

Cadherin-6B is rapidly depleted from premigratory neural crest cells during EMT, suggestive of posttranslational mechanisms. ADAM10, ADAM19, and γ-secretase cleave cadherin-6B, and ADAM perturbation alters the premigratory neural crest cell domain. The study provides the first evidence for cadherin-6B proteolysis in neural crest cells during EMT.
Abstract
The epithelial-to-mesenchymal transition (EMT) is a highly coordinated process underlying both development and disease. Premigratory neural crest cells undergo EMT, migrate away from the neural tube, and differentiate into diverse cell types during vertebrate embryogenesis. Adherens junction disassembly within premigratory neural crest cells is one component of EMT and, in chick cranial neural crest cells, involves cadherin-6B (Cad6B) down-regulation. Whereas Cad6B transcription is repressed by Snail2, the rapid loss of Cad6B protein during EMT is suggestive of posttranslational mechanisms that promote Cad6B turnover. For the first time in vivo, we demonstrate Cad6B proteolysis during neural crest cell EMT, which generates a Cad6B N-terminal fragment (NTF) and two C-terminal fragments (CTF1/2). Coexpression of relevant proteases with Cad6B in vitro shows that a disintegrin and metalloproteinases (ADAMs) ADAM10 and ADAM19, together with γ-secretase, cleave Cad6B to produce the NTF and CTFs previously observed in vivo. Of importance, both ADAMs and γ-secretase are expressed in the appropriate spatiotemporal pattern in vivo to proteolytically process Cad6B. Overexpression or depletion of either ADAM within premigratory neural crest cells prematurely reduces or maintains Cad6B, respectively. Collectively these results suggest a dual mechanism for Cad6B proteolysis involving two ADAMs, along with γ-secretase, during cranial neural crest cell EMT.
INTRODUCTION
The cellular steps comprising the epithelial-to-mesenchymal transition (EMT), in which stationary epithelial cells become migratory, are highly coordinated and regulated at multiple levels. Several critical processes requiring cell movement during embryogenesis, along with many human diseases, involve EMTs (Micalizzi and Ford, 2009; Lim and Thiery, 2012). The generation of migratory neural crest cells from immotile precursors in the embryonic dorsal neural tube is one important example of an EMT that is necessary for proper embryonic patterning during development. Premigratory neural crest cells undergo EMT to give rise to migratory neural crest cells that differentiate to form many specialized cell types, including neurons and glia of peripheral and sensory ganglia, odontoblasts, craniofacial tissues, adrenal cells, portions of the heart, and melanocytes. During EMT, premigratory neural crest cells lose apicobasal polarity, down-regulate junctional complexes, and reorganize their cytoskeleton to facilitate emigration from the neural tube (Hay, 1995; Lim and Thiery, 2012). Dismantling of premigratory neural crest cell adherens junctions alone requires proper coordination of many mechanisms, from transcriptional to posttranslational regulation of junction molecules (Pla et al., 2001; Taneyhill, 2008). Furthermore, the removal of adherens junctions in epithelial cells is a universal hallmark of EMTs occurring during development or pathologies, including organ fibrosis and tumor metastasis (Acloque et al., 2009; Thiery et al., 2009).
In the chick, premigratory cranial neural crest cells express N-cadherin and cadherin-6B (Cad6B), both of which are drastically reduced before and during neural crest cell EMT (Hatta and Takeichi, 1986; Duband et al., 1988; Nakagawa and Takeichi, 1995, 1998; Taneyhill et al., 2007). Down-regulation of N-cadherin is also important for proper neural crest cell EMT and migration in the chick trunk (Shoval et al., 2007). Previous studies demonstrated that Cad6B transcriptional repression is achieved, in part, through direct binding of the Snail2 repressor to E boxes (Snail2-binding sites) within the Cad6B regulatory region (Taneyhill et al., 2007) and via the deacetylation of the Cad6B promoter by a Snail2-PHD12-Sin3a complex (Strobl-Mazzulla and Bronner, 2012). Intriguingly, Cad6B protein persists in the chick cranial midbrain region at stages when Cad6B transcription is actively repressed during early EMT (seven-somite stage [7ss]). It is not until 90 min or one developmental stage later (8ss), however, that Cad6B protein is rapidly depleted (Taneyhill et al., 2007). This 90-min discrepancy between transcriptional repression and Cad6B protein turnover during EMT is considerably less than the 4- to 6-h half-life reported for other cadherins in vitro (Ireton et al., 2002; Pujuguet et al., 2003; Pon et al., 2005). Moreover, the cellular changes accompanying EMTs, such as cadherin down-regulation, include rapid processes that cannot be explained solely by transcriptional repression (Hay, 1995). These results point to the possibility that posttranslational regulation of Cad6B may occur to reduce Cad6B levels and facilitate neural crest cell EMT. No molecular data exist delineating mechanisms underlying Cad6B posttranslational regulation in vivo during chick cranial neural crest cell EMT.
Removal of cadherin ectodomains by sheddases such as a disintegrin and metalloproteinases (ADAMs) and matrix metalloproteinases (MMPs) is the first step in a series of proteolytic processing events that are used to deplete cellular cadherin protein levels (McCusker and Alfandari, 2009; Weber and Saftig, 2012). ADAM-mediated cleavage of the cadherin extracellular domain releases an N-terminal fragment (NTF), leaving behind a membrane-bound peptide known as C-terminal fragment 1 (CTF1). This fragment is often subjected to additional proteolysis at the cadherin juxtamembrane domain via a γ-secretase complex containing the catalytic presenilin subunit (PS-1 or PS-2; Marambaud et al., 2002, 2003; Uemura et al., 2006). The newly generated cleavage product, or CTF2, and any interacting proteins, such as catenins, are liberated from the membrane and remain in the cytoplasm or are imported into the nucleus (Ito et al., 1999; Marambaud et al., 2002, 2003; Reiss et al., 2005; Uemura et al., 2006). Although cadherin processing has been documented in vivo in other systems (Shoval et al., 2007; McCusker et al., 2009), whether ADAM/γ-secretase proteolysis of Cad6B occurs during cranial neural crest cell EMT has not been explored.
Here we identify ADAM10 and ADAM19, along with γ-secretase, as the enzymes responsible for Cad6B proteolytic processing in vitro and in vivo during cranial neural crest cell EMT. All three enzymes are expressed in the proper spatiotemporal pattern in vivo to mediate Cad6B cleavage. In keeping with these findings, we detect Cad6B proteolytic fragments during cranial neural crest cell EMT. Of importance, the appearance of these fragments is abrogated upon inhibition of protease activity in vitro and in vivo. We also show that ADAM perturbation affects neural crest cell EMT, with Cad6B lost prematurely upon ADAM overexpression, whereas Cad6B, and subsequently premigratory neural crest cells, are retained within the dorsal neural tube upon ADAM depletion. Collectively our biochemical data define a molecular mechanism for Cad6B posttranslational processing during neural crest cell EMT in the chick midbrain.
RESULTS
Cad6B protein down-regulation in the chick midbrain is temporally distinct from its transcriptional repression
Prior work alluded to the possibility that Cad6B protein turnover was coordinated independently of its transcriptional repression (Taneyhill et al., 2007). We examined this further by delineating Cad6B transcript and protein distribution in chick midbrain transverse sections before and during neural crest cell EMT. We observe Cad6B transcripts within premigratory neural crest cells up to the 6ss, after which time transcripts are down-regulated and no longer detectable 3 h later at the 8ss (Figure 1A, black arrows), in keeping with previous studies (Taneyhill et al., 2007; Fairchild and Gammill, 2013). Cad6B protein, however, is still maintained at the 7ss (∼1.5 h later) in premigratory neural crest cells and then begins to decline from the 8ss onward, with low levels of Cad6B maintained in the apical region of the dorsal neural tube (Figure 1A, white arrows). Taken together, these results corroborate our earlier work in the chick head and demonstrate a disparity between Cad6B RNA and protein expression.
FIGURE 1:

Cad6B protein levels decline during EMT due to proteolysis, yielding a Cad6B NTF and CTFs. (A) Representative transverse sections taken through embryos that underwent immunohistochemistry for Cad6B protein (top, green) from the 4ss to the 8ss, with merge images with DAPI shown (middle). Cad6B protein is localized to the dorsal neural folds containing premigratory neural crest cells in all stages, with protein concentrated within the fusing neural folds and peaking dorsally around the 6ss (arrows). During EMT stages (7ss and 8ss), Cad6B protein is down-regulated and is retained only at low levels in the most apical region of the dorsal neural tube. Bottom, representative transverse sections taken through embryos after whole-mount in situ hybridization for Cad6B transcripts. Arrows denote Cad6B transcripts in premigratory neural crest cells of the dorsal neural tube from the 4ss to the 6ss, with notable transcript down-regulation by the 7ss and 8ss. The duration between somite stages is ∼1.5 h. Scale bars, 50 μm (all section images). (B) Immunoblot showing Cad6B protein turnover in Flp-In Cad6B stably transfected cells treated with cycloheximide, with a t1/2 of 5.5 h. (C) Immunoblot reveals a decrease in full-length (FL) Cad6B during embryonic development, with high levels seen pre-EMT (5ss and 6ss) and lower levels observed during EMT (7ss and 8ss). This reduction inversely correlates with the appearance of a putative 91-kDa Cad6B NTF during EMT isolated through concanavalin A treatment of embryonic lysates. (D) Dorsal/ventral electroporation assay to detect Cad6B CTFs by immunoblotting reveals Cad6B CTFs during EMT, with a substantial reduction in CTFs observed during EMT in the presence of the protease inhibitor GM6001, leading to retention of FL Cad6B. For all immunoblots, β-actin was performed as a loading control.
This difference between Cad6B RNA and protein levels at the 7ss is perhaps not surprising, given that cadherins in general possess long half-lives (Ireton et al., 2002; Pujuguet et al., 2003; Pon et al., 2005), but is unlikely to explain the rapid depletion of Cad6B protein we observe after this point. To confirm this, we assessed the half-life of Cad6B in vitro using Chinese hamster ovary cells expressing a single, stably-integrated copy of chick Cad6B. Chinese hamster ovary cells were chosen for stable Cad6B expression, as they are a common in vitro model for studying cadherins due to their minimal expression of them (Niessen and Gumbiner, 2002; Paterson et al., 2003). Our data indicate that, like other cadherins, Cad6B possesses a half-life of 5.5 h (Figure 1B), which is much longer than its persistence in vivo in the chick midbrain (3 h). Taken together with our immunohistochemical data at different embryonic stages, these results suggest active posttranslational mechanisms that promote Cad6B turnover during cranial neural crest cell EMT.
Cad6B proteolytic cleavage fragments are present in vivo during cranial neural crest cell EMT
To evaluate potential posttranslational processing of Cad6B through proteolysis, we examined Cad6B protein in lysates prepared from chick midbrains both before and during neural crest cell EMT. Premigratory neural crest/dorsal neural fold tissue from chick midbrains was dissected out of embryos from the 5ss to the 8ss (EMT at 7ss), and immunoblotting experiments were performed using an antibody that recognizes the Cad6B N-terminus. Our results reveal a gradual decrease in full-length Cad6B at each embryonic stage examined (Figure 1C), in keeping with our immunohistochemical data. Concanavalin A–Sepharose beads were then used to isolate putative Cad6B NTFs (Alfandari et al., 1997; McCusker et al., 2009). We note the presence of a 91-kDa Cad6B NTF, which is of the appropriate relative molecular mass (Mr) after presumptive proteolysis. This NTF is found at very low levels pre-EMT (5–6ss) but is more abundant during EMT stages (7–8ss) as full-length Cad6B levels decline (Figure 1C). These data led us to surmise that perhaps Cad6B was indeed a substrate for proteolysis and this proteolytic cleavage event was correlated with neural crest cell EMT.
To address this hypothesis directly and circumvent the absence of an available antibody that recognizes the endogenous Cad6B C-terminus, we used an electroporation-based assay to detect Cad6B CTFs in the chick midbrain during neural crest cell EMT (see Materials and Methods). A low-efficiency electroporation protocol was performed at the 3–4ss to express trace amounts of C-terminal hemagglutinin (HA)-tagged Cad6B in the premigratory neural crest cell population. This protocol targets the fusing neural folds and does not elicit any neural crest cell developmental phenotypes (Supplemental Figure S1; Coles et al., 2007). Immunoblotting of pre-EMT–stage (4ss) or EMT-stage (8ss) embryos reveals the existence of a putative Cad6B CTF1 (∼33 kDa) and CTF2 (∼30 kDa) during EMT (Figure 1D, lane 4). These CTFs are the expected Mr, given extracellular and juxtamembrane cleavage of Cad6B. Furthermore, treatment of embryos with GM6001, a broad-spectrum inhibitor of ADAMs and MMPs (Kang et al., 2004), results in the loss of CTF1 and CTF2 and the accumulation of full-length Cad6B (Figure 1D, lane 5). Collectively these data provide additional evidence that Cad6B is proteolytically processed in vivo during cranial neural crest cell EMT.
ADAM10, ADAM19, and γ-secretase process Cad6B in vitro
To define the protease(s) involved in Cad6B down-regulation, we performed in vitro transient transfection assays in Chinese hamster ovary cells. We chose to examine a battery of different chick ADAMs and MMPs due to their published spatiotemporal expression profile and/or their known role in processing other cadherins in vitro and in vivo (Hall and Erickson, 2003; McGuire et al., 2003; Duong and Erickson, 2004; Lewis et al., 2004; Maretzky et al., 2005; Reiss et al., 2005; Lin et al., 2007, 2010; Shoval et al., 2007; Yan et al., 2011). All constructs (proteases and Cad6B) were transfected at comparable levels, and HA-tagged mature ADAMs were detected in all transfections involving ADAMs (Figure 2).
FIGURE 2:
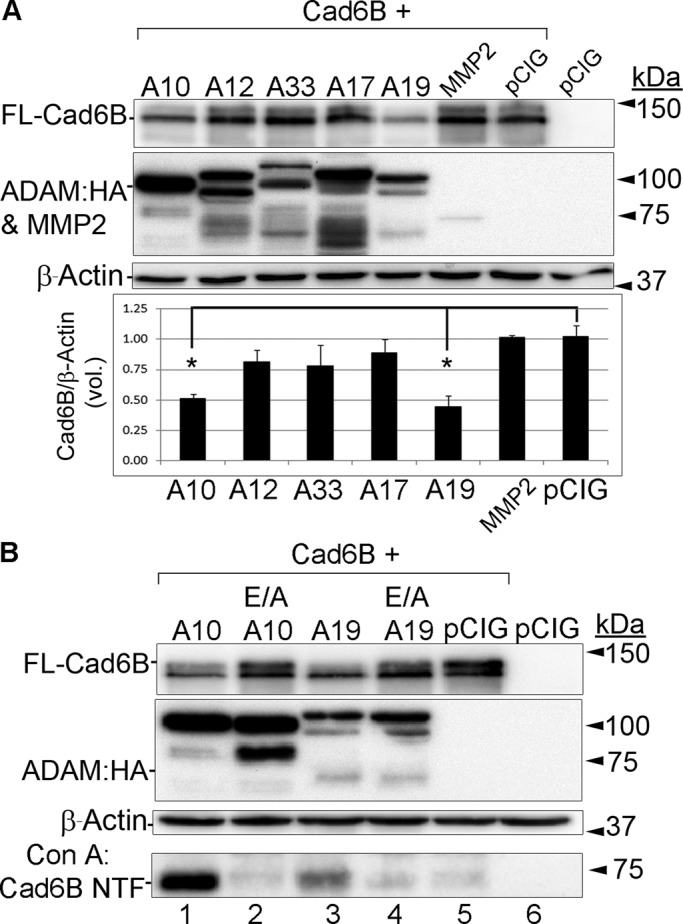
Cad6B is proteolytically cleaved by ADAM10, ADAM19, and γ-secretase in vitro. Transient transfection experiments in Chinese hamster ovary cells, followed by immunoblotting for FL-Cad6B, NTF, and CTFs. (A) An approximate twofold reduction in FL-Cad6B is observed upon cotransfection of Cad6B with either ADAM10 or ADAM19, with no statistically significant difference observed for other ADAMs, MMP2, or the control pCIG construct (*p < 0.05, n = 2). (B) Catalytically inactive (E/A) ADAM10 and ADAM19 mutants do not cleave Cad6B as efficiently as wild-type ADAM10 and ADAM19, resulting in the presence of more FL-Cad6B, comparable to what is observed in the pCIG control. Cotransfection of the mutant ADAMs also leads to a concomitant reduction in the appearance of the Cad6B NTF. For all immunoblots, β-actin was performed as a loading control.
Cotransfection of Cad6B with either ADAM10 or ADAM19 leads to an approximate twofold reduction in full-length Cad6B (p < 0.05), with no significant change observed with the control pCIG construct or with other proteases (Figure 2A). Moreover, cotransfection of Cad6B with catalytically inactive ADAM10 or ADAM19 mutant (E385A or E335A; Black and White, 1998; Smith et al., 2002; Chesneau et al., 2003) precludes Cad6B processing compared with cotransfection of wild-type ADAMs (Figure 2B, compare lanes 1 vs. 2, and 3 vs. 4). In addition, concanavalin A-Sepharose bead–mediated isolation of the putative Cad6B NTF from media after cotransfection of Cad6B with wild-type or catalytically inactive ADAM10 or ADAM19 mutant shows a marked reduction in the Cad6B NTF in the presence of the mutant ADAMs (Figure 2B, lanes 2 and 4), demonstrating specificity of these ADAMs for Cad6B proteolysis in vitro. Additional proof of transfected protease function/specificity is indicated by N-cadherin proteolysis, which occurs in the presence of ADAM10 only and leads to a reduction in full-length N-cadherin (Supplemental Figure S2), as reported previously (Shoval et al., 2007). As expected, Cad6B is also processed by γ-secretase to generate Cad6B CTF2 after production of the Cad6B NTF and CTF1 (Supplemental Figure S3). These results indicate that Cad6B is a substrate for ADAM10-, ADAM19-, and γ-secretase-mediated proteolysis in vitro.
ADAM10, ADAM19, and PS-1 are expressed in the proper spatiotemporal pattern to process Cad6B in vivo
We examined the distribution of ADAM10, ADAM19, and PS-1 (the catalytic subunit of γ-secretase; Tolia and De Strooper, 2009) in the developing chick embryo midbrain both before and during cranial neural crest cell EMT. Before EMT, we observe ADAM10 in non–neural crest tissues (such as the nonneural ectoderm and neural tube basal lamina) and in premigratory neural crest cells (Figure 3, A–C and F–H, asterisks and arrowheads, respectively). At the 6ss, ADAM10 protein levels increase in the apical region of the neural tube, including in the dorsal neural folds/premigratory neural crest cell region (Figure 3, C and H, arrowheads). At the 7ss and 8ss, expression is still apparent in the apical region of the neural tube in premigratory neural crest cells (Figure 3, D, E, I, and J, arrowheads) and in the basal lamina associated with the neural tube and ectoderm. Migratory neural crest cells, however, are devoid of ADAM10 protein (Figure 3, D, E, I, and J). PS-1 is observed throughout the premigratory (Figure 3, K–M and P–R, arrowheads) and migratory (Figure 3, N, O, S, and T, arrows) neural crest cell populations, at the plasma membrane, and in puncta. Owing to the lack of an available antibody, we were unable to document ADAM19 protein distribution. We instead performed whole-mount in situ hybridization for ADAM19 transcripts and noted ADAM19 in premigratory neural crest cells of the dorsal neural folds before EMT (Figure 4, A–H, arrowheads), as well as in migratory neural crest cells (Figure 4, I–N, arrows). These results indicate that proteases that process Cad6B in vitro are expressed in the correct spatiotemporal pattern to cleave Cad6B in vivo.
