

Abstract
α-Tocopherol is a lipid-soluble antioxidant that is specifically required for reproduction and embryogenesis. However, since its discovery, α-tocopherol’s specific biologic functions, other than as an antioxidant, and the mechanism(s) mediating its requirement for embryogenesis, remain unknown. As an antioxidant, α-tocopherol protects polyunsaturated fatty acids (PUFAs) from lipid peroxidation. α-Tocopherol is likely required during embryonic development to protect PUFAs that are crucial to development, specifically arachidonic (ARA) and docosahexaenoic (DHA) acids. Additionally, ARA and DHA are metabolized to bioactive lipid mediators via lipoxygenase enzymes and α-tocopherol may directly protect, or it may mediate the production and/or actions of these lipid mediators. In this review, we discuss how α-tocopherol 1) prevents the nonspecific, radical-mediated peroxidation of PUFAs, 2) functions within a greater antioxidant network to modulate the production and/or function of lipid mediators derived from 12- and 12/15-lipoxygenase and 3) modulates 5-lipoxygenase activity. The application and implication of such interactions with be discussed in the context α-tocopherol requirements during embryogenesis.
α-Tocopherol and Lipid Peroxidation
α-Tocopherol, a lipid-soluble antioxidant, is one of the eight vitamin E forms synthesized by plants [1], and is the only form that meets human vitamin E requirements [2]. α-Tocopherol scavenges peroxyl radicals during the propagation of lipid peroxidation (FIGURE 1), and is termed a chain-breaking antioxidant because it prevents the chain reaction of lipid peroxidation, but it does not prevent the formation of the initial lipid peroxyl radical [3]. α-Tocopherol is particularly enriched in neuronal tissue, especially the brain, where it is tenaciously retained during inadequate vitamin E intake even after the peripheral tissues become α-tocopherol-depleted [4]. Overt vitamin E deficiency occurs rarely in humans, but does occur in patients with fat malabsorption syndromes or genetic defects in the hepatic α-tocopherol transfer protein (α-TTP) [5] and in severe malnutrition [6, 7]. Humans with vitamin E deficiency present initially with a mild sensory neuropathy, which leads to a progressive, peripheral neuropathy caused by a dying back of large-caliber, sensory neurons, that advances to a spinocerebellar ataxia and ultimately death [5].
Figure 1. Lipid peroxidation and the antioxidant network.
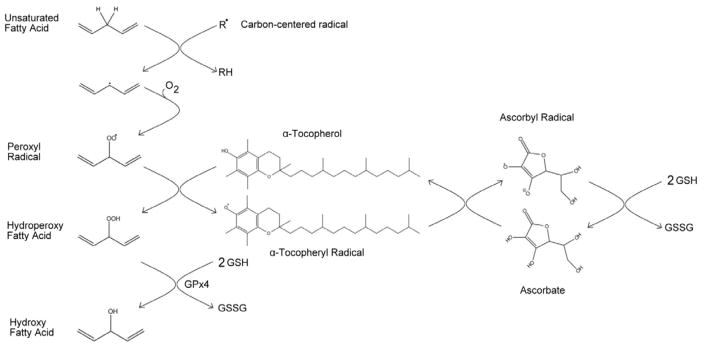
During the propagation stage of lipid peroxidation, a carbon-centered radical (R·) abstracts an allylic hydrogen from a neighboring, unsaturated fatty acid. Molecular oxygen reacts with the fatty acid radical, generating a peroxyl radical, which can be reduced by α-tocopherol, creating a hydroperoxy fatty acid and the α-tocopheryl radical. The hydroperoxy fatty acids are converted to hydroxy fatty acids via phospholipid hydroperoxide glutathione peroxidase (GPx4), using two glutathiones (GSH) as the reducing agents and creating oxidized gluthatione disulfide (GSSG). To replenish the α-tocopherol, the α-tocopheryl radical is reduced by ascorbate. The oxidized ascorbyl radical is subsequently reduced back to ascorbate via GSH.
α-Tocopherol protects cellular membranes from lipid peroxidation in association with a larger antioxidant network (FIGURE 1). Once α-tocopherol reduces lipid peroxyl radicals to lipid hydroperoxides, the selenium-dependent enzyme, phospholipid hydroperoxide glutathione peroxidase (GPx4), converts the hydroperoxides to the less toxic lipid hydroxides at the expense of glutathione. Ascorbate (vitamin C) reduces the α-tocopherol radical, regenerating active α-tocopherol [8]. Subsequently, ascorbate is regenerated at the expense of glutathione. Maintenance of this antioxidant network is crucial to protect cellular membranes against radical-mediated degradation [9]. For example, vitamin E disappears faster from plasma in individuals who smoke, but vitamin C supplementation corrects the rapid α-tocopherol disappearance [10, 11]. In adult zebrafish, chronic vitamin E deficiency causes a secondary depletion of vitamin C, and concomitantly, a severe degeneration of skeletal muscle [12].
α-Tocopherol protects polyunsaturated fatty acids (PUFAs), notably arachidonic acid (ARA, 20:4 ω-6) and docosahexaenoic acid (DHA, 22:6 ω-3). Indeed, human α-tocopherol requirements increase in parallel with dietary PUFA consumption or with an increasing index of fatty acid unsaturation [13]. α-Tocopherol is postulated to co-localize with PUFA-enriched phospholipid domains of the cell membrane [14]. Moreover, a peroxyl radical generated from the PUFA will localize to the air-water interface, where the hydroxyl group of α-tocopherol intercepts the radical and reduces it [15]. Studies in animals fed experimental diets have demonstrated the importance of α-tocopherol in protecting PUFAs. For example, when zebrafish are fed diets that require them to synthesize ARA and DHA from their respective precursors, the vitamin E deficient fish have decreased percentages of total ω-6 and ω-3 PUFAs compared with those fed a vitamin E-sufficient diets [16], suggesting that vitamin E protects these long chain PUFAs. Similarly, feeding fish oil to pregnant rats decreased fetal brain α-tocopherol concentrations [17].
Is vitamin E deficiency a significant cause of spontaneous embryonic death?
In 1922, α-tocopherol was discovered because vitamin E deficient, pregnant rats fed rancid fat failed to carry their offspring to term [18]. α-Tocopherol and the α-tocopherol transfer protein (α-TTP) have critical roles in embryonic development [19]. α-TTP is expressed in the human yolk sac [20]; therefore, we used zebrafish (Danio rerio) embryos, which abundantly express α-TTP by 48 hour post-fertilization (hpf) and up-regulate α-TTP in response to oxidative stress [21]. Remarkably, adult α-tocopherol deficient zebrafish could spawn and produce viable eggs, but within days the embryos suffered developmental impairment and increased mortality [22]. Similar findings have been reported for α-TTP knockout mice [23]. Importantly, we found that the zebrafish embryonic brain accumulates α-tocopherol and expresses α-TTP. α-TTP knockdown caused head and eye malformations prior to 15 hpf [19]. Intriguingly, the α-tocopherol requirement for neurologic development in zebrafish coincides with increased synthesis of highly peroxidizable lipids by the zebrafish embryo, evidenced by increased Elovl4 [24] and Elovl5 [25] expression in the head/brain region.
Our knowledge of how vitamin E is delivered to the brain is very limited. α-Tocopherol is transported in the circulation by all lipoproteins and is delivered by pathways that deliver other lipids to cells [26]. α-Tocopherol is readily exchanged between HDL and apoB-containing lipoproteins [27]. HDL, however, is likely more important in brain development, because the central nervous system (CNS) does not contain apoB, but rather large apoE particles serve this function [28]. Scavenger receptor class B, type I (SR-BI) facilitates selective uptake of HDL-associated α-tocopherol by the blood brain barrier in vitro [29]. In vivo the cerebrospinal fluid (CSF) only contains HDL [30] and CSF-HDL contain α-tocopherol [31].
The importance of both HDL and apoB-containing lipoproteins is illustrated in patients with the autosomal recessive disorder, abetalipoproteinemia. These patients have extraordinarily low circulating lipids because they have only high density lipoproteins (HDL) and no lipoproteins containing apolipoprotein B (apoB; e.g. chylomicrons, very low density (VLDL), or low density (LDL)) [32]. Vitamin E deficiency in humans was first described in abetalipoproteinemic patients [33]. When these patients were supplemented with large vitamin E doses (150 mg/kg body weight), they did not experience the neurologic defects seen in unsupplemented patients; moreover, they were able to bear normal children [34, 35].
Abetalipoproteinemia is distinct from homozygous hypobetalipoproteinemia [36]. The latter is caused by a defect in the apoB gene [36], while abetalipoproteinemia is caused by a defect in the gene for the microsomal triglyceride transfer protein (MTP) [37]. MTP lipidates apoB and is critical in humans for normal secretion of chylomicrons and VLDL [38]. Importantly, both cause the virtual absence of circulating apoB-containing lipoproteins, which then results in vitamin E deficiency. Knockdown of either apoB [39, 40] or MTP [41] is embryonically lethal in mice.
Importantly, mouse apoB or MTP knockout embryos had neural tube defects, which were proposed to result from insufficient α-tocopherol [39–41], while mice expressing either apoB100 or apoB48 had normal embryos and did not have vitamin E deficiency [42]. Similarly, α-TTP knockout in mice was also embryonically lethal, but the pregnancy could be rescued by feeding the mother large amounts of vitamin E or synthetic antioxidant (presumably salvaging the minor amounts of remaining tissue α-tocopherol fed) [23]. Thus, there is a close relationship between α-tocopherol and normal embryonic development.
Human vitamin E deficiency caused by inadequate diets is largely assumed to be non-existent in the US. The USDA’s 2010 Dietary Guidelines (http://www.cnpp.usda.gov/dietaryguidelines.htm) gave short shrift to the observation that 96% of women do not consume sufficient α-tocopherol to meet the estimated average requirement (EAR) [43]. The expectation from the nutrition community is that requirements are too high and that the amount of α-tocopherol needed by humans to prevent deficiency is very low, thus vitamin E deficiency as a result of inadequate intake in humans is unlikely. However, our studies using the embryonic zebrafish model shows that the embryo with inadequate α-tocopherol undergoes severe developmental defects [22], and the α-TTP knockdown causes even more severe defects, especially impaired head/brain and eye formation [22]. Likely, a vitamin E continuum exists, where sufficient α-tocopherol at critical time points allows the embryo to progress to the next developmental stage.
Our discoveries in zebrafish embryos also suggest that α-tocopherol is critical in humans for embryonic development prior to when a woman knows she is pregnant (e.g. hours post fertilization (hpf) for zebrafish [44], and days for rats [45] or humans [46–48]). This early requirement for vitamin E is analogous to the situation in humans where there is an early requirement for folic acid to prevent neural tube defects. Surprisingly, folic acid supplements were not as effective in preventing neural tube defects as were the combination of folic acid and multivitamin, as a review of five human trials showed [49]. Specifically, the Hungarian trial to evaluate neural tube defects contained 15 mg vitamin E and various other vitamins [50]. Hypothetically, some of the neural tube defects observed in humans are due to inadequate α-tocopherol status. Studies in China suggest, based on maternal and cord blood vitamin E levels, that higher infant vitamin E status at birth improves cognitive function assessed at age 2 y [51], again emphasizing the importance of good vitamin E status during pregnancy for brain development.
PUFAs and Lipid mediators derived from PUFAs
Since its discovery more than 90 years ago [18], α-tocopherol’s specific in vivo biologic functions, and the mechanism(s) mediating its requirement, remain unknown. The unclear requirement of α-tocopherol for reproduction exemplifies our present gap in knowledge. It is possible that α-tocopherol is required during embryonic development specifically to protect ARA and DHA, or mediate the production and/or actions of bioactive lipid mediators derived from these PUFAs.
ARA and DHA are required for proper embryonic development. DHA is highly enriched in the central nervous system (CNS), comprising upwards of 50% of CNS PUFA [52]. DHA deficiency during pregnancy distinctly and adversely affects neurodevelopment. For example, DHA deficiency inhibits fetal neurogenesis [53, 54] and synaptogenesis [53], alters the synaptic proteome [55], serotoninergic neurotransmission [56], dopaminergic regulatory protein composition [57], neuronal phospholipid composition [58] and signaling [59], and impairs neuronal migration [60]. Adverse developmental outcomes caused by DHA inadequacy persist even after repletion with DHA [61, 62], demonstrating long-lasting effects of embryonic DHA deficiency regardless of later restitution with an adequate diet.
ARA is the most abundant 3–6 neuronal fatty acid throughout gestation and postnatal development [63]. An appropriate balance between DHA and ARA is required during neonatal development, as infant formula supplemented with DHA, but lacking ARA, impaired infant growth [64]. Indeed, higher infant ARA concentrations are positively correlated with infant birth weight and length [65]. Conversely, ARA inadequacy is associated with delayed postnatal development and reduced growth [17, 66].
Production of bioactive lipid mediators derived from ARA and DHA can occur through enzymatic peroxidation or non-enzymatic radical-mediated peroxidation. The three known enzymatic pathways that act upon PUFAs include the cyclooxygenase (COX; two major isoforms: COX-1 and COX-2), lipoxygenase (LOX; four major isoforms: 5-LOX, platelet-type 12-LOX (12-LOX), leukocyte-type 12-LOX (12/15-LOX), and 15-LOX [67]), and cytochrome P450 (CYP450) pathways. The oxidation of ARA gives rise to hydroxyeicosatetraenoic acids (HETEs) and the eicosanoids, a class of lipids, which encompasses the prostaglandins, prostacyclins, thromboxanes, leuokotrienes, lipoxins, and isoprostanes (FIGURE 2). Similarly, the oxidation of DHA gives rise to the docosanoids, which include the resolvins (D-series), neuroprotectins, and maresins, as well as intermediary monohydroxy lipids termed HDHAs. The synthesis of docosanoids requires the complex coordination of multiple enzymes, including LOXs and COXs [68].
Figure 2.

Lipid mediators derived from arachidonic acid.
Arachidonic acid (ARA) is cleaved from membrane phospholipids by phospholipase A2. The lipoxygenase (LOX) pathways convert ARA to 5-hydroperoxyeicosatetraenoic acid (5-HpETE) via 5-LOX, 12-hydroperoxyeicosatetraenoic acid (12-HpETE) via 12-LOX, or 15-hydroperoxyeicosatetraenoic acid (15-HpETE) via 15-LOX (not pictured; LOXs can also oxidize membrane ARA without prior release by phospholipase A2 [110]). 12/15-LOX can generate 12- or 15-HpETE (not pictured). The HpETEs are reduced by a peroxidase to produce 5-HETE and 12-HETE. 5-LOX converts the peroxide of 5-HpETE to an epoxide, generating leukotriene A4 (LTA4). LTA4 is the precursor for leukotriene B4 (LTB4), lipoxins, and cysteinyl leukotrienes (CysLTs). The cyclooxygenase (COX) enzymes, which are either inducible (COX-2) or constitutively expressed (COX-1), convert ARA to prostaglandin G2 (PGG2), which is a precursor for other prostaglandins, prostacyclins, and thromboxanes. Non-enzymatic, radical-mediated peroxidation of ARA yields the isoprostane class of signaling molecules (8-F2-isoprostane pictured). Finally, cytochrome P450s (CYP450) convert ARA to 20-HETE or the epoxyeicosatrienoic acids (-EETs; 8,9-EET pictured).
An emerging body of evidence indicates that oxidation of ARA (and potentially DHA) is needed during embryogenesis. For example, knockdown of COX-1 in zebrafish embryos led to gastrulation arrest [69], while inhibition of COX-1 after completion of gastrulation caused defective vascular tube formation [70]. In mice, COX-2 regulates ovulation and embryonic implantation [71], however no effect of COX-2 knockdown was noted in zebrafish embryos [69, 70]. In zebrafish embryos, inhibition of five lipoxygenase-activating protein (FLAP), a membrane protein required for 5-LOX function, resulted in pericardial edema and reduced intersegmental vasculature and vessel/axial blood flow [72]. Neuronal growth-cone collapse requires functional 12/15-LOX [73, 74], as does hematopoietic stem cell function [75] and epidermal barrier formation [76]. Knockdown of 12/15-LOX in zebrafish embryos diminished the production of ARA and DHA lipid mediators, altered PUFA profiles, and caused abnormal brain, eye, and tail development by 24 hpf [76, 77]. Interestingly, studies in 12/15-LOX knockout mice are somewhat different than the findings generated in zebrafish. In one study, deletion of 12/15-LOX altered bone mass in developing mice [78]; however, in another study 12/15-LOX deficient mice had no gross morphologic defects and were born at the expected Mendelian ratios [79], suggesting that 12/15-LOX is not needed for mouse embryonic development. Similarly, deletion of platelet 12-LOX in mice resulted in expected Mendelian ratios in offspring [80], also suggesting that platelet 12-LOX is also not needed for mouse embryonic development. However, platelet 12-LOX knockout 6–10 wk old mice have abnormal epidermal permeability barrier function [81].
α-Tocopherol modulates the actions of lipid mediators
Programmed cell death is a key function throughout embryonic development, regulating the formation and remodeling of complex multicellular tissues, but it must be closely regulated. In numerous experiments, PUFAs, LOX, and the antioxidant network have been implicated in the induction and control of apoptosis. Mechanisms from such studies could be applied to the processes occurring during embryogenesis and lend insight into the requirement of α-tocopherol for reproduction. For example, glutathione depletion has long-been recognized to induce neuronal cell death. Specifically, glutathione depletion leads to an increase in 12-LOX activity, which is a requisite step for neuronal apoptosis induced by glutathione depletion [82–84]. Furthermore, neuronal apoptosis following knockdown of GPx4 requires 12/15-LOX activity and induces the apoptosis-inducing factor (AIF) translocation from the inner mitochondrial membrane to the nucleus [85]. Other mechanisms by which LOX activity and its products may induce apoptosis include altering membrane fluidity and permeability, increasing calcium influx into the cell, disruption of mitochondrial integrity, and release of cytochrome C [86]. Notably, α-tocopherol entirely prevents 12/15-LOX mediated cell death [85]. In neuroblastoma cells, 15-LOX converts DHA to a series of hydroperoxy DHAs, including 4-HpDHA, 7-HpDHA, 14-HpDHA and 17-HpDHA [87]. Exposure to either DHA or 17-HpDHA, but not the monohydroxy DHA product 17-HDHA, potentiates apoptosis in neuroblastoma cells [87, 88]. Notably, co-treatment of neuroblastoma cells with α-tocopherol prevents apoptosis induced by DHA [88] or 17-HpDHA [87]. Similarly, ARA depletes intracellular glutathione and induces apoptosis in cultured cortical neurons, which is attenuated by LOX inhibitors or the α-Tocopherol analog, trolox [89], suggesting that ARA-derived 12-LOX products are responsible for apoptosis. Indeed, 12-HETE, the major ARA derived 12-LOX product, mimics ARA-induced apoptosis [89]. Given that neuronal tissue is highly enriched with DHA, it is notable that DHA hydroperoxides are more cytotoxic in neuroblastoma cells than are hydroperoxides derived from either linoleic acid or ARA [90]. This evidence illustrates important interactions between α-tocopherol, ARA and DHA, LOX, and hydroperoxy fatty acids, suggesting that these interactions mediate neuronal function. The implications of these findings are that regulation of lipid mediators that control apoptosis are essential for embryonic development and thus may explain the requirement for α-tocopherol to protect these key regulators. Notably, when the α-tocopherol transfer protein was knocked down in zebrafish embryos, the brain failed to form appropriately, suggesting that apoptosis was dysregulated [91].
12/15-LOX, 5-LOX and α-tocopherol
Vitamin E must be administered to the mother on post-fertilization days 5 to 9 to prevent fetal resorption in vitamin E-deficient rodents [92, 93]. Interestingly, this is the same critical period when the 12/15-LOX pathway appears to mediate implantation [94] and when GPx4-knockout mice embryos are resorbed [95]. Importantly, in the studies discussed above, 5-LOX and COX inhibitors did not prevent neuronal cell death induced by ARA or DHA in vitro [82, 83, 85, 89]; only 12/15-LOX inhibitors and α-tocopherol rescued the cells from death. These findings suggest that neuronal α-tocopherol is required for protection against excessive 12/15-LOX activity and PUFA peroxidation. Additionally, given the involvement of the larger antioxidant network (i.e. GPx4 and glutathione), the above studies suggest that this protection is mediated through an oxidative stress mechanism.
In contrast to the role of α-tocopherol mediating 12/15-LOX-dependent apoptosis in neuronal cells, the interactions of α-tocopherol and 5-LOX activity appears to be important in inflammation, but the mechanisms are unclear. Using 5-LOX isolated from potato tubers, α-tocopherol inhibited the enzyme with an IC50 of 5 micromolar and was apparently irreversibly bound to the enzyme [96]; however, these were studies using plant material. More recently, the inhibitory effect of α-tocopherol on human 5-LOX activity has been confirmed [97]. Additionally, α-tocopherol supplementation was found to suppress 5-LOX activity in peripheral blood mononuclear cells from end-stage renal disease patients undergoing hemodialysis [98–100]. Leukotriene B(4) (LTB(4)) is one of the products of the 5-LOX pathway, and has been demonstrated to increase tumor necrosis factor-alpha (TNF-alpha) and interleukin-1 beta (IL-1 beta) protein levels in fibroblasts [101]. α-Tocopherol inhibits LTB(4) production by human blood neutrophils or differentiated HL-60 cells, as does δ;-tocopherol and 13′-carboxy-δ;-Tocopherol [97]. The various vitamin E forms did not inhibit 5-LOX directly, but rather impaired intracellular calcium increase and influx, as well as the 5-LOX translocation from cytosol to the nucleus, a key event for 5-LOX activation [97]. α-Tocopherol also reduced the release of TNF-alpha and IL-1 beta from human activated monocytes [102, 103], an effect that was abrogated by addition of LTB(4). DHA also inhibits the production of IL-1 beta and TNF-alpha by monocytes, via inhibition of 5-LOX activity and LTB(4) synthesis [104], likely through substrate competition with ARA for 5-LOX [105], or by decreasing the availability of ARA. Thus, it is clear that α-tocopherol inhibits 5-LOX, however the mechanisms remain unclear. 5-LOX may be directly inhibited by α-tocopherol, other forms of vitamin E or their metabolites. It is also equally likely that α-tocopherol inhibits the production of various oxidized lipid products and it is the oxidized lipids that stimulate 5-LOX activity.
α-Tocopherol-deficient zebrafish embryos have increased levels of nonspecific (i.e. malondialdehde) lipid peroxidation products as compared with α-tocopherol sufficient embryos, confirming the role of α-tocopherol in radical-mediated lipid peroxidation. Interestingly, α-tocopherol deficient embryos have reduced mRNA expression of 5-LOX (Miller GM et al, unpublished data), suggesting this pathway is important and that α-tocopherol deficiency during embryogenesis may affect the transcription and/or mRNA processing of 5-LOX in addition to its activity. However, specific oxidized lipids produced via 5-LOX need to be investigated in α-tocopherol deficient embryos to determine if this pathway mediates the requirement of α-tocopherol for embryogenesis.
Implications and Future Directions
A complex interaction takes place between α-tocopherol, PUFAs, and lipid mediators. Apparently, α-tocopherol 1) prevents the nonspecific, radical-mediated peroxidation of PUFAs, 2) functions within a greater antioxidant network to modulate the production and/or function of lipid mediators derived from 12-LOX and 12/15-LOX, and 3) from 5-LOX. All of these mechanisms likely mediate the requirement of α-tocopherol for embryogenesis. The above evidence of the interaction between α-tocopherol, PUFAs, and lipid mediators warrants a new perspective in vitamin E research. Viewing oxidized PUFAs as only an outcome of α-tocopherol deficiency is too simplistic; oxidation of PUFAs gives rise to lipid mediators with potent physiological roles that are not simply non-specific by-products of lipid peroxidation. Furthermore, current guidelines recommend increasing ω-3 consumption during pregnancy and as a preventative/treatment measure for other diseases (ex: cardiovascular health [106]). The recommendations to increase PUFA intake are occurring concurrent with reports regarding the negative effects of α-tocopherol on health outcomes (ex: prostate [107] and bone health [108]). It has been estimated by the USDA that 96% of women do not meet the dietary recommended intake for vitamin E [109]. It is concerning given the interaction between α-tocopherol, PUFAs, and lipid mediators, and the possible applications to embryonic development, that PUFA intake in humans may increase while α-tocopherol intake may decrease. Nutritional recommendations must take into account nutrient-nutrient interactions and whether or not supplementation with one nutrient during critical life stages, such as pregnancy and embryogenesis, may alter the requirements for another nutrient.
HIGHLIGHTS.
We discuss how α-tocopherol:
prevents the nonspecific, radical-mediated peroxidation of PUFAs,
functions within a greater antioxidant network to modulate the production and/or function of lipid mediators derived from 12- and 12/15-lipoxygenase,
modulates 5-lipoxygenase activity, within the context of embryogenesis.
Acknowledgments
Funding sources: NIEHS (P30 ES000210), NICHD (HD062109)
Grant support: NICHD HD062109 (MGT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mene-Saffrane L, DellaPenna D. Biosynthesis, regulation and functions of tocochromanols in plants. Plant Physiol Biochem. 2010;48:301–309. doi: 10.1016/j.plaphy.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Food and Nutrition Board; Institute of Medicine. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. Washington: National Academy Press; 2000. [PubMed] [Google Scholar]
- 3.Burton GW, Ingold KU. Autoxidation of biological molecules. I. The antioxidant activity of vitamin E and related chain-breaking phenolic antioxidants in vitro. J Amer Chem Soc. 1981;103:6472–6477. [Google Scholar]
- 4.Vatassery GT, Angerhofer CK, Peterson FJ. Vitamin E concentrations in the brains and some selected peripheral tissues of selenium-deficient and vitamin E-deficient mice. J Neurochem. 1984;42:554–558. doi: 10.1111/j.1471-4159.1984.tb02713.x. [DOI] [PubMed] [Google Scholar]
- 5.Traber MG. Vitamin E. In: Shils ME, Shike M, Ross AC, Caballero B, Cousins R, editors. Modern Nutrition in Health and Disease. Baltimore: Lippincott Williams & Wilkins; 2006. pp. 396–411. [Google Scholar]
- 6.Kalra V, Grover J, Ahuja GK, Rathi S, Khurana DS. Vitamin E deficiency and associated neurological deficits in children with protein-energy malnutrition. J Trop Pediatr. 1998;44:291–295. doi: 10.1093/tropej/44.5.291. [DOI] [PubMed] [Google Scholar]
- 7.Kalra V, Grover JK, Ahuja GK, Rathi S, Gulati S, Kalra N. Vitamin E administration and reversal of neurological deficits in protein-energy malnutrition. J Trop Pediatr. 2001;47:39–45. doi: 10.1093/tropej/47.1.39. [DOI] [PubMed] [Google Scholar]
- 8.Buettner GR. The pecking order of free radicals and antioxidants: lipid peroxidation, alpha-tocopherol, and ascorbate. Arch Biochem Biophys. 1993;300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 9.Traber MG, Stevens JF. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic Biol Med. 2011;51:1000–1013. doi: 10.1016/j.freeradbiomed.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruno RS, Leonard SW, Atkinson J, Montine TJ, Ramakrishnan R, Bray TM, Traber MG. Faster plasma vitamin E disappearance in smokers is normalized by vitamin C supplementation. Free Radic Biol Med. 2006;40:689–697. doi: 10.1016/j.freeradbiomed.2005.10.051. [DOI] [PubMed] [Google Scholar]
- 11.Bruno RS, Rainakrishnan R, Montine TJ, Bray TM, Traber MG. alpha-Tocopherol disappearance is faster in cigarette smokers and is inversely related to their ascorbic acid status. American Journal of Clinical Nutrition. 2005;81:95–103. doi: 10.1093/ajcn/81.1.95. [DOI] [PubMed] [Google Scholar]
- 12.Lebold KM, Lohr CV, Barton CL, Miller GW, Labut EM, Tanguay RL, Traber MG. Chronic vitamin E deficiency promotes vitamin C deficiency in zebrafish leading to degenerative myopathy and impaired swimming behavior. Comp Biochem Physiol C Toxicol Pharmacol. 2013;157:382–389. doi: 10.1016/j.cbpc.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valk EE, Hornstra G. Relationship between vitamin E requirement and polyunsaturated fatty acid intake in man: a review. Int J Vitam Nutr Res. 2000;70:31–42. doi: 10.1024/0300-9831.70.2.31. [DOI] [PubMed] [Google Scholar]
- 14.Atkinson J, Harroun T, Wassall SR, Stillwell W, Katsaras J. The location and behavior of alpha-tocopherol in membranes. Mol Nutr Food Res. 2010;54:641–651. doi: 10.1002/mnfr.200900439. [DOI] [PubMed] [Google Scholar]
- 15.Marquardt D, Williams JA, Kucerka N, Atkinson J, Wassall SR, Katsaras J, Harroun TA. Tocopherol activity correlates with its location in a membrane: a new perspective on the antioxidant vitamin E. J Am Chem Soc. 2013;135:7523–7533. doi: 10.1021/ja312665r. [DOI] [PubMed] [Google Scholar]
- 16.Lebold KM, Jump DB, Miller GW, Wright CL, Labut EM, Barton CL, Tanguay RL, Traber MG. Vitamin E deficiency decreases long-chain PUFA in zebrafish (Danio rerio) J Nutr. 2011;141:2113–2118. doi: 10.3945/jn.111.144279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amusquivar E, Ruperez FJ, Barbas C, Herrera E. Low arachidonic acid rather than alpha-tocopherol is responsible for the delayed postnatal development in offspring of rats fed fish oil instead of olive oil during pregnancy and lactation. J Nutr. 2000;130:2855–2865. doi: 10.1093/jn/130.11.2855. [DOI] [PubMed] [Google Scholar]
- 18.Evans HM, Bishop KS. On the Existence of a Hitherto Unrecognized Dietary Factor Essential for Reproduction. Science. 1922;56:650–651. doi: 10.1126/science.56.1458.650. [DOI] [PubMed] [Google Scholar]
- 19.Miller GW, Ulatowski L, Labut EM, Lebold KM, Manor D, Atkinson J, Barton CL, Tanguay RL, Traber MG. The alpha-tocopherol transfer protein is essential for vertebrate embryogenesis. PLoS One. 2012;7:e47402. doi: 10.1371/journal.pone.0047402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jauniaux E, Cindrova-Davies T, Johns J, Dunster C, Hempstock J, Kelly FJ, Burton GJ. Distribution and transfer pathways of antioxidant molecules inside the first trimester human gestational sac. J Clin Endocrinol Metab. 2004;89:1452–1458. doi: 10.1210/jc.2003-031332. [DOI] [PubMed] [Google Scholar]
- 21.Usenko CY, Harper SL, Tanguay RL. Fullerene C60 exposure elicits an oxidative stress response in embryonic zebrafish. Toxicol Appl Pharmacol. 2008;229:44–55. doi: 10.1016/j.taap.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller GW, Labut EM, Lebold KM, Floeter A, Tanguay RL, Traber MG. Zebrafish (Danio rerio) fed vitamin E-deficient diets produce embryos with increased morphologic abnormalities and mortality. J Nutr Biochem. 2012;23:478–486. doi: 10.1016/j.jnutbio.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jishage K, Arita M, Igarashi K, Iwata T, Watanabe M, Ogawa M, Ueda O, Kamada N, Inoue K, Arai H, Suzuki H. alpha-tocopherol transfer protein is important for the normal development of placental labyrinthine trophoblasts in mice. Journal of Biological Chemistry. 2001;276:1669–1672. doi: 10.1074/jbc.C000676200. [DOI] [PubMed] [Google Scholar]
- 24.Monroig O, Rotllant J, Cerda-Reverter JM, Dick JR, Figueras A, Tocher DR. Expression and role of Elovl4 elongases in biosynthesis of very long-chain fatty acids during zebrafish Danio rerio early embryonic development. Biochim Biophys Acta. 2010;1801:1145–1154. doi: 10.1016/j.bbalip.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Tan SH, Chung HH, Shu-Chien AC. Distinct developmental expression of two elongase family members in zebrafish. Biochem Biophys Res Commun. 2010;393:397–403. doi: 10.1016/j.bbrc.2010.01.130. [DOI] [PubMed] [Google Scholar]
- 26.Traber MG. Mechanisms for the Prevention of Vitamin E Excess. J Lipid Res. 2013 doi: 10.1194/jlr.R032946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traber MG, Lane JC, Lagmay NR, Kayden HJ. Studies on the transfer of tocopherol between lipoproteins. Lipids. 1992;27:657–663. doi: 10.1007/BF02536020. [DOI] [PubMed] [Google Scholar]
- 28.Vance JE, Hayashi H. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim Biophys Acta. 2010;1801:806–818. doi: 10.1016/j.bbalip.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 29.Balazs Z, Panzenboeck U, Hammer A, Sovic A, Quehenberge rO, Malle E, Sattler W. Uptake and transport of high-density lipoprotein (HDL) and HDL-associated alpha-tocopherol by an in vitro blood-brain barrier model. J Neurochem. 2004;89:939–950. doi: 10.1111/j.1471-4159.2004.02373.x. [DOI] [PubMed] [Google Scholar]
- 30.LaDu MJ, Gilligan SM, Lukens JR, Cabana VG, Reardon CA, Van Eldik LJ, Holtzman DM. Nascent astrocyte particles differ from lipoproteins in CSF. J Neurochem. 1998;70:2070–2081. doi: 10.1046/j.1471-4159.1998.70052070.x. [DOI] [PubMed] [Google Scholar]
- 31.Hensley K, Barnes LL, Christov A, Tangney C, Honer WG, Schneider JA, Bennett DA, Morris MC. Analysis of postmortem ventricular cerebrospinal fluid from patients with and without dementia indicates association of vitamin E with neuritic plaques and specific measures of cognitive performance. J Alzheimers Dis. 2011;24:767–774. doi: 10.3233/JAD-2011-101995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scanu AM, Aggerbeck LP, Kruski AW, Lim CT, Kayden HJ. A study of the abnormal lipoproteins in abetalipoproteinemia. J Clin Invest. 1974;53:440–453. doi: 10.1172/JCI107578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kayden HJ. Vitamin E deficiency in patients with abetalipoproteinemia. In: von Kress HF, Blum KU, editors. Vitamine A, E und K. Klinische und physiologisch-chemische probleme. Stuttgard, Germany: F.K. Schattauer Verlag; 1967. pp. 301–308. [Google Scholar]
- 34.Zamel R, Khan R, Pollex RL, Hegele RA. Abetalipoproteinemia: two case reports and literature review. Orphanet J Rare Dis. 2008;3:19. doi: 10.1186/1750-1172-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hegele RA, Angel A. Arrest of neuropathy and myopathy in abetalipoproteinemia with high-dose vitamin E therapy. Can Med Assoc J. 1985;132:41–44. [PMC free article] [PubMed] [Google Scholar]
- 36.Ross RS, Gregg RE, Law SW, Monge JC, Grant SM, Higuchi K, Triche TJ, Jefferson J. Homozygous Hypobetalipoproteinemia - a Disease Distinct from Abetalipoproproteinemia at the Molecular-Level. Journal of Clinical Investigation. 1988;81:590–595. doi: 10.1172/JCI113357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wetterau JR, Aggerbeck LP, Bouma ME, Eisenberg C, Munck A, Hermier M, Schmitz J, Gay G, Rader DJ, Gregg RE. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999–1001. doi: 10.1126/science.1439810. [DOI] [PubMed] [Google Scholar]
- 38.Davidson NO, Shelness GS. APOLIPOPROTEIN B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annu Rev Nutr. 2000;20:169–193. doi: 10.1146/annurev.nutr.20.1.169. [DOI] [PubMed] [Google Scholar]
- 39.Farese RV, Jr, Ruland SL, Flynn LM, Stokowski RP, Young SG. Knockout of the mouse apolipoprotein B gene results in embryonic lethality in homozygotes and protection against diet-induced hypercholesterolemia in heterozygotes. Proc Natl Acad Sci U S A. 1995;92:1774–1778. doi: 10.1073/pnas.92.5.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farese RV, Jr, Cases S, Ruland SL, Kayden HJ, Wong JS, Young SG, Hamilton RL. A novel function for apolipoprotein B: lipoprotein synthesis in the yolk sac is critical for maternal-fetal lipid transport in mice. J Lipid Res. 1996;37:347–360. [PubMed] [Google Scholar]
- 41.Raabe M, Flynn LM, Zlot CH, Wong JS, Veniant MM, Hamilton RL, Young SG. Knockout of the abetalipoproteinemia gene in mice: reduced lipoprotein secretion in heterozygotes and embryonic lethality in homozygotes. Proc Natl Acad Sci U S A. 1998;95:8686–8691. doi: 10.1073/pnas.95.15.8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farese RV, Jr, Véniant MM, Cham CM, Flynn LM, Pierotti V, Loring JF, Traber MG, Ruland S, Stokowski RS, Huszar D, Young SG. Phenotypic analysis of mice expressing exclusively apolipoprotein B48 or apolipoprotein B100. Proc Natl Acad Sci USA. 1996;93:6393–6398. doi: 10.1073/pnas.93.13.6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maras JE, Bermudez OI, Qiao N, Bakun PJ, Boody-Alter EL, Tucker KL. Intake of alpha-tocopherol is limited among US adults. J Am Diet Assoc. 2004;104:567–575. doi: 10.1016/j.jada.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 44.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 45.Altman PL Federation of American Societies for Experimental Biology. Committee on Biological Handbooks. Growth, including reproduction and morphological development. Washington; 1962. [Google Scholar]
- 46.O’Rahilly R. Early human development and the chief sources of information on staged human embryos. Eur J Obstet Gynecol Reprod Biol. 1979;9:273–280. doi: 10.1016/0028-2243(79)90068-6. [DOI] [PubMed] [Google Scholar]
- 47.Wilcox AJ, Baird DD, Weinberg CR. Time of implantation of the conceptus and loss of pregnancy. N Engl J Med. 1999;340:1796–1799. doi: 10.1056/NEJM199906103402304. [DOI] [PubMed] [Google Scholar]
- 48.Gilbert SF. Developmental Biology. Sunderland, MA: Sinauer Associates, Inc; 2010. [Google Scholar]
- 49.Czeizel AE, Dudas I, Paput L, Banhidy F. Prevention of neural-tube defects with periconceptional folic Acid, methylfolate, or multivitamins? Ann Nutr Metab. 2011;58:263–271. doi: 10.1159/000330776. [DOI] [PubMed] [Google Scholar]
- 50.Czeizel AE, Dudas I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327:1832–1835. doi: 10.1056/NEJM199212243272602. [DOI] [PubMed] [Google Scholar]
- 51.Chen K, Zhang X, Wei XP, Qu P, Liu YX, Li TY. Antioxidant vitamin status during pregnancy in relation to cognitive development in the first two years of life. Early Hum Dev. 2009;85:421–427. doi: 10.1016/j.earlhumdev.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 52.Bazan NG, Molina MF, Gordon WC. Docosahexaenoic acid signalolipidomics in nutrition: significance in aging, neuroinflammation, macular degeneration, Alzheimer’s, and other neurodegenerative diseases. Annu Rev Nutr. 2011;31:321–351. doi: 10.1146/annurev.nutr.012809.104635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao D, Kevala K, Kim J, Moon HS, Jun SB, Lovinger D, Kim HY. Docosahexaenoic acid promotes hippocampal neuronal development and synaptic function. J Neurochem. 2009;111:510–521. doi: 10.1111/j.1471-4159.2009.06335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coti Bertrand P, O’Kusky JR, Innis SM. Maternal dietary (n-3) fatty acid deficiency alters neurogenesis in the embryonic rat brain. J Nutr. 2006;136:1570–1575. doi: 10.1093/jn/136.6.1570. [DOI] [PubMed] [Google Scholar]
- 55.Sidhu VK, Huang BX, Kim HY. Effects of docosahexaenoic acid on mouse brain synaptic plasma membrane proteome analyzed by mass spectrometry and (16)O/(18)O labeling. J Proteome Res. 2011;10:5472–5480. doi: 10.1021/pr2007285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kodas E, Galineau L, Bodard S, Vancassel S, Guilloteau D, Besnard JC, Chalon S. Serotoninergic neurotransmission is affected by n-3 polyunsaturated fatty acids in the rat. J Neurochem. 2004;89:695–702. doi: 10.1111/j.1471-4159.2004.02401.x. [DOI] [PubMed] [Google Scholar]
- 57.Kuperstein F, Eilam R, Yavin E. Altered expression of key dopaminergic regulatory proteins in the postnatal brain following perinatal n-3 fatty acid dietary deficiency. J Neurochem. 2008;106:662–671. doi: 10.1111/j.1471-4159.2008.05418.x. [DOI] [PubMed] [Google Scholar]
- 58.Hamilton L, Greiner R, Salem N, Jr, Kim HY. n-3 fatty acid deficiency decreases phosphatidylserine accumulation selectively in neuronal tissues. Lipids. 2000;35:863–869. doi: 10.1007/s11745-000-0595-x. [DOI] [PubMed] [Google Scholar]
- 59.Akbar M, Calderon F, Wen Z, Kim HY. Docosahexaenoic acid: a positive modulator of Akt signaling in neuronal survival. Proc Natl Acad Sci U S A. 2005;102:10858–10863. doi: 10.1073/pnas.0502903102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yavin E, Himovichi E, Eilam R. Delayed cell migration in the developing rat brain following maternal omega 3 alpha linolenic acid dietary deficiency. Neuroscience. 2009;162:1011–1022. doi: 10.1016/j.neuroscience.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 61.Anderson GJ, Neuringer M, Lin DS, Connor WE. Can prenatal N-3 fatty acid deficiency be completely reversed after birth? Effects on retinal and brain biochemistry and visual function in rhesus monkeys. Pediatr Res. 2005;58:865–872. doi: 10.1203/01.pdr.0000182188.31596.5a. [DOI] [PubMed] [Google Scholar]
- 62.Harauma A, Salem N, Jr, Moriguchi T. Repletion of n-3 fatty acid deficient dams with alpha-linolenic acid: effects on fetal brain and liver fatty acid composition. Lipids. 2010;45:659–668. doi: 10.1007/s11745-010-3443-y. [DOI] [PubMed] [Google Scholar]
- 63.Martinez M. Tissue levels of polyunsaturated fatty acids during early human development. J Pediatr. 1992;120:S129–138. doi: 10.1016/s0022-3476(05)81247-8. [DOI] [PubMed] [Google Scholar]
- 64.Carlson SE, Cooke RJ, Werkman SH, Tolley EA. First year growth of preterm infants fed standard compared to marine oil n-3 supplemented formula. Lipids. 1992;27:901–907. doi: 10.1007/BF02535870. [DOI] [PubMed] [Google Scholar]
- 65.Elias SL, Innis SM. Infant plasma trans, n-6, and n-3 fatty acids and conjugated linoleic acids are related to maternal plasma fatty acids, length of gestation, and birth weight and length. Am J Clin Nutr. 2001;73:807–814. doi: 10.1093/ajcn/73.4.807. [DOI] [PubMed] [Google Scholar]
- 66.Food and Nutrition Board. Institute of Medicine. Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids. Washington, D.C: The National Academies Press; 2005. [Google Scholar]
- 67.Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111:5866–5898. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 68.Bannenberg G, Serhan CN. Specialized pro-resolving lipid mediators in the inflammatory response: An update. Biochim Biophys Acta. 2010;1801:1260–1273. doi: 10.1016/j.bbalip.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grosser T, Yusuff S, Cheskis E, Pack MA, FitzGerald GA. Developmental expression of functional cyclooxygenases in zebrafish. Proc Natl Acad Sci U S A. 2002;99:8418–8423. doi: 10.1073/pnas.112217799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cha YI, Kim SH, Solnica-Krezel L, Dubois RN. Cyclooxygenase-1 signaling is required for vascular tube formation during development. Dev Biol. 2005;282:274–283. doi: 10.1016/j.ydbio.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 71.Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, Dey SK. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell. 1997;91:197–208. doi: 10.1016/s0092-8674(00)80402-x. [DOI] [PubMed] [Google Scholar]
- 72.Kalen M, Wallgard E, Asker N, Nasevicius A, Athley E, Billgren E, Larson JD, Wadman SA, Norseng E, Clark KJ, He L, Karlsson-Lindahl L, Hager AK, Weber H, Augustin H, Samuelsson T, Kemmet CK, Utesch CM, Essner JJ, Hackett PB, Hellstrom M. Combination of reverse and chemical genetic screens reveals angiogenesis inhibitors and targets. Chem Biol. 2009;16:432–441. doi: 10.1016/j.chembiol.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de La Houssaye BA, Mikule K, Nikolic D, Pfenninger KH. Thrombin-induced growth cone collapse: involvement of phospholipase A(2) and eicosanoid generation. J Neurosci. 1999;19:10843–10855. doi: 10.1523/JNEUROSCI.19-24-10843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mikule K, Gatlin JC, de la Houssaye BA, Pfenninger KH. Growth cone collapse induced by semaphorin 3A requires 12/15-lipoxygenase. J Neurosci. 2002;22:4932–4941. doi: 10.1523/JNEUROSCI.22-12-04932.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kinder M, Wei C, Shelat SG, Kundu M, Zhao L, Blair IA, Pure E. Hematopoietic stem cell function requires 12/15-lipoxygenase-dependent fatty acid metabolism. Blood. 2010;115:5012–5022. doi: 10.1182/blood-2009-09-243139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Epp N, Furstenberger G, Muller K, de Juanes S, Leitges M, Hausser I, Thieme F, Liebisch G, Schmitz G, Krieg P. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J Cell Biol. 2007;177:173–182. doi: 10.1083/jcb.200612116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haas U, Raschperger E, Hamberg M, Samuelsson B, Tryggvason K, Haeggstrom JZ. Targeted knock-down of a structurally atypical zebrafish 12S-lipoxygenase leads to severe impairment of embryonic development. Proc Natl Acad Sci U S A. 2011;108:20479–20484. doi: 10.1073/pnas.1117094108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klein RF, Allard J, Avnur Z, Nikolcheva T, Rotstein D, Carlos AS, Shea M, Waters RV, Belknap JK, Peltz G, Orwoll ES. Regulation of bone mass in mice by the lipoxygenase gene Alox15. Science. 2004;303:229–232. doi: 10.1126/science.1090985. [DOI] [PubMed] [Google Scholar]
- 79.Sun D, Funk CD. Disruption of 12/15-lipoxygenase expression in peritoneal macrophages. Enhanced utilization of the 5-lipoxygenase pathway and diminished oxidation of low density lipoprotein. J Biol Chem. 1996;271:24055–24062. [PubMed] [Google Scholar]
- 80.Johnson EN, Brass LF, Funk CD. Increased platelet sensitivity to ADP in mice lacking platelet-type 12-lipoxygenase. Proc Natl Acad Sci U S A. 1998;95:3100–3105. doi: 10.1073/pnas.95.6.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johnson EN, Nanney LB, Virmani J, Lawson JA, Funk CD. Basal transepidermal water loss is increased in platelet-type 12-lipoxygenase deficient mice. J Invest Dermatol. 1999;112:861–865. doi: 10.1046/j.1523-1747.1999.00595.x. [DOI] [PubMed] [Google Scholar]
- 82.Tobaben S, Grohm J, Seiler A, Conrad M, Plesnila N, Culmsee C. Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell Death Differ. 2011;18:282–292. doi: 10.1038/cdd.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- 84.Pallast S, Arai K, Pekcec A, Yigitkanli K, Yu Z, Wang X, Lo EH, van Leyen K. Increased nuclear apoptosis-inducing factor after transient focal ischemia: a 12/15-lipoxygenase-dependent organelle damage pathway. J Cereb Blood Flow Metab. 2010;30:1157–1167. doi: 10.1038/jcbfm.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 86.Maccarrone M, Melino G, Finazzi-Agro A. Lipoxygenases and their involvement in programmed cell death. Cell Death Differ. 2001;8:776–784. doi: 10.1038/sj.cdd.4400908. [DOI] [PubMed] [Google Scholar]
- 87.Gleissman H, Yang R, Martinod K, Lindskog M, Serhan CN, Johnsen JI, Kogner P. Docosahexaenoic acid metabolome in neural tumors: identification of cytotoxic intermediates. FASEB J. 2010;24:906–915. doi: 10.1096/fj.09-137919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lindskog M, Gleissman H, Ponthan F, Castro J, Kogner P, Johnsen JI. Neuroblastoma cell death in response to docosahexaenoic acid: sensitization to chemotherapy and arsenic-induced oxidative stress. Int J Cancer. 2006;118:2584–2593. doi: 10.1002/ijc.21555. [DOI] [PubMed] [Google Scholar]
- 89.Kwon KJ, Jung YS, Lee SH, Moon CH, Baik EJ. Arachidonic acid induces neuronal death through lipoxygenase and cytochrome P450 rather than cyclooxygenase. J Neurosci Res. 2005;81:73–84. doi: 10.1002/jnr.20520. [DOI] [PubMed] [Google Scholar]
- 90.Liu X, Shibata T, Hisaka S, Kawai Y, Osawa T. DHA Hydroperoxides as a Potential Inducer of Neuronal Cell Death: a Mitochondrial Dysfunction-Mediated Pathway. J Clin Biochem Nutr. 2008;43:26–33. doi: 10.3164/jcbn.2008040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kirkwood JS, Lebold KM, Miranda CL, Wright CL, Miller GW, Tanguay RL, Barton CL, Traber MG, Stevens JF. Vitamin C deficiency activates the purine nucleotide cycle in zebrafish. J Biol Chem. 2012;287:3833–3841. doi: 10.1074/jbc.M111.316018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leth T, Sondergaard H. Biological activity of vitamin E compounds and natural materials by the resorption-gestation test, and chemical determination of the vitamin E activity in foods and feeds. J Nutr. 1977;107:2236–2243. doi: 10.1093/jn/107.12.2236. [DOI] [PubMed] [Google Scholar]
- 93.Ames SR. Biopotencies in rats of several forms of alpha-tocopherol. J Nutr. 1979;109:2198–2204. doi: 10.1093/jn/109.12.2198. [DOI] [PubMed] [Google Scholar]
- 94.Li Q, Cheon YP, Kannan A, Shanker S, Bagchi IC, Bagchi MK. A novel pathway involving progesterone receptor, 12/15-lipoxygenase-derived eicosanoids, and peroxisome proliferator-activated receptor gamma regulates implantation in mice. J Biol Chem. 2004;279:11570–11581. doi: 10.1074/jbc.M311773200. [DOI] [PubMed] [Google Scholar]
- 95.Imai H, Hirao F, Sakamoto T, Sekine K, Mizukura Y, Saito M, Kitamoto T, Hayasaka M, Hanaoka K, Nakagawa Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun. 2003;305:278–286. doi: 10.1016/s0006-291x(03)00734-4. [DOI] [PubMed] [Google Scholar]
- 96.Reddanna P, Rao MK, Reddy CC. Inhibition of 5-lipoxygenase by vitamin E. FEBS Lett. 1985;193:39–43. doi: 10.1016/0014-5793(85)80075-2. [DOI] [PubMed] [Google Scholar]
- 97.Jiang Z, Yin X, Jiang Q. Natural forms of vitamin E and 13′-carboxychromanol, a long-chain vitamin E metabolite, inhibit leukotriene generation from stimulated neutrophils by blocking calcium influx and suppressing 5-lipoxygenase activity, respectively. J Immunol. 2011;186:1173–1179. doi: 10.4049/jimmunol.1002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maccarrone M, Manca-di-Villahermosa S, Meloni C, Massoud R, Mascali A, Guarina R, Finazzi-Agro A, Taccone-Gallucci M. Arachidonate cascade, apoptosis, and vitamin E in peripheral blood mononuclear cells from hemodialysis patients. Am J Kidney Dis. 2002;40:600–610. doi: 10.1053/ajkd.2002.34920. [DOI] [PubMed] [Google Scholar]
- 99.Maccarrone M, Meloni C, Manca-di-Villahermosa S, Cococcetta N, Casciani CU, Finazzi-Agro A, Taccone-Gallucci M. Vitamin E suppresses 5-lipoxygenase-mediated oxidative stress in peripheral blood mononuclear cells of hemodialysis patients regardless of administration route. Am J Kidney Dis. 2001;37:964–969. doi: 10.1016/s0272-6386(05)80012-5. [DOI] [PubMed] [Google Scholar]
- 100.Maccarrone M, Taccone-Gallucci M, Finazzi-Agro A. 5-Lipoxygenase-mediated mitochondrial damage and apoptosis of mononuclear cells in ESRD patients. Kidney Int Suppl. 2003:S33–36. doi: 10.1046/j.1523-1755.63.s84.26.x. [DOI] [PubMed] [Google Scholar]
- 101.Xu S, Lu H, Lin J, Chen Z, Jiang D. Regulation of TNFalpha and IL1beta in rheumatoid arthritis synovial fibroblasts by leukotriene B4. Rheumatol Int. 2010;30:1183–1189. doi: 10.1007/s00296-009-1125-y. [DOI] [PubMed] [Google Scholar]
- 102.Devaraj S, Jialal I. alpha-Tocopherol decreases interleukin-1beta release from activated human monocytes by inhibition of 5-lipoxygenase. Arterioscler Thromb Vasc Biol. 1999;19:1125–1133. doi: 10.1161/01.atv.19.4.1125. [DOI] [PubMed] [Google Scholar]
- 103.Devaraj S, Jialal I. Alpha-tocopherol decreases tumor necrosis factor-alpha mRNA and protein from activated human monocytes by inhibition of 5-lipoxygenase. Free Radic Biol Med. 2005;38:1212–1220. doi: 10.1016/j.freeradbiomed.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 104.Taccone-Gallucci M, Manca-di-Villahermosa S, Battistini L, Stuffler RG, Tedesco M, Maccarrone M. N-3 PUFAs reduce oxidative stress in ESRD patients on maintenance HD by inhibiting 5-lipoxygenase activity. Kidney Int. 2006;69:1450–1454. doi: 10.1038/sj.ki.5000291. [DOI] [PubMed] [Google Scholar]
- 105.Calder PC. Polyunsaturated fatty acids and inflammation. Prostaglandins Leukot Essent Fatty Acids. 2006;75:197–202. doi: 10.1016/j.plefa.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 106.Kris-Etherton PM, Harris WS, Appel LJ. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation. 2002;106:2747–2757. doi: 10.1161/01.cir.0000038493.65177.94. [DOI] [PubMed] [Google Scholar]
- 107.Klein EA, Thompson IM, Jr, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, Minasian LM, Ford LG, Parnes HL, Gaziano JM, Karp DD, Lieber MM, Walther PJ, Klotz L, Parsons JK, Chin JL, Darke AK, Lippman SM, Goodman GE, Meyskens FL, Jr, Baker LH. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2011;306:1549–1556. doi: 10.1001/jama.2011.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fujita K, Iwasaki M, Ochi H, Fukuda T, Ma C, Miyamoto T, Takitani K, Negishi-Koga T, Sunamura S, Kodama T, Takayanagi H, Tamai H, Kato S, Arai H, Shinomiya K, Itoh H, Okawa A, Takeda S. Vitamin E decreases bone mass by stimulating osteoclast fusion. Nat Med. 2012;18:589–594. doi: 10.1038/nm.2659. [DOI] [PubMed] [Google Scholar]
- 109.Moshfegh A, Goldman J, Cleveland L. What we eat in America, NHANES 2001–2002: usual nutrient intakes from food compared to dietary reference intakes. United States Department of Agriculture, Agricultural Research Service; 2005. [Google Scholar]
- 110.Takahashi Y, Glasgow WC, Suzuki H, Taketani Y, Yamamoto S, Anton M, Kuhn H, Brash AR. Investigation of the oxygenation of phospholipids by the porcine leukocyte and human platelet arachidonate 12-lipoxygenases. Eur J Biochem. 1993;218:165–171. doi: 10.1111/j.1432-1033.1993.tb18362.x. [DOI] [PubMed] [Google Scholar]