

Abstract
It is generally accepted that gene regulation serves an important role in determining the phenotype. To shed light on the evolutionary forces operating on gene regulation, previous studies mainly focused on the expression differences between species and their inter-specific hybrids. Here, we use RNA-Seq to study the intra-specific distribution of cis- and trans-regulatory variation in Drosophila pseudoobscura. Consistent with previous results, we find almost twice as many genes (26%) with significant trans-effects than genes with significant cis-effects (18%). While this result supports the previous suggestion of a larger mutational target of trans-effects, we also show that trans-effects may be subjected to purifying selection. Our results underline the importance of intra-specific analyses for the understanding of the evolution of gene expression.
Introduction
It is well understood that variation in gene expression is an important source of phenotypic differences. Hence, it has been a long-term goal in biology to understand the regulation of gene expression. The genetic basis of variation in gene expression can be divided into two classes: 1) Variation in regulatory domains (cis-regulatory elements) that modulates gene expression or polymorphisms that influence the stability of mRNA. 2) Variation in trans-acting proteins, e.g. transcription factors that regulate the expression of a set of target genes. The combined effects of cis- and trans-regulation determine the expression of every gene. Different approaches ranging from diallel crosses [1] to eQTLs [2] have been pursued to understand the regulation of gene expression, but the highest level of resolution has been obtained from allele-specific gene expression measurements in parents and their offspring. Since offspring share the trans-acting factors from both parents the contrast of gene expression in parents and offspring provides an estimate for the magnitude of cis- and trans-effects [3].
The contribution of cis- and trans-effects to the evolution of gene expression has been studied in several species (e.g. in Drosophila: [3], [4], [5], [6], yeast: [7], [8], Arabidopsis: [9]). Independent of the species studied, frequently more cis- than trans-effects were found when two closely related species and their hybrids were analyzed. This observation is particularly interesting since the mutational target of trans-effects (i.e.: all trans-regulating factors) is larger than the one of cis-effects [10], [11]. The interpretation of the greater amounts of cis-effects found is that trans-effects are more pleiotropic than cis-effects. Thus, trans-mutations are more difficult to establish and become fixed even if they have a beneficial effect on one target gene as other target genes can be negatively affected by the mutation. In contrast, cis-mutations affect a single gene only. Therefore, they tend to be less deleterious than trans-effects and are more likely to become fixed [4].
The comparison of mutation accumulation lines, with very low selection efficacy due to a small effective population size, to natural isolates also showed that trans-effects are subject to strong purifying selection [11]. Furthermore, D. sechellia, a species with a very small effective population size (and thus a lower selection efficacy) shows more trans-effects than the cosmopolitan D. simulans [6], [12], [13].
For intra-specific studies, the relative importance of cis- and trans-effects is still controversial. Early attempts to understand the contribution of cis- and trans-regulatory components within Drosophila species showed more trans- than cis-effects in D. simulans males [14]. A study in D. melanogaster identified 90% cis-effects and only very few trans-effects, but it was not clear to what extent this observation results from low power to detect trans-effects [5]. A comparison of the two D. melanogaster strains with strong assortative mating, Z and M, suggested that the expression differences between them could be largely attributed to cis-regulatory variation [15]. In yeast, trans-effects were found to contribute more to intra-specific expression variation [7]. Among 863 of differentially expressed genes 558 (∼64%) had been influenced by trans-regulation, whereas 421 (∼49%) genes were found to have significant cis- regulation [7]. In Arabidopsis thaliana, more pure trans-effects than pure cis-effects were found [16], which contrasts with an earlier study that found an excess of cis-effects in A. thaliana [1].
Since it is not clear to what extent the contrasting observations reflect differences in the experimental design and applied technologies, we studied the relative contribution of cis- and trans-effects to the intra-specific expression variation in D. pseudoobscura. Using the latest next generation sequencing technology we show that trans-effects are more abundant than cis-effects, probably reflecting their larger mutational target size. Furthermore, we show that the distribution of effect sizes differs between cis- and trans-effects, most likely caused by different selective forces operating on them.
Materials and Methods
Sample preparation and sequencing
The D. pseudoobscura strains ps94 (stock number 14011-0121.94) and ps88 (stock number 14011-0121.88) were obtained from the UC San Diego Drosophila Species Stock Center. Libraries of the parental strains were those used by Palmieri et al. [17]. Reciprocal hybrids were reared in parallel and under the same conditions as the parental strains by either crossing a virgin ps94 female with a ps88 male or a virgin ps88 female with a ps94 male. Flies were reared on standard cornmeal-molasse-yeast-agar medium and maintained at 19°C under constant dark conditions. Virgin female offspring of 15–20 replicate crosses from either direction were collected, allowed to age for three to seven days and shock-frozen in liquid nitrogen. For extraction of total RNA, we used equal numbers of females from the two directions of crosses, and performed two replicate RNA extractions that were pooled for library construction. This strategy aimed to reduce possible imprinting effects and the biological variation among crosses. Paired-end Illumina mRNA libraries were generated from 10 ug total RNA using the mRNA Sample Prep Kit (Illumina, San Diego, CA) as previously described [17]. All libraries were sequenced on the same flow cell as the female libraries of the two parental strains used in [17]. We did not sequence replicate libraries. The RNA-Seq read libraries were deposited in the ArrayExpress database https://www.ebi.ac.uk/arrayexpress/under the accession number E-MTAB-1424.
Read mapping
Reads were trimmed using the Mott algorithm implemented in PoPoolation [18] (minimum read length = 40, quality threshold = 20). We mapped three RNA-Seq paired-end (2×100 bp) libraries derived from ps88 females, ps94 females and hybrid females, against the D. pseudoobscura reference genome (FlyBase, assembly r2.23) using GSNAP [19]. Only non-ambiguously mapping read pairs were retained. To minimize the mapping bias, we first mapped the reads from the parental lines to the reference and identified SNPs fixed between the two strains. Then, we remapped the reads including the SNP information with GSNAP. Using an improved D. pseudoobscura annotation [17] we identified SNPs that were fixed (allele frequency = 1) for different alleles in the two parental lines. Based on these SNPs we generated two distinct reference genomes, one for each parental strain. Allele specific gene expression was measured by mapping the RNA-Seq reads to both genomes simultaneously and counting the number of reads mapping unambiguously to one of the two genomes (i.e. read pairs which covered at least one of the SNPs distinguishing the two genomes). The generation of two parental genomes not only identifies reads from either parental allele but also aims to avoid a well described mapping bias for different allelelic variants [20], [21]. Moreover, it has been shown that after the adjustment for the different parental alleles via two reference genomes a mapping bias can remain for some genes [21]. To quantify and account for this remaining mapping bias we determined an empirical correction factor through simulations. These simulations included the following steps: 1) For each gene the same number of RNA-Seq reads (paired ends) were generated for the identical positions in the two genomes matching the read length and insert size of the empirical data. (i.e.: differing only by the identified SNPs). 2) The simulated RNA-Seq read libraries were mapped against both genomes simultaneously retaining only non-ambiguously mapped reads. 3) For genes with a different number of reads mapping to each of the genomes, we determined a correction factor, which is the inverse of the ratio of the mapped reads. This correction factor was then included in the G-test to correct the Null-expectation for allele specific expression in hybrids.
Finally, we normalized the expression data using the TMM method [22]. The pipeline described above to identify allele-specific gene expression analysis is implemented in the Allim software package (http://code.google.com/p/allim/, [21]).
Data analysis
Cis- and trans-effects were estimated by contrasting gene expression in the parental and F1 libraries. In order to test for significance of the regulatory scenarios we used the G-test of independence followed by correction using the Benjamini-Hochberg method at a false discovery rate cutoff (FDR) of 0.05. Testing was performed in a gene-wise manner. Total differential expression (TDE) between parents was estimated by comparing the ratio of the counts in the two parents to the ratio of total counts (i.e.: ratio of library sizes). For allele specific differential expression in F1 individuals (cis), we applied the same logic to the F1 library and compared the ratio of allelic counts at a given gene to the ratio of the allele counts summed over all genes. For the trans-test, we compared the ratio of parental gene expression counts to the ratio of allele specific counts in the F1 library. We note that this testing strategy results in a lower power to detect differences in trans compared to cis-effects since the total number of counts is higher in our tests for cis-effects than in our tests for trans-effects (see below). Following previous studies [6], [23], we distinguish between different types of regulation namely cis, trans, cis-by-trans, cis+trans, compensatory, conserved and ambiguous (Table S1). Modes of inheritance were classified as additive, dominant, over-dominant, under-dominant and conserved expression as described by Landry et al. [23] and McManus et al. [6] (Table S2).
Statistical power estimation for cis- and trans-tests through simulations
We hypothesized that statistical power of the G-test for allele-specific differential expression (cis) is higher than for trans-test due to the fact that only one “noisy” allele-specific expression measurements were included into the cis-tests whereas two such measurements appeared in a trans-test. To support this hypothesis we performed computer simulations, in which we compared the power for different effect sizes. Since the power critically depends on the expression level of a given gene, we based our simulations on the observed expression level n (averaged over the three libraries) of randomly selected genes. The allelic imbalance γ was modulated on a grid of values ranging from 1 to 10 with the step size of 0.2. We simulated allele expression counts taken from a Poisson (λ) distribution for the parental lines (P1 and P2) and from a Poisson (λ/2) distribution for the hybrid lines (H1 and H2). For each gene two different values λ 1 and λ 2 were computed according to λ 1 = 2n/(γ+1), λ 2 = 2n γ/(γ+1). For pure cis-effects we simulated expression counts from H1 according to Poisson (λ 1/2) and for H2 according to Poisson (λ 2/2), and Poisson (λ 1) for P1 and Poisson (λ 2) for P2. For pure trans-effects, we took Poisson (λ 1) for P1 and Poisson (λ 2) for P2, and Poisson (n/2) for H1 and H2. Combined cis- and trans-effects can be simulated by taking λ P1 = 2n/(γ P+1), λ P2 = 2n γ P/(γ P+1), λ H1 = 2n/(γ H+1) and λ H2 = 2n γ H/(γ H+1) with allelic imbalances γ P and γ H for parents and hybrids. Using the proportion of rejections, the statistical power (1-Type II error) of the tests was estimated by simulating expression counts for 10000 genes under each imbalance parameter value (γ, and (γ P, γ H)). All statistical analyses were performed using R.
GO analysis
GO analysis were performed with FuncAssociate 2.0 [24] using the gene IDs of the D. melanogaster orthologs of D. pseudoobscura.
Results
We performed RNA-Seq for two highly inbred parental lines (ps88 and ps94) and F1 individuals obtained from bi-directional crosses of the two inbred lines. On average, we obtained about 41×106 reads for each library (Table S3), which resulted on average in about 4×106 unambiguously mapping read pairs for each library after all filtering steps. Out of 16743 annotated genes in D. pseudoobscura [17], we identified 8116 (48.5%) genes with at least one fixed difference between the two lines. On average, we detected 4.4 fixed SNPs per gene between the two lines. 7631 genes with more than 20 reads in both parental lines (ps88+ps94≥20) were included in the subsequent analysis.
Classification of cis- and trans-regulation
Gene expression is determined by cis- and trans-regulatory variation. Since variation in cis-regulatory elements results in allele specific gene expression, it is possible to measure cis-effects in F1 individuals. We detected significant (FDR≤0.05) differences in gene expression due to cis-effects between the two parental alleles for 1359 genes (including 154 genes from the ambiguous category, Table S1), which correspond to 18% of the expressed genes. Trans-effects were estimated by a significant (FDR≤0.05) difference in gene expression for the same allele in the parental and F1 background [3], [4], [5], [6]. We identified 1982 (26%) genes (including 89 genes from the ambiguous category, Table S1) with significant trans-regulation.
Following the classification introduced by [23] we grouped the expressed genes in further sub-categories (Table S4). We identified 604 genes for which only cis-regulation was significant. More than twice as many genes (1292) had only significant trans-effects. We used computer simulations to test if the large excess of genes with trans-effects could be an artifact of our method. Interestingly, our simulations showed a slightly lower power to identify trans-effects over a wide range of imbalances (Fig. 1). For genes with combined cis- and trans-effects the power of the testing procedure was substantially reduced on average, as shown by the total area under the power curve that is smaller for combined effects than for pure cis and trans effects (Fig. 1). This lower power to detect trans- than cis-effects makes the observed excess of genes with trans-effects particularly relevant since it is most likely an underestimate. For 601 genes we identified combined cis- and trans-effects: cis-by-trans (176), cis+trans (183) and compensatory (242). For 3187 (42%) genes the gene expression pattern was conserved among the three samples. The remaining 1947 genes had an ambiguous expression pattern with no clear biological interpretation, i.e. the genes that cannot be placed in any category of regulation based on their patterns of parental and hybrid expression (Figure 2A,B). Using more stringent significance FDR thresholds of 0.01 and 0.005 did not change the overall pattern (Figure S1).
Figure 1. Power analysis of cis- and trans-tests.

We used computer simulations to determine the power to detect cis-effects (black), pure trans-effects (red), and trans-effects in genes with combined regulatory modes (blue). The power was determined for 10000 randomly selected genes over a range of imbalance indices (γ) and the averages are shown. The imbalance indices for genes with combined effects were calculated as |γ P – γ H|+1).
Figure 2. Cis- and trans-regulatory differences.

A) Distribution of regulatory effects depending on parental expression and allele specific expression in the hybrid. B) Relative distribution of the regulatory types (cis: 604 (7.9%); trans: 1292 (16.9%); cis–by–trans: 176 (2.3%); cis+trans: 183 (2.4%); compensatory: 242 (3.2%); conserved: 3187 (41.8%); ambiguous: 1947 (25.5%)).
Our observation of more genes with trans-effects than cis-effects is consistent with the larger mutational target of trans-effects. To further scrutinize this hypothesis we determined the frequency distribution of cis- and trans-regulatory effect sizes (cis-magnitude = |log2(ps88 hybrid/ps94 hybrid)|, trans-magnitude = |log2(ps88/ps94) – log2(ps88 hybrid/ps94 hybrid)| and noticed a striking difference. In the small effect size class we detected a significant excess of genes with trans-effect. In all classes with a larger effect size, we found an excess of genes with cis-effects (Figure 3). Qualitatively the same result was obtained when genes with combined effects were also included (data not shown). This marked difference between effect size classes is consistent with stronger purifying selection operating on genes with trans-effects, possibly due to their pleiotropic nature.
Figure 3. Frequency distribution of genes with significant cis- and trans- regulation.

Genes with pure trans (red) and cis (black) effects were grouped according to the magnitude of their effect.
Dissecting modes of inheritance
The comparison of expression intensities in F1 individuals relative to the one in their parents provides information about the mode of inheritance (Table S2). Out of 7631 genes analyzed, 607 genes were classified as additive and 3120 genes as dominant. 1326 genes were misexpressed in the F1 individuals, with 369 genes being over-dominant (higher expression in the F1) and 957 genes being under-dominant (lower expression in the F1 than in both parents). 2578 genes had no significant difference between parents and the F1 (conserved, Figure 4). We note, that the high fraction of genes with conserved inheritance reflects statistical power of our experiment rather than a true biological signal since the genes with conserved mode of inheritance have lower expression intensities (ps88 parent+ps94 parent+F1) than those genes with additive, dominant or misexpressed modes of inheritance (Wilcoxon's one-tailed rank-sum test, P<2.2e-16). As expected, we found no indication of one parent being over-represented among the genes with a dominant mode of inheritance: among 3 120 genes 1518 (∼49%) and 1602 (∼51%) genes have ps88-like and ps94-like expression.
Figure 4. Modes of inheritance.
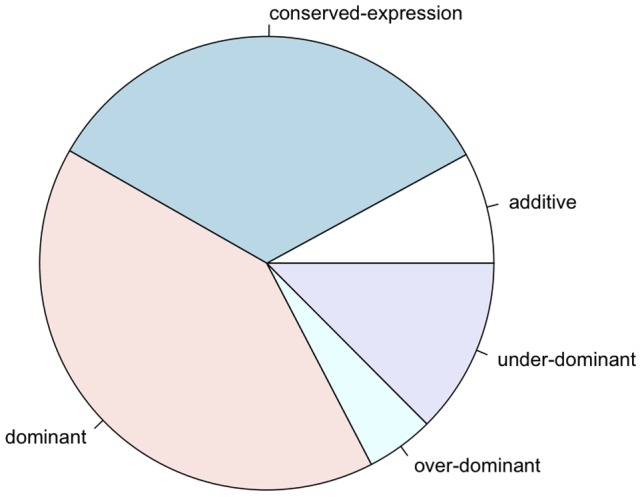
Relative proportions of the inheritance modes (dominant: 3120 (40.9%); additive: 607 (8%); over-dominant: 369 (4.8%); under-dominant: 957 (12.5%); conserved-expression: 2578 (33.8%)).
Intra-specific comparisons suggested that cis-regulation tends towards additivity whereas trans-regulation shows higher degrees of dominance [25]. Consistent with these previous results, we found that cis-magnitude (|log2(ps88 hybrid/ps94 hybrid)|) within genes with additive mode of inheritance was significantly greater than the cis-magnitude of genes with other significant inheritance types (Wilcoxon's one-tailed rank-sum test, P = 0.007738) (Figure S2).
GO analysis
We tested for an enrichment of functional categories among the different modes of gene regulation using a gene ontology (GO) analysis. While some marginal significance was detected for some modes of gene regulation, none of them survived a correction for multiple testing. Similarly, no significant enrichment of a GO category was observed for the different modes of inheritance. Hence, we conclude that our data do not support a functional differentiation among the categories studied.
Discussion
This report is the first study of intra-specific cis-and trans-effects using RNA-Seq in Drosophila and the first report for D. pseudoobscura. We observe about twice as many genes with pure trans-effects (1292) as genes with pure cis-effects (604). This result is consistent with earlier studies in Drosophila [4], Arabidopsis [16] and yeast [7]. Hence, we suggest that this pattern is most likely general and contrasting results [5] probably reflect more methodological differences than an alternative biological phenomenon.
Of particular interest is the contrast to studies of cis- and trans-effects relying on the analysis of expression differences between closely related species and their corresponding hybrids. These analyses typically observed a clear excess of genes with cis-effects [4], [8], [9], [26]. The prevailing explanation for this apparent discrepancy is that cis-effects affect only a single gene, while trans-effects are typically pleiotropic [3], [4], [27]. Hence, cis-effects become more easily fixed between species [28], [29]. Within species, however, the larger mutational target of trans-effects results in the observed excess of trans-effects [4], [7]. In our report, we have further scrutinized the hypothesis of differential selection operating on cis- and trans-effects by contrasting the frequency distribution of different effect sizes. Reasoning that trans-effect mutations with a large effect size are more effectively purged than cis-effect ones, we predicted an underrepresentation of genes with trans-effects in the large effect size classes. In fact, our data show a trend of a more pronounced underrepresentation of genes with trans-effects with an increasing effect size (Figure 3). Furthermore, the larger number of genes with trans-effects is consistent with them being a larger mutational target. Alternatively, the observed distribution of cis- and trans-effects (Figure 3) could be also obtained if new trans-mutations have, in general, smaller regulatory effect sizes than mutations in regulatory regions.
Overall, we conclude that the patterns of intra-specific and inter-specific cis- and trans-effects discussed above are compatible with the possibility that most regulatory variation is deleterious and their distribution reflects the balance between the occurrence of new mutations and their different fixation probabilities. This idea is in line with other results showing that purifying selection is the major force affecting the evolution of gene expression [30]. Nevertheless, if purifying selection shapes the pattern of trans-effects while positive selection contributes largely to cis-effects [31], the distribution of intra-specific cis- and trans-effect sizes may also differ.
We caution that the evolution of gene expression is probably more complex. One indication for this could be obtained from the comparison of the effect sizes in intra- and inter-specific comparisons. While we found an underrepresentation of genes with large trans-effects, inter-specific comparisons suggest large effect sizes of trans-effects [6]. We hypothesize that this discrepancy could be due to species specific trans-regulatory mutations that may, for example, be involved in compensatory changes.
To obtain a complete picture of the evolutionary dynamics of cis- and trans-effects it is important to analyze their context specific effects. Depending on the developmental stage, tissue, and environment the regulatory landscape may differ and consequently also the effects of cis- and trans-regulatory variation. More experiments are needed to understand to what extent the patterns described for a single sex, developmental stage and environment can be generalized. Furthermore, future studies may benefit from replication, which will improve the accuracy of the estimated effects beyond the already high repeatability of RNA-Seq studies [32].
Supporting Information
Effect of different FDR threshold on the identification of regulatory effects. A) FDR < 0.05. B) FDR < 0.01. C) FDR <0.005.
(PDF)
Cis -magnitude comparison of genes with additive and non-additive modes of inheritance. Genes with additive mode (A) of inheritance exhibit higher cis-magnitude vs. all other genes with non-additive modes (NA) excluding genes with conserved expression mode.
(PDF)
Determination of regulatory categories based on total differential expression between parents (TDE), allele-specific differential expression in F1 hybrid (cis) and trans-test. All the statistical tests were performed using G-test, followed by FDR correction (q≤0.05).
(DOC)
Identification of inheritance modes based on differential expression and directionality of expression between parental and total expression in F1 hybrids.
(DOC)
Library size information.
(DOC)
Gene summary table. This table provides information about raw and normalized expression counts for parents and hybrids, statistical tests, regulatory types, inheritance modes and cis and trans magnitudes for each gene revealed by RNA-seq.
(XLS)
Acknowledgments
We are grateful to N. Palmieri for his support throughout the entire study.
Funding Statement
The laboratory of CS is supported by Austrian Science Fund (FWF, P22834, W1225) and the European Research Council (ERC, ArchAdapt). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kiekens R, Vercauteren A, Moerkerke B, Goetghebeur E, Van Den Daele H, et al. (2006) Genome-wide screening for cis-regulatory variation using a classical diallel crossing scheme. Nucleic Acids Res 34: 3677–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doss S, Schadt EE, Drake TA, Lusis AJ (2005) Cis-acting expression quantitative trait loci in mice. Genome Res 15: 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wittkopp PJ, Haerum BK, Clark AG (2004) Evolutionary changes in cis and trans gene regulation. Nature 430: 85–88. [DOI] [PubMed] [Google Scholar]
- 4. Wittkopp PJ, Haerum BK, Clark AG (2008) Regulatory changes underlying expression differences within and between Drosophila species. Nat Genet 40: 346–350. [DOI] [PubMed] [Google Scholar]
- 5. Genissel A, McIntyre LM, Wayne ML, Nuzhdin SV (2008) Cis and trans regulatory effects contribute to natural variation in transcriptome of Drosophila melanogaster . Mol Biol Evol 25: 101–110. [DOI] [PubMed] [Google Scholar]
- 6. McManus CJ, Coolon JD, Duff MO, Eipper-Mains J, Graveley BR, et al. (2010) Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res 20: 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Emerson JJ, Hsieh LC, Sung HM, Wang TY, Huang CJ, et al. (2010) Natural selection on cis and trans regulation in yeasts. Genome Res 20: 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tirosh I, Reikhav S, Levy AA, Barkai N (2009) A yeast hybrid provides insight into the evolution of gene expression regulation. Science 324: 659–662. [DOI] [PubMed] [Google Scholar]
- 9. Shi X, Ng DW, Zhang C, Comai L, Ye W, et al. (2012) Cis- and trans-regulatory divergence between progenitor species determines gene-expression novelty in Arabidopsis allopolyploids. Nat Commun 3: 950. [DOI] [PubMed] [Google Scholar]
- 10. Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL (2007) Genetic properties influencing the evolvability of gene expression. Science 317: 118–121. [DOI] [PubMed] [Google Scholar]
- 11. Denver DR, Morris K, Streelman JT, Kim SK, Lynch M, et al. (2005) The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans . Nat Genet 37: 544–548. [DOI] [PubMed] [Google Scholar]
- 12. Kliman RM, Andolfatto P, Coyne JA, Depaulis F, Kreitman M, et al. (2000) The population genetics of the origin and divergence of the Drosophila simulans complex species. Genetics 156: 1913–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Legrand D, Tenaillon MI, Matyot P, Gerlach J, Lachaise D, et al. (2009) Species-wide genetic variation and demographic history of Drosophila sechellia, a species lacking population structure. Genetics 182: 1197–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wayne ML, Pan YJ, Nuzhdin SV, McIntyre LM (2004) Additivity and trans-acting effects on gene expression in male Drosophila simulans . Genetics 168: 1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Osada N, Kohn MH, Wu CI (2006) Genomic inferences of the cis-regulatory nucleotide polymorphisms underlying gene expression differences between Drosophila melanogaster mating races. Mol Biol Evol 23: 1585–1591. [DOI] [PubMed] [Google Scholar]
- 16. Zhang X, Borevitz JO (2009) Global analysis of allele-specific expression in Arabidopsis thaliana . Genetics 182: 943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palmieri N, Nolte V, Suvorov A, Kosiol C, Schlotterer C (2012) Evaluation of different reference based annotation strategies using RNA-Seq – a case study in Drososphila pseudoobscura . PLoS One 7: e46415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kofler R, Orozco-terWengel P, De Maio N, Pandey RV, Nolte V, et al. (2011) PoPoolation: a toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS One 6: e15925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu TD, Nacu S (2010) Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, et al. (2009) Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25: 3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pandey RV, Franssen SU, Futschik A, Schlotterer C (2013) Allelic imbalance metre (Allim), a new tool for measuring allele-specific gene expression with RNA-seq data. Mol Ecol Resour 13: 740–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Landry CR, Wittkopp PJ, Taubes CH, Ranz JM, Clark AG, et al. (2005) Compensatory cis-trans evolution and the dysregulation of gene expression in interspecific hybrids of Drosophila . Genetics 171: 1813–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berriz GF, Beaver JE, Cenik C, Tasan M, Roth FP (2009) Next generation software for functional trend analysis. Bioinformatics 25: 3043–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lemos B, Araripe LO, Fontanillas P, Hartl DL (2008) Dominance and the evolutionary accumulation of cis- and trans-effects on gene expression. Proc Natl Acad Sci U S A 105: 14471–14476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graze RM, McIntyre LM, Main BJ, Wayne ML, Nuzhdin SV (2009) Regulatory divergence in Drosophila melanogaster and D. simulans, a genomewide analysis of allele-specific expression. Genetics 183: 547–561, 541SI–521SI. [DOI] [PMC free article] [PubMed]
- 27. Wittkopp PJ (2005) Genomic sources of regulatory variation in cis and in trans . Cell Mol Life Sci 62: 1779–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wray GA (2007) The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8: 206–216. [DOI] [PubMed] [Google Scholar]
- 29. Wittkopp PJ, Kalay G (2012) Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet 13: 59–69. [DOI] [PubMed] [Google Scholar]
- 30. Bedford T, Hartl DL (2009) Optimization of gene expression by natural selection. Proc Natl Acad Sci U S A 106: 1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schaefke B, Emerson JJ, Wang TY, Lu MY, Hsieh LC, et al. (2013) Inheritance of gene expression level and selective constraints on trans- and cis-regulatory changes in yeast. Mol Biol Evol 30: 2121–2133. [DOI] [PubMed] [Google Scholar]
- 32. Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y (2008) RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18: 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of different FDR threshold on the identification of regulatory effects. A) FDR < 0.05. B) FDR < 0.01. C) FDR <0.005.
(PDF)
Cis -magnitude comparison of genes with additive and non-additive modes of inheritance. Genes with additive mode (A) of inheritance exhibit higher cis-magnitude vs. all other genes with non-additive modes (NA) excluding genes with conserved expression mode.
(PDF)
Determination of regulatory categories based on total differential expression between parents (TDE), allele-specific differential expression in F1 hybrid (cis) and trans-test. All the statistical tests were performed using G-test, followed by FDR correction (q≤0.05).
(DOC)
Identification of inheritance modes based on differential expression and directionality of expression between parental and total expression in F1 hybrids.
(DOC)
Library size information.
(DOC)
Gene summary table. This table provides information about raw and normalized expression counts for parents and hybrids, statistical tests, regulatory types, inheritance modes and cis and trans magnitudes for each gene revealed by RNA-seq.
(XLS)