FIGURE 3:

ADAM10 and PS-1 are expressed in the appropriate spatiotemporal pattern to cleave Cad6B in vivo. Representative transverse sections taken through the midbrain region of embryos at various stages and processed for immunohistochemistry for ADAM10 (A–E, red) and PS-1 (K–O, red), with corresponding merge images (F–J and P–T, respectively). Arrowheads and arrows indicate expression in premigratory and migratory neural crest cells, respectively. Asterisk denotes expression in non–neural crest tissues, such as the ectoderm and neural tube basal lamina (A, F). In all stages examined, ADAM10 is observed in premigratory neural crest cells (A–E, F–J) but is not detected in migratory neural crest cells (D, E, I, J). Expression is also noted in the ectoderm and basal lamina. PS-1 is observed in puncta in premigratory neural crest cells (K–M, P–R), as well as in migratory neural crest cells (N, O, S, T). Scale bar, 50 μm (A, all section images). DAPI (blue) labels cell nuclei.
FIGURE 4:

ADAM19 is expressed in the relevant spatiotemporal pattern to cleave Cad6B in vivo. (A, C, E, G, I, K, M) Whole-mount in situ hybridization for ADAM19 using an ADAM19 antisense riboprobe, followed by indicated transverse sections (B, D, F, H, J, L, N) through the chick midbrain. Arrowheads and arrows indicate expression in premigratory and migratory neural crest cells, respectively. ADAM19 is noted as early as the 3ss (A) in the dorsal neural folds (B) and later on in the dorsalmost region of the neural folds as neural fold fusion is occurring (C–H). At the onset of EMT and neural crest cell migration, ADAM19 is expressed by migratory neural crest cells and is absent in the dorsal neural tube (I–N). ADAM19 is also present at low levels in the mesoderm, ectoderm, and neural tube. ADAM19 sense probe (O, P) shows no signal and serves as a negative control. Scale bar, 250 μm (A) and 50 μm (B), all whole-mount and section images, respectively.
ADAM10 and ADAM19 both regulate Cad6B expression in vivo
Using the same constructs from our in vitro experiments, we overexpressed ADAM10 or ADAM19 in chick premigratory cranial neural crest cells using the technique of in ovo electroporation (Itasaki et al., 1999; Jhingory et al., 2010; Wu et al., 2011; Wu and Taneyhill, 2012). ADAM expression was verified in vivo on transverse midbrain sections using immunohistochemistry (unpublished data). Embryos were then fixed at stages before normal Cad6B down-regulation in order to assess Cad6B distribution upon ADAM10 or ADAM19 overexpression. We observe precocious loss of Cad6B protein upon overexpression of either ADAM protease (ADAM10: Figure 5, A, A′, and D, nine of 10 embryos, arrows; ADAM19: Figure 5, E, E′, and H, six of six embryos, arrows), with no change observed in the presence of the pCIG control construct (Figure 5, I, I′, and L, 10 of 10 embryos, arrows). These data provide strong evidence that ADAM10 and ADAM19 are likely responsible for processing Cad6B in premigratory cranial neural crest cells in vivo.
FIGURE 5:

Overexpression of either ADAM10 or ADAM19 leads to a premature reduction in Cad6B protein in premigratory neural crest cells. Representative transverse sections taken through the midbrain region of 7ss embryos that were electroporated unilaterally with pCIG.ADAM10 (A–D), pCIG.ADAM19 (E–H), or the pCIG control construct (I–L), followed by immunohistochemistry for Cad6B (A, A′, E, E′, I, I′). Arrows indicate Cad6B protein, which is absent upon ADAM overexpression. GFP expression (B, F, J) points to electroporated neural tube cells, with merge image shown in D, H, and L. Scale bar, 100 μm (A′, applicable to E′, I′), 50 μm (A, applicable to remaining section images). DAPI (blue) labels cell nuclei (C, G, K).
Depletion of ADAM10 and/or ADAM19 leads to maintenance of Cad6B in premigratory neural crest cells undergoing EMT
We used translation-blocking morpholinos (MOs) to deplete ADAM10 and/or ADAM19 in premigratory neural crest cells. To validate MOs for knockdown efficiency, we first electroporated the ADAM10 or ADAM19 MO (or a 5–base pair mismatch control MO) independently into premigratory midbrain neural crest cells and collected embryos at the 8ss, when endogenous levels of Cad6B are significantly reduced (Figure 6), and performed immunoblots for each ADAM, as carried out previously (Taneyhill et al., 2007; Wu et al., 2011). These MOs achieve significant reduction of ADAM10 (Figure 6A; 59%) and ADAM19 (Figure 6B; 70%) while also increasing overall Cad6B levels within the dorsal neural tube (Figure 6, A and B; 46 and 25%, respectively). Depletion of ADAM10 results in marked increase in the levels of apical and lateral Cad6B expression (Figure 7, A and A′, lines and arrowhead, respectively) and maintenance of the premigratory neural crest domain in comparison to the contralateral control side (Figure 7, A–D and M; five of five embryos, 1.4-fold increase, p < 0.001). Retention of the Cad6B-positive premigratory neural crest domain and increased apicolateral Cad6B were also evident in embryos electroporated with ADAM19 MO (Figure 7, E–H, M, and E′, lines and arrowhead, respectively; four of four embryos, 1.2-fold increase, p < 0.05). Similarly, electroporation of both MOs results in maintenance of Cad6B and, subsequently, the premigratory neural crest cell domain (Figure 7, I–L, M, and I′, arrowhead and lines, respectively, five of five embryos, 1.4-fold increase, p < 0.05). We detect no qualitative changes in the migratory neural crest cell domain, however, as evidenced by immunostaining for FoxD3 (Figure 7, B, F, and J). Electroporation of ADAM10 or ADAM19 control MOs, either singly or together, has no statistically significant effect on the size of the Cad6B-positive premigratory neural crest domain (ADAM10 control MO, ADAM19 control MO, ADAM10 + ADAM19 control MOs: fold difference, 1.0; three of three, five of five, and three of three embryos, respectively; Supplemental Figure S4). These results demonstrate that endogenous ADAM10 and ADAM19 work in concert to proteolytically process Cad6B in cranial neural crest cells during neural crest cell EMT in vivo.
FIGURE 6:
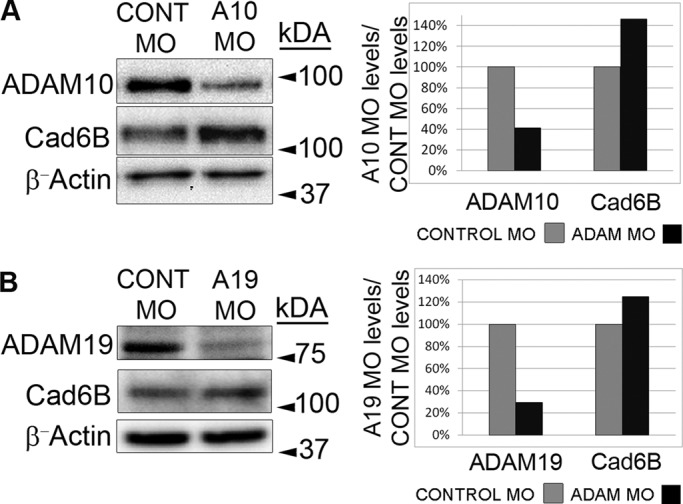
ADAM MOs efficiently block ADAM translation and lead to a concomitant increase in Cad6B protein levels. ADAM and ADAM control MOs were electroporated into fusing dorsal neural folds at the 3–4ss, and then embryos were reincubated for ∼6 h until the 8ss. Dorsal neural tubes were excised and pooled in lysis buffer (30 per lysate), and 30 μg of lysate was loaded per treatment to examine ADAM protein levels by immunoblotting. (A) Immunoblot analysis for ADAM10 reveals 59% reduction in ADAM10 protein as compared with the control MO–treated lysate. This decrease in ADAM10 levels correlates with 46% increase in Cad6B levels. (B) Immunoblot analysis for ADAM19 reveals 70% reduction in ADAM19 protein as compared with the control MO–treated dorsal neural tube lysate. The reduction in ADAM19 results in 25% increase in Cad6B levels. ADAM and Cad6B levels for each treatment were normalized to β-actin levels before comparison.
FIGURE 7:

MO-mediated depletion of ADAM10 and/or ADAM19 leads to maintenance of the Cad6B-positive premigratory neural crest domain. Representative transverse sections taken through the midbrain region of 8ss embryos after unilateral electroporation of ADAM10 MO (A–D), ADAM19 MO (E–H), or an equimolar mixture of ADAM10 + ADAM19 MOs (I–L), followed by whole-mount immunohistochemistry for Cad6B (A, E, I; magnified images in A′, E′, I′). Retained Cad6B apicolateral expression is indicated by the arrowheads, with dotted lines demarcating the dorsal neural tube midline and solid lines marking the width of the left and right Cad6B-positive premigratory neural crest cell domains in A ′, E′, and I′. Extent of MO electroporation shown in C, G, and K, with FoxD3 expression documented in B, F, and J. (M) Graphical representation showing statistically significant 1.4-, 1.2-, and 1.4-fold increase in the Cad6B-positive premigratory neural crest domain. Means that share letter superscripts are not significantly different (p < 0.05). Scale bar, 100 μm (A′, applicable to E′, I′), 50 μm (A, applicable to remaining section images. DAPI (blue) labels cell nuclei (D, H, L).
Ectopic expression of Cad6B CTF2 negatively regulates Cad6B levels in premigratory neural crest cells
To examine a possible function for Cad6B CTF2 during EMT, we overexpressed CTF2 in premigratory neural crest cells and examined Cad6B levels and effects on early neural crest emigration. Embryos were fixed during early EMT (6–8ss) and immunostained for Cad6B and FoxD3. Similar to our results obtained after ADAM10 and ADAM19 overexpression, we observe precocious loss of Cad6B in electroporated premigratory neural crest cells that have not completed EMT (Figure 8, A and A′, arrowhead; nine of nine embryos). The loss of Cad6B in premigratory neural crest cells overexpressing Cad6B CTF2 is cell autonomous and evident from both premigratory neural crest cell lateral membranes and apical regions. In addition, the size of the Cad6B-positive premigratory neural crest cell domain was significantly diminished (29%) compared with the Cad6B-positive domain observed on the contralateral control side (p < 0.001). FoxD3 immunostaining did not reveal any obvious effects of Cad6B CTF2 overexpression on early neural crest cell emigration at the stages analyzed (Figure 8B, 7ss). No significant differences in Cad6B reduction within individual cells or the Cad6B-positive premigratory neural crest domain were observed in embryos electroporated with the pCIG control construct (Figure 8, E and E′, arrowhead; three of three embryos). These results suggest that the Cad6B CTF2 generated via ADAM-mediated proteolysis may possess a pro-EMT role by facilitating loss of Cad6B in premigratory neural crest cells.
FIGURE 8:
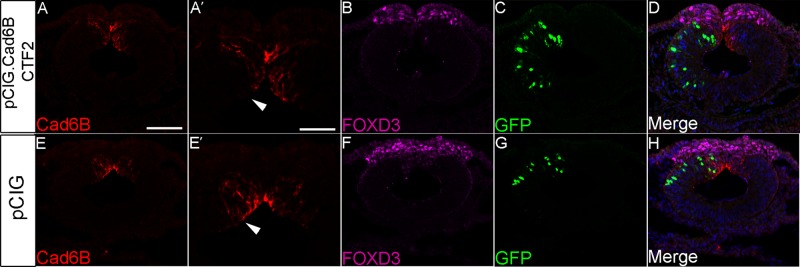
Overexpression of Cad6B CTF2 leads to precocious reduction of Cad6B in premigratory neural crest cells. Representative transverse sections taken through the midbrain region of 6–7ss embryos that were electroporated unilaterally with pCIG.Cad6B.CTF2 (A–D) or the pCIG control construct (E–H), followed by immunohistochemistry for Cad6B (A, A′, E, E′) and FoxD3 (B, F). Arrowheads indicate the Cad6B-positive premigratory neural crest cell domain, which is reduced upon Cad6B CTF2 overexpression. GFP expression (C, G) points to electroporated neural tube cells, with merge images shown in D and H. Scale bar, 100 μm (A′, applicable to E′), 50 μm (A, applicable to remaining section images). DAPI (blue) labels cell nuclei (D, H).
DISCUSSION
ADAM function, including proteolysis of cadherins, is important during neural crest cell ontogeny
Precise regulation of cellular EMTs is critical during embryonic development and disease and involves the use of both transcriptional and posttranscriptional means by which to modulate gene expression. The dissolution of cellular junctions is one example of an event associated with EMT that is controlled in a tight spatiotemporal manner. For instance, down-regulation of E-cadherin occurs during the EMT that underlies gastrulation (Ciruna and Rossant, 2001) and is essential for mesoderm formation. During migratory neural crest cell formation, premigratory neural crest cells reduce levels of N-cadherin (Shoval et al., 2007; Monsonego-Ornan et al., 2012) and Cad6B before and/or during EMT to allow them to emigrate from the dorsal neural tube (Nakagawa and Takeichi, 1995, 1998; Coles et al., 2007; Shoval et al., 2007; Taneyhill et al., 2007). Of importance, cadherin down-regulation can occur aberrantly and lead to conditions such as organ fibrosis and cancer metastasis (Lim and Thiery, 2012). As such, elucidating the molecular mechanisms that regulate cadherin levels is crucial to shedding light on these normal developmental processes and during diseases in which EMT is coopted.
ADAMs and MMPs play vital roles in facilitating proper neural crest cell development in a variety of systems across all stages of neural crest ontogeny. During Xenopus neural crest cell induction, ADAM19 (adamalysin 19, metrin-β) regulates levels of the neural crest specifiers Sox8 and Slug through an unknown mechanism (Neuner et al., 2009), whereas ADAM13 processes ephrins B1 and B2 and subsequently upregulates Wnt signaling and Slug (Wei et al., 2010). Furthermore, ADAM13 is the major ADAM required for proteolytic processing of cadherin-11, the predominant type II cadherin expressed in Xenopus migratory cranial neural crest cells. Multiple meltrin ADAMs (9, 13, and 19) are expressed in Xenopus migratory cranial neural crest cells, and individual knockdown of each ADAM affects neural crest cell migration (McCusker et al., 2009). Of interest, this disruption in migration can be rescued by overexpressing the cadherin-11 NTF, which may antagonize homophilic interactions of intact cadherin-11 dimers and reduce migratory cranial neural crest cell–cell adhesion. In the chick, the gelatinase subfamily members MMP2 and MMP9 are expressed in premigratory and migratory neural crest cells and are essential for proper neural crest cell delamination and migration (Duong and Erickson, 2004; Monsonego-Ornan et al., 2012). Within the chick trunk dorsal neural tube, N-cadherin is depleted from neural crest cells undergoing EMT via BMP4-activated ADAM10 processing (Shoval et al., 2007). Taken together, these findings underscore the importance of protease function in the generation of migratory neural crest cells in multiple systems.
Our results provide insight into the regulation of cadherins during cranial neural crest cell EMT. These results are significant in light of the axial-level differences in neural crest cell EMT (Theveneau et al., 2007), and in particular cadherin expression and regulation (Coles et al., 2007; Taneyhill et al., 2007; Park and Gumbiner, 2010, 2012), that have been noted in the chick. Chick embryos express two primary cadherins that mark the premigratory neural crest cell population, N-cadherin and Cad6B. In the chick head, Cad6B levels increase until the 6ss. After this point, neural crest cell EMT commences, and we observe a concomitant reduction in Cad6B protein, even though Cad6B transcripts are absent at the 7ss due to transcriptional repression by Snail2 (Taneyhill et al., 2007). This rapid clearing of Cad6B after only 90 min is suggestive of an active degradation mechanism due to the long half-life of Cad6B protein. Our work now uncovers a role for posttranslational mechanisms to deplete Cad6B protein from premigratory cranial neural crest cells, with Cad6B protein loss important for proper neural crest cell EMT.
Cad6B proteolysis occurs in vitro and in vivo during chick cranial neural crest cell EMT
Prior studies showed a functional role for Cad6B during both cranial (Coles et al., 2007) and trunk (Nakagawa and Takeichi, 1998; Park and Gumbiner, 2010) neural crest cell migration. Overexpression of Cad6B in chick premigratory cranial neural crest cells leads to a disruption in migration, as evidenced by neural crest cell aggregation adjacent to the neural tube, a phenotype been observed previously after overexpression of other cadherins within the neural crest (Nakagawa and Takeichi, 1998; Coles et al., 2007; Shoval et al., 2007). More important, Cad6B knockdown within the midbrain neural crest cell domain results in precocious neural crest cell emigration, indicating that Cad6B levels within neural crest cells undergoing EMT en masse dictate the timing and rate of delamination and emigration, respectively (Coles et al., 2007). Posttranscriptional molecular mechanisms underlying Cad6B regulation, however, had not been deciphered until our present work.
We demonstrate for the first time that Cad6B undergoes proteolytic processing in vivo in neural crest cells undergoing EMT. Our findings provide definitive biochemical evidence to explain how Cad6B protein, which possesses a long half-life like other cadherins, is rapidly depleted from premigratory neural crest cells over a short time frame (3 h). These results implicate both transcriptional and posttranslational mechanisms of Cad6B down-regulation during neural crest cell EMT. Cad6B proteolysis leads to the formation of lower–molecular weight products that correspond to a Cad6B NTF and CTFs 1 and 2, the appearance of which correlates with the onset of neural crest cell EMT. Of importance, these products are sensitive to metalloproteinase inhibition, lending further credence to the hypothesis that they are bona fide Cad6B cleavage products. In vitro studies revealed that the generation of E- or N-cadherin CTF2 depends on prior N-terminal cleavage to create E- or N-cadherin NTF and CTF1 (Marambaud et al., 2002, 2003). This finding is also true for Cad6B as demonstrated in our in vivo experiments using the metalloproteinase inhibitor GM6001. Of interest, specific roles for cadherin NTFs and CTFs have been described in various in vitro and in vivo systems (Maretzky et al., 2005; Reiss et al., 2005; Uemura et al., 2006; Lyon et al., 2009; McCusker and Alfandari, 2009; McCusker et al., 2009). Our results demonstrate for the first time a unique pro-EMT function for a cadherin CTF in vivo, in which the Cad6B CTF2 negatively regulates levels of full-length Cad6B in premigratory cranial neural crest cells undergoing EMT. As such, it is plausible that Cad6B cleavage fragments will perform other functions during chick cranial neural crest cell EMT and migration, a possibility that is being explored.
In vitro transient transfection experiments corroborate our in vivo data and reveal that ADAM10 and ADAM19, along with γ-secretase, proteolytically process Cad6B, generating a Cad6B NTF and CTFs 1 and 2. Furthermore, Cad6B proteolysis is specific to ADAM10, ADAM19, and γ-secretase function, as inhibition of these proteases (through the use of either catalytically inactive mutants or a potent γ-secretase inhibitor) abrogates Cad6B cleavage and leads to the subsequent loss of the Cad6B NTF and CTFs in vitro. The catalytically inactive ADAM mutants we generated are hypothesized to act as dominant negatives due to their function in Xenopus (McCusker et al., 2009) and their inhibition of Cad6B processing upon introduction into Chinese hamster ovary cells in the absence of any exogenous ADAMs (Figure 2B, lanes 2 and 4, and Supplemental Figure S3, lane 2). These data imply that the catalytically inactive ADAMs are likely reducing the activity of an endogenous Chinese hamster ovary cell sheddase(s) toward Cad6B (LaVoie and Selkoe, 2003; Dyczynska et al., 2007), which, along with the known expression of γ-secretase in Chinese hamster ovary cells (Magold et al., 2009), leads to some Cad6B processing in vitro into CTF1 and CTF2 (Supplemental Figure S3, lane 3). This proteolysis, however, is not as extensive as that observed upon inclusion of wild-type ADAM10 or ADAM19 and PS-1 (unpublished observations).
ADAM10 and ADAM19: a dual mechanism to down-regulate Cad6B in cranial neural crest cells
To examine the functional significance of Cad6B proteolysis during neural crest cell EMT and migration, we first overexpressed ADAM10 or ADAM19 in cranial premigratory neural crest cells. Processing of Cad6B within premigratory cranial neural crest cells is specific to ADAM10 and ADAM19 because overexpression of each ADAM leads to precocious loss of Cad6B. Loss-of-function data reveal that ADAM10 and/or ADAM19 knockdown in the dorsal neural tube permits retention of Cad6B on apicolateral membranes and, as such, maintenance of premigratory neural crest cells. These results are statistically significant and further emphasize the requirement for both ADAMs during Cad6B proteolysis, albeit potentially at different times and locations during development due to their expression patterns. ADAM10 is localized exclusively to premigratory neural crest cells, whereas ADAM19 and PS-1 are found in both premigratory and migratory neural crest cells. As such, these enzymes are all present at the correct time and location to modulate Cad6B levels. To our knowledge, this is the first report to document ADAM10, ADAM19, and PS-1 expression in the early chick embryo during cranial neural crest cell EMT and early migration. Of interest, although ADAM knockdown directly impinges upon Cad6B and the premigratory neural crest cell population, it does not appear to perturb overt neural crest cell migration, at least at the embryo stages that we examined. This result could be due to the mosaic nature of the MO electroporation and is a limitation of our system, in that we cannot achieve true knockout of each ADAM. At 8ss, the majority of FoxD3-positive migratory neural crest cells in our knockdown assays are not MO positive. This result suggests that there may be a subpopulation of premigratory neural crest cells in the neural folds at the stages at which we are electroporating that may not be as accessible to morpholino uptake due to their position, in comparison to those premigratory neural crest cells localized closer to the dorsal midline and thus the MO solution. These cells would undergo EMT on time and confound any significant migration delay that would be observed in morpholino-positive cells. Alternatively, there may be different pools of premigratory neural crest cells within the dorsal neural folds, the first of which are more refractory to the effects of the MO, perhaps due to alternative mechanisms (see later discussion) functioning therein for down-regulating Cad6B and allowing for their successful emigration. Effects on neural crest cell EMT and migration may only be evident at later stages of EMT and migration, which is being explored. In addition, although we achieved >59 and 70% reduction in ADAM10 and ADAM19 levels, respectively, the residual ADAM activity may be sufficient to proteolytically process Cad6B such that levels drop to those required for premigratory neural crest cells to undergo EMT and migrate normally. It is important to also note that Cad6B transcriptional repression is still occurring during our assays, thereby preventing formation of new Cad6B protein and contributing to an overall decline in Cad6B protein levels. Finally, it is likely that redundant mechanisms exist to clear any remaining Cad6B from premigratory and/or emigrating neural crest cell membranes in the absence of (reduced) ADAM function. This is particularly apparent in the ADAM10 + ADAM19 double MO knockdown, for which the migratory neural crest cell population is unaffected. Such mechanisms may include Cad6B endocytosis (unpublished observations), cleavage by additional proteases within migratory neural crest cells, and mechanical separation of Cad6B-containing apical tails of premigratory neural crest cells undergoing EMT, as observed in a subpopulation of chick trunk neural crest cells (Ahlstrom and Erickson, 2009).
Prior work demonstrating functional roles for cadherin fragments in other neural crest model systems (Shoval et al., 2007; McCusker et al., 2009) led us to surmise that Cad6B proteolytic fragments might function to regulate EMT in chick cranial neural crest cells. To maximize the amount of endogenous Cad6B CTF2 that would be generated in cranial neural crest cells, we focused our analysis on early EMT (6–8ss). At these stages, we observed that Cad6B CTF2 overexpression prematurely reduces the level of preexisting membrane pools of Cad6B. At such early EMT stages, transcriptional repression and ADAM-mediated proteolysis have yet to fully clear Cad6B levels in premigratory neural crest cells undergoing EMT. These results suggest a possible feedback mechanism by which the reduction of Cad6B-containing adherens junctions during EMT is, at a minimum, a two-part mechanism that includes loss of cell–cell adhesion due to ADAM10/ADAM19 proteolysis and an unknown mechanism involving the γ-secretase–generated Cad6B CTF2. Future studies will characterize this latter mechanism.
Overexpression of Cad6B CTF2 did not result in neural crest cell aggregation and disruption of migration as observed after overexpression of full-length Cad6B (Coles et al., 2007). This is not surprising, given that premigratory neural crest cells constitutively expressing high levels of Cad6B exhibit increased cell–cell adhesion via the Cad6B N-terminal domain. This increase in premigratory neural crest cell adhesion will counteract any potential pro-EMT effects attributable to the presence of additional Cad6B CTF2. Furthermore, it is likely that this ectopic Cad6B is not proteolytically cleaved due to competition with endogenous Cad6B, thereby effectively saturating the proteolysis machinery and giving the observed adhesion and migration phenotypes. In addition, the mechanism in which Cad6B CTF2 regulates levels of Cad6B may be coordinated with, or downstream of, other mechanisms, such as ADAM-mediated proteolysis itself. Nonetheless, Cad6B CTFs may promote EMT by facilitating the dismantling of Cad6B adherens junctions, which is essential for generating migratory neural crest cells. Finally, the lack of an apparent emigration phenotype (as determined by FoxD3 immunostaining) may be explained by the short incubation period (5 h) that we used in our experiments. Any overt effects on neural crest cell migration may not be discernible until later stages. Further examination is warranted to determine the exact effect of Cad6B CTF2 in facilitating EMT in chick cranial neural crest cells.
It is intriguing that two ADAMs appear to regulate Cad6B protein levels in chick cranial neural crest cells. This finding is not without precedent, as ADAM13 and ADAM19 exhibit functional redundancy and play an important role in Xenopus cranial neural crest cell migration (McCusker et al., 2009). Given the spatiotemporal distribution of ADAM10 and ADAM19 during chick neural crest cell EMT and the differential increase in Cad6B protein observed upon immunoblotting after individual ADAM knockdown, we hypothesize that rapid clearance of Cad6B protein occurs primarily through the activity of ADAM10 in premigratory neural crest cells, with ADAM19 playing a more minor role in the premigratory and newly emigrating neural crest cell populations. Additional evidence for this stems from our MO immunohistochemical experiments, in which ADAM10 depletion can lead at times to the presence of a few newly emigrating neural crest cells that express Cad6B. Conversely, ADAM19 knockdown rarely gives rise to emigrating and migrating neural crest cells that are Cad6B positive. In the former situation, ADAM19 activity may not be as efficient as ADAM10 in removing Cad6B from either premigratory or newly emigrating neural crest cells, thereby leaving some Cad6B remaining in these cells as they emigrate/migrate. In the absence of ADAM19, however, the majority of premigratory neural crest cell Cad6B is cleared by ADAM10, so that little to no Cad6B remains in emigrating neural crest cells, with only the rare cell retaining Cad6B. As such, a strong correlation exists between the requirement for ADAM10 and ADAM19 proteolytic processing of Cad6B and the ability of neural crest cells to undergo EMT and migrate.
It is also possible that ADAM10 and ADAM19 are preferentially used to cleave different pools of Cad6B, with membrane-bound ADAM10 processing membrane-localized Cad6B, whereas internal pools of Cad6B are depleted through the function of ADAM19, which is found in the Golgi and cleaves proproteins such as prorenin (Yoshikawa et al., 2011). The proteolytic activity of both of these ADAMs would then be sufficient to reduce Cad6B protein levels in premigratory and early migratory cranial neural crest cells, thereby promoting neural crest cell EMT. Alternatively, our data may indicate that both ADAMs, and most notably ADAM10, play a role in neural crest segregation from the neural tube by modulating Cad6B levels in premigratory neural crest cells to maintain a normal domain size and boundary. Moreover, the presence of ADAM MO–positive migratory neural crest cells that are devoid of Cad6B indicates that additional mechanisms are in place to remove Cad6B pools from emigrating neural crest cell membranes, including endocytic trafficking as observed for N-cadherin (Lock and Stow, 2005; Linford et al., 2012) and/or processing by other, as-yet-unidentified proteases.
Our results provide a molecular basis for Cad6B protein down-regulation during cranial neural crest cell EMT. These findings describe an additional level of Cad6B regulation that is cranial specific and offer further evidence for axial level differences in the control of Cad6B expression, as observed previously in the chick head (Coles et al., 2007; Taneyhill et al., 2007) and trunk (Park and Gumbiner, 2010, 2012). Although both ADAM10 and ADAM19 are expressed in the chick trunk (Shoval et al., 2007; unpublished observations), the maintenance of Cad6B protein within the early migratory trunk neural crest cell population suggests that Cad6B is likely not a substrate for protease cleavage within premigratory neural crest cells at this axial level. The later down-regulation of Cad6B observed within trunk migratory neural crest cells (Nakagawa and Takeichi, 1995; unpublished observations) might then be due to the activity of ADAM19 and/or another protease present in migratory neural crest cells. Finally, our data provide insight into recent findings ascribing a role for tetraspanin proteins in regulating Cad6B protein in neural crest cells (Fairchild and Gammill, 2013). Collectively our results further underscore the need to carefully modulate Cad6B levels in the premigratory cranial neural crest cell population so that EMT occurs at the appropriate time, generating a migratory neural crest cell population to build and pattern the embryo.
MATERIALS AND METHODS
Chicken embryo culture
Fertilized chicken eggs were obtained from Hy-Line North America (Elizabethtown, PA) or B & E Farms (York, PA) and incubated at 38°C in humidified incubators (EggCartons.com, Manchaug, MA). Embryos were staged either according to the number of pairs of somites or by Hamburger and Hamilton staging (Hamburger and Hamilton, 1992).
Cloning of proteases and Cad6B for in vitro and in vivo expression
Full-length cDNAs for chick ADAMs 10, 12, 33, 17, and 19, MMP2, and presenilin-1 (PS-1) were PCR amplified from 5–10ss chick midbrain cDNA and cloned into pCR-II (Life Technologies, Carlsbad, CA). ADAM coding sequences were PCR amplified, HA epitope tagged at their C-terminus, and directionally cloned into the pCIG expression construct. MMP2 and PS-1 coding sequences were also subcloned into pCIG. For trace expression of Cad6B (Coles et al., 2007) in vivo, Cad6B was HA epitope tagged and then recloned into pCIG. To generate “catalytically inactive/dead” E/A ADAM mutant expression constructs (Black and White, 1998; Smith et al., 2002; Chesneau et al., 2003), pCIG.ADAM10 and pCIG.ADAM19 were mutagenized (E385A and E335A, respectively) using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). For Cad6B CTF2 experiments, the predicted CTF2 cytoplasmic product generated after γ-secretase cleavage (based on amino acid sequence comparisons to data obtained in Uemura et al., 2006) was PCR amplified as described and directionally cloned into pCIG. All constructs were sequenced to confirm accuracy (Genewiz, South Plainfield, NJ).
In vitro transient transfection assays
Chinese hamster ovary cells (CCL-61; American Type Culture Collection, Manassas, VA) were cultured according to the provider's instructions. Transient transfection assays were carried out in Chinese hamster ovary cells using the Lipofectamine 2000 reagent (Life Technologies). Cells were grown to 90% confluency, and transfections were performed according to the manufacturer's protocol with the following modifications. Cotransfections of single protease expression constructs (2.1 μg) and pCIG.Cad6B or pCI.N-cad constructs (0.7 μg) were performed at a 2-to-1 ratio (microliters of Lipofectamine 2000/micrograms of DNA) in 12-well plates or scaled up in six-well plates. Cotransfections involving three constructs were performed using equivalent DNA concentrations (2 μg) at a 2-to-1 ratio (microliters of Lipofectamine 2000/micrograms of total DNA) in six-well plates. At 48 h posttransfection, cells were harvested and lysed for immunoblotting analyses. For detection of C-terminal Cad6B fragments in cell lysates, transfected cell cultures were supplemented with any of the following reagents during the 48-h incubation: 10 mM NH4Cl for the last 5 h, 10 μM L-685,458 (Tocris Biosciences, Bristol, United Kingdom) for the last 24 h, and/or 1 μM ionomycin (EMD Millipore, Billerica, MA) for the last 30 min. To enhance detection of CTF levels in cell lysates in Supplemental Figure 3, 10 mM NH4Cl was added to all transfection wells, as NH4Cl is a known disruptor of the lysosomal protein degradation pathway. In addition to 10 mM NH4Cl supplementation, transfected cells in Supplemental Figure 3 were also treated with ionomycin, a known inducer of ADAM10 activity (Maretzky et al., 2005), or L-685,458, a γ-secretase inhibitor (Shearman et al., 2000).
Protein extraction and immunoblotting
Glycosylated N-terminal Cad6B fragments were enriched from cell culture media as previously described (Alfandari et al., 1997). Briefly, 1.3 ml of clarified supernatants was supplemented with the following reagents in Tris-buffered saline (TBS) to achieve the following final concentrations: 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 5 mM EDTA, pH 8. Samples were incubated with 30 μl of concanavalin A–Sepharose beads (Sigma-Aldrich, St. Louis, MO) overnight at 4°C and washed twice in TBS and 1% Triton X-100. After complete wash buffer removal, beads were suspended in 2× reducing Laemmli sample buffer and boiled at 100˚C for 5 min, and supernatants were then loaded into SDS–PAGE gels.
Chinese hamster ovary cells were harvested by gentle mechanical scraping in ice-cold phosphate-buffered saline (PBS) and pelleted at 4°C for 5 min at 500 × g. Pellets were then flash-frozen in liquid nitrogen and stored at −80°C until needed for immunoblot analysis. Embryo protein extracts were collected in the following manner. Embryos were excised from eggs using tungsten needles and cleaned in Ringer's solution. Whole midbrains or dorsal neural tubes of midbrains were extracted and pooled on ice and centrifuged at 500 × g for 5 min at 4°C. Pellets were then snap-frozen in liquid nitrogen. Tissue and Chinese hamster ovary pellets were thawed on ice and lysed in lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% IGEPAL CA-630) supplemented with cOmplete Protease Inhibitor Cocktail (Roche, Basel, Switzerland) and 1 mM PMSF for 30 min at 4˚C with periodic mixing. Soluble fractions were collected after removal of cell debris by centrifugation at maximum g for 15 min at 4°C, and protein concentration was quantified by Bradford assay (Thermo Scientific, Rockford, IL). Equivalent amounts of protein per sample were boiled at 100°C for 4 min in 4× reducing Laemmli sample buffer and then centrifuged at maximum speed for 5 min at room temperature. Supernatants were processed by SDS–PAGE (Bio-Rad, Hercules, CA) and then transferred to 0.45-μm BioTrace PVDF membrane (Pall, Port Washington, NY). Membranes were incubated in blocking solution (5% donkey serum or 5% dry milk in 1× PBS + 0.1% Tween-20 [PTW]) for 1 h at room temperature and then incubated overnight at 4°C with the following primary antibodies diluted in blocking solution: Cad6B (CC6DB-1, 1:80; Developmental Studies Hybridoma Bank, Iowa City, IA); β-actin (1:1000, sc-47778; Santa Cruz Biotechnology, Dallas, TX); HA (1:1000, 3F10; Roche); CDH6 (1:1000, ab64917; Abcam, Cambridge); N-cad (BD Biosciences, San Jose, CA ); sheep anti-quail ADAM19 antiserum (a gift from Donald Newgreen [Murdoch Children's Research Institute, Royal Children's Hospital, Parkville, Australia]; Lewis et al., 2004); and MMP-2 (1:1000, MAB13405; EMD Millipore). Membranes were washed in PTW and then incubated with species- and isotype-specific horseradish peroxidase–conjugated secondary antibodies (40 ng/ml; Jackson ImmunoResearch, West Grove, PA) in 5% blocking solution for 1 h at room temperature. Membranes were washed again in PTW, and antibody detection was performed using the Supersignal West Pico or Femto chemiluminescent substrates (Thermo Scientific) and visualized using a ChemiDoc XRS system (Bio-Rad). Immunoblots were probed either simultaneously with two antibodies (HA and MMP2; Figure 2A) or serially between antibody stripping with Restore Western Blot Stripping Buffer according to manufacturer's instructions (Thermo Scientific). Immunoblot images for figures were gamma-modified and processed using Photoshop 9.0 (Adobe Systems, San Jose, CA). Immunoblot band volumes (intensities) were calculated from unmodified immunoblot images using Image Lab software (Bio-Rad), and relative expression levels were generated by normalizing Cad6B and protease band volumes to β-actin band volumes. Fold changes in protein expression are presented as means and SEMs of these ratios. Immunoblots were analyzed by analysis of variance (ANOVA) using the PROC MIXED model in SAS statistical software (SAS Institute, Cary, NC), and levels were deemed significantly different (p < 0.05) using the PDIFF procedure.
Generation of Flp-In Cad6B–stable cell lines
Flp-In Chinese hamster ovary cells (Life Technologies) were grown in F12 media supplemented with 1 mM l-glutamine and 100 μg/ml Zeocin (CellGro, Manassas, VA). The C-terminal HA-tagged Cad6B was cloned into the pcDNA5/FRT expression vector and cotransfected with the pOG44 plasmid, at a ratio of 1:9, into the Flp-In Chinese hamster ovary cell line using Lipofectamine 2000 (Life Technologies). After 24 h posttransfection, media was replaced with antibiotic-free F12 media, and then 24 h later, cells were trypsinized and plated at 20–25% confluency in F12 media supplemented with 1 mM l-glutamine and 500 μg/ml hygromycin to select for positive transfectants (cells were subsequently grown in this medium). Fresh medium was added every 2 d until small colonies of stable cells were observed. Individual colonies were transferred to 96 wells and cultured to confluency. Cells from individual wells were then passaged into 48-, 24-, and 12-plate wells. Colonies were screened for Cad6B expression by immunohistochemistry, and those with maximum transfection efficiency were eventually passaged to 10-cm plates. Immunoblotting was then performed to confirm Cad6B expression.
Cycloheximide half-life assays
F12 medium containing 500 μg/ml hygromycin and 50 μg/ml cycloheximide (dissolved in DMSO) was added to confluent Flp-In Cad6B-HA cells cultured in 6-cm dishes and incubated at 37°C for 2, 4, 6, 8, 10, and 12 h. Cells were then collected at each time point, pelleted, and flash-frozen for immunoblot analysis. Relative protein levels were measured as described and analyzed by linear regression (y = −0.1883x + 1.2149, R2 = 0.99) using Excel software (Microsoft, Redmond, WA).
In ovo electroporation
Expression constructs were introduced into premigratory midbrain neural crest cells in developing 3–4ss chick embryos using a modified version of in ovo electroporation (Itasaki et al., 1999). For unilateral electroporation of the neural tube, constructs (2.5 μg/μl) were injected into the neural tube lumen and two 30-ms pulses at 25 V were applied across the embryo (Jhingory et al., 2010; Wu et al., 2011; Wu and Taneyhill, 2012). For Cad6B:HA electroporations (which produce trace amounts so as to not elicit a neural crest phenotype; see Coles et al., 2007), 2 μg/μl construct was injected into the midbrain neural tube lumen and dorsal/ventrally electroporated using three 30-ms pulses at 10 V. Successfully electroporated embryos were selected based on green fluorescent protein (GFP) fluorescence for further analyses. After electroporation, inhibition of Cad6B proteolysis in vivo was immediately performed by applying pluronic gel (Lutrol F127; BASF, Florham Park, NJ) saturated with 50 μM protease inhibitor GM6001 (Tocris) to the top of the embryo midbrain neural folds at the 3–4ss as in Hall and Erickson (2003). For ADAM10 and ADAM19 knockdown, a 3′ lissamine-labeled antisense MO (ADAM10, 5′-ATCGTCCGCTAGATCCATCTTCTCC-3′; ADAM19, 5′-TCGTCACCGCAACCCCTCGTATATA-3′) was designed to target mRNA and block translation according to the manufacturer's software (GeneTools, Philomath, OR). A 5′ base pair mismatch, lissamine-labeled antisense control MO (ADAM10, 5′-ATCcTCCcCTAGATgCATgTTgTCC-3′; ADAM19, 5′-TCcTgACCGgAACCCgTCcTATATA-3′) was used that does not target the ADAM mRNA (mutated bases are in lowercase; GeneTools). MOs were unilaterally electroporated into the neural tube as described. Briefly, MOs were injected at a final concentration of 500 μM (Taneyhill et al., 2007; Jhingory et al., 2010) into the neural tube lumen at the desired axial level, and two 25-V, 3-ms pulses were applied across the embryo. For double-MO experiments, equimolar concentrations of each MO were used (450 μM) to achieve a final concentration of 900 μM.
Whole-mount in situ hybridization
Whole-mount in situ hybridization was performed as described previously (Wilkinson, 1992; Jhingory et al., 2010; Wu et al., 2011; Wu and Taneyhill, 2012). A riboprobe sequence to ADAM19 was generated to encompass 1082 nucleotides of the chick ADAM19 coding sequence (Accession number NM_001195122.1), beginning in the immediate upstream 5′ untranslated region (sense primer, 5′-AGTCCAGCGTGGAGCCATG-3′) and ending within the coding sequence (antisense primer, 5′-CCTCCGCACGGGTGGTACAG-3′). Stained embryos were viewed in whole mount at room temperature in 70% glycerol using a Zeiss SteREO Discovery V8 compound fluorescence microscope (Carl Zeiss, Oberkochen, Germany). Images were captured using Zeiss AxioVision, release 4.6, software with the Zeiss AxioCam MRc5 camera. Transverse sections were obtained by cryostat sectioning gelatin-embedded embryos at 14 μm in a Leica Frigocut (Buffalo Grove, IL) or Fisher Microm cryostat (Thermo Scientific), and coverslips were mounted on processed sections using Fluoromount G (Southern Biotech, Birmingham, AL). Sections were viewed at room temperature using a Zeiss AxioObserver Z1 inverted microscope, and images were acquired using the Zeiss AxioVision, release 4.6, software with the Zeiss AxioCam HRC camera. All exported images were processed in Photoshop 9.0.
Immunohistochemistry
Immunohistochemical detection of various proteins was performed in whole mount or on transverse sections after 4% paraformaldehyde fixation of embryos and cryostat sectioning (Jhingory et al., 2010; Wu et al., 2011; Wu and Taneyhill, 2012). The following primary antibodies and concentrations were used: Cad6B (CC6DB-1, 1:100; Developmental Studies Hybridoma Bank); GFP (clone 3E6, 1:500; Life Technologies); HNK-1 (clone 3H5, 1:100; Developmental Studies Hybridoma Bank); ADAM10 (ab1997, 1:200; Abcam); PS-1 (ab71181, 1:1000; Abcam); FoxD3 (1:1000; a gift from David Raible [Department of Biological Structure, University of Washington, Seattle, WA]; Lister et al., 2006); and HA (71-500, 1:1000; Life Technologies). Immunostaining for Cad6B was performed in whole mount or on sections as described previously (Coles et al., 2007; Jhingory et al., 2010). Appropriate fluorescently conjugated secondary antibodies (Alexa Fluor 488, 594, 647; Life Technologies) were used at a concentration of 1:500. Imaging of sections and data processing were carried out as described for in situ hybridizations using the Zeiss AxioObserver Z1 inverted microscope and Photoshop 9.0, respectively. Sections were stained with 4′,6-diamidino-2-phenylindole (DAPI) to mark cell nuclei and mounted using Fluoromount G.
Premigratory neural crest cell domain measurements
To capture changes in the Cad6B apicolateral domain after ADAM depletion or Cad6B CTF2 overexpression, an area analysis of the Cad6B-positive premigratory neural crest cell domain (both the treated and contralateral control side) was performed on at least four serial sections from a minimum of three embryos at the same magnification, using Photoshop to calculate arbitrary square pixel units. Arbitrary pixel areas of MO-electroporated and contralateral sides were first analyzed via unpaired Student's t test (p < 0.05) using Excel software. Results obtained from the treated side were then normalized to the contralateral control side and reported as a fold change. Calculated means of fold change per MO treatment were then analyzed by 1-way ANOVA using the PROC MIXED model in SAS statistical software, and levels were deemed significantly different (p < 0.05) using the PDIFF procedure.
Supplementary Material
Acknowledgments
We thank Donald Newgreen for generously supplying the goat anti-quail ADAM19 antiserum and David Raible for his gift of the FoxD3 antibody. This work was supported by National Institutes of Health grants to L.A.T. (R00HD044535) and A.T.S. (F32DE022990).
Abbreviations used:
- ADAM
a disintegrin and metalloproteinase
- Cad6B
cadherin-6B
- CTF
C-terminal fragment
- DAPI
4′,6-diamidino-2-phenylindole
- EMT
epithelial-to-mesenchymal transition
- GFP
green fluorescent protein
- HA
hemagglutinin
- MMP
matrix metalloproteinase
- MO
morpholino
- NTF
N-terminal fragment
- PS
presenilin
- SS
somite stage
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-08-0459) on November 6, 2013.
REFERENCES
- Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlstrom JD, Erickson CA. The neural crest epithelial-mesenchymal transition in 4D: a “tail” of multiple non-obligatory cellular mechanisms. Development. 2009;136:1801–1812. doi: 10.1242/dev.034785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfandari D, Wolfsberg TG, White JM, DeSimone DW. ADAM 13: a novel ADAM expressed in somitic mesoderm and neural crest cells during Xenopus laevis development. Dev Biol. 1997;182:314–330. doi: 10.1006/dbio.1996.8458. [DOI] [PubMed] [Google Scholar]
- Black RA, White JM. ADAMs: focus on the protease domain. Curr Opin Cell Biol. 1998;10:654–659. doi: 10.1016/s0955-0674(98)80042-2. [DOI] [PubMed] [Google Scholar]
- Chesneau V, Becherer JD, Zheng Y, Erdjument-Bromage H, Tempst P, Blobel CP. Catalytic properties of ADAM19. J Biol Chem. 2003;278:22331–22340. doi: 10.1074/jbc.M302781200. [DOI] [PubMed] [Google Scholar]
- Ciruna B, Rossant J. FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev Cell. 2001;1:37–49. doi: 10.1016/s1534-5807(01)00017-x. [DOI] [PubMed] [Google Scholar]
- Coles EG, Taneyhill LA, Bronner-Fraser M. A critical role for Cadherin6B in regulating avian neural crest emigration. Dev Biol. 2007;312:533–544. doi: 10.1016/j.ydbio.2007.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duband JL, Volberg T, Sabanay I, Thiery JP, Geiger B. Spatial and temporal distribution of the adherens-junction-associated adhesion molecule A-CAM during avian embryogenesis. Development. 1988;103:325–344. doi: 10.1242/dev.103.2.325. [DOI] [PubMed] [Google Scholar]
- Duong TD, Erickson CA. MMP-2 plays an essential role in producing epithelial-mesenchymal transformations in the avian embryo. Dev Dyn. 2004;229:42–53. doi: 10.1002/dvdy.10465. [DOI] [PubMed] [Google Scholar]
- Dyczynska E, Sun D, Yi H, Sehara-Fujisawa A, Blobel CP, Zolkiewska A. Proteolytic processing of delta-like 1 by ADAM proteases. J Biol Chem. 2007;282:436–444. doi: 10.1074/jbc.M605451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairchild CL, Gammill LS. Tetraspanin18 is a FoxD3-responsive antagonist of cranial neural crest epithelial to mesenchymal transition that maintains cadherin6B protein. J Cell Sci 126. 2013. pp. 1464–1476. [DOI] [PMC free article] [PubMed]
- Hall RJ, Erickson CA. ADAM 10: an active metalloprotease expressed during avian epithelial morphogenesis. Dev Biol. 2003;256:146–159. doi: 10.1016/s0012-1606(02)00133-1. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. 1951. Dev Dyn. 1992;195:231–272. doi: 10.1002/aja.1001950404. [DOI] [PubMed] [Google Scholar]
- Hatta K, Takeichi M. Expression of N-cadherin adhesion molecules associated with early morphogenetic events in chick development. Nature. 1986;320:447–449. doi: 10.1038/320447a0. [DOI] [PubMed] [Google Scholar]
- Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- Ireton RC, et al. A novel role for p120 catenin in E-cadherin function. J Cell Biol. 2002;159:465–476. doi: 10.1083/jcb.200205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itasaki N, Bel-Vialar S, Krumlauf R. “Shocking” developments in chick embryology: electroporation and in ovo gene expression. Nat Cell Biol. 1999;1:E203–E207. doi: 10.1038/70231. [DOI] [PubMed] [Google Scholar]
- Ito K, Okamoto I, Araki N, Kawano Y, Nakao M, Fujiyama S, Tomita K, Mimori T, Saya H. Calcium influx triggers the sequential proteolysis of extracellular and cytoplasmic domains of E-cadherin, leading to loss of beta-catenin from cell-cell contacts. Oncogene. 1999;18:7080–7090. doi: 10.1038/sj.onc.1203191. [DOI] [PubMed] [Google Scholar]
- Jhingory S, Wu CY, Taneyhill LA. Novel insight into the function and regulation of alphaN-catenin by Snail2 during chick neural crest cell migration. Dev Biol. 2010;344:896–910. doi: 10.1016/j.ydbio.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T, Tschesche H, Amy Sang QX. Evidence for disulfide involvement in the regulation of intramolecular autolytic processing by human adamalysin19/ADAM19. Exp Cell Res. 2004;298:285–295. doi: 10.1016/j.yexcr.2004.04.022. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Selkoe DJ. The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J Biol Chem. 2003;278:34427–34437. doi: 10.1074/jbc.M302659200. [DOI] [PubMed] [Google Scholar]
- Lewis SL, Farlie PG, Newgreen DF. Isolation and embryonic expression of avian ADAM 12 and ADAM 19. Gene Expr Patterns. 2004;5:75–79. doi: 10.1016/j.modgep.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Lim J, Thiery JP. Epithelial-mesenchymal transitions: insights from development. Development. 2012;139:3471–3486. doi: 10.1242/dev.071209. [DOI] [PubMed] [Google Scholar]
- Lin J, Redies C, Luo J. Regionalized expression of ADAM13 during chicken embryonic development. Dev Dyn. 2007;236:862–870. doi: 10.1002/dvdy.21071. [DOI] [PubMed] [Google Scholar]
- Lin J, Yan X, Markus A, Redies C, Rolfs A, Luo J. Expression of seven members of the ADAM family in developing chicken spinal cord. Dev Dyn. 2010;239:1246–1254. doi: 10.1002/dvdy.22272. [DOI] [PubMed] [Google Scholar]
- Linford A, Yoshimura S, Nunes Bastos R, Langemeyer L, Gerondopoulos A, Rigden DJ, Barr FA. Rab14 and its exchange factor FAM116 link endocytic recycling and adherens junction stability in migrating cells. Dev Cell. 2012;22:952–966. doi: 10.1016/j.devcel.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister JA, Cooper C, Nguyen K, Modrell M, Grant K, Raible DW. Zebrafish Foxd3 is required for development of a subset of neural crest derivatives. Dev Biol. 2006;290:92–104. doi: 10.1016/j.ydbio.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Lock JG, Stow JL. Rab11 in recycling endosomes regulates the sorting and basolateral transport of E-cadherin. Mol Biol Cell. 2005;16:1744–1755. doi: 10.1091/mbc.E04-10-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon CA, Johnson JL, Williams H, Sala-Newby GB, George SJ. Soluble N-cadherin overexpression reduces features of atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2009;29:195–201. doi: 10.1161/ATVBAHA.108.178087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magold AI, Cacquevel M, Fraering PC. Gene expression profiling in cells with enhanced gamma-secretase activity. PLoS One. 2009;4:e6952. doi: 10.1371/journal.pone.0006952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, et al. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002;21:1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–645. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Maretzky T, Reiss K, Ludwig A, Buchholz J, Scholz F, Proksch E, de Strooper B, Hartmann D, Saftig P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc Natl Acad Sci USA. 2005;102:9182–9187. doi: 10.1073/pnas.0500918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker C, Cousin H, Neuner R, Alfandari D. Extracellular cleavage of cadherin-11 by ADAM metalloproteases is essential for Xenopus cranial neural crest cell migration. Mol Biol Cell. 2009;20:78–89. doi: 10.1091/mbc.E08-05-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker CD, Alfandari D. Life after proteolysis: exploring the signaling capabilities of classical cadherin cleavage fragments. Commun Integr Biol. 2009;2:155–157. doi: 10.4161/cib.7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JK, Li Q, Parks WC. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am J Pathol. 2003;162:1831–1843. doi: 10.1016/S0002-9440(10)64318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micalizzi DS, Ford HL. Epithelial-mesenchymal transition in development and cancer. Future Oncol. 2009;5:1129–1143. doi: 10.2217/fon.09.94. [DOI] [PubMed] [Google Scholar]
- Monsonego-Ornan E, Kosonovsky J, Bar A, Roth L, Fraggi-Rankis V, Simsa S, Kohl A, Sela-Donenfeld D. Matrix metalloproteinase 9/gelatinase B is required for neural crest cell migration. Dev Biol. 2012;364:162–177. doi: 10.1016/j.ydbio.2012.01.028. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Takeichi M. Neural crest cell-cell adhesion controlled by sequential and subpopulation-specific expression of novel cadherins. Development. 1995;121:1321–1332. doi: 10.1242/dev.121.5.1321. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Takeichi M. Neural crest emigration from the neural tube depends on regulated cadherin expression. Development. 1998;125:2963–2971. doi: 10.1242/dev.125.15.2963. [DOI] [PubMed] [Google Scholar]
- Neuner R, Cousin H, McCusker C, Coyne M, Alfandari D. Xenopus ADAM19 is involved in neural, neural crest and muscle development. Mech Dev. 2009;126:240–255. doi: 10.1016/j.mod.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen CM, Gumbiner BM. Cadherin-mediated cell sorting not determined by binding or adhesion specificity. J Cell Biol. 2002;156:389–399. doi: 10.1083/jcb.200108040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Gumbiner BM. Cadherin 6B induces BMP signaling and de-epithelialization during the epithelial mesenchymal transition of the neural crest. Development. 2010;137:2691–2701. doi: 10.1242/dev.050096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Gumbiner BM. Cadherin-6B stimulates an epithelial mesenchymal transition and the delamination of cells from the neural ectoderm via LIMK/cofilin mediated non-canonical BMP receptor signaling. Dev Biol. 2012;366:232–243. doi: 10.1016/j.ydbio.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson AD, Parton RG, Ferguson C, Stow JL, Yap AS. Characterization of E-cadherin endocytosis in isolated MCF-7 and Chinese hamster ovary cells: the initial fate of unbound E-cadherin. J Biol Chem. 2003;278:21050–21057. doi: 10.1074/jbc.M300082200. [DOI] [PubMed] [Google Scholar]
- Pla P, Moore R, Morali OG, Grille S, Martinozzi S, Delmas V, Larue L. Cadherins in neural crest cell development and transformation. J Cell Physiol. 2001;189:121–132. doi: 10.1002/jcp.10008. [DOI] [PubMed] [Google Scholar]
- Pon YL, Auersperg N, Wong AS. Gonadotropins regulate N-cadherin-mediated human ovarian surface epithelial cell survival at both post-translational and transcriptional levels through a cyclic AMP/protein kinase A pathway. J Biol Chem. 2005;280:15438–15448. doi: 10.1074/jbc.M410766200. [DOI] [PubMed] [Google Scholar]
- Pujuguet P, Del Maestro L, Gautreau A, Louvard D, Arpin M. Ezrin regulates E-cadherin-dependent adherens junction assembly through Rac1 activation. Mol Biol Cell. 2003;14:2181–2191. doi: 10.1091/mbc.E02-07-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss K, Maretzky T, Ludwig A, Tousseyn T, de Strooper B, Hartmann D, Saftig P. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. EMBO J. 2005;24:742–752. doi: 10.1038/sj.emboj.7600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman MS, Beher D, Clarke EE, Lewis HD, Harrison T, Hunt P, Nadin A, Smith AL, Stevenson G, Castro JL. L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid beta-protein precursor gamma-secretase activity. Biochemistry. 2000;39:8698–8704. doi: 10.1021/bi0005456. [DOI] [PubMed] [Google Scholar]
- Shoval I, Ludwig A, Kalcheim C. Antagonistic roles of full-length N-cadherin and its soluble BMP cleavage product in neural crest delamination. Development. 2007;134:491–501. doi: 10.1242/dev.02742. [DOI] [PubMed] [Google Scholar]
- Smith KM, Gaultier A, Cousin H, Alfandari D, White JM, DeSimone DW. The cysteine-rich domain regulates ADAM protease function in vivo. J Cell Biol. 2002;159:893–902. doi: 10.1083/jcb.200206023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl-Mazzulla PH, Bronner ME. A PHD12-Snail2 repressive complex epigenetically mediates neural crest epithelial-to-mesenchymal transition. J Cell Biol. 2012;198:999–1010. doi: 10.1083/jcb.201203098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneyhill LA. To adhere or not to adhere: the role of cadherins in neural crest development. Cell Adh Migr. 2008;2:223–230. doi: 10.4161/cam.2.4.6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneyhill LA, Coles EG, Bronner-Fraser M. Snail2 directly represses cadherin6B during epithelial-to-mesenchymal transitions of the neural crest. Development. 2007;134:1481–1490. doi: 10.1242/dev.02834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E, Duband JL, Altabef M. Ets-1 confers cranial features on neural crest delamination. PLoS One. 2007;2:e1142. doi: 10.1371/journal.pone.0001142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Tolia A, De Strooper B. Structure and function of gamma-secretase. Semin Cell Dev Biol. 2009;20:211–218. doi: 10.1016/j.semcdb.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Uemura K, Kihara T, Kuzuya A, Okawa K, Nishimoto T, Ninomiya H, Sugimoto H, Kinoshita A, Shimohama S. Characterization of sequential N-cadherin cleavage by ADAM10 and PS1. Neurosci Lett. 2006;402:278–283. doi: 10.1016/j.neulet.2006.04.018. [DOI] [PubMed] [Google Scholar]
- Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development. 2012;139:3693–3709. doi: 10.1242/dev.076398. [DOI] [PubMed] [Google Scholar]
- Wei S, Xu G, Bridges LC, Williams P, White JM, DeSimone DW. ADAM13 induces cranial neural crest by cleaving class B Ephrins and regulating Wnt signaling. Dev Cell. 2010;19:345–352. doi: 10.1016/j.devcel.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DG. Whole mount in situ hybridization of vertebrate embroys. In: Wilkinson DG, editor. In: In Situ Hybridization. Oxford, UK: Oxford University Press; 1992. pp. 75–83. [Google Scholar]
- Wu CY, Jhingory S, Taneyhill LA. The tight junction scaffolding protein cingulin regulates neural crest cell migration. Dev Dyn. 2011;240:2309–2323. doi: 10.1002/dvdy.22735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CY, Taneyhill LA. Annexin A6 modulates chick cranial neural crest cell emigration. PLoS One. 2012;7:e44903. doi: 10.1371/journal.pone.0044903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Lin J, Markus A, Rolfs A, Luo J. Regional expression of ADAM19 during chicken embryonic development. Dev Growth Differ. 2011;53:333–346. doi: 10.1111/j.1440-169X.2010.01238.x. [DOI] [PubMed] [Google Scholar]
- Yoshikawa A, Aizaki Y, Kusano K, Kishi F, Susumu T, Iida S, Ishiura S, Nishimura S, Shichiri M, Senbonmatsu T. The (pro)renin receptor is cleaved by ADAM19 in the Golgi leading to its secretion into extracellular space. Hypertens Res. 2011;34:599–605. doi: 10.1038/hr.2010.284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.