

Interindividual variation was screened for inflammatory cell death—pyroptosis. Natural variation in expression of the tubulin isoform TUBB6 or experimental manipulation of expression altered microtubule stability and susceptibility of cells to pyroptosis. Diversity in microtubule stability regulates pyroptosis and likely other human traits.
Abstract
Pyroptosis is proinflammatory cell death that occurs in response to certain microbes. Activation of the protease caspase-1 by molecular platforms called inflammasomes is required for pyroptosis. We performed a cellular genome-wide association study (GWAS) using Salmonella typhimurium infection of human lymphoblastoid cell lines as a means of dissecting the genetic architecture of susceptibility to pyroptosis and identifying unknown regulatory mechanisms. Cellular GWAS revealed that a common human genetic difference that regulates pyroptosis also alters microtubule stability. An intergenic single-nucleotide polymorphism on chromosome 18 is associated with decreased pyroptosis and increased expression of TUBB6 (tubulin, β 6 class V). TUBB6 is unique among tubulin isoforms in that its overexpression can completely disrupt the microtubule network. Cells from individuals with higher levels of TUBB6 expression have lower microtubule stability and less pyroptosis. Reducing TUBB6 expression or stabilizing microtubules pharmacologically with paclitaxel (Taxol) increases pyroptosis without affecting the other major readout of caspase-1 activation, interleukin-1β secretion. The results reveal a new role for microtubules and possibly specific tubulin isoforms in the execution of pyroptosis. Furthermore, the finding that there is common diversity in TUBB6 expression and microtubule stability could have broad consequences for other microtubule-dependent phenotypes, diseases, and pharmacological responses.
INTRODUCTION
Microbial pathogens manipulate the host cell cytoskeleton during infection to promote cellular invasion, intracellular movement, and replication (Haglund and Welch, 2011). Bacterial effects on the actin cytoskeleton are well defined and include Salmonella-induced actin-based macropinocytosis to promote bacterial epithelial invasion (Francis et al., 1993) and actin-based cytoplasmic motility by Shigella spp. and Listeria monocytogenes (Tilney and Portnoy, 1989). For Salmonella, a precisely orchestrated remodeling of the actin cytoskeleton promotes invasion. These dramatic changes are directed by multiple effectors injected through a type III secretion system (SPI-1; Haraga et al., 2008). Later in Salmonella infection, a different type III secretion system (SPI-2) injects other effectors across the phagosome membrane, including SifA and PipB2, which regulate microtubule-dependent positioning and vesicular transport from Salmonella’s intracellular niche (Leone and Meresse, 2011).
Host cells direct their own dramatic changes in response to pathogens. One rapid response to intracellular bacteria is the recognition of pathogen-associated molecular patterns and the formation of molecular scaffolds called inflammasomes (Martinon et al., 2002; Lamkanfi and Dixit, 2011). Inflammasomes activate the caspase-1 protease, which can trigger a unique cell death process called pyroptosis and also lead to cleavage and secretion of the proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 (Fink and Cookson, 2006). Pyroptosis is a rapid, proinflammatory process that likely serves as a means for destroying the niche for intracellular bacteria (Miao et al., 2010). Although microtubules have been demonstrated to undergo substantial remodeling during the noninflammatory cell death process of apoptosis (Moss and Lane, 2006), their role in pyroptosis has not been examined. Intriguingly, the microtubule-depolymerizing drug colchicine is used to treat both common and rare inflammatory diseases that involve activation of caspase-1, such as gout and familial Mediterranean fever (Terkeltaub, 2009). However, a mechanism linking microtubules directly to pyroptosis is unknown.
Previously we used a functional cellular genome-wide association screen (high-throughput human in vitro susceptibility testing [Hi-HOST]) to identify common genetic differences that are associated with variation in susceptibility of cells to Salmonella typhimurium–induced pyroptosis. We discovered that a nonsense mutation in a known caspase-1 inhibitor (CARD8; Ko et al., 2009) and a single-nucleotide polymorphism (SNP) associated with expression of a gene encoding a methionine salvage enzyme (apoptotic protease activating factor 1–interacting protein [APIP]; Ko et al., 2012) were associated with the pyroptosis phenotype. As would be expected for a complex polygenic trait, each of these variants contributes a small fraction to the overall genetically inherited variance (heritability) of the trait. For pyroptosis in human lymphoblastoid cell lines (LCLs), we previously estimated a heritability of 15% (Ko et al., 2009), and the SNP affecting expression of the methionine salvage enzyme APIP accounts for ∼2% of the total variation (Ko et al., 2012). Although such differences may seem small, the same SNP near APIP cut the odds of mortality in systemic inflammatory response syndrome in half (Ko et al., 2012). Thus variants that have small but reproducible effects in cellular phenotypes can have profound consequences for human health.
We now report that a previously uncharacterized human SNP, found to have an even stronger association with the pyroptosis phenotype, affects the expression of a novel isoform of β-tubulin (TUBB6; GenBank accession NM_032525). TUBB6 is unique among tubulin isoforms in that even modest overexpression destabilizes microtubules and results in resistance to the microtubule-stabilizing drug paclitaxel (Bhattacharya and Cabral, 2004, 2009). We demonstrate that natural variation for higher expression of TUBB6 is associated with reduced microtubule stability and lower susceptibility to pyroptosis. Consistent with a role for microtubules affecting pyroptosis downstream of caspase-1, TUBB6 knockdown or paclitaxel treatment of cells to pharmacologically stabilize microtubules increased pyroptosis without affecting IL-1β. Our results demonstrate that common genetic variation altering microtubule stability can regulate the inflammatory cell death response independent of cytokine secretion.
RESULTS
SNP associated with TUBB6 expression is associated with pyroptosis
We previously carried out a genome-wide association screen of naturally occurring human genetic variation in Salmonella-induced cell death, or pyroptosis (Ko et al., 2009, 2012). Briefly, >400 LCLs from different people were measured for susceptibility to cell death in response to S. typhimurium. Because all of these LCLs had been genotyped by the HapMap project (International HapMap et al., 2010), we were able to identify human genetic differences that correlated with the percentage of cells that underwent pyroptosis. In an earlier study, we demonstrated that SNPs associated with pyroptosis with p < 0.001 and also associated with the expression of nearby genes (i.e., cis–expression quantitative trait loci [cis-EQTL]) were more likely to be true positives (Ko et al., 2012). We characterized SNP rs514182 as meeting these criteria and functionally validated the effect of this SNP and the affected gene (APIP) through multiple approaches (Ko et al., 2012). Two of the four uncharacterized cis-EQTL with p < 0.001 for association with pyroptosis are <400 base pairs apart in an intergenic region between the cell death-inducing DFFA-like effector a (CIDEA) and TUBB6 genes on chromosome 18 (rs9807444 and rs9303768; Figure 1A). The chromosomal location of these genes is well conserved in placental mammals, including mice. The two SNPs are in strong linkage disequilibrium (LD; R2 = 1.0 in Utah residents with ancestry from northern and western Europe [CEU]; R2 = 0.84 in Yoruba from Ibadan, Nigeria [YRI]) and are likely signals due to the same causal SNP. The rest of this article focuses on rs9807444.
