

Abstract
We report herein an operationally simple method for the direct conversion of benzylic C–H groups to C–F. We show that visible light can activate diarylketones to abstract a benzylic hydrogen atom selectively. Adding a fluorine radical donor yields the benzylic fluoride and regenerates the catalyst. The selective formation of mono- and difluorination products can be achieved by catalyst-control. 9-Fluorenone catalyzes benzylic C–H monofluorination while xanthone catalyzes benzylic C–H difluorination. The scope and efficiency of this new C–H fluorination method are significantly better than those of the existing methods. This is also the first report of selective C–H gem-difluorination.
Fluorine-containing molecules are of particular interest in the fields of biomedicine, agriculture, and material sciences.1 They possess unique physical and chemical properties because of fluorine’s small size and high electro-negativity. Incorporating fluorine atoms into small-molecule drugs often improves their lipophilicity and metabolic stability. However, fluorine’s special characteristics also make its incorporation difficult.2 Most fluorination methods rely on functional group transformation or metal-catalyzed C–H insertion/abstraction (Figure 1a). We now show that visible light can be used to promote a metal-free catalytic reaction to directly incorporate a fluorine atom onto the benzylic position of aromatic compounds under mild conditions (Figure 1b). No special photochemical equipment is required. 9-Fluorenone catalyzes benzylic C–H monofluorination with significantly improved scope and efficiency compared to existing C–H fluorination methods. In contrast, xanthone catalyzes selective benzylic C–H difluorinaton with good efficiency. This is the first report of catalytic C–H gem-difluorination.3
Figure 1.

Metal-free, photolytic C–H activation as a new method for direct C–H fluorination.
INTRODUCTION
Several new fluorination reactions for facile synthesis of organofluorides have been developed recently.4–12 However, prior installation of a functional group at the reaction site is typically required. There are only a few reports of direct C–H fluorination.13 Sanford and Yu developed palladium-catalyzed directed C–H fluorination methods in 2006 and 2009, respectively.14,15 Groves reported a manganese porphyrin-catalyzed C–H fluorination method in 2012;16 Lectka described copper- and iron-catalyzed methods in 2012;17 and Inoue disclosed a thermal free radical method in 2013.18
We have long-term interest in radical chemistry and photochemistry.19 Innate C–H activation reactions that involve a radical species for C–H abstraction are a focus of our research.19g During the study of vanadium-catalyzed C–H fluorination in 2012,19h we found that photoexcited arylketones abstract a hydrogen atom more efficiently than the vanadium catalysts we examined. This observation inspired us to develop a metal-free, photolytic C–H fluorination reaction. We show here that this operationally simple method has broader scope than the recently reported C–H fluorination methods.16–18 Particularly, it can be used for both monofluorination and gem-difluorination.
Yang reported in 1958 that irradiating a mixture of acetone and cyclohexane with short-wavelength UV light gave 2-cyclohexylpropan-2-ol.20 The photoexcited acetone abstracted a hydrogen atom from cyclohexane and the two resulting radicals combined to form a new C–C bond. This reaction is significantly more efficient when carried out intramolecularly, and is referred to as Norrish Yang cyclization when forming a cyclobutane ring. Breslow later developed a remote oxidation strategy for fatty alcohols and steroids based on this reaction.21 This photoreaction22 typically requires at least a substoichiometric amount of benzophenone and the use of UV light for photoexcitation.23 It is also limited to C–C bond formation.24 We demonstrate herein that visible light can drive an intermolecular C–H activation reaction with a low loading of a ketone catalyst to introduce a heteroatom to a specific position of aromatic compounds.
RESULTS AND DISCUSSION
Research Design
We hypothesized that the reactivity of photo-activated ketones could be modulated to achieve selective C–H abstraction. We first targeted the weakly activated benzylic position because it is an ideal site for strategic fluorination in medicinal chemistry for metabolic reasons.3b We envisioned that an intermolecular benzylic C–H abstraction of I by a photoexcited ketone would give benzylic radical II (Figure 1c). Subsequent atom transfer from a fluorine atom donor would provide benzylic fluoride III. Photoexcitation of ketones has traditionally been carried out by UV irradiation. We wondered if the less energetic visible light could be used instead to provide better functional group tolerance. Given the recent success of MacMillan, Yoon, Stephenson, and others in using visible light to promote photoredox reactions,25–28 we believed that visible light could also energize IV bearing a suitable chromophore.
