcb.201005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala S, Guo Q, Kalia R, Bui HT, Eckert DM, Frost A, Shaw JM. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc Natl Acad Sci USA. 2013;110:E1342–E1351. doi: 10.1073/pnas.1300855110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. 2013;339:464–467. doi: 10.1126/science.1228360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrousse AM, Zapaterra M, Rube DA, van der Bliek AM. C. elegans dynamin-related protein drp-1 controls severing of the mitochondrial outer membrane. Mol Cell. 1999;4:815–826. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–667. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- Miller JC, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Montessuit S, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine Simmen K, Cooper TJ, Thomas G, Simmen T. The subcellular distribution of calnexin is mediated by PACS-2. Mol Biol Cell. 2008;19:2777–2788. doi: 10.1091/mbc.E07-10-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoue K, et al. Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J Cell Sci. 2013;126:176–185. doi: 10.1242/jcs.111211. [DOI] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT. MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem. 2013;288:27584–27593. doi: 10.1074/jbc.M113.479873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–573. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature. 2007;447:550–555. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Disatnik MH, Shen N, Sobel RA, Mochly-Rosen D. Aberrant mitochondrial fission in neurons induced by protein kinase Cδ under oxidative stress conditions in vivo. Mol Biol Cell. 2011;22:256–265. doi: 10.1091/mbc.E10-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakovic A, Grunewald A, Seibler P, Ramirez A, Kock N, Orolicki S, Lohmann K, Klein C. Effect of endogenous mutant and wild-type PINK1 on Parkin in fibroblasts from Parkinson disease patients. Hum Mol Genet. 2010;19:3124–3137. doi: 10.1093/hmg/ddq215. [DOI] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the Parkin-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner VA, Ploug T, Tao-Cheng JH. Subcellular localization of SV2 and other secretory vesicle components in PC12 cells by an efficient method of preembedding EM immunocytochemistry for cell cultures. J Histochem Cytochem. 1996;44:1481–1488. doi: 10.1177/44.12.8985140. [DOI] [PubMed] [Google Scholar]
- Tieu Q, Nunnari J. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol. 2000;151:353–366. doi: 10.1083/jcb.151.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- Topaloglu O, Hurley PJ, Yildirim O, Civin CI, Bunz F. Improved methods for the generation of human gene knockout and knockin cell lines. Nucleic Acids Res. 2005;33:e158. doi: 10.1093/nar/gni160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, Chang BH, Schumacker PT, Danesh FR. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15:186–200. doi: 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762–2778. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.