

Abstract
Purpose
To identify the disease-causing mutation in a five-generation Chinese family affected with bilateral congenital nuclear cataract.
Methods
Linkage analysis was performed for the known candidate genes and whole-exome sequencing was used in two affected family members to screen for potential genetic mutations; Sanger sequencing was used to verify the mutations throughout family.
Results
A novel beaded filament structural protein 1 (BFSP1) gene missense mutation was identified. Direct sequencing revealed a heterozygous G>A transversion at c.1042 of the coding sequence in exon 7 of BFSP1 (c.1042G>A) in all affected members, which resulted in the substitution of a wild-type aspartate to an asparagine (D348N). This mutation was neither seen in unaffected family members nor in 200 unrelated people as controls.
Conclusions
A novel mutation (c.1042G>A) at exon 7 of BFSP1, which creates a substitution of an aspartate to an asparagine (p.D348N) was identified to be associated with autosomal dominant congenital cataract in a Chinese family. This is the first report of autosomal dominant congenital cataract being associated with a mutation in BFSP1, highlighting the important role of BFSP1 for physiological lens function and optical properties.
Introduction
Congenital cataract is a major cause of visual impairment and childhood blindness [1]. Nonsyndromic congenital cataracts have an estimated frequency of 1–6 per 10,000 live births, with 1/3 of cases having a positive family history [2]. Among these families, an autosomal dominant genotype is the most prevalent [3]. To date, at least 19 genes have been reported to be linked with different modes of nonsyndromic autosomal dominant congenital cataract. These genes encode for numerous crystallins: alpha A crystallin (CRYAA) [4], alpha B crystallin (CRYAB) [5], beta A1/A3 crystallin (CRYBA1/A3) [6], beta A4 crystallin (CRYBA4) [7], beta B1 crystallin (CRYBB1) [8], beta B2 crystallin (CRYBB2) [9], gamma C crystallin (CRYGC) [10], gamma D crystallin (CRYGD) [11], gamma Scrystallin (CRYGS) [12], alpha 3 gap junction protein (GJA3) [13], alpha 8 gap junction protein (GJA8) [14], major intrinsic protein (MIP) [15], beaded structural filament protein-2 (BFSP2) [16], paired box gene 6 (PAX6) [17], paired-like homeodomain 3 (PITX3) [18], heat shock transcription factor 4 (HSF4) [19], v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog (MAF) [20], charged multivesicular body protein 4B (CHMP4B) [21], and EPH receptor A2 (EPHA2) [22].
BFSPs are lens-specific proteins belonging to the intermediate filament (IF) protein family. BFSP1 (filesin) and BFSP2 (phakinin) are the principal components of beaded filaments, which are unique cytoskeletal lens structures [23]. The distinctive sequence characteristics of filesin and phakinin make it difficult to classify them into any present group of cytoplasmic IFs. Although the biological functions of filesin and phakinin are not clear, evidence indicates that they play an important role in maintaining lens transparency during fetal development and fiber cell differentiation [23,24]. Three different mutations of BFSP2 are known to comprise autosomal dominant inherited cataract (E233△; R287W; G109A) [16,25,26]. Currently, only one study has reported recessive inheritance associated with BFSP1 mutation [27].
A five-generation Chinese family with individuals affected with bilateral congenital cataract was studied. Microsatellite markers and linkage analysis were excluded candidate genes at 1p, 1q, 2q, 3q21–25, 3q27, 10q, 11q, 11p, 12q, 13q, 16q, 17q, 20q, 21q, and 22q. Whole-exome sequencing was applied to determine mutations and a BFSP1 mutation (c.1042G>A) was found in two affected family members. Sequencing showed that this mutation was observed in all affected members of this family, but not in unaffected family members or in 200 ethnically matched controls. Here, for the first time, we report that BFSP1 mutation is associated with autosomal dominant inherited cataract.
Methods
Subjects and sample collection
Data on a five-generation Chinese family, consisting of 48 individuals, were collected at the Eye Hospital of China Medical University (China Medical University, Shenyang, China). Two hundred unrelated subjects without eye diseases were also recruited from the Eye Hospital of China Medical University as normal controls. The family history revealed 15 affected members in total (Figure 1). All available members underwent detailed ophthalmic examination, including visual acuity (Snellen chart), slit-lamp examination, and fundus and retinoscopy examination after pupil dilation. Clinical details were recorded using a standard questionnaire. Blood specimens were obtained from 20 available members (10 affected, 4 unaffected, 6 spouses). This study adhered to the provisions of the Declaration of Helsinki and was approved by the Ethics Review Board of China Medical University.
Figure 1.
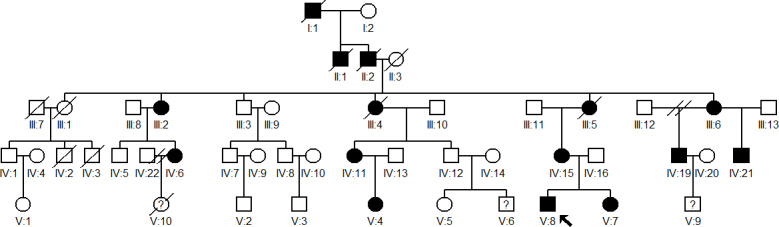
Pedigree of a five-generation Chinese family with autosomal dominant congenital cataract is shown. The proband is indicated by a black arrow. Except for the proband, all affected individuals had undergone cataract surgery within the first decade.
Genotyping and linkage analysis
Blood samples (5–10 ml) of 20 available members were collected and genomic DNA was extracted with a TIANamp DNA Blood Mini Kit (Tiangen Ltd., Beijing, China). Two or three microsatellite markers of each candidate gene were chosen based on details from the Marshfield map of the NCBI database. Ultimately, 50 microsatellite markers were analyzed for 19 known candidate genes. Microsatellites were amplified by polymerase chain reaction with a PCR-cycler (Biorad, Herculas, CA). Primers were designed with Primer 3. Amplifications were performed in 20 μl reactions containing 100 ng of genomic DNA, 0.1 μmol of each forward and reverse primer, 0.1 mM of deoxyribonucleotide, 1X PCR buffer, 0.1 mM MgSO4, and 0.5 U Taq DNA Polymerase (Life Technologies Corporation, Carlsbad, CA). The thermocycler program was as follows: initial denaturation at 94 °C for 30 s followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 54–60 °C for 30 s, and extension at 72 °C for 1 min. PCR products were pooled, mixed with loading dye containing internal size standards, denatured at 99 °C for 15 min and electrophoresed on 8% denaturing polyacrylamide gels. Finally, genotypes for each individual are documented. Two-point linkage analysis was performed by MLINK from the LINKAGE (Version 5.1) program package. Autosomal dominant inheritance with penetrance of 0.9999 and a disease gene frequency of 0.0001 were assumed. Recombination frequencies were considered to be equal between males and females for all markers.
Whole-exome sequencing
Because there was no evidence of linkage in any known loci after analysis, whole-exome sequencing was undertaken on two genomic DNA samples (IV6 and V8). Three micrograms of genomic DNA were sheared with Covaris S2 (Covaris; Woburn, MA) to achieve fragments with a mean size of 300 bp. Exome enrichment was conducted with TruSeq Exome Enrichment Kit, which is designed to target 62 Mb genome regions, including 20,794 targeted genes. Appropriate amounts of enrichment samples were pooled and sequenced on a HiSeq2000 instrument (Illumina; San Diego, CA). Sequencing data were aligned to the human genome reference (hg19) sequence using the Burrows Wheeler Alignment tool (BWA 0.5.9) [28] with default parameters. PCR duplicates were excluded with the Samtools package [16], while regional realignment and quality score recalibration were determined with the Genome Analysis Toolkit (GaTK v1.3) [27]. Variants were filtered with single nucleotide polymorphism database (dbSNPs) and the 1000 Genome database.
Mutation analysis
BFSP1, the candidate gene, which is located at 20p12.1 (GenBank ref. no. NM_001195), was analyzed with Sanger sequencing. Primers were designed according to the mutation following whole-exome sequencing analysis to amplify the mutated base and flanking sequences of exon 7 (forward primer: 5′-CAC TTC CTT TAT GTC CTC ATC CA-3′; reverse primer: 5′-GTA GCC TAG CCC CCT AGT CG-3′). Amplifications were performed in 20 μl reactions as described above. The thermal cycler program protocol was as follows: initial denaturation at 94 °C for 30 s followed by 35 cycles of denaturing at 94 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 1 min. PCR products were purified with a gel extraction kit (TaKaRa, Japan) and sequenced with an ABI 3730×l DNA analyzer (Applied Biosystems). All family members and 200 unrelated normal subjects were screened.
Bioinformatics analysis
The amino acid sequences of BFSP1 from different species (humans, mice, rats, cows, and chickens) were obtained from the NCBI GenBank, and conservation analysis was performed with the webpage Uniprot. The impact and physicochemical characteristics of mutation were predicted with PolyPhen 2.
Results
Clinical evaluation
All the affected family members had bilateral cataract. Opacities were visible in early childhood, which had a great influence on visual acuity. The proband, a 32-year-old woman, was diagnosed with bilateral cataract and presented with nuclear opacity (Figure 2). Except for the proband, all of the affected individuals underwent cataract surgery within the first decade; the proband had undergone iridectomy in childhood. There was no evidence of other systemic or ocular defects.
Figure 2.

Slit-lamp photograph of the lens of an affected individual (IV6) with nuclear cataract. The individual underwent iridectomy in childhood in both eyes.
Mutation analysis
There was no evidence of linkage at any known loci following linkage analysis, so whole-exome sequencing was used. On average, 85% of the exomal regions of genomic DNA samples (IV6 and V8) were covered with a sequencing depth of more than 61×. After all the candidate genes were screened, a novel mutation at exon 7 of BFSP1 (c.1042G>A) was found (Figure 3). The mutation was not found in any unaffected member or among 200 unrelated normal subjects.
Figure 3.
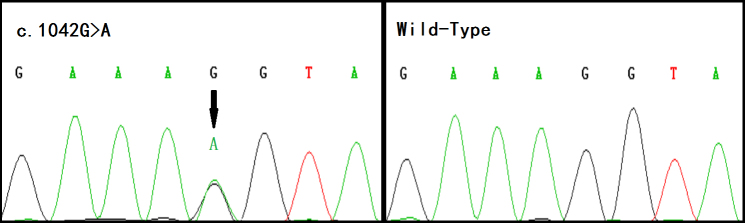
Sequence chromatogram from an affected individual shows the heterozygous c.1042G>A missense mutation in beaded structural filament protein-1 (BFSP1). The arrow indicates the mutation spot in the sequence of the affected individual.
Bioinformatics analysis
The results indicated a heterozygous G→A transversion at c.1042 in exon 7 of BFSP1 in all affected family members that resulted in the substitution of a wild-type aspartate (Asp) to an asparagine (Asn, p.D348N). After alignment of the BFSP1, the protein sequence revealed that the Asp at the 348th amino acid position was highly conserved among many species (Figure 4). Moreover, the functional effects of the D348N mutation were predicted by PolyPhen 2. The results indicated that the mutation may possibly be damaging, with a score of 0.980.
Figure 4.
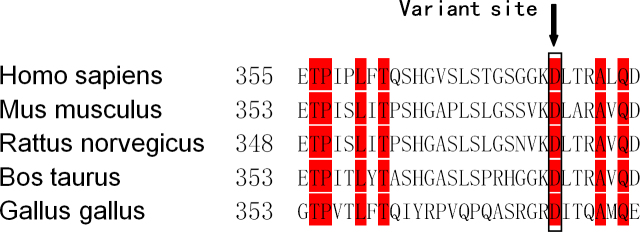
The multiple-sequence alignment of BFSP1 from different species is shown. The Asp348 residue (highlighted with red boxes) is located within a highly conserved region.
Discussion
Here, we report for the first time that a BFSP1 mutation is associated with autosomal dominant congenital cataract in humans. In this study, a novel mutation (c.1042G>A) at exon 7 of BFSP1 was detected in a Chinese family. The mutation created a substitution of an Asp to an Asn (p.D348N). This sequence change cosegregated with a nuclear-cataract phenotype in the family and was not observed in 200 unrelated normal individuals. In the literature, there is currently only one BFSP1 mutation study (c.736–1384_c.957–66 del) involving an autosomal recessive cataract family [27]. In that study, homozygous affected members of an Indian family presented with cotton-like cortical opacities in the anterior cortex. The difference in cataract phenotype between this family and the Indian family may demonstrate that genes or environmental factors modify the expression of the primary mutation associated within cataracts [23]. Considering the development of early onset age-related nuclear cataract in the Ramachandran et al. study, the c.1042G>A mutation discovered here may play a more important role in maintaining lens transparency and physiological function.
Filesin and phakinin are intermediate filaments and form beaded filament protein, which is exclusively expressed in lens fiber cells. Like other IFs, filesin consists of a central α-helical rod flanked by nonhelical head and tail domains. Studies from BFSP1 and BFSP2 knockout mice suggest that the beaded filament is required to maintain cell morphology and three-dimensional membrane architecture [23,24].
There are three types of BFSP2 mutation reported as the genetic base of inherited cataract. A deletion mutation in exon 3 (E233Δ) causes autosomal dominant congenital cataract [26] and other families with the mutation (R287W; G1019A) showed childhood cataract [16,25]. Another mutation, E233Δ, was reported in a Chinese family and was associated with childhood cataract and myopia [24]. However, in the present study there was no evidence of myopia in affected members. Despite phenotypic differences in the BFSP1 and BFSP2 mutations, the present results further confirm that beaded filaments are essential for lens transparency and lens homeostasis.
According to the GenBank (Q12934), the mutation (c.1042G>A) highlighted in the present study family is localized to the tail region of filesin, which along with phakinin, has an important effect on beaded filament formation. The mutation found by Ramachandran et al. is also predicted to result in the loss of the tail region of the protein. Moreover, a study of BFSP1 gene knockout mice [23] found that the lenses of knockout mice scatter more light than those of the wild type. Alignment of the BFSP1 protein sequence among different species revealed that the Asp residue at position 348 is highly conserved. Furthermore, PolyPhen 2 analysis indicated that D348N may possibly be damaging. In addition, with the substitution of Asp for Asn in the protein sequence, Asp-Leu-Thr becomes Asn-Leu-Thr, forming a potential N-glycosylation site (N-X-S/T, where X is any amino acid except for proline). As such, filesin may be glycosylated, hindering BFSP1 beaded filament assembly, ultimately leading to cataract. Because wild-type human filesin has not been crystallized and there is no homologous sequence of filesin, a three-dimensional model to predict effects on protein function cannot be made with Swissmodel software. However, taken together with previous research, the results of the present study add to the suspected pathogenicity of the BFSP1 mutation in human autosomal dominant congenital cataract.
Whole-exome sequencing was used in this study. Compared with genome-wide scans or other genetic methods, there are definite advantages to whole-exome sequencing, including its high output, ease of use, and cost effectiveness [28]. In this study, after sequencing data were aligned to the human genome reference (hg19) sequence, the suspect BFSP1 mutation (c.1042G>A) associated with congenital cataract was analyzed first because a study indicated that BFSP1 mutation is associated with autosomal recessive congenital cataract; this strategy of mutation screening proved time-saving and effective.
In conclusion, the novel missense BFSP1 (c.1042G>A) mutation in a Chinese family is associated with isolated autosomal dominant inherited cataract. The present study also further highlighted the congenital cataract genotype and phenotype in humans. This is the first report of a BFSP1 mutation associated with autosomal dominant inherited cataract and underscores the important role BFSP1 has in maintaining lens opacity.
Acknowledgments
We thank the members for their participation for analysis in this study. Thanks are also due to Tianxiao Zhang and Di Wu for their technical assistance. Contract grant sponsor: The National Natural Science Foundation of China (to Tianxiao Zhang); Contract grant number: 81100700.
References
- 1.Gilbert C, Foster A. Childhood blindness in the context of VISION 2020–the right to sight. Bull World Health Organ. 2001;79:227–32. [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert SR, Drack AV. Infantile cataracts. Surv Ophthalmol. 1996;40:427–58. doi: 10.1016/s0039-6257(96)82011-x. [DOI] [PubMed] [Google Scholar]
- 3.François J. Genetics of cataract. Ophthalmologica. 1982;184:61–71. doi: 10.1159/000309186. [DOI] [PubMed] [Google Scholar]
- 4.Litt M, Kramer P, LaMorticella DM, Murphey W, Lovrien EW, Weleber RG. Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA. Hum Mol Genet. 1998;7:471–4. doi: 10.1093/hmg/7.3.471. [DOI] [PubMed] [Google Scholar]
- 5.Berry V, Francis P, Reddy MA, Collyer D, Vithana E, MacKay I, Dawson G, Carey AH, Moore A, Bhattacharya SS, Quinlan RA. Alpha-B crystallin gene (CRYAB) mutation causes dominant congenital posterior polar cataract in humans. Am J Hum Genet. 2001;69:1141–5. doi: 10.1086/324158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reddy MA, Bateman OA, Chakarova C, Ferris J, Berry V, Lomas E, Sarra R, Smith MA, Moore AT, Bhattacharya SS, Slingsby C. Characterization of the G91del CRYBA1/3-crystallin protein: a cause of human inherited cataract. Hum Mol Genet. 2004;13:945–53. doi: 10.1093/hmg/ddh110. [DOI] [PubMed] [Google Scholar]
- 7.Billingsley G, Santhiya ST, Paterson AD, Ogata K, Wodak S, Hosseini SM, Manisastry SM, Vijayalakshmi P, Gopinath PM, Graw J, Heon E. CRYBA4, a novel human cataract gene, is also involved in microphthalmia. Am J Hum Genet. 2006;79:702–9. doi: 10.1086/507712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackay DS, Boskovska OB, Knopf HL, Lampi KJ, Shiels A. A nonsense mutation in CRYBB1 associated with autosomal dominant cataract linked to human chromosome 22q. Am J Hum Genet. 2002;71:1216–21. doi: 10.1086/344212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gill D, Klose R, Munier FL, McFadden M, Priston M, Billingsley G, Ducrey N, Schorderet DF, Heon E. Genetic heterogeneity of the Coppock-like cataract: a mutation in CRYBB2 on chromosome 22q11.2. Invest Ophthalmol Vis Sci. 2000;41:159–65. [PubMed] [Google Scholar]
- 10.Li XQ, Cai HC, Zhou SY, Yang JH, Xi YB, Gao XB, Zhao WJ, Li P, Zhao GY, Tong Y, Bao FC, Ma Y, Wang S, Yan YB, Lu CL, Ma X. A novel mutation impairing the tertiary structure and stability of gammaC-crystallin (CRYGC) leads to cataract formation in humans and zebrafish lens. Hum Mutat. 2012;33:391–401. doi: 10.1002/humu.21648. [DOI] [PubMed] [Google Scholar]
- 11.Gu F, Li R, Ma XX, Shi LS, Huang SZ, Ma X. A missense mutation in the gammaD-crystallin gene CRYGD associated with autosomal dominant congenital cataract in a Chinese family. Mol Vis. 2006;12:26–31. [PubMed] [Google Scholar]
- 12.Sun H, Ma Z, Li Y, Liu B, Li Z, Ding X, Gao Y, Ma W, Tang X, Li X, Shen Y. Gamma-S crystallin gene (CRYGS) mutation causes dominant progressive cortical cataract in humans. J Med Genet. 2005;42:706–10. doi: 10.1136/jmg.2004.028274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackay D, Ionides A, Kibar Z, Rouleau G, Berry V, Moore A, Shiels A, Bhattacharya S. Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet. 1999;64:1357–64. doi: 10.1086/302383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shiels A, Mackay D, Ionides A, Berry V, Moore A, Bhattacharya S. A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant “zonular pulverulent” cataract, on chromosome 1q. Am J Hum Genet. 1998;62:526–32. doi: 10.1086/301762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berry V, Francis P, Kaushal S, Moore A, Bhattacharya S. Missense mutations in MIP underlie autosomal dominant 'polymorphic' and lamellar cataracts linked to 12q. Nat Genet. 2000;25:15–7. doi: 10.1038/75538. [DOI] [PubMed] [Google Scholar]
- 16.Ma X, Li FF, Wang SZ, Gao C, Zhang M, Zhu SQ. A new mutation in BFSP2 (G1091A) causes autosomal dominant congenital lamellar cataracts. Mol Vis. 2008;14:1906–11. [PMC free article] [PubMed] [Google Scholar]
- 17.Dansault A, David G, Schwartz C, Jaliffa C, Vieira V, de la Houssaye G, Bigot K, Catin F, Tattu L, Chopin C, Halimi P, Roche O, Van Regemorter N, Munier F, Schorderet D, Dufier JL, Marsac C, Ricquier D, Menasche M, Penfornis A, Abitbol M. Three new PAX6 mutations including one causing an unusual ophthalmic phenotype associated with neurodevelopmental abnormalities. Mol Vis. 2007;13:511–23. [PMC free article] [PubMed] [Google Scholar]
- 18.Semina EV, Ferrell RE, Mintz-Hittner HA, Bitoun P, Alward WL, Reiter RS, Funkhauser C, Daack-Hirsch S, Murray JC. A novel homeobox gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD. Nat Genet. 1998;19:167–70. doi: 10.1038/527. [DOI] [PubMed] [Google Scholar]
- 19.Forshew T, Johnson CA, Khaliq S, Pasha S, Willis C, Abbasi R, Tee L, Smith U, Trembath RC, Mehdi SQ, Moore AT, Maher ER. Locus heterogeneity in autosomal recessive congenital cataracts: linkage to 9q and germline HSF4 mutations. Hum Genet. 2005;117:452–9. doi: 10.1007/s00439-005-1309-9. [DOI] [PubMed] [Google Scholar]
- 20.Jamieson RV, Perveen R, Kerr B, Carette M, Yardley J, Heon E, Wirth MG, van Heyningen V, Donnai D, Munier F, Black GC. Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum Mol Genet. 2002;11:33–42. doi: 10.1093/hmg/11.1.33. [DOI] [PubMed] [Google Scholar]
- 21.Shiels A, Bennett TM, Knopf HL, Yamada K, Yoshiura K, Niikawa N, Shim S, Hanson PI. CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am J Hum Genet. 2007;81:596–606. doi: 10.1086/519980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiels A, Bennett TM, Knopf HL, Maraini G, Li A, Jiao X, Hejtmancik JF. The EPHA2 gene is associated with cataracts linked to chromosome 1p. Mol Vis. 2008;14:2042–55. [PMC free article] [PubMed] [Google Scholar]
- 23.Alizadeh A, Clark J, Seeberger T, Hess J, Blankenship T, FitzGerald PG. Targeted deletion of the lens fiber cell-specific intermediate filament protein filensin. Invest Ophthalmol Vis Sci. 2003;44:5252–8. doi: 10.1167/iovs.03-0224. [DOI] [PubMed] [Google Scholar]
- 24.Alizadeh A, Clark JI, Seeberger T, Hess J, Blankenship T, Spicer A, FitzGerald PG. Targeted genomic deletion of the lens-specific intermediate filament protein CP49. Invest Ophthalmol Vis Sci. 2002;43:3722–7. [PubMed] [Google Scholar]
- 25.Conley YP, Erturk D, Keverline A, Mah TS, Keravala A, Barnes LR, Bruchis A, Hess JF, FitzGerald PG, Weeks DE, Ferrell RE, Gorin MB. A juvenile-onset, progressive cataract locus on chromosome 3q21-q22 is associated with a missense mutation in the beaded filament structural protein-2. Am J Hum Genet. 2000;66:1426–31. doi: 10.1086/302871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cui X, Gao L, Jin Y, Zhang Y, Bai J, Feng G, Gao W, Liu P, He L, Fu S. The E233del mutation in BFSP2 causes a progressive autosomal dominant congenital cataract in a Chinese family. Mol Vis. 2007;13:2023–9. [PubMed] [Google Scholar]
- 27.Ramachandran RD, Perumalsamy V, Hejtmancik JF. Autosomal recessive juvenile onset cataract associated with mutation in BFSP1. Hum Genet. 2007;121:475–82. doi: 10.1007/s00439-006-0319-6. [DOI] [PubMed] [Google Scholar]
- 28.Jakobs PM, Hess JF, FitzGerald PG, Kramer P, Weleber RG, Litt M. Autosomal-dominant congenital cataract associated with a deletion mutation in the human beaded filament protein gene BFSP2. Am J Hum Genet. 2000;66:1432–6. doi: 10.1086/302872. [DOI] [PMC free article] [PubMed] [Google Scholar]