FIGURE 1:

An intergenic SNP between the TUBB6 and CIDEA genes (rs9807444) is associated with Salmonella-induced cell death and TUBB6 expression. (A) rs9807444 is associated with pyroptosis. The p values for family-based association are given for the original screen (Ko et al., 2009), the replication screen, and the combined data sets (Ko et al., 2012). Coordinates are given for human genome release hg18. The genomic region around rs9807444 and rs9303768 (both SNPs indicated by green circle) shows extensive linkage disequilibrium (LD) encompassing the SNP and extending into the TUBB6 gene. LD is shown for CEU population. (B) The derived (C) allele of rs9807444 is associated with higher levels of Salmonella-induced cell death assessed by 7-AAD. Mean-centered percentage dead cells are plotted for HapMap 2 and 3 CEU LCLs, with mean and SD in red. The table lists mean-centered genotypic values for CEU and YRI populations analyzed separately, with p values from family-based association analysis (QFAM-parents). (C) The derived allele of rs9807444 is associated with higher TUBB6 RNA levels. TUBB6 expression from the CEU LCLs is plotted from the Stranger et al. (2007) data set. An association in the same direction is verified in YRI LCLs (although not statistically significant) and in a German monocyte data set (Zeller et al., 2010). For CEU and YRI, genotypic means and p values (analysis of variance [ANOVA]) are from parents only. (D) rs9807444 is only associated with expression of TUBB6 and not other genes in the region. The −log p values are plotted from Stranger et al. (2012) using Genevar. (E) The derived allele of rs9807444 is associated with higher TUBB6 protein levels. TUBB6 protein levels are plotted from measurements of LCLs by Wu et al. (2013) by isobaric tag-based mass spectrometry. The p value is from ANOVA.
The derived, minor allele of rs980744 (C) was associated with less pyroptosis. This was observed in the combined family-based association of CEU and YRI samples (table within Figure 1A), as well as when each population was examined individually (Figure 1B). How might this genetic difference be altering pyroptosis? The previous characterization of CIDEA as a regulator of cell death (Inohara et al., 1998) made it appear to be the likeliest candidate gene in the region. However, the SNP was associated with the expression level of TUBB6 (Figure 1C) based on mining expression data from the same LCLs (Stranger et al., 2007), as well as a separate German primary monocyte data set (Zeller et al., 2010). In addition, examination of a recent analysis of HapMap phase 3 LCLs (Stranger et al., 2012) uncovered an association between rs9807444 and TUBB6 expression to varying degrees in eight different populations but not with any of the other nearby genes, including CIDEA (Figure 1D). Finally, TUBB6 protein levels from a recent proteomics study of some of the same LCLs (Wu et al., 2013) demonstrated that rs9807444 is associated with the level of TUBB6 protein (Figure 1E). Thus the expression data suggested that rs9807444 (or another genetic change in LD) could be affecting pyroptosis by altering the expression of TUBB6.
TUBB6 is an inhibitor of pyroptosis
The fact that the minor allele of rs9807444 is associated with higher levels of TUBB6 expression and lower levels of pyroptosis suggested that TUBB6 is an inhibitor of pyroptosis. Experiments decreasing expression of TUBB6 by RNA interference (RNAi) or its overexpression by transfection supported this hypothesis. Reducing TUBB6 expression (Figure 2A) had no effect on viability of uninfected cells but increased S. typhimurium-induced pyroptosis in both LCLs (Figure 2B) and THP-1 monocytes (Figure 2C). In contrast, no effect was seen with small interfering RNAs (siRNAs) directed against the neighboring CIDEA gene. THP-1 monocytes allow for the measurement of IL-1β release as a second measure of caspase-1 activation. TUBB6 knockdown increased pyroptosis while not altering IL-1β secretion (Figure 2D), indicating that TUBB6 only affects the cell death aspect of the pathway and likely acts downstream of caspase-1 activation.
FIGURE 2:
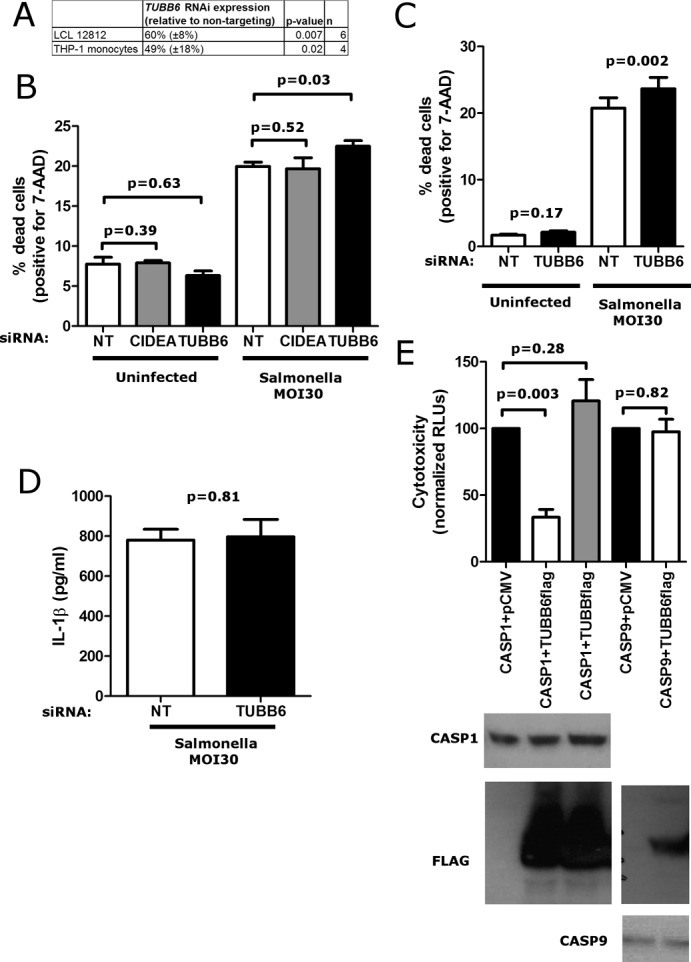
TUBB6 is an inhibitor of pyroptosis. (A) Knockdown of TUBB6 expression in LCL 12812 and THP-1 quantified by quantitative PCR. Expression of TUBB6 RNA is relative to cells treated with nontargeting siRNA. (B) RNAi against TUBB6 but not CIDEA in LCL 12812 increases cell death in response to S. typhimurium (MOI30) assessed at 3 h by 7-AAD staining. Increased cell death with TUBB6 RNAi was also noted in LCL 19239 (p = 0.001). (C) RNAi against TUBB6 in THP-1 monocytes increases cell death in response to S. typhimurium (MOI30) assessed at 3 h by 7-AAD staining. (D) RNAi against TUBB6 in THP-1 monocytes has no effect on secreted IL-1β in response to S. typhimurium (MOI30). (E) Overexpression of TUBB6 in HEK-293T cells inhibits cell death induced by caspase-1 but not caspase-9. Cells were transfected with the indicated human caspase constructs and TUBB6-Flag, TUBB-Flag, or empty vector. At 23 h posttransfection, supernatants were assayed for cytotoxicity. Western blots demonstrated equal expression of caspases with or without TUBB6-flag coexpression. For A–E, mean, SEM, and p values (paired t tests) are from duplicate readings from four to six independent experiments.
TUBB6 also inhibited caspase-1–mediated cell death in an overexpression system. HEK-293 cells have been used as a cell line for inflammasome reconstitution as they are easily transfected and do not express caspase-1 or other inflammasome components (Agostini et al., 2004). We previously demonstrated that another inhibitor of pyroptosis identified by our Hi-HOST screen, APIP, inhibited caspase-1–mediated cell death, whereas overexpression of inflammasome components ASC and NLCR4 increased cell death (Ko et al., 2012). Coexpression of TUBB6 with caspase-1 strongly inhibited caspase-1–mediated cell death without affecting the expression of caspase-1 (Figure 2E). The effect of TUBB6 is highly specific—overexpression of a different β-tubulin gene, TUBB (tubulin, β class I), was unable to inhibit caspase-1–mediated cell death. Similar specificity was seen regarding the caspase. Caspase-9, an initiator caspase central to apoptosis, also induced cell death in HEK-293s when overexpressed, but TUBB6 coexpression had no effect (Figure 2E). Thus the overexpression studies agree with the RNAi experiments and the association data, providing multiple independent lines of evidence that TUBB6 inhibits pyroptosis.
Variation in TUBB6 expression alters stability of microtubules
Our data demonstrate that TUBB6 regulates pyroptosis and that there is common variation affecting the expression of this gene. Previous studies examining the effects of the different isoforms of β-tubulin found that although TUBB6 is a minor β-tubulin component (making up ∼5% of total β-tubulin), it is essential for cell cycle progression (Bhattacharya et al., 2008). Furthermore, TUBB6 is functionally distinct as the only β-tubulin isoform that severely destabilizes microtubules when overexpressed (Bhattacharya and Cabral, 2004). More recent studies indicate that this microtubule destabilization is due to increased rates of microtubule shortening with prolonged phases of uninterrupted depolymerization and increased detachment of microtubules from the centrosome (Bhattacharya et al., 2011). We verified the microtubule-destabilizing effect of TUBB6 overexpression and examined how naturally occurring variation in expression of TUBB6 among LCLs affected this phenotype.
In agreement with previous studies (Bhattacharya et al., 2008), overexpression of TUBB6 destabilized microtubules based on the level of acetylated tubulin (Figure 3A). This posttranslational modification on lysine 40 of α-tubulin occurs preferentially on stable polymerized microtubules and has been used as a marker of microtubule stability (Maruta et al., 1986; Bhattacharya et al., 2011). To determine whether similar changes in microtubule stability are also observed with interindividual variation in TUBB6 expression, we measured both the expression of TUBB6 and the level of acetylated tubulin in LCLs (Figure 3B). Cells with higher TUBB6 expression contained lower amounts of acetylated tubulin (Figure 3C). As a more direct measure of microtubule stability, we used a flow cytometric assay for measuring relative levels of polymerized microtubule mass (Morrison and Hergenrother, 2012). As predicted, polymerized microtubule mass was reduced in LCLs that had less acetylated tubulin and higher TUBB6 expression (Figure 3D). To confirm these results in a second cell type, we measured TUBB6 mRNA and acetylated tubulin levels in primary fibroblasts from different individuals and observed an even more significant inverse correlation between TUBB6 expression and microtubule acetylation than in the LCLs (Figure 3E). Our data provide strong evidence that natural variation in TUBB6 expression, which can partially be attributed to the rs9807444 SNP, results in differences in microtubule stability among people.
FIGURE 3:

Variation in TUBB6 expression alters microtubule stability. (A) Overexpression of TUBB6 in HEK-293 cells reduces microtubule stability based on amount of acetylated α-tubulin. Cells were transfected to overexpress the indicated protein or treated with 50 nM paclitaxel. After 40 h, cell extracts were harvested and immunoblotted for acetylated α-tubulin and total α-tubulin. TUBB6 overexpression decreased acetylated tubulin. Blots are representative of three different experiments. Values below the blots show the relative amount of acetylated tubulin, normalizing for the amount of total α-tubulin and setting the GFP transfected cells to a value of 1. Mean, SEM, and p value are calculated from quantification of blots from three different experiments. (B) TUBB6 expression and acetylated tubulin in LCLs. TUBB6 mRNA was measured by Taqman real-time PCR assay using 18S expression as the control. Data are the mean and SEM from three experiments. Bar graphs are color coded by genotype: TT, white; CT, gray; and CC, black. From the same LCLs, the amounts of acetylated α-tubulin and total α-tubulin are shown below the bar graph. Blots are representative of three experiments. (C) TUBB6 expression and acetylated tubulin are inversely related in LCLs. Mean relative TUBB6 expression was plotted vs. the mean amount of acetylated α-tubulin normalized for total α-tubulin. The points are the mean TUBB6 expression and mean acetylated tubulin values from three separate experiments for eight LCLs. The plot displays an inverse relationship, with p = 0.005 for significantly nonzero slope. Dashed lines show 95% confidence intervals. Higher levels of TUBB6 are correlated with lower levels of acetylated tubulin. (D) The amount of polymerized tubulin is reduced in LCLs that have less acetylated tubulin and higher TUBB6 expression. The two LCLs with the lowest and highest levels of acetylated tubulin in C were stained for anti–tubulin-FITC using a method that stains preferentially for polymerized tubulin by permeabilizing at the same time as fixation (Morrison and Hergenrother, 2012). Geometric mean intensity for the four LCLs is normalized to 100 for LCL 7056. LCL 7357 gave similar staining intensity, whereas the two LCLs with the lowest level of acetylated tubulin (10838 and 12812) had lower staining intensity. Mean, SEM, and p values (paired t tests) are from three independent experiments. As a control for severely reduced polymerized tubulin, the intensity of colchicine treated LCLs is also shown (gray) and found to be ∼40% of the untreated level for 7357. (E) TUBB6 expression and acetylated tubulin are inversely related in primary fibroblasts. Mean relative TUBB6 expression was plotted vs. the mean amount of acetylated α-tubulin normalized for total α-tubulin. The points are the mean TUBB6 expression and mean acetylated tubulin values from three separate experiments for fibroblasts from eight individuals. The plot displays an inverse relationship, with p = 0.001 for significantly nonzero slope. Dashed lines show 95% confidence intervals. Higher levels of TUBB6 are correlated with lower levels of acetylated tubulin.
Microtubules are rapidly broken down during pyroptosis
Our finding that natural variation in the expression of a microtubule-destabilizing β-tubulin isoform regulates pyroptosis raised the question of how microtubules are altered during induction of the inflammatory cell death process. Given that the formation of a stable subplasma membrane microtubule “cocoon” has been noted during late stages of apoptosis (Sanchez-Alcazar et al., 2007), we asked whether a similar reorganization occurred during pyroptosis and could perhaps explain the effects of TUBB6 on pyroptosis. Instead, we observed a rapid dissolution of the microtubule network within minutes of Salmonella infection. This was observed by both flow cytometry of stained THP-1 monocytes (Figure 4A) and by fluorescence microscopy of mouse bone marrow–derived macrophages (BMMs), which allowed for examination of morphology over time and assessment of the role of caspase-1 in this process (Figure 4, B and C). By 15 min, cells with active caspase-1 (based on the appearance of “inflammasome foci” staining; Fink et al., 2008) were a mixed population of cells with intact, disrupted/diffuse, or absent microtubule staining. By 60 min, nearly all cells with inflammasome foci had absent microtubule staining. The observed loss of microtubules required caspase-1, as it was not observed in casp1−/− BMMs (Figure 4D). Casp1−/− mice lack both casp1 and neighboring casp11, but it has been demonstrated that Salmonella-induced pyroptosis requires only caspase-1 (Broz et al., 2010). Furthermore, this dissolution of the microtubule network was also observed using a different inducer of caspase-1, Francisella novicida (Figure 4E).
FIGURE 4:
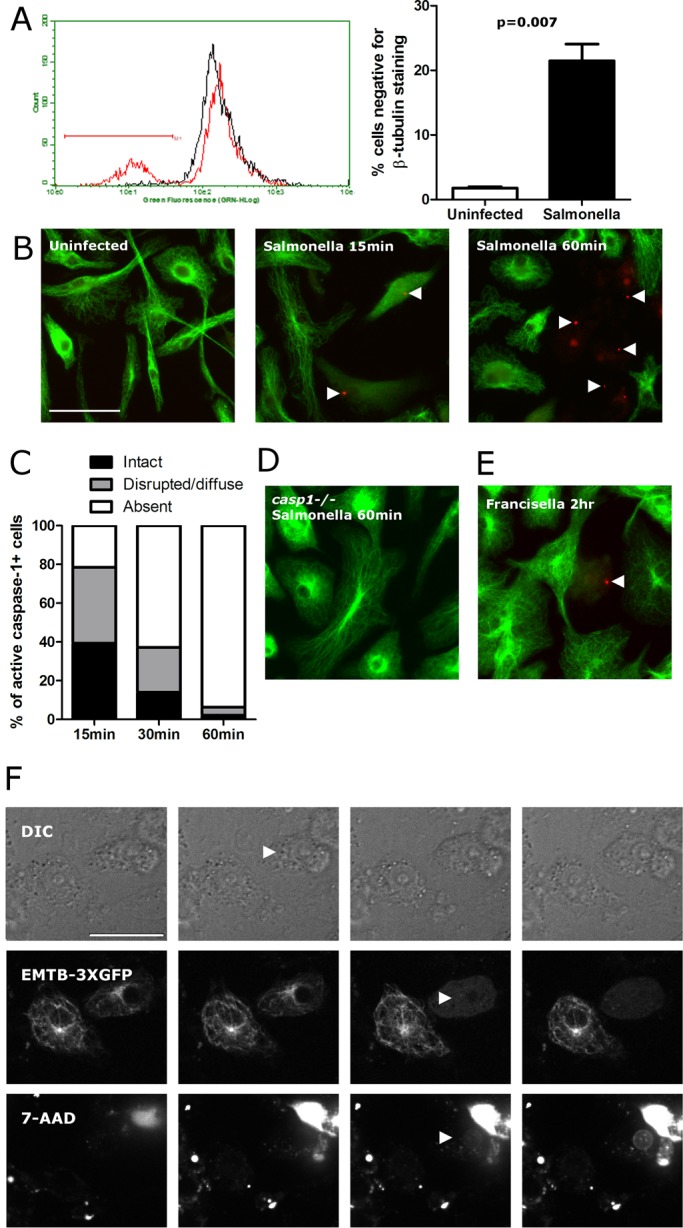
Microtubules are disrupted during the execution of pyroptosis. (A) Staining of THP-1 monocytes after S. typhimurium infection demonstrates a loss of microtubule staining. THP-1 cells were stained for β-tubulin 2 h after S. typhimurium infection (MOI 30), and fluorescence was measured by flow cytometry and plotted on the histogram. Uninfected cells are in black, and S. typhimurium–infected cells are in red. Bar graphs display mean percentage cells with absent microtubule staining and SEM from four independently infected and stained samples. (B) Microtubule staining in Salmonella-infected BMMs shows rapid breakdown of microtubules. Cells were incubated with FLICA reagent for active caspase-1 (red), infected with S. typhimurium (MOI10), fixed at the indicated time, and stained for microtubules (green). Cells with active caspase-1 at 15 min postinfection have disrupted microtubule staining that proceeds to a complete loss of staining by 60 min. (C) Quantification of BMM staining as imaged in C. At least 50 cells with active caspase-1 (FLICA+) were examined for each condition. (D) Microtubule staining of Salmonella-infected casp1−/− BMMs at 60 min show no signs of microtubule disruption. (E). Microtubule staining in Francisella-infected BMMs demonstrates microtubule breakdown by 2 h. All microtubule staining shown was conducted with an α-tubulin antibody, but equivalent staining was observed using a β-tubulin antibody. (F) Live-cell imaging of RAW264.7 macrophages during S. typhimurium infection demonstrates temporal order of events during pyroptosis. Images shown are 2 min apart. Arrowheads show that cellular swelling (DIC, second panel and increasing in subsequent panels) initiates before microtubule disruption (EMTB-3xGFP, third panel) and membrane lysis (nuclear 7-AAD, third panel). Bar, 30 μm. For the accompanying movie, see Supplemental Movie S1.
The data indicate that microtubules undergo rapid caspase-1–dependent breakdown during pyroptosis. However, we found it puzzling that cells with less TUBB6 and more stable microtubules displayed increased pyroptosis. Live-cell imaging of morphology (by differential interference contrast [DIC]), microtubules (EMTB-3xGFP), and membrane integrity (7-aminoactinomycin D [7-AAD]) during Salmonella infection in macrophages was used to determine the temporal relationship of microtubule breakdown to other events during pyroptosis (Figure 4F and Supplemental Movie S1). We observed that microtubule breakdown occurred at the same time as initial 7-AAD staining. Thus loss of microtubules could be secondary to loss of nucleotides or other cellular components with membrane rupture, although caspase-1 could also play an active role, as α-tubulin is reported to be a caspase-1 substrate (Shao et al., 2007). By DIC, the cells began to appear rounder and gradually swell in size by 6.0 min (±5.1 min) before microtubule loss and 7-AAD staining. This swelling continued after membrane rupture for 11.2 min (±2.3 min). These microscopic observations are consistent with pyroptosis being dependent on the formation of caspase-1–dependent pores resulting in cellular swelling followed by osmotic lysis (Fink and Cookson, 2006). Consideration of the microscopic observations in light of the functional TUBB6 RNAi and overexpression assays suggested that intact microtubules promote pyroptosis downstream of caspase-1 activation but before membrane lysis, when they are rapidly dismantled. To test the model that more-stable microtubules promote pyroptosis, we examined the effects of well-characterized drugs that alter microtubule stability.
Stabilization of microtubules with paclitaxel increases pyroptosis likely subsequent to pore formation
On the basis of our observations with TUBB6, we predicted that stabilizing microtubules pharmacologically would also increase pyroptosis. Addition of paclitaxel to stabilize microtubules increased Salmonella-induced pyroptosis in both THP-1s and LCLs (Figure 5A and Supplemental Figure S1A). This was observed whether cell death was measured with a membrane-impermeable DNA dye (7-AAD) or through release of a cytosolic protein (Figure 5B). Also consistent with what was observed with TUBB6 depletion, IL-1β secretion was unaltered by paclitaxel treatment (Figure 5C). The effect of paclitaxel was also observed with Francisella-induced pyroptosis, although the overall level of cell death observed was less than with Salmonella (Figure 5D). Thus, enhanced microtubule stability, either by paclitaxel or reduced expression of TUBB6, increased pyroptosis.
FIGURE 5:
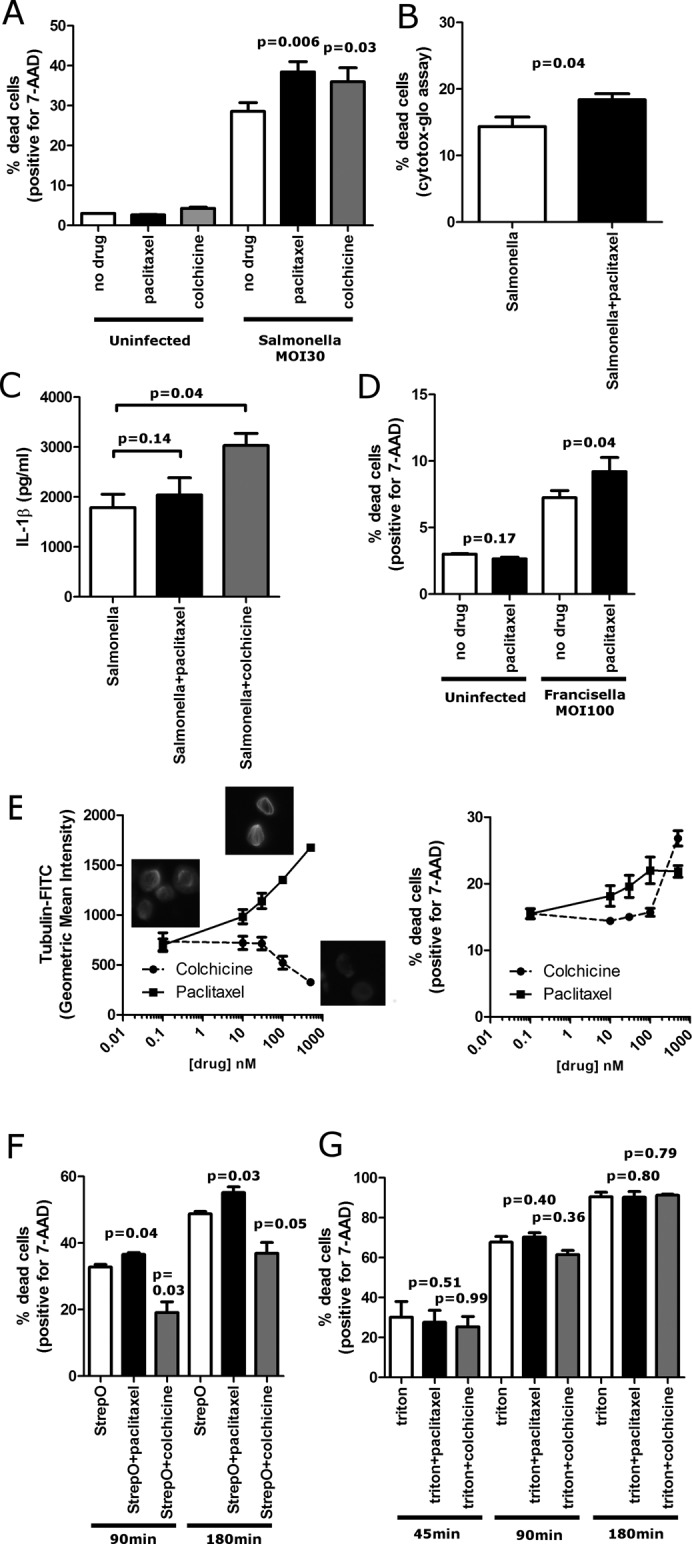
Paclitaxel stabilization of microtubules increases Salmonella-induced pyroptosis and cell death due to a pore-forming toxin. (A) Paclitaxel and colchicine both increase Salmonella-induced pyroptosis. THP-1s were treated for 30 min with 500 nM paclitaxel or colchicine before infection with S. typhimurium MOI30. Cells were assayed with 7-AAD staining and flow cytometry at 3 h after initial infection. (B) Paclitaxel increases pyroptosis as measured by release of cytosolic protein. THP-1s were treated for 30 min with 500 nM paclitaxel before infection with S. typhimurium MOI30. Cells were assayed with Cytotox-Glo at 3 h after initial infection. (C) Paclitaxel does not alter active IL-1β release from THP-1s presimulated overnight with LPS (20 ng/ml) and then infected with S. typhimurium (MOI30, 3 h), whereas colchicine results in a substantial increase. (D) Paclitaxel increases F. novicida–induced pyroptosis. THP-1s were incubated overnight with 20 ng/ml LPS and treated with paclitaxel (500 nM) for 30 min before infection. (E) Paclitaxel and colchicine dose–response curves for polymerized microtubule mass and pyroptosis indicate that microtubule stabilization progressively increases pyroptosis. Paclitaxel or colchicine was added to cells 30 min before infection at the indicated doses. Colchicine had no effect on pyroptosis except at the highest dose of colchicine (500 nM), which resulted in near-complete loss of microtubules. Paclitaxel gradually increased polymerized microtubule mass and also caused a gradual increase in pyroptosis until 100 nM, at which further stabilization of microtubules had no further effect on pyroptosis. Inset micrographs are of microtubule staining at 0 and 500 nM paclitaxel and 500 nM colchicine. (F) Microtubules regulate cell death induced by the pore-forming toxin streptolysin O. THP-1s were treated for 30 min with 500 nM paclitaxel, 500 nM colchicine, or buffer before addition of streptolysin O (200 U). Cells were assayed with 7-AAD staining and flow cytometry at 90 min and 3 h after addition of streptolysin O. Paclitaxel increased cell death, whereas colchicine decreased cell death. (G) Microtubules have no effect on cell death induced by the detergent Triton X-100. THP-1s were treated for 30 min with 500 nM paclitaxel, 500 nM colchicine, or buffer before addition of Triton X-100 (250 μM). Cells were assayed with 7-AAD staining and flow cytometry at 45 min, 90 min, and 3 h after addition of Triton X-100. No effect on cell death was seen with either paclitaxel or colchicine. For A–F data are mean ± SEM for three or four separate experiments, and p value is for paired t test.
We suspected that destabilizing microtubules with colchicine might have the opposite effect on pyroptosis, but colchicine (or nocodazole; unpublished data) also resulted in an increase in pyroptosis (Figure 5A). This effect is likely secondary to an increased interaction of Salmonella with the host cells that obscures the effects of microtubule destabilization on other subsequent events. In contrast to what was observed with paclitaxel, colchicine increased Salmonella-dependent IL-1β secretion in the THP-1 cells (Figure 5C), suggesting that microtubule depolymerization affects an early event before caspase-1 activation. More important, colchicine increased Salmonella invasion of HeLa cells (which do not undergo pyroptosis), whereas paclitaxel had no effect (Supplemental Figure S1B). These results suggest that disruption of microtubules by colchicine increases Salmonella interaction with host cells, resulting in increased caspase-1 activation and causing both increased pyroptosis and IL-1B secretion. In contrast, the microtubule stabilization effects of the TUBB6 SNP or paclitaxel are downstream of caspase-1 activation, resulting in only increased pyroptosis without altering IL-1β secretion.
Because colchicine and paclitaxel can both inhibit microtubule dynamics without disrupting stability at lower doses (Jordan et al., 1993; Yvon et al., 1999), we also considered the possibility that the increase in pyroptosis observed with both drugs could be due to inhibition of microtubule dynamics as opposed to alteration of levels of polymerized microtubules. However, we observed that effects on pyroptosis only occurred with doses of the drugs that changed microtubule stability, based on quantitative flow cytometric measurements of polymerized microtubule mass (Figure 5E). Colchicine completely disrupted microtubules at 500 nM (Figure 5E, inset) but not at the lower doses tested, and we saw increased pyroptosis only at this highest dose—lower doses that inhibited dynamics but did not completely disrupt microtubules had no effect on pyroptosis. In contrast, increasing paclitaxel resulted in a gradual increase in polymerized microtubule mass that mirrored the gradual increase in pyroptosis. However, there is an upper limit to this effect on pyroptosis. Doses >100 nM continued to increase polymerized microtubule mass without further increasing pyroptosis. Thus, although the colchicine data indicate that the presence of intact microtubules is not an absolute requirement for pyroptosis, microtubule stabilization with paclitaxel clearly increases pyroptosis in a dose-dependent but saturable manner.
Pyroptosis proceeds by the formation of caspase-1–dependent pores followed by osmotic lysis (Fink and Cookson, 2006). If microtubule stabilization exerted its effects subsequent to pore formation, then microtubule stabilization would be predicted to increase cell death regardless of the mechanism of pore formation. Therefore, we tested the bacterial pore-forming toxin streptolysin O, which forms 30-nm pores through direct protein insertion into the plasma membrane (Bhakdi et al., 1985). Remarkably, we observed that paclitaxel increased cell death caused by streptolysin O. (Figure 5F). Destabilizing microtubules with colchicine had the opposite effect and decreased cell death due to streptolysin O (Figure 5F). Thus, when we bypassed caspase-1 activation and simply inserted pores into the plasma membrane, microtubule stabilization still increased cell death, and microtubule depolymerization had the opposite effect and decreased cell death. In contrast, no effect of paclitaxel was observed when cell death was induced with Triton X-100 (Figure 5G), a nonionic detergent that does not form pores but instead causes lysis by disrupting hydrogen bonding within the plasma membrane (Koley and Bard, 2010). Our results demonstrate that microtubule stabilization renders the plasma membrane more susceptible to lysis regardless of the mechanism of pore formation. We conclude that stable microtubules are most likely affecting a step in the execution of pyroptosis during or after pore formation but before cellular lysis.
Variation in microtubule stability during differentiation
Unexpectedly, a paclitaxel-dependent increase in pyroptosis was not observed in THP-1s differentiated into macrophages with phorbol-12-myristate-13-acetate (PMA) or in mouse BMMs (Figure 6A). Previous studies demonstrated that alveolar macrophages have much greater microtubule stability than monocytes (Allen et al., 1997). Consistent with this, we noted increased tubulin acetylation after PMA treatment in THP1s (Figure 6, B and C). Remarkably, this was accompanied by a clear decrease in TUBB6 mRNA levels (Figure 6C). These data suggest that during monocyte-to-macrophage differentiation, TUBB6 gene expression is reduced to promote stabilization of microtubule networks. That stabilization appears to push them above a threshold level, such that further stabilization with paclitaxel has no effect on pyroptosis. This is also consistent with the paclitaxel dose–response data showing no further increase in pyroptosis past a certain threshold in microtubule stabilization (Figure 5B). Thus variation in TUBB6 expression occurs both in cells from different individuals and during differentiation to regulate microtubule stability.
FIGURE 6:

Differentiation of THP-1 monocytes decreases TUBB6 expression and increases microtubule stability. (A) Pyroptosis is unaffected by paclitaxel in PMA-differentiated THP-1 cells and mouse BMMs. THP-1s were treated for 4 h with 20 ng/ml PMA and then incubated for an additional 24 h without PMA to allow attachment and differentiation. THP-1s or BMMs were treated for 30 min with 500 nM paclitaxel before infection with S. typhimurium MOI30 (THP-1s + PMA) or MOI10 (BMMs). Cells were assayed with Cytotox-Glo at 3 h (THP-1s + PMA) or 2 h (BMMs) after initial infection. Data are mean ± SEM for three separate experiments, and p value is for paired t test. (B) PMA differentiation of THP-1 cells increases microtubule stability as assessed by acetylation of α-tubulin. THP-1s were mock treated or differentiated into macrophage-like cells using PMA, and cell extracts were blotted for acetylated and total α-tubulin. Values below the blots show the relative amount of acetylated tubulin, normalizing for the amount of total α-tubulin and setting the undifferentiated THP-1s to a value of 1. (C) PMA-differentiation of THP-1 cells increases microtubule stability and decreases TUBB6 mRNA expression. Normalized acetylated tubulin was quantified from three different experiments as in B. TUBB6 mRNA was measured by Taqman real-time PCR assay using 18S expression as the control. Data are the mean and SEM from four biological replicates from two experiments. The p values are from paired t tests.
DISCUSSION
Cellular genome-wide association study (GWAS) using pathogens as probes is a novel approach to understanding human variation in basic cellular traits (Ko and Urban, 2013). In this study, we found that a common human variant affects expression of the TUBB6 gene and that this leads to altered microtubule stability. Although the nearby CIDEA gene, a known regulator of cell death, was initially viewed as a more plausible candidate gene in the region, no association was observed with CIDEA expression, and RNAi against this gene had no effect on pyroptosis. In contrast, cells from individuals with the minor allele of rs9807444 had higher TUBB6 gene expression and less pyroptosis, consistent with TUBB6 acting as an inhibitor of pyroptosis. We verified the inhibitory activity of TUBB6 on caspase-1–mediated cell death both by RNAi and overexpression. Furthermore, stabilizing microtubules with paclitaxel also increased pyroptosis. Thus genetic association data for TUBB6, experimental manipulation of TUBB6 expression, and drug treatment all support our findings that microtubule stability regulates execution of pyroptosis and there is interindividual variation in this proinflammatory phenotype.
Although activation of inflammasomes by various stimuli through NLR oligomerization and caspase-1 recruitment has been extensively characterized (Lamkanfi and Dixit, 2011), how caspase-1 activation causes cell death is still not well understood. Fink and Cookson (2006) demonstrated that execution of pyroptosis after caspase-1 activation involved the formation of discrete pores (diameter 1.1–2.4 nm) followed by DNA cleavage and plasma membrane rupture. What roles do TUBB6 and microtubules play in pyroptosis? Our findings indicate that microtubules have dynamic roles during inflammasome activation and execution of pyroptosis. Before or during inflammasome activation, disruption of microtubules can have varying effects depending on the stimuli used. In our data, colchicine increased pyroptosis and IL-1β secretion in response to Salmonella, likely secondary to enhanced uptake by macropinocytosis. In contrast, Jurg Tschopp's lab demonstrated that colchicine could inhibit IL-1β processing induced by monosodium urate (gout) crystals (Martinon et al., 2006). They noted no inhibition by colchicine when a different stimulator, ATP, was used to activate the NLRP3 inflammasome. Given that Salmonella, gout crystals, and ATP all enter cells through different means, microtubule depolymerization may be affecting early upstream steps of caspase-1 activation to affect both pyroptosis and IL-1β secretion. A recent study also suggested that the effect of colchicine on NLRP3 inflammasome activation might involve transport of mitochondria along microtubules to facilitate NLRP3 inflammasome assembly (Misawa et al., 2013).
In contrast, both TUBB6 RNAi and stabilization of microtubules with paclitaxel increased pyroptosis without altering IL-1β secretion, indicating a distinct mechanism of microtubule involvement downstream of caspase-1 activation in the execution of pyroptosis. Furthermore, the effect of microtubule stabilization on cell death was noted even when a pore-forming toxin, streptolysin O, was used to create pores at the plasma membrane. Thus, stabilizing microtubules increased cell death regardless of whether pores were formed subsequent to caspase-1 activation or through direct insertion of a bacterial protein. The pore-forming toxin studies also suggest that when the complexities of host–pathogen interactions and inflammasome activation are bypassed and pores are simply inserted into the plasma membrane, colchicine has the opposite effect of paclitaxel on cell death. Microtubules have long been known to be closely associated with the plasma membrane and affect membrane microviscosity (Aszalos et al., 1985; Wolff, 2009). Depolymerization of microtubules by colchicine increases membrane fluidity (Aszalos et al., 1985), which could allow for repair of the plasma membrane before rupture. In contrast, stabilization by paclitaxel or reduction in TUBB6 expression may restrict membrane fluidity, rendering cells more prone to lysis after pore formation. Microtubules likely play diverse roles before inflammasome activation, during induction of pyroptosis, and then are dismantled during the late stages of pyroptosis. Our findings provide a framework for further elucidation of the mechanisms by which microtubules are involved in pyroptosis (Figure 7).
FIGURE 7:
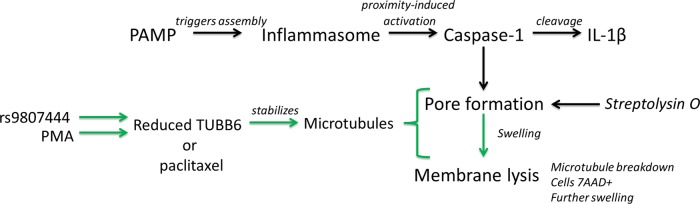
Model for the role of microtubule stabilization in execution of pyroptosis. Pathogen-associated molecular patterns trigger inflammasome assembly, leading to caspase-1 activation. Caspase-1 activation causes both pyroptosis and cleavage of IL-1β. Pyroptosis proceeds by the formation of discrete pores, followed by cellular swelling and membrane lysis. Human genetic variation (rs9807444 or another genetic change in LD) or PMA treatment causes a reduction in TUBB6 expression that leads to microtubule stabilization. Microtubule stabilization has an effect downstream of caspase-1 in the pyroptotic branch of the pathway (green). This effect, which may involve rendering the plasma membrane more prone to lysis due to increased rigidity, occurs during or after pore formation but before membrane lysis.
In addition to implicating microtubules in the regulation of caspase-1–mediated cell death, human variation in microtubule stability could have other profound consequences for human health. Because microtubules are involved in diverse processes ranging from neuronal transport to cell division, the TUBB6 variant may have pleiotropic effects on numerous human phenotypes. Although rs9807444 has not yet shown genome-wide significance for risk in any published GWAS, our cellular GWAS findings prompt closer examination for possible non–genome-wide significant associations that still may be biologically relevant. Human variation affecting microtubule stability could also affect drug responsiveness, as microtubule-stabilizing and destabilizing drugs are used extensively. Although there is a statistically significant inverse correlation between TUBB6 expression and paclitaxel sensitivity in LCLs (p = 0.05), there is no association between the rs9807444 SNP and paclitaxel sensitivity in this data set (E. R. Gamazon and M. E. Dolan, personal communication). Perhaps paclitaxel uptake through solute transporters is playing a large role in susceptibility of these cells, as recently suggested (Njiaju et al., 2012), and obscuring an effect of TUBB6 genotype. The role of the rs9807444 variant and TUBB6 expression variation on sensitivity to paclitaxel will require more extensive study in cells and cancer patient populations.
This work further demonstrates the value of cellular GWAS approaches using pathogens as probes for both uncovering common human variation in basic cellular processes and elucidating mechanism in host–pathogen interactions. Expanding this approach to additional pathogens and readouts will be useful in increasing the understanding of human variation in basic cell biological processes.
MATERIALS AND METHODS
Cells
HapMap LCLs and primary fibroblasts (GM05756, GM17101, GM17151, GM17152, GM17153, GM17169, GM17347, and GM17379) were purchased from the Coriell Institute (Camden, NJ). THP-1 cells were purchased from the American Type Culture Collection (Manassas, VA). Cells were maintained at 37°C in a 5% CO2 atmosphere. LCLs and THP-1s were grown in RPMI 1640 medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) or 15% for the carboplatin screen, 2 mM glutamine, 100 U/ml penicillin-G, and 100 mg/ml streptomycin. Primary fibroblasts were grown in DMEM 15% FBS, 1 mM pyruvate, 1× nonessential amino acids, 2 mM glutamine, 100 U/ml penicillin-G, and 100 mg/ml streptomycin. BMMs were prepared from the femurs of C57BL/6 mice and casp1−/− (Kuida et al., 1995) mice by culture with 85% supplemented RPMI + 15% L929-cell supernatant. HEK-293T and HeLa cells were grown in DMEM supplemented with 10% FBS, 1 mM glutamine, 100 U/ml penicillin-G, and 100 mg/ml streptomycin. Where indicated, THP-1s were differentiated using 20 ng/ml PMA for 4 h followed by 24 h in the absence of PMA to allow for adhesion and differentiation before assay.
Bacterial infection
Assaying LCLs, THP-1s, and BMMs for Salmonella-induced cell death was conducted as previously described using S. typhimurium 14028s tagged with an inducible green fluorescent protein (GFP) plasmid (Ko et al., 2009). Briefly, overnight bacterial cultures were subcultured 1:33 and grown for 2 h, 40 min at 37°C. Bacterial invasion was conducted for 1 h at a multiplicity of infection (MOI) of 30 (or 10 where indicated), followed by gentamicin (50 μg/ml) treatment. Cell death was assessed at 3 h after initial infection by staining with 7-AAD (Enzo Life Sciences, Farmingdale, NY) and measurement on a Guava Easycyte Plus flow cytometer (Millipore, Billerica, MA). When IL-1β was measured from THP-1s, cells were prestimulated either for 2 h, 10 min with 20 ng/ml lipopolysaccharide (LPS) for S. typhimurium or overnight with 20 ng/ml LPS for F. novicida U112. When paclitaxel or colchicine was used to treat cells, the drug or solution minus drug was added 30 min before infection at a dose of 500 nM. F. novicida was spun onto cells for 10 min at 700 × g, and infection was allowed to proceed for 2 h before addition of gentamicin (50 μg/ml). Cytotoxicity was measured by Cytotox-Glo 5 h after addition of Francisella. Cell death with this assay was reported as percentage cytotoxicity, with cells lysed by addition of digitonin to obtain a 100% reading.
Streptolysin O (Sigma, St. Louis, MO) was reduced with 10 mM dithiothreitol to increase activity before addition to cells.
RNAi experiments
LCLs or THP1s (2 × 105 cells) were treated for 3 d in 500 μl of Accell media (ThermoScientific, Waltham, MA) with either nontargeting Accell siRNA #1 or an Accell SmartPool directed against human TUBB6 (1μM total siRNA; ThermoScientific). Before infection, cells were plated at 1 × 105 in 100 μl of RPMI complete medium (without antibiotics) in 96-well plates. Salmonella infections were conducted as described. Cell death was assessed after 3 h for S. typhimurium.
HEK-293T overexpression experiments
HEK-293T cells were plated at 4 × 105 cells in six-well plates and transfected using Fugene6 according to manufacturer's instructions (Promega, Madison, WI). Plasmids used included human TUBB6-Flag and TUBB-Flag (ThermoScientific), human caspase-1-Flag (variant alpha; OriGene, Rockville, MD), and human caspase-9 (variant alpha; OriGene). For assessing inhibition of caspase-induced cytotoxicity, 1 μg of total plasmid was used for each transfection, with 400 ng of caspase plasmid and 300 ng of TUBB6-flag plasmid and the remainder made up with empty vector. Twenty-three hours posttransfection, cytotoxicity was measured using Cytotox-Glo. Cytotoxicity measurements were background subtracted (cells transfected with empty vector) and expressed as a percentage relative to caspase-1 or caspase-9 plus empty vector set to 100 to obtain “normalized RLUs.” Expression levels were assessed by Western blot for human caspase-1 (1:200, SC-515; Santa Cruz Biotechnology, Dallas, TX), caspase-9 (1:1000, C9; Cell Signaling Technology, Danvers, MA), and Flag (1:1000, M2; Sigma).
Quantitative PCR
Total RNA was purified from cells using RNeasy (Qiagen, Venlo, Netherlands) and treated with DNase, and cDNA was prepared with iScript (Bio-Rad, Hercules, CA). Relative expression of TUBB6 was measured using Taqman gene expression assays (Life Technologies) normalized for 18S rRNA.
Acetylated tubulin immunoblotting
Protein extracts were prepared in RIPA buffer (50 mM Tris 7.4, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100) with protease inhibitor table (Roche). Blots were probed with anti–acetylated α-tubulin (1:1000, D20G3; Cell Signaling Technologies) and anti–α-tubulin (1:1000; Cell Signaling Technologies) and developed using either horseradish peroxidase–conjugated secondary antibody and WestPico Reagent (ThermoScientific) or IRDye infrared secondary antibody (LI-COR, Lincoln, NE). Quantitation was by scanning x-ray film and quantifying bands in ImageJ (National Institutes of Health, Bethesda, MD) or by integrated intensity using an Odyssey Infrared Imaging System (LI-COR).
Flow cytometric measurement of polymerized microtubules
Microtubules were fixed and stained as previously described (Morrison and Hergenrother, 2012). Briefly, cells were fixed and permeabilized in 100 μl of 0.5% glutaraldehyde in 80 mM 1,4-piperazinediethanesulfonic acid (pH 6.8), 1 mM MgCl2, 5 mM EDTA, and 0.5% Triton X-100 for 10 min, followed by quenching with 1 mg/ml NaBH4. Cells were pelleted at 400 × g for 5 min, washed with phosphate-buffered saline (PBS) and 1% bovine serum albumin (BSA), and then blocked in PBS, 0.2% Triton X-100, and 2% BSA for 1 h, with staining with anti–α-tubulin- fluorescein isothiocyanate (FITC) for 3 h (1:250; Sigma). Samples were diluted fivefold in PBS, and fluorescence was quantified using a Guava Easycyte Plus flow cytometer (Millipore).
Microscopy of fixed cells
BMMs were stained with FAM-YVAD-fmk (caspase-1 FLICA; Immunochemistry Technologies, Bloomington, MN) beginning 5 min before infection with S. typhimurium or F. novicida at an MOI of 30. Cells were fixed at the indicated times with 4% paraformaldehyde for 20 min at 37°C. Blocking and permeabilization were carried out for 30 min with 0.1% Triton X-100 and 5% normal donkey serum in PBS. Primary antibody staining was carried out against β-tubulin (1:100; Developmental Studies Hybridoma Bank, Iowa City, IA; E7) or α-tubulin (1:100; Cell Signaling Technologies). Secondary antibodies conjugated to Alexa Fluor 555 and Alexa Fluor 647 (Life Technologies) were used for staining. Imaging was done with a Nikon TE2000 inverted fluorescence microscope and an EVOS fluorescence microscope.
Live-cell imaging
RAW 264.7 macrophages were transfected with EMTB-3xGFP (Faire et al., 1999) using FuGENE HD (Promega) according to the manufacturer's instructions. At 48 h, cells were washed with PBS, scraped into new media, and replated on 35-mm dishes (no. 1.5 thickness; MatTek, Ashland, MA) coated with poly-d-lysine. Seventy-two hours posttransfection, cells were imaged using an Andor XD revolution spinning disk confocal microscope with 60× silicon oil objective maintained at 37°C and 5% CO2. Images were taken every 2 min for 1 h. Analysis was done with MetaMorph and Fiji.
Computational analysis and statistics
Descriptive statistics were performed with GraphPad Prism 5. Linkage disequilibrium was examined with HaploView (Barrett et al., 2005).
Supplementary Material
Acknowledgments
We thank Edward Miao and Emily Ray Ko for valuable discussion. This project was funded by an award from the National Institute of Allergy and Infectious Diseases to the Northwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (U54 AI057141), a National Institute of Allergy and Infectious Diseases Research Scholar Development Award (K22 AI093595), and a Duke School of Medicine Whitehead Scholarship.
Abbreviations used:
- BMM
bone marrow–derived macrophage
- eQTL
expression quantitative trait loci
- GWAS
genome-wide association study
- LCL
lymphoblastoid cell line
- LD
linkage disequilibrium
- SNP
single-nucleotide polymorphism
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-06-0294) on October 30, 2013.
REFERENCES
- Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–325. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- Allen JN, Moore SA, Liao Z, Wewers MD. Changes in mononuclear phagocyte microtubules after endotoxin stimulation. I. Changes in microtubule stability. Am J Respir Cell Mol Biol. 1997;16:119–126. doi: 10.1165/ajrcmb.16.2.9032118. [DOI] [PubMed] [Google Scholar]
- Aszalos A, Yang GC, Gottesman MM. Depolymerization of microtubules increases the motional freedom of molecular probes in cellular plasma membranes. J Cell Biol. 1985;100:1357–1362. doi: 10.1083/jcb.100.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Bhakdi S, Tranum-Jensen J, Sziegoleit A. Mechanism of membrane damage by streptolysin-O. Infect Immun. 1985;47:52–60. doi: 10.1128/iai.47.1.52-60.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya R, Cabral F. A ubiquitous beta-tubulin disrupts microtubule assembly and inhibits cell proliferation. Mol Biol Cell. 2004;15:3123–3131. doi: 10.1091/mbc.E04-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya R, Cabral F. Molecular basis for class V beta-tubulin effects on microtubule assembly and paclitaxel resistance. J Biol Chem. 2009;284:13023–13032. doi: 10.1074/jbc.M900167200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya R, Frankfurter A, Cabral F. A minor beta-tubulin essential for mammalian cell proliferation. Cell Motil Cytoskeleton. 2008;65:708–720. doi: 10.1002/cm.20292. [DOI] [PubMed] [Google Scholar]
- Bhattacharya R, Yang H, Cabral F. Class V beta-tubulin alters dynamic instability and stimulates microtubule detachment from centrosomes. Mol Biol Cell. 2011;22:1025–1034. doi: 10.1091/mbc.E10-10-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8:471–483. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faire K, Waterman-Storer CM, Gruber D, Masson D, Salmon ED, Bulinski JC. E-MAP-115 (ensconsin) associates dynamically with microtubules in vivo and is not a physiological modulator of microtubule dynamics. J Cell Sci. 1999;112:4243–4255. doi: 10.1242/jcs.112.23.4243. [DOI] [PubMed] [Google Scholar]
- Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci USA. 2008;105:4312–4317. doi: 10.1073/pnas.0707370105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8:1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- Francis CL, Ryan TA, Jones BD, Smith SJ, Falkow S. Ruffles induced by Salmonella and other stimuli direct macropinocytosis of bacteria. Nature. 1993;364:639–642. doi: 10.1038/364639a0. [DOI] [PubMed] [Google Scholar]
- Haglund CM, Welch MD. Pathogens and polymers: microbe-host interactions illuminate the cytoskeleton. J Cell Biol. 2011;195:7–17. doi: 10.1083/jcb.201103148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraga A, Ohlson MB, Miller SI. Salmonellae interplay with host cells. Nat Rev Microbiol. 2008;6:53–66. doi: 10.1038/nrmicro1788. [DOI] [PubMed] [Google Scholar]
- Inohara N, Koseki T, Chen S, Wu X, Nunez G. CIDE, a novel family of cell death activators with homology to the 45 kDa subunit of the DNA fragmentation factor. EMBO J. 1998;17:2526–2533. doi: 10.1093/emboj/17.9.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap C, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA. 1993;90:9552–9556. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko DC, et al. A genome-wide in vitro bacterial-infection screen reveals human variation in the host response associated with inflammatory disease. Am J Hum Genet. 2009;85:214–227. doi: 10.1016/j.ajhg.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko DC, et al. Functional genetic screen of human diversity reveals that a methionine salvage enzyme regulates inflammatory cell death. Proc Natl Acad Sci USA. 2012;109:E2343–E2352. doi: 10.1073/pnas.1206701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko DC, Urban TJ. Understanding human variation in infectious disease susceptibility through clinical and cellular GWAS. PLoS Pathog. 2013;9:e1003424. doi: 10.1371/journal.ppat.1003424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koley D, Bard AJ. Triton X-100 concentration effects on membrane permeability of a single HeLa cell by scanning electrochemical microscopy (SECM) Proc Natl Acad Sci USA. 2010;107:16783–16787. doi: 10.1073/pnas.1011614107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Modulation of inflammasome pathways by bacterial and viral pathogens. J Immunol. 2011;187:597–602. doi: 10.4049/jimmunol.1100229. [DOI] [PubMed] [Google Scholar]
- Leone P, Meresse S. Kinesin regulation by Salmonella. Virulence. 2011;2:63–66. doi: 10.4161/viru.2.1.14603. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Maruta H, Greer K, Rosenbaum JL. The acetylation of alpha-tubulin and its relationship to the assembly and disassembly of microtubules. J Cell Biol. 1986;103:571–579. doi: 10.1083/jcb.103.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14:454–460. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- Morrison KC, Hergenrother PJ. Whole cell microtubule analysis by flow cytometry. Analyt Biochem. 2012;420:26–32. doi: 10.1016/j.ab.2011.08.020. [DOI] [PubMed] [Google Scholar]
- Moss DK, Lane JD. Microtubules: forgotten players in the apoptotic execution phase. Trends Cell Biol. 2006;16:330–338. doi: 10.1016/j.tcb.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Njiaju UO, Gamazon ER, Gorsic LK, Delaney SM, Wheeler HE, Im HK, Dolan ME. Whole-genome studies identify solute carrier transporters in cellular susceptibility to paclitaxel. Pharmacogenet Genomics. 2012;22:498–507. doi: 10.1097/FPC.0b013e328352f436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Alcazar JA, Rodriguez-Hernandez A, Cordero MD, Fernandez-Ayala DJ, Brea-Calvo G, Garcia K, Navas P. The apoptotic microtubule network preserves plasma membrane integrity during the execution phase of apoptosis. Apoptosis. 2007;12:1195–1208. doi: 10.1007/s10495-006-0044-6. [DOI] [PubMed] [Google Scholar]
- Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem. 2007;282:36321–36329. doi: 10.1074/jbc.M708182200. [DOI] [PubMed] [Google Scholar]
- Stranger BE, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8:e1002639. doi: 10.1371/journal.pgen.1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terkeltaub RA. Colchicine update: 2008. Semin Arthritis Rheum. 2009;38:411–419. doi: 10.1016/j.semarthrit.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Tilney LG, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff J. Plasma membrane tubulin. Biochim Biophys Acta. 2009;1788:1415–1433. doi: 10.1016/j.bbamem.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, Tang H, Snyder M. Variation and genetic control of protein abundance in humans. Nature. 2013;499:79–82. doi: 10.1038/nature12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvon AM, Wadsworth P, Jordan MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol Biol Cell. 1999;10:947–959. doi: 10.1091/mbc.10.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller T, et al. Genetics and beyond—the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.