Because the photoexcited ketone V is short-lived and can potentially react with all accessible hydrogen atoms, Breslow covalently linked benzophenone to a remote position of the substrate to enhance the reaction efficiency and regioselectivity.21 The photoexcited benzophenone is known to preferentially abstract the hydrogen atom adjacent to a heteroatom.22 We therefore hypothesized that V could selectively and effectively abstract the weakly activated benzylic hydrogen atom from I. We reasoned that the resulting benzylic radical II would be rather stable and might diffuse away from ketyl radical VI to react intermolecularly with a fluorine atom donor, for example, VII. Selectfluor is known to react through radical manifolds, and thus could represent an ideal fluorine atom donor.29 The back-hydrogen transfer from stable ketyl radical VI is usually slow. Accumulation of VI would lead to its dimerization and loss of the catalyst, giving low or no catalyst turnover. We envisioned that aminium radical cation VIII would be highly energetic and could effectively turn over the catalyst. A low loading of IV might therefore be used for this photolytic C–H activation reaction.
Monofluorination
We started our study by searching for a suitable ketone catalyst at 5 mol % loading (Table 1). We used ethylbenzene (1) as the substrate, Selectfluor (I) as the fluorine donor, and an 11 W household CFL as the light source. As expected, no reaction occurred when acetone was used as the catalyst (entry 1). However, we found that benzophenone and 9-fluorenone30 catalyzed the benzylic fluorination cleanly to give 2 in 83 and 86% yield, respectively (entries 2 and 3). Replacing the fluorine donor Selectfluor (A) with Selectfluor II (B), NFSI (C) or N-fluoropyridinium salts (D and E) led to a significantly lower conversion or no reaction (entries 4–7). An LED lamp could be used as an alternative light source (entry 8), but no reaction occurred in dark at 50 °C (entry 9). The commonly used visible-light active photocatalyst Ir(ppy)3 did not promote the fluorination of 1 (entry 10). This C–H fluorination reaction is oxygen- but not moisture-sensitive. However, the reaction rate was significantly lower when using water as a co-solvent.
Table 1.
Effects of the catalyst, fluorine donor, and light source on the C–H fluorination of 1.
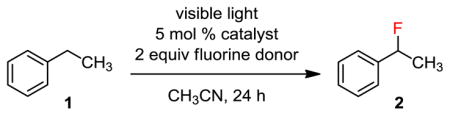
| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Fluorine source | Light sourcea | Temp | Yieldb |
| 1 | Acetone | A | CFL | 27 °C | 0% |
| 2 | Benzophenone | A | CFL | 27 °C | 83% |
| 3 | 9-Fluorenone | A | CFL | 27 °C | 86% |
| 4 | 9-Fluorenone | B | CFL | 27 °C | 40% |
| 5 | 9-Fluorenone | C | CFL | 27 °C | 9% |
| 6 | 9-Fluorenone | D | CFL | 27 °C | 0% |
| 7 | 9-Fluorenone | E | CFL | 27 °C | 0% |
| 8 | 9-Fluorenone | A | LED | 27 °C | 78% |
| 9 | 9-Fluorenone | A | in dark | 50 °C | 0% |
| 10 | Ir(ppy)3 | A | CFL | 27 °C | 0% |
CFL: 11 W IKEA E26 CFL Bulb, LED: 10.5 W Philips A19 LED Bright White Bulb.
Determined by 19F NMR using C6H5F as an external standard.
We used 9-fluorenone as the standard catalyst to explore the scope of this benzylic C–H fluorination reaction (Figure 2). Both electron-rich (2–5) and deficient (6–9) substrates reacted well. In general, substrates with a non-positive Hammett constant31 reacted faster. For example, it took only 3–6 h for 2–5 to react completely, but an extended reaction time was needed for 6–9. Anisyl and phenolic substrates are highly electron-rