

PDLIM2 integrates cytoskeletal signaling with gene expression to enable reversible differentiation of epithelial cancer cells. PDLIM2 associates with the COP9 signalosome and controls its nuclear translocation and the stability of key transcription factors necessary for either a mesenchymal or an epithelial phenotype.
Abstract
Epithelial cell differentiation and polarized migration associated with epithelial-to-mesenchymal transition (EMT) in cancer requires integration of gene expression with cytoskeletal dynamics. Here we show that the PDZ-LIM domain protein PDLIM2 (Mystique/SLIM), a known cytoskeletal protein and promoter of nuclear nuclear factor κB (NFκB) and signal transducer and activator of transcription (STAT) degradation, regulates transcription factor activity and gene expression through the COP9 signalosome (CSN). Although repressed in certain cancers, PDLIM2 is highly expressed in invasive cancer cells. Here we show that PDLIM2 suppression causes loss of directional migration, inability to polarize the cytoskeleton, and reversal of the EMT phenotype. This is accompanied by altered activity of several transcription factor families, including β-catenin, Ap-1, NFκB, interferon regulatory factors, STATs, JUN, and p53. We also show that PDLIM2 associates with CSN5, and cells with suppressed PDLIM2 exhibit reduced nuclear accumulation and deneddylation activity of the CSN toward the cullin 1 and cullin 3 subunits of cullin-RING ubiquitin ligases. Thus PDLIM2 integrates cytoskeleton signaling with gene expression in epithelial differentiation by controlling the stability of key transcription factors and CSN activity.
INTRODUCTION
Epithelial-to-mesenchymal transition (EMT) involves a complex series of molecular and cellular events by which epithelial cells acquire a migratory and invasive phenotype during embryonic development and in cancer progression (reviewed in Thiery et al., 2009; Nieto and Cano, 2012). EMT is associated with suppression of epithelial genes such as E-cadherin, loss of cell polarity, and loss of cell–cell contacts enabling cell migration and invasion. In turn, reverse differentiation facilitates colonization and secondary growth at new sites and requires reexpression of E-cadherin, a process referred to as mesenchymal-to-epithelial reverting transition (MErT; Chao et al., 2010; Tsai et al., 2012). Although much evidence exists for reversible epithelial differentiation in metastasis, the mechanism of its regulation at the cellular level is not well understood.
The PDZ-LIM domain protein PDLIM2 (also known as Mystique or SLIM) is a cytoskeletal and nuclear protein that was identified in cornea epithelial cells (Torrado et al., 2004), in fibroblasts transformed by overexpression of the Insulin-like Growth Factor 1 (IGF-1) Receptor (Mystique), and in epithelial cancer cells, where it is required for cell adhesion and migration (Loughran et al., 2005a, b). In T-lymphocytes, PDLIM2 (SLIM) regulates the stability of signal transducer and activator of transcription (STAT) and nuclear factor κB (NFκB)-p65 transcription factors in the nucleus (Tanaka et al., 2005, 2007, 2011; Guo et al., 2010), and in macrophages, differentiation causes increased PDLIM2 expression and sequestration in the cytoplasm (Healy and O'Connor, 2009).
Several studies implicate PDLIM2 expression with tumorigenesis and potentially with tumor suppression. PDLIM2 is located on chromosome 8p21 (Loughran et al., 2005a), a region that is disrupted in many cancers (Macartney-Coxson et al., 2008). In colon cancer, PDLIM2 expression is slightly decreased in tumor compared with normal colon tissue, and although a single nucleotide polymorphism was detected in metastasis, no significant difference in expression was observed between tumor and liver metastasis (Macartney-Coxson et al., 2008). The PDLIM2 gene is methylated in lung cancer (Pfeifer and Rauch, 2009) and in several cell lines (Qu et al., 2010a, b). PDLIM2 promotes degradation of the human T-cell virus type 1 (HTLV-1) Tax oncoprotein and is repressed in response to HTLV1-mediated transformation of T lymphocytes (Yan et al., 2009; Zhao et al., 2009; Fu et al., 2010). Overexpression of PDLIM2 in cancer cell lines decreases anchorage-independent growth and reduces tumor growth in vivo, again suggesting a tumor-suppressive function (Loughran et al., 2005a; Qu et al., 2010a, b). However, PDLIM2 is highly expressed in cell lines derived from metastatic cancer, including androgen-independent prostate cancer cell lines (Loughran et al., 2005b).
PDZ- and LIM-domain proteins have important functions in controlling cell fate (Zheng et al., 2010). We hypothesized that PDLIM2 expression is associated with the differentiation status of epithelial cancer cells during transformation or reversible EMT. We tested this by stably suppressing PDLIM2 expression in DU145 and MDA-MB-231 cells that express high levels of endogenous PDLIM2. Our findings demonstrate that PDLIM2 controls the stability and activity of key transcription factors and activity of the COP9 signalosome (CSN) in reversible epithelial cell differentiation.
RESULTS
PDLIM2 suppression increases proliferation but decreases clonogenic growth
Previously we showed that PDLIM2 expression is higher in androgen-independent, migratory prostate carcinoma cell lines DU145 and PC-3 than in androgen-dependent PPC1 and Alva31 cell lines (Loughran et al., 2005b). PDLIM2 is located at the cytoskeleton, at points of focal adhesions, and in the nuclei of DU145 cells (Figure 1A). Moreover, it is present in the soluble nuclear fraction but not in the insoluble nuclear fraction (Figure 1B), in agreement with observations in T-lymphocytes (Tanaka et al., 2007). To investigate whether PDLIM2 is required for the migratory or EMT phenotype, we used short hairpin RNA (shRNA)–encoding vectors to stably suppress PDLIM2 in DU145 cells. Three clones of control (shScramble) cells or cells with PDLIM2 suppressed by at least 60% (shPDLIM2) were isolated (Figure 1C), and two clones of each were used for subsequent experiments. ShPDLIM2 DU145 cells displayed a noticeably altered morphology compared with shScramble cells, with increased cell–cell contact and less-pronounced fibroblastoid morphology, including decreased ability to scatter (Figure 1D). ShPDLIM2 DU145 cells also exhibited significantly increased proliferation rates compared with control cells (Figure 1E). PDLIM2 can suppress anchorage-independent growth (Loughran et al., 2005a; Qu et al., 2010a, b), but we found that shPDLIM2 DU145 cells formed significantly fewer colonies in soft agarose than shScramble cells (Figure 1F). We conclude that suppression of PDLIM2 in DU145 cells increases cell–cell contact and anchorage-dependent cell proliferation but decreases anchorage-independent cell proliferation.
FIGURE 1:

PDLIM2 suppression alters morphology, proliferation, and clonogenic growth. (A) DU145 cells were immunostained for PDLIM2 (gray/green), counterstained with Hoechst (blue; N), and photographed. Scale bar, 10 μm. Arrows indicate points of focal adhesion. (B) Subcellular fractionations of cytoplasmic (CY), soluble nuclear (SN), and insoluble nuclear (IN) fractions of DU145 cells analyzed by Western blotting for PDLIM2 expression. Purity of the fractions was confirmed using markers of each fraction: CY (HSP90), SN (PARP), and IN (lamin B). (C) Clones of DU145 cells stably expressing scrambled shRNA (shScramble) or PDLIM2 shRNA (shPDLIM2) were analyzed by Western blotting. PDLIM2 expression normalized to tubulin loading control, relative to shScramble clone2, quantified by densitometry using Odyssey software. (D) Cells were plated at low densities on collagen for 5 d, and colonies were photographed with phase microscopy. Representative micrographs of three separate experiments. Scale bar, 50 μm. (E) Cell cultures seeded at equal densities were monitored by crystal violet staining over 5 d. Data represent an average of six wells from a representative experiment. (F) Cells seeded at equal densities in soft agarose were cultured for 4 wk, when colonies were stained with crystal violet and counted. Data represent mean colonies per well from triplicate cultures (*p < 0.05, **p < 0.005, ***p < 0.0005).
Suppression of PDLIM2 decreases polarized cell migration
We next investigated the migratory capacity of shPDLIM2 DU145 cells using wound-healing and electrical cell-substrate impedance sensing (ECIS) assays, which allow real-time quantification of cell migration rates. Cells were grown to confluency on collagen-coated electrodes that monitor the capacitance of the culture (typically 0–2 nF), which increases sharply to approximately 6 nF when an electric current is applied to generate the wound (Figure 2A). Migration of cells to fill the wound is represented by a progressive decrease in capacitance over time. As can be seen in Figure 2A, shScramble cells display a time-dependent decrease in capacitance starting at approximately 8 h postwounding, and they reach 1–3 nF by 25 h, indicating that these cells “sense” the wound and move directionally to fill it. However, after an initial slight decrease of approximately 1 nF at 8 h, shPDLIM2 DU145 cells show little change in capacitance up to 25 h postwounding, never reaching capacitance levels <4 nF. This indicates that suppression of PDLIM2 greatly inhibits directional cell migration in DU145 cells. In addition, time-lapse movies of wound-healing assays confirm that in contrast to controls, shPDLIM2 cells do not migrate directionally to fill the wound (Supplemental Movies S1 and S2).
FIGURE 2:
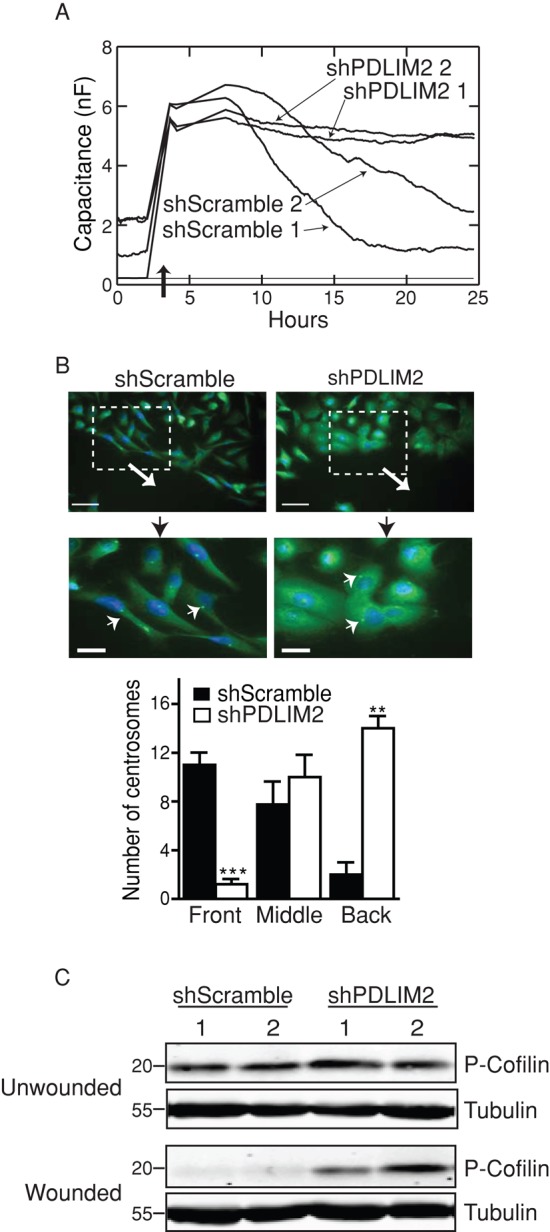
Suppression of PDLIM2 decreases polarized, directional cell migration. (A) Two clones each of shScramble and shPDLIM2 DU145 cells were electrically wounded by applying a voltage using the ECIS system (dark arrow). Capacitance of cells migrating on the electrode was measured over 25 h as described in Materials and Methods. (B) Confluent shScramble and shPDLIM2 DU145 cells were wounded with a pipette tip, and cells were allowed to migrate for 6 h. Cells were fixed and immunostained for γ-Tubulin to label MTOC (centrosomes; green, small arrows). Nuclei were counterstained with Hoechst (blue). Large arrows indicate the direction of migration. Scale bars, 50 μm (top), 20 μm (bottom). Centrosomes were scored according to position relative to the nucleus (front, middle, and back), as described in Materials and Methods (**p < 0.005, ***p < 0.0005). (C) Confluent monolayers of two clones of each cell type were scored with several wounds using a multichannel pipette. At 6 h postwounding, cells were lysed and probed for phospho-cofilin serine 3 expression by Western blotting. Results represent one of three independent experiments with similar results. See also Supplemental Movies S1 and S2.
To establish whether the disruption in directional migration of shPDLIM2 DU145 cells was associated with an inability to polarize the actin cytoskeleton, we analyzed the orientation of microtubule-organizing centers (MTOCs), whose orientation toward the front of the cell is critical for migrating cells to “sense” the wound and move directionally (Cuschieri et al., 2007). Immunofluorescence imaging of γ-tubulin within centrosomes of wounded DU145 cell monolayers (Cuschieri et al., 2007) demonstrated that in shScramble cells, the centrosomes were located predominantly at the anterior edge of the cell nucleus, toward the leading edge of migrating cells (Figure 2B). In contrast, centrosomes in shPDLIM2 DU145 cells were located behind the nucleus, away from the direction of migration (Figure 2B). The numbers of cells with centrosomes at the front, middle, or behind the nucleus indicated significant reduction in directional MTOC localization in shPDLIM2 cells (Figure 2B). Thus suppression of PDLIM2 results in reduced ability of cells to polarize and migrate directionally in response to wounding. This conclusion is supported by sustained phosphorylation of the actin-depolymerizing protein cofilin in shPDLIM2 cells at 6 h postwounding, whereas decreased phospho-cofilin in wounded shScramble cells indicates turnover of actin filaments necessary for directed migration (Figure 2C).
Thus PDLIM2 is required to maintain the mesenchymal phenotype and allow cells to polarize and migrate directionally.
PDLIM2 suppression reverses EMT and suppresses β-catenin activity
To test whether PDLIM2 suppression reverses EMT, we investigated the status of the epithelial marker E-cadherin and the transcriptional regulators of EMT, Snail and β-catenin (Schmalhofer et al., 2009). E-cadherin protein and mRNA levels were significantly increased in shPDLIM2 DU145 cells (Figure 3, A and B), and this was accompanied by decreased protein expression of the mesenchymal marker, vimentin (Supplemental Figure S1, A and B). Of interest, Snail protein expression was markedly decreased, and overexpression of PDLIM2 in other cell types caused reexpression of Snail protein without any changes in mRNA expression (Supplemental Figure S1, C and D). β-Catenin protein levels were increased in shPDLIM2 DU145 cells, although mRNA levels were unaltered (Figure 3, A-C; Supplemental Figure S1B). Increased β-catenin was prominently located at cell–cell junctions in association with E-cadherin, and there was a concomitant decrease in nuclear β-catenin levels (Supplemental Figure S1, E and F). Reduced nuclear expression of β-catenin correlated with decreased transcriptional activity indicated by reduced mRNA expression of its target genes, c-Myc, c-Jun, and Cyclin D1, compared to controls (Supplemental Figure S1G).
FIGURE 3:

Reversal of EMT with increased E cadherin and reduced β-catenin activity. (A) Cell lysates of shScramble and shPDLIM2 DU145 cells were probed for expression of E-cadherin, β-catenin, and PDLIM2, with tubulin as loading control. (B) Total RNA was extracted from shScramble and shPDLIM2 DU145 cells, and quantitative PCR was performed for E-cadherin, β-catenin, and PDLIM2 expression. β-2-Microglobulin (β2M) was used as control. Gene expression levels are normalized to shScramble samples and expressed as fold change. (C) Subcellular cytoplasmic (CY) and nuclear (N) fractions of shScramble and shPDLIM2 DU145 cells were analyzed by Western blotting for Snail expression. Purity of the fractions was confirmed by probing for markers of each fraction: CY (tubulin) and N (PARP). (D) Western blot analysis of β-catenin and tubulin expression in shScramble and shPDLIM2 DU145 cells treated with MG132 (10 μM). (E) Clones of MDA-MB-231 cells transfected with shScramble or shPDLIM2 photographed under phase contrast microscopy. Scale bar, 100 μm. (F) Western blotting of E-cadherin and PDLIM2 in shScramble and shPDLIM2 MDA-MB-231 cells. Loading control is actin. (G) Quantitative PCR for E-cadherin and PDLIM2 on total RNA extracted from shScramble and shPDLIM2 MDA-MB-231 cells. β2M was used as the control, and gene expression levels normalized to shScramble samples are expressed as fold change. (H) Lysates of shScramble and shPDLIM2 MDA-MB-231 cells were probed for expression of β-catenin, Snail, PDLIM2, and tubulin as loading control. Results represent one of three similar independent experiments. *p < 0.05, ***p < 0.0005. See also Supplemental Figures S1 and S2.
We also tested whether the increased cell junction expression of β-catenin was due to protein stabilization. After exposure to the proteasome inhibitor MG132 for up to 24 h, control cells displayed a marked increase in β-catenin levels, indicating that β-catenin is normally degraded in these cells. In contrast, shPDLIM2 DU145 cells displayed high basal levels of β-catenin (time 0) and no further accumulation in the presence of MG132, indicating resistance to proteasomal degradation (Figure 3D). Thus PDLIM2 suppression reduces β-catenin activity and reverses EMT.
To test whether PDLIM2 suppression reverses EMT in other cells, we stably expressed PDLIM2 shRNA in MDA-MB-231 breast cells and observed a similar reversal of the EMT phenotype. MDA-MB-231 cells with suppressed PDLIM2 exhibited increased cell–cell contact (Figure 3E) and increased E-cadherin mRNA and protein expression (Figure 3, F and G), whereas Snail expression was markedly reduced compared with controls (Figure 3H). ShPDLIM2 MDA-MB-231 cells also exhibited increased proliferation and decreased anchorage-independent growth (Supplemental Figure S2). A role for EMT in breast cancer is supported by data from public databases (http://glados.ucd.ie/BreastMark/index.html), indicating that PDLIM2 expression is associated with poor outcome in basal breast cancers.
Altered NFκB target gene expression in cells with suppressed PDLIM2
Overexpressed PDLIM2 can target the p65 subunit of NFκB for degradation, thus suppressing its transcriptional activity (Tanaka et al., 2007; Qu et al., 2010a, b). NFκB activity is associated with tumorigenesis and EMT in different cancer types (Maier et al., 2010). Therefore we investigated whether NFκB activity was altered in DU145 cells with suppressed PDLIM2.
First, we observed a slight but significant increase in total level of p65 protein in shPDLIM2 DU145 compared with control cells (+1.24-fold; Figure 4A). We next assessed the mRNA expression of selected NFκB target genes (Figure 4B). PLAU, CXCL10, and interleukin 8 (IL-8) mRNA levels were suppressed, whereas ID1 and vascular endothelial growth factor C (VEGF-C) mRNA levels were increased in shPDLIM2 DU145 cells. This indicates that suppression of PDLIM2 differentially regulates NFκB target genes. Of interest, we found that there was no significant difference in basal NFκB promoter activity in shScramble and shPDLIM2 cells (Figure 4C). However, when p65 was coexpressed with the NFκB reporter, there was a moderate but significant increase in tumor necrosis factor-α (TNF-α)–mediated NFκB promoter activity in shPDLIM2 cells (Figure 4D). Taken together, these data indicate that although PDLIM2 suppression alters expression of several NFκB target genes, the effect on basal NFκB activity in DU145 cells is minimal.
FIGURE 4:

Suppression of PDLIM2 does not alter basal NFκB activity in DU145 cells. (A) Western blotting for NFκB p65 expression in shScramble and shPDLIM2 DU145 cells. Densitometry measurements of the average fold difference in p65 expression in shPDLIM2 cells compared with shScramble controls from six independent experiments. (B) Quantitative PCR on total RNA extracted from shScramble and shPDLIM2 cells for PLAU, CXCL10, IL8, ID1, VEGF-C, and PDLIM2 mRNA expression. β2M mRNA levels were used as a control; expression levels were compared to shScramble mRNA levels and expressed as fold change. (C) shScramble or shPDLIM2 DU145 cells were transfected with a dual-luciferase plasmid containing an NFκB promoter and cotransfected with pCMV-empty vector (pCMV-EV) or pCMV-p65 construct. (D) Cells were transfected as in C and treated with 20 ng/ml TNF-α for 4 h before analysis. Luciferase activity in p65-transfected cells normalized to that of Renilla luciferase is presented as mean fold change of activity in shScramble cells expressing the empty vector ± SEM in triplicate samples and three independent experiments. *p < 0.05, **p < 0.005.
PDLIM2 regulates a broad transcriptional program and transcription factors
Our data suggest that the effect of PDLIM2 suppression on EMT is not entirely mediated by NFκB. To gain insight into the mechanisms by which PDLIM2 acts, we next determined the overall gene expression profile of DU145 cells with suppressed PDLIM2. We identified 457 genes that had altered expression in shPDLIM2 compared with shScramble cells. These included 213 down-regulated genes and 244 up-regulated genes (fold change, ≥1.5; p ≤ 0.05; Supplemental Table S1). Figure 5 shows a basic heat map of the top 100 differentially expressed genes (50 most up-regulated and 50 most down-regulated) based on fold changes.
FIGURE 5:
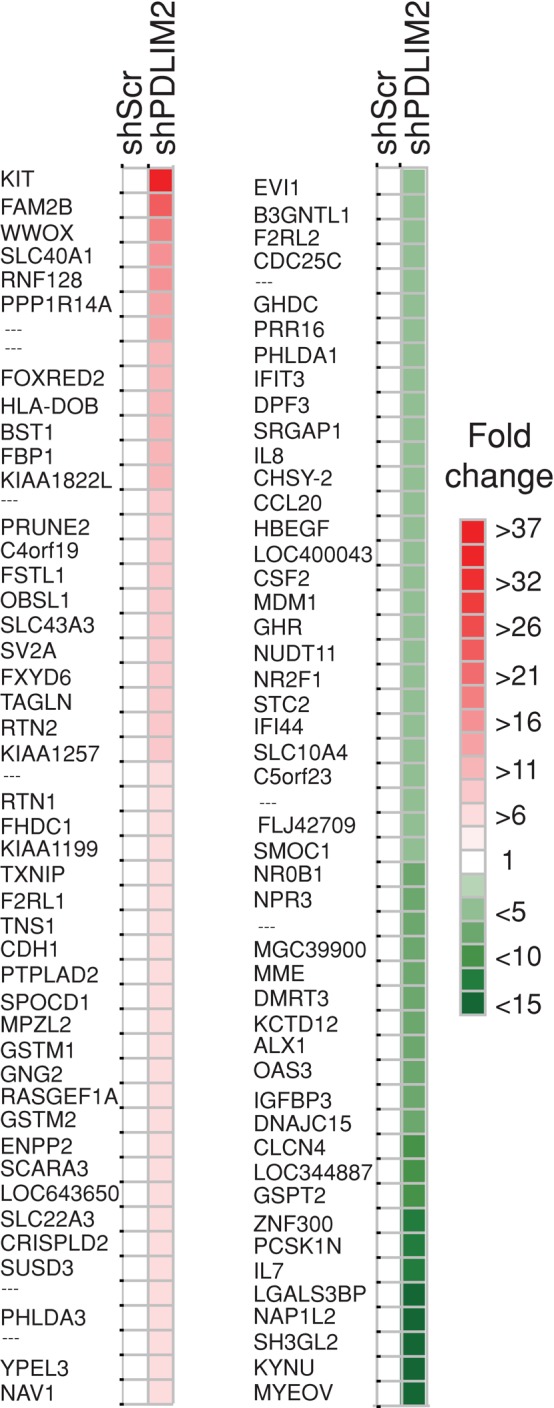
Suppression of PDLIM2 alters global gene expression in DU145 cells. A basic heat map demonstrating the top 100 differentially expressed genes (50 most up-regulated and 50 most down-regulated), based on fold changes generated using JColorGrid software. See also Supplemental Table S1.
The functional significance of the gene expression profile was objectively assessed used Ingenuity Pathway Analysis (IPA), which associates known signaling pathways and networks with the differentially expressed genes (Supplemental Table S2). This analysis revealed that the most affected biofunction in shPDLIM2 cells was “cancer” (Figure 6A), followed by “reproductive system disease,” both of which are consistent with reversion of EMT in a prostate cancer cell line. Other altered biofunctions consistent with the phenotype of shPDLIM2 cells are “cellular movement,” “cellular growth and proliferation,” and “cell morphology.” The IPA software also predicted that PDLIM2 suppression would alter “cellular development,” “hematological system development and function,” and “hematopoiesis” (Figure 6A). These may reflect PDLIM2 function in lymphocytes and macrophages (Tanaka et al., 2007, 2011; Healy and O'Connor, 2009; Qu et al., 2012).
FIGURE 6:

PDLIM2 suppression alters activity of key transcription factor families. (A) The top 20 biofunctions from the IPA of genes differentially expressed in shPDLIM2-DU145, based on most-significant p values. Data are presented as –log(p value) for the probability that the specific function is affected. A significance threshold of p = 0.05 is represented by –log(p value) = –1.301 (gray dashed line). (B) Changes in the activation status of transcription factors were plotted based on their activation z-score from IPA. (C–E) Quantitative PCR analysis of selected genes differentially expressed in the microarray gene list performed in two separate shPDLIM2-DU145 clones. Gene expression was normalized to levels in shScramble clones, using β2M as control and expressed as mean fold change ± SEM from three independent experiments (*p < 0.05, **p < 0.005, ***p < 0.0005). See also Supplemental Tables S1–S4 and Supplemental Figure S3.
Using IPA, we could also predict the “activation state” of specific cellular functions included within each biofunction (Figure 6A). This analysis, shown in Supplemental Table S3, indicates several functions consistent with the phenotype we observed, including “genital tumor,” “invasion of cells,” and “invasion of tumor cell lines.” Again, other decreased functions predicted by IPA are consistent with the role of PDLIM2 function in hematopoietic cells, such as “mobilization of hematopoietic progenitor cells” and “attraction of neutrophils and phagocytes” (Supplemental Table S3). Of interest, the predicted activation state of several transcription factor families was altered, including TRIM24, Gli1, Ap-1, NFκB, and interferon regulatory factors (Figure 6B and Supplemental Table S4). Moreover, IPA predicted a mechanistic network in which NFκB, STATs, and JUN transcriptional activity is inhibited, which is consistent with the roles of these transcription factors in transformation and metastasis (Supplemental Figure S3). This network also predicts the inhibition of β-catenin activity (CTNNB1), even though its mRNA levels are unchanged, which is in agreement with data shown in Figure 3 and Supplemental Figure S1. On the other hand, acetyltransferase EP300, p53, and the glucocorticoid receptor NR3C1 are predicted to be activated (Supplemental Table S4 and Figure 6B). Overall these predicted events correlate well with the observed effects on the EMT phenotype when PDLIM2 is suppressed.
We next validated expression of selected genes associated with the top biofunctions based on their known involvement in EMT/MErT or their frequent appearance in the biofunction groups. As shown in Figure 6, C and D, the expression of WWOX1, CITED2, FPN1, GNG2, and MYL9 mRNA was greatly increased in shPDLIM2 DU145 cells, whereas IGFBP3 expression was decreased. Thus these data and also the mRNA levels of several NFκB target genes shown in Figure 4B are consistent with the expression profiles determined by microarray analysis.
Overall the results indicate that PDLIM2 suppression alters the expression of a large number of genes and the activity of key transcriptional regulators of epithelial cell differentiation and malignancy.
PDLIM2 regulates CSN5 nuclear translocation and activity
On the basis of our observation that PDLIM2 suppression alters key transcriptional regulators of epithelial cell differentiation and on its previously described function in promoting degradation of NFκB and STATs (Tanaka et al., 2005; Guo et al., 2010), we hypothesized that PDLIM2 regulates the cellular protein degradation machinery at a level that impinges on multiple transcriptional regulators. To address this, we searched for PDLIM2-interacting proteins and identified subunits of the CSN complex, which controls protein stability and activity of several transcription factors associated with EMT (reviewed in Lee et al., 2011). The CSN consists of eight subunits (CSN1–8), several of which form smaller complexes and may also function as monomers (Kato and Yoneda-Kato, 2009). PDLIM2 could be coimmunoprecipitated with CSN5 (Jab1), the catalytic subunit of the CSN (Shackleford and Claret, 2010), in DU145 cell lysates (Figure 7A), MCF7 cells overexpressing PDLIM2, and MDA-MB-231 cells (Supplemental Figure S4). Nuclear levels of both CSN5 and CSN2 were reduced in shPDLIM2 DU145 cells (Figure 7,B and C).
FIGURE 7:

PDLIM2 interacts with CSN subunits CSN5 and CSN8 and regulates CSN5 localization. (A) CSN5 immunoprecipitates from DU145 shScramble cells were assessed by Western blotting for PDLIM2 interaction. Beads and lysate without antibody (–Ab) is a negative control. (B) Western blotting for CSN5 and CSN2 expression in cytoplasmic (CY) and nuclear (N) extracts of DU145 shScramble or shPDLIM2 cells. Loading controls are tubulin (CY) and PARP (N). (C) Subcellular localization of CSN5 was assessed in shScramble and shPDLIM2 DU145 cells by immunostaining. Scale bars, 10 μM. (D) Peptide arrays were generated for all CSN subunits and overlaid with lysate from DU145 shScramble and DU145 shPDLIM2 cells. Spots that showed less reactivity with the anti-PDLIM2 antibody on PDLIM2 lysates were considered positive. CSN5 and CSN8 arrays showed intense spots of interaction with PDLIM2. Positive peptides were then synthesized with successive amino acid substitution (bottom, with graph and substitution arrays). Graphs represent intensity of interaction of peptides with PDLIM2 expressed as percentage of the intensity of the original interaction peptide. Alanines were substituted by glutamic acids; all other amino acids were substituted with alanines. (E, F) Immunoprecipitations (IPs) for CSN5 (E; top), PDLIM2 (E; bottom), or CSN8 (F), were performed from lysates of MCF7 cells stably transfected with GFP-empty vector (EV) or GFP-PDLIM2 as described in Materials and Methods. Immunocomplexes were probed for each protein by Western blotting. Controls for IPs included no lysate (–Ly) and beads plus lysate only, no antibody (–Ab). See also Supplemental Figure S4 and Supplemental Table S5.
To test whether PDLIM2 associates with individual subunits or potentially the entire CSN complex, we used peptide arrays encompassing the entire sequence of each CSN subunit (spot synthesized as 18-mer peptides overlapping by five amino acids). Each array was overlaid with cell lysate from DU145 shScramble or DU145 shPDLIM2 cells. Peptide spots that showed less reactivity with the anti-PDLIM2 antibody on shPDLIM2 lysates were considered positive. From this we observed PDLIM2 binding to the CSN5 and CSN8 peptides, slightly to CSN3, but not to CSN1, 2, 4, 6, or 7 (Figure 7 and Supplemental Table S5). To identify more specific sites of interaction, we further analyzed positively reacting peptides from CSN5 and CSN8 using amino acid substitution arrays (alanine for all amino acids and glutamic acid for alanine; Supplemental Table S5). From this analysis we identified one peptide for CSN5 (166-VIDPTRTISAGKVNLGAF-183) for which binding was greatly disrupted by substitution of Lys-180. For CSN8, binding to one peptide (-ADSTTRMVLPRKPVAGAL-) was disrupted by substitution of Arg-160 and 165, Lys-166, and Ala-169 and 171. These amino acids are found in three peptides from the ones interacting, with increased fold of 3.9, 2.4, and 7.3, respectively (Figure 7 and Supplemental Table S5). The other peptides tested did not lose binding with substitution of a single amino acid, suggesting that they are not essential but may be involved as indirect binding sites. These results confirm that PDLIM2 binds to CSN5 and CSN8.
Coimmunoprecipitation of PDLIM2 with CSN5 and enhanced nuclear location of CSN5 could also be observed when PDLIM2 was overexpressed in MCF-7 cells. Taken together, these data indicating that the PDLIM2-CSN5 interaction is functional in other cells (Supplemental Figure S4).
The CSN deconjugates Nedd8 (deneddylation) from the cullin family of proteins, thereby regulating protein degradation complexes, including the Skp1-cullin-F box (SCF) complex (Kato and Yoneda-Kato, 2009). When cullin1 (Cul1) is deneddylated the target substrate levels may be stabilized. Therefore, to test whether CSN activity is regulated by PDLIM2, we compared Cul1 neddylation in shScramble and shPDLIM2 DU145 cells. As can be seen in Figure 8A, more neddylated Cul1 accumulates in the nucleus with shPDLIM2 than in controls. This is accompanied by greatly increased stability of the cell cycle regulator p27, a key target of CSN5 activity (Shackleford and Claret, 2010; Figure 8B). Similarly, nuclear cullin3 neddylation was increased in shPDLIM2-DU145, and p27 was increased in the cytoplasm of these cells (Figure 8C). The effect of PDLIM2 on CSN5-mediated regulation of p27 is further supported by the observation that in cells in which PDLIM2 is overexpressed, P27 protein levels are decreased (Figure 8D). Taken together, these data indicate that PDLIM2 associates with CSN5 and enhances its nuclear localization and activity. Reduced CSN activity is also consistent with the effects of PDLIM2 suppression on enhanced β-catenin stability (Figure 3D) and with the altered activity of several transcriptional regulators whose stability and activity is regulated by the cullin-RING ubiquitin ligases (p53, Smad4, NFκB, JUN, AP-1; Figure 6B; Kato and Yoneda-Kato, 2009; Shackleford and Claret, 2010).
FIGURE 8:
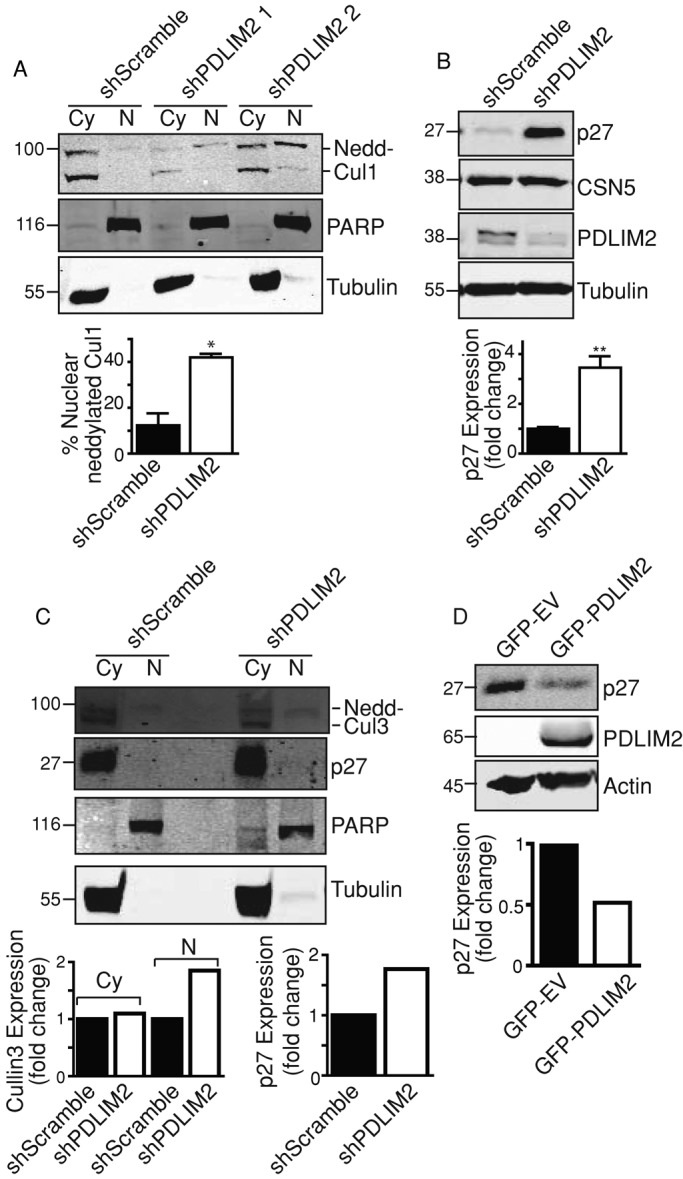
PDLIM2 regulates CSN5 AND CSN activity. (A) Neddylated Cul1 was detected by Western blotting of cytoplasmic (CY) and nuclear (N) fractions from shScramble and two clones of shPDLIM2 DU145 cells with cullin 1 antibody. Cell fraction loading controls are tubulin (CY) and PARP (N). Quantification of nuclear neddylated Cul1 measured as percentage of total Cul1 using Odyssey software. (B) Western blotting of P27, CSN5, PDLIM2, and tubulin in DU145 shScramble and shPDLIM2 cells. Quantification of P27 normalized to tubulin using Odyssey software is shown as mean fold change ±SEM from three independent experiments. (C) Neddylated Cul3 was detected by Western blotting of cytoplasmic (CY) and nuclear (N) fractions from shScramble shPDLIM2 DU145 cells. Cell fraction loading controls are tubulin (CY) and PARP (N). Quantification of total cullin 3 in cytoplasmic fractions normalized to tubulin and neddylated cullin 3 in nuclear fractions normalized to PARP (left graph). Quantification of P27 normalized to tubulin (right graph) using Odyssey software. (D) Whole-cell lysates were prepared from MCF7 cells expressing GFP-EV or GFP-PDLIM2 and probed for p27, PDLIM2, and actin as loading control. Quantification of P27 normalized to tubulin using Odyssey software.
DISCUSSION
The findings of this study, combined with the documented functions in T-lymphocytes and macrophage differentiation, demonstrate that PDLIM2 is a key regulator of cellular differentiation. We show that suppression of PDLIM2 is sufficient to suppress a gene expression program necessary for maintaining a mesenchymal phenotype in cancer cells and activate an epithelial gene expression program. Of importance, we show that PDLIM2 regulates the activity of several key transcription factor families, as well as the nuclear localization and activity of the CSN subunit CSN5.
PDLIM2 has one PDZ domain that associates with the actin cytoskeleton via α-actinin (Loughran et al., 2005a) and one LIM domain that has potential to associate with different protein partners (Krcmery et al., 2010). PDLIM2 shuttles between the cytoskeleton and nucleus (Healy and O'Connor, 2009; Krcmery et al., 2010) and thus is well suited for integration of cytoskeleton signals with gene expression in cell polarization and migration. Other LIM-domain proteins, such as LASP-1 and Four and a Half LIM domain (FHL) 2 protein, have been described as courier proteins that connect genome activity with cytoskeleton dynamics (Olson and Nordheim, 2010). Of interest, FHL-2 enhances nuclear β-catenin transcriptional activity in cardiac stem cells (Olson and Nordheim, 2010). Our observation of decreased nuclear β-catenin and increased stabilization at cell–cell junctions in shPDLIM2 cells suggests a similar role for PDLIM2 in enhancing β-catenin transcriptional activity to promote EMT. Similarly, the LIM domain–only proteins (LMO1–4) are proposed to act as coactivators or repressors in nucleating or disrupting transcriptional complexes (Matthews et al., 2013). Indeed, overexpression of LMO2 in androgen-dependent LNCAP prostate cancer cells is sufficient to induce EMT (Ma et al., 2007).
PDLIM2 suppression in DU145 cells altered the expression of many genes and transcription factors associated with the EMT phenotype. Genes associated with tumorigenesis were suppressed, and genes associated with tumor suppression were increased. These included WWOX1, which is frequently suppressed in prostate and other cancers (Del Mare et al., 2009); CITED2, a key transcription factor in mouse embryonic cells primed to undergo MErT (Bard et al., 2008) and whose suppression increases colon cancer invasiveness (Bai and Merchant, 2007); FPN1, which is suppressed in metastatic breast cancer (Pinnix et al., 2010); and IGFBP-3, which enhances transforming growth factor β–promoted EMT (Natsuizaka et al., 2010). Genes associated with cellular movement were also altered by PDLIM2 suppression. These include GNG2, which regulates the Rho family of small GTPases (Ueda et al., 2008), and MYL9, which is implicated in actin cytoskeleton contractility (Medjkane et al., 2009). Overall the profile indicates that PDLIM2 has an essential function in integrating signals from the actin cytoskeleton with gene expression to determine cell phenotype.
Our data strongly suggest that PDLIM2 mediates its effects on transcription factors by associating with the CSN and enhancing its activity. Cells with suppressed PDLIM2 have reduced nuclear targeting of CSN subunits and reduced deneddylation of cullin 1, whereas overexpression of PDLIM2 is sufficient to promote nuclear accumulation of CSN5. The CSN has a complex role in regulating protein stability, which is determined by expression levels of its subunits and the cellular signaling context (Lee et al., 2011). Its actions can be mediated through its regulation of cullin-RING ligases, RING-containing ubiquitin ligases, and deubiquitinating proteins (Kato and Yoneda-Kato, 2009). Several oncogenes and tumor suppressors are known to have modified CSN activity (β-catenin, NFκB, p53, p27, MDM2; Huang et al., 2009; Kato and Yoneda-Kato, 2009; Schweitzer and Naumann, 2010; Lee et al., 2011). Moreover, CSN subunits may act as monomers, and CSN5 can positively regulate several transcription factors, including NFκB, JUN/AP-1, Hif1α, and E2F1 (reviewed in Kato and Yoneda-Kato, 2009; Shackleford and Claret, 2010). Of note, the mechanistic network generated by IPA with the DU145 RNA profile indicates inhibition of CTNNB1 (β-catenin), NFκB, and JUN, whereas p53 activity is activated. This further supports the conclusion that reduced CSN activity mediates the effects of PDLIM2 suppression on transcription factor activity and gene expression. Of interest, CSN5 and CSN8 have no common sequence, interact not directly but through CSN4, and have opposite location inside the CSN complex (Kapelari et al., 2000). Because we did not detect interaction of PDLIM2 with CSN4, it is more likely that PDLIM2 is interacting directly with the CSN complex and thereby regulating its deneddylation activity toward cullins.
Reduced CSN activity is also intriguing from the perspective of PDLIM2’s effects on NFκB activity. PDLIM2 promotes degradation of STATs and NFκB in T-lymphocytes (Tanaka et al., 2005, 2007, 2011; Guo et al., 2010). In DU145 cells with suppressed PDLIM2, we noted no effect on basal NFκB activity, and a subset of NFκB target genes was down-regulated, whereas other target genes were up-regulated. Several of the down-regulated genes (PLAU, IL-8, and CXCL10) are known NFκB-target genes that promote cancer progression (reviewed in Baud and Karin, 2009). NFκB-target genes that were increased include VEGF-C, which is associated with angiogenesis (Watari et al., 2008), and ID1, which represses stem cell differentiation (Romero-Lanman et al., 2012). Of interest, IPA predicts overall inhibition of the NFκB complex activity (z score, −2.901, p < 0.0001) rather than activation. This indicates that PDLIM2 does not simply promote NFκB degradation and may instead reflect the cellular context in determining PDLIM2’s effects on CSN activity.
PDLIM2 may now be added to a small group of LIM-domain proteins that regulate phenotypic changes in cancer progression (Craene and Berx, 2013). This has important consequences for tumorigenesis and cancer progression, for which PDLIM2 expression could determine the potential of cancer cells to undergo reversible EMT, a process necessary for tumor growth (Tsai et al., 2012). PDLIM2 is highly expressed in certain cells with an EMT phenotype (this study; Loughran et al., 2005a); however, its expression is suppressed by methylation in breast and lung cancer cells (Pfeifer and Rauch, 2009; Qu et al., 2010a). Suppression of PDLIM2 by methylation would facilitate cellular transformation and tumor growth, whereas subsequent up-regulation in cells primed for invasion would facilitate the mesenchymal, migratory (EMT) phenotype required for metastasis. Our data demonstrate that if PDLIM2 were again repressed, this would facilitate MErT and potential tumor growth at a secondary site. A study showing vitamin D–mediated regulation of PDLIM2 expression in breast cancer further illustrates how PDLIM2 expression may determine phenotype (Vanoirbeek et al., 2013).
In summary, we showed that PDLIM2 is required for maintenance of a gene expression program that supports the mesenchymal phenotype associated with EMT. We propose that PDLIM2 is an essential courier protein linking cytoskeleton signaling to CSN-mediated control of stability and activation of key transcriptional regulators of EMT. The function of PDLIM2 and the possibilities for regulating both its expression and subcellular localization reflect the potential for plasticity in epithelial cell differentiation in cancer progression.
Materials and Methods
Cell culture and generation of shPDLIM2-expressing cell lines
DU145 and MDA-MB-231 cells were cultured in RPMI-1640 or DMEM, respectively, supplemented with 10% fetal bovine serum (FBS), 1 mM l-glutamine, and 5 mg/ml penicillin/streptomycin at 37°C with 5% CO2. Cell lines stably expressing shRNA were generated by transfection with pSUPER vectors encoding shRNA targeting PDLIM2 (shPDLIM2, ACATAATCGTGGCCATCAA) or a control shRNA (shScramble, TGACATGATAATACTCTCT) using Lipofectamine (DU145; Invitrogen, Paisley, United Kingdom) or Lipofectamine 2000 (MDA-MB-231), following the manufacturer's protocol. Cells were cultured in the presence of 1 mg/ml G418 (Calbiochem, La Jolla, CA) for 2–4 wk, at which time individual clones were screened for expression of PDLIM2 by Western blotting.
Generation of MCF7–GFP PDLIM2 cell lines
MCF7 cells were transfected with pEGFP–empty vector or pEGFP-PDLIM2 constructs using Lipofectamine, and stable clones were generated after selection in 1 mg/ml G418 as described above.
RNA isolation, cDNA synthesis, and real-time semiquantitative reverse transcription PCR
Total RNA was extracted from cells using the TRIzol method (Invitrogen) and cDNA synthesized from 2 μg using Moloney murine leukemia virus reverse transcriptase (MLTV) with an oligo-dT12-18 primer (Invitrogen). For real-time quantitative reverse transcription PCR analysis, equal amounts of cDNA were amplified using Quantitect SYBR Green PCR Kit (Qiagen Sciences, Germantown, MD) in an ABI Prism 7900HT instrument (Applied Biosystems, Foster City, CA). The Supplemental Experimental Procedures lists primer sequences.
Cell lysates, subcellular fractions, Western blotting, and immunoprecipitation
Total cellular protein extracts and cytoplasmic, soluble, and insoluble nuclear extracts were prepared as described previously (Healy and O'Connor, 2009). The purity of the fractions was confirmed by Western blotting with anti-HSP90 (cytoplasm marker), anti-PARP (soluble nuclear fraction marker), and anti–lamin B (insoluble nuclear fraction marker).
For immunoprecipitations (IPs), cell lysates prepared in 50 mM Tris-HCl, pH 7.4, 0.2% Triton X-100, and 10% glycerol (supplemented with phosphatase and protease inhibitors) were incubated with 2–3 μg of antibody overnight, followed by incubation with protein agarose G beads for 3 h on a rotator at 4°C. Beads were washed three times with ice-cold lysis buffer and proteins eluted by boiling in SDS–PAGE loading buffer. Protein input controls are 40–50 μg of protein from lysates used for IPs.
All protein samples for Western blot analysis were separated by 10% SDS–PAGE and transferred to a nitrocellulose membrane. After overnight incubation with primary antibody, membranes were probed with IRdye700 or 800–conjugated secondary antibodies, and proteins were detected using the Odyssey Image scanner system (LI-COR Biosciences, Cambridge, United Kingdom). The approximate protein molecular weight in kilodaltons is indicated on the left of each Western blot panel.
Immunofluorescence and microscopy
For cell morphology analysis by phase microscopy, cells were cultured for 48 h on 10-cm tissue culture plates. Multiple fields of live cells were photographed with a Nikon TE300 inverted microscope equipped with a Nikon DXM1200F digital camera (Micron Optical, Wexford, Ireland). Images were captured and processed using MetaMorph software, version 6.2r6 (Molecular Devices, Sunnyvale, CA).
For immunofluorescence, cells were cultured on collagen-coated glass coverslips, washed, and then fixed using 3.75% formaldehyde. Cells were probed with primary antibodies, followed by Cy2- or Cy3-conjugated secondary antibodies, and nuclei were visualized with Hoechst dye as described previously (Loughran et al., 2005a). Cells were photographed using a Nikon Eclipse E600 microscope equipped with a SPOT digital camera and SPOT software, version 4.6 (Diagnostic Instruments, Sterling Heights, MI). For analysis of polarization of MTOCs, confluent monolayers of cells cultured on collagen-coated glass coverslips were wounded, washed, reincubated with medium, and allowed to migrate for 6 h. Cells were then fixed in ice-cold methanol/5 mM ethylene glycol tetraacetic acid and processed for immunofluorescence detection of γ-tubulin. All images were processed using Photoshop CS2, version 9.0.2 (Adobe, San Jose, CA).
Proliferation, colony scattering, and soft agarose assays
Cells were seeded at equal densities in RPMI/10% FBS in wells of a 24-well plate. Cells were fixed and stained with 0.5% crystal violet/20% ethanol solution every 24 h for 5 d. Stained cells were scanned and quantified on the Odyssey Image scanner system (LI-COR Biosciences).
Colony scattering assays were performed by plating cells at low density on collagen-coated plates. Anchorage-independent growth was determined by assessing colony formation in soft agarose, as described previously (Loughran et al., 2005a). After 4–7 wk, colonies were stained with crystal violet and counted.
Electric cell-substrate impedance sensing and wound-healing assays
For ECIS migration assays (Applied BioPhysics, Troy, NY), cells were plated at high densities on collagen-coated eight-well ECIS array slides containing one electrode per well. Confluent monolayers were electrically wounded with 6 V for 30 s. Capacitance of the cells on the wound site was immediately and consistently recorded every 30 s for a period of 15–25 h.
For analysis of cofilin phosphorylation status, multiple wounds were scored on confluent cell cultures with sterile pipette tips. Cultures were washed, reincubated with growth medium, and allowed to migrate for 6 h before cell lysis and Western blotting.
NFκB-p65 activation and dual-luciferase assay
NFκB activity was measured using a dual-luciferase assay (Cignal NFκB reporter (luc) Kit; SABiosciences, Qiagen, Crawley, UK) as described previously (Healy and O'Connor, 2009). Cells were transfected with a NFκB dual-luciferase reporter plasmid (Super Array, Madison, WI) and pCMV-p65 plasmid. Cells were cultured for 48 h to assess basal activity, or after 36 h, cells were stimulated with 20 ng/ml TNF-α for 4 h. Cell lysates were analyzed for luciferase activity of the reporter plasmid using a dual-luciferase reporter assay system kit (Promega, Madison, WI) and a TD20/20 luminometer (Promega).
Statistical analysis
Data were analyzed for statistical significance using Student's t test to compare samples where appropriate. p < 0.05 was deemed significant, and graded p values are represented as follows: *p < 0.05, **p < 0.005, and ***p < 0.0005.
Gene expression profiling and data analysis
RNA was extracted from three separate cultures of the DU145 cell lines (shScramble or shPDLIM2 DU145) and prepared by reverse transcription as probes for hybridization to HGU133 Plus 2.0 Affymetrix gene chips at Almac Diagnostics (Craigavon, Northern Ireland) using three chips for each cell line. The full, normalized gene list (54,675 genes) was filtered based on confidence values using Student's t test to generate a stringent list. Gene expression differences of ≥1.5-fold, with p ≤ 0.05, were considered significantly different from expression in the control samples. This list of 663 differentially expressed genes was further refined to remove duplicates and nondescribed proteins, and the most statistically significant (lowest p value <0.05) of multiple transcripts was used to represent the target gene.
The final list consisting of 457 genes differentially expressed between the shScramble and shPDLIM2 DU145 cells was analyzed using IPA software (Ingenuity Systems, Redwood City, CA) “Core Analysis” to assess which signaling pathways, cellular processes, and transcription factors were affected by PDLIM2 suppression. For cellular function “Activation State” predictions, the IPA software considers a regulation z-score value of >2.0 for activation and <−2.0 for inhibition of cellular functions as statistically significant. A heat map was generated using JColorGrid software for visualization of biological measurements (Joachimiak et al., 2006).
Spot synthesis of peptides and overlay analysis
Peptides arrays of CSN subunits in nitrocellulose were generated as previously described (Kiely et al., 2008, 2009). Essentially, scanning libraries of overlapping 18-mer peptides covering the entire sequence of CSN subunits were produced by automatic SPOT synthesis and synthesized on continuous cellulose membrane supports on Whatman 50 cellulose using 9-fluorenylmethyloxycarbonyl chemistry with the MultiPep RS system (Intavis Bioanalytical Instruments, Cologne, Germany). The interaction of PDLIM2 with the CSN subunits arrays was investigated by overlaying the cellulose membranes with lysates of DU145 shScramble (100 μg/ml protein in NP-40 lysis buffer) at 37°C for 3 h. Bound PDLIM2 was detected using PDLIM2 antibody and enhanced chemiluminescence (GE Healthcare, Amersham, Little Chalfont, United Kingdom). Intensity was determined by densitometric analysis using ImageJ software (National Institutes of Health, Bethesda, MD). Substitution arrays were performed in a similar manner, except that alanines were substituted by glutamic acids and all other amino acids were substituted by alanines.
Supplementary Material
Acknowledgments
We are grateful to our colleagues in the Cell Biology Laboratory, to the Andalusian Bioinformatics Platform at the University of Malaga for access to Ingenuity software, and to Kurt Tidmore for preparing illustrations. This work was funded by Science Foundation Ireland (PI awards 07/INI/B107 and 11/PI/1136), the Health Research Board (PD/2007/29 to V.A.), and the Breast Cancer Campaign (2008NovPR24).
Abbreviations used:
- CSN
Cop9 signalosome
- CSN1–8
Cop9 signalosome subunit 1–8
- Cul1
cullin1
- EMT
epithelial-to-mesenchymal transition
- MErT
mesenchymal-to-epithelial reverting transition
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-06-0306) on November 6, 2013.
*These two authors contributed equally to this work.
†These two authors contributed equally to this work.
REFERENCES
- Bai L, Merchant JL. A role for CITED2, a CBP/p300 interacting protein, in colon cancer cell invasion. FEBS Lett. 2007;581:5904–5910. doi: 10.1016/j.febslet.2007.11.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard JB, Lam MS, Aitken S. A bioinformatics approach for identifying candidate transcriptional regulators of mesenchyme-to-epithelium transitions in mouse embryos. Dev Dyn. 2008;237:2748–2754. doi: 10.1002/dvdy.21652. [DOI] [PubMed] [Google Scholar]
- Baud V, Karin M. Is NF-[kappa]B a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao Y, Shepard C, Wells A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer. 2010;9:179. doi: 10.1186/1476-4598-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craene BD, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- Cuschieri L, Nguyen T, Vogel J. Control at the cell center: the role of spindle poles in cytoskeletal organization and cell cycle regulation. Cell Cycle. 2007;6:2788–2794. doi: 10.4161/cc.6.22.4941. [DOI] [PubMed] [Google Scholar]
- Del Mare S, Salah Z, Aqeilan RI. WWOX: its genomics, partners, and functions. J Cell Biochem. 2009;108:737–745. doi: 10.1002/jcb.22298. [DOI] [PubMed] [Google Scholar]
- Fu J, Yan P, Li S, Qu Z, Xiao G. Molecular determinants of PDLIM2 in suppressing HTLV-I Tax-mediated tumorigenesis. Oncogene. 2010;29:6499–507. doi: 10.1038/onc.2010.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Mi Z, Bowles DE, Bhattacharya SD, Kuo PC. Osteopontin and protein kinase C regulate PDLIM2 activation and STAT1 ubiquitination in LPS-treated murine macrophages. J Biol Chem. 2010;285:37787–37796. doi: 10.1074/jbc.M110.161869. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Healy NC, O'Connor R. Sequestration of PDLIM2 in the cytoplasm of monocytic/macrophage cells is associated with adhesion and increased nuclear activity of NF-κB. J Leukoc Biol. 2009;85:481–490. doi: 10.1189/jlb.0408238. [DOI] [PubMed] [Google Scholar]
- Huang X, Langelotz C, Hetfeld-Pechoc BK, Schwenk W, Dubiel W. The COP9 signalosome mediates beta-catenin degradation by deneddylation and blocks adenomatous polyposis coli destruction via USP15. J Mol Biol. 2009;391:691–702. doi: 10.1016/j.jmb.2009.06.066. [DOI] [PubMed] [Google Scholar]
- Joachimiak M, Weisman J, May B. JColorGrid: software for the visualization of biological measurements. BMC Bioinformat. 2006;7:225. doi: 10.1186/1471-2105-7-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapelari B, Bech-Otschir D, Hegerl R, Schade R, Dumdey R, Dubiel W. Electron microscopy and subunit-subunit interaction studies reveal a first architecture of COP9 signalosome. J Mol Biol. 2000;300:1169–1178. doi: 10.1006/jmbi.2000.3912. [DOI] [PubMed] [Google Scholar]
- Kato J-Y, Yoneda-Kato N. Mammalian COP9 signalosome. Genes Cells. 2009;14:1209–1225. doi: 10.1111/j.1365-2443.2009.01349.x. [DOI] [PubMed] [Google Scholar]
- Kiely PA, Baillie GS, Barrett R, Buckley DA, Adams DR, Houslay MD, O'Connor R. Phosphorylation of RACK1 on tyrosine 52 by c-Abl is required for insulin-like growth factor I-mediated regulation of focal adhesion kinase. J Biol Chem. 2009;284:20263–20274. doi: 10.1074/jbc.M109.017640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiely PA, Baillie GS, Lynch MJ, Houslay MD, O'Connor R. Tyrosine 302 in RACK1 is essential for insulin-like growth factor-I-mediated competitive binding of PP2A and beta1 integrin and for tumor cell proliferation and migration. J Biol Chem. 2008;283:22952–22961. doi: 10.1074/jbc.M800802200. [DOI] [PubMed] [Google Scholar]
- Krcmery J, Camarata T, Kulisz A, Simon H-G. Nucleocytoplasmic functions of the PDZ-LIM protein family: new insights into organ development. BioEssays. 2010;32:100–108. doi: 10.1002/bies.200900148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Zhao R, Phan L, Yeung SC. Roles of COP9 signalosome in cancer. Cell Cycle. 2011;10:3057–3066. doi: 10.4161/cc.10.18.17320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughran G, Healy NC, Kiely PA, Huigsloot M, Kedersha NL, O'Connor R. Mystique is a new insulin-like growth factor-I-regulated PDZ-LIM domain protein that promotes cell attachment and migration and suppresses anchorage-independent growth. Mol Biol Cell. 2005a;16:1811–1822. doi: 10.1091/mbc.E04-12-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughran G, Huigsloot M, Kiely PA, Smith LM, Floyd S, Ayllon V, O'Connor R. Gene expression profiles in cells transformed by overexpression of the IGF-I receptor. Oncogene. 2005b;24:6185–6193. doi: 10.1038/sj.onc.1208772. [DOI] [PubMed] [Google Scholar]
- Ma S, Guan XY, Beh PSL, Wong KY, Chan YP, Yuen HF, Vielkind J, Chan KW. The significance of LMO2 expression in the progression of prostate cancer. J Pathol. 2007;211:278–285. doi: 10.1002/path.2109. [DOI] [PubMed] [Google Scholar]
- Macartney-Coxson D, et al. Metastatic susceptibility locus, an 8p hot-spot for tumour progression disrupted in colorectal liver metastases: 13 candidate genes examined at the DNA, mRNA and protein level. BMC Cancer. 2008;8:187. doi: 10.1186/1471-2407-8-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier HJ, Schmidt-Strassburger U, Huber MA, Wiedemann EM, Beug H, Wirth T. NF-κB promotes epithelial–mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010;295:214–228. doi: 10.1016/j.canlet.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Matthews JM, Lester K, Joseph S, Curtis DJ. LIM-domain-only proteins in cancer. Nat Rev Cancer. 2013;13:111–122. doi: 10.1038/nrc3418. [DOI] [PubMed] [Google Scholar]
- Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol. 2009;11:257–268. doi: 10.1038/ncb1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsuizaka M, Ohashi S, Wong GS, Ahmadi A, Kalman RA, Budo D, Klein-Szanto AJ, Herlyn M, Diehl JA, Nakagawa H. Insulin-like growth factor-binding protein-3 promotes transforming growth factor-β1-mediated epithelial-to-mesenchymal transition and motility in transformed human esophageal cells. Carcinogenesis. 2010;31:1344–1353. doi: 10.1093/carcin/bgq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Cano A. The epithelial-mesenchymal transition under control: global programs to regulate epithelial plasticity. Semin Cancer Biol. 2012;22:361–368. doi: 10.1016/j.semcancer.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11:353–365. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer GP, Rauch TA. DNA methylation patterns in lung carcinomas. Semin Cancer Biol. 2009;19:181–187. doi: 10.1016/j.semcancer.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinnix ZK, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Translational Med. 2010;2:43ra56. doi: 10.1126/scisignal.3001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Fu J, Ma H, Zhou J, Jin M, Mapara MY, Grusby MJ, Xiao G. PDLIM2 restricts Th1 and Th17 differentiation and prevents autoimmune disease. Cell Biosci. 2012;2:23. doi: 10.1186/2045-3701-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Fu J, Yan P, Hu J, Cheng S-Y, Xiao G. Epigenetic repression of PDZ-LIM domain-containing protein 2. J Biol Chem. 2010a;285:11786–11792. doi: 10.1074/jbc.M109.086561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Yan P, Fu J, Jiang J, Grusby MJ, Smithgall TE, Xiao G. DNA methylation-dependent repression of PDZ-LIM domain-containing protein 2 in colon cancer and its role as a potential therapeutic target. Cancer Res. 2010b;70:1766–1772. doi: 10.1158/0008-5472.CAN-09-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Lanman EE, Pavlovic S, Amlani B, Chin Y, Benezra R. Id1 maintains embryonic stem cell self-renewal by up-regulation of Nanog and repression of Brachyury expression. Stem Cells Dev. 2012;21:384–393. doi: 10.1089/scd.2011.0428. [DOI] [PubMed] [Google Scholar]
- Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- Schweitzer K, Naumann M. Control of NF-kappaB activation by the COP9 signalosome. Biochem Society Trans. 2010;38:156–161. doi: 10.1042/BST0380156. [DOI] [PubMed] [Google Scholar]
- Shackleford T, Claret F. JAB1/CSN5: a new player in cell cycle control and cancer. Cell Div. 2010;5:26. doi: 10.1186/1747-1028-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Grusby M, Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. 2007;8:584–591. doi: 10.1038/ni1464. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Soriano MA, Grusby MJ. SLIM is a nuclear ubiquitin E3 ligase that negatively regulates STAT signaling. Immunity. 2005;22:729–736. doi: 10.1016/j.immuni.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yamamoto Y, Muromoto R, Ikeda O, Sekine Y, Grusby MJ, Kaisho T, Matsuda T. PDLIM2 inhibits T helper 17 cell development and granulomatous inflammation through degradation of STAT3. Sci Signal. 2011;4:ra85. doi: 10.1126/scisignal.2001637. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Torrado M, Senatorov VV, Trivedi R, Fariss RN, Tomarev SI. Pdlim2, a novel PDZ-LIM domain protein, interacts with α-actinins and filamin A. Invest Ophthalmol Vis Sci. 2004;45:3955–3963. doi: 10.1167/iovs.04-0721. [DOI] [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–736. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Nagae R, Kozawa M, Morishita R, Kimura S, Nagase T, Ohara O, Yoshida S, Asano T. Heterotrimeric G protein βγ subunits stimulate FLJ00018, a guanine nucleotide exchange factor for Rac1 and Cdc42. J Biol Chem. 2008;283:1946–1953. doi: 10.1074/jbc.M707037200. [DOI] [PubMed] [Google Scholar]
- Vanoirbeek E, Eelen G, Verlinden L, Carmeliet G, Mathieu C, Bouillon R, O'Connor R, Xiao G, Verstuyf A. PDLIM2 expression is driven by vitamin D and is involved in the pro-adhesion, and anti-migration and -invasion activity of vitamin D. Oncogene. 2013. doi: 10.1038/onc.2013. [DOI] [PubMed]
- Watari K, Nakao S, Fotovati A, Basaki Y, Hosoi F, Bereczky B, Higuchi R, Miyamoto T, Kuwano M, Ono M. Role of macrophages in inflammatory lymphangiogenesis: enhanced production of vascular endothelial growth factor C and D through NF-[kappa]B activation. Biochem Biophys Res Commun. 2008;377:826–831. doi: 10.1016/j.bbrc.2008.10.077. [DOI] [PubMed] [Google Scholar]
- Yan P, Fu J, Qu Z, Li S, Tanaka T, Grusby MJ, Xiao G. PDLIM2 suppresses human T-cell leukemia virus type I Tax-mediated tumorigenesis by targeting Tax into the nuclear matrix for proteasomal degradation. Blood. 2009;113:4370–4380. doi: 10.1182/blood-2008-10-185660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Yasunaga J-I, Satou Y, Nakao M, Takahashi M, Fujii M, Matsuoka M. Human T-cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF-κB. Blood. 2009;113:2755–2764. doi: 10.1182/blood-2008-06-161729. [DOI] [PubMed] [Google Scholar]
- Zheng M, Cheng H, Banerjee I, Chen J. ALP/Enigma PDZ-LIM domain proteins in the heart. J Mol Cell Biol. 2010;2:96–102. doi: 10.1093/jmcb/mjp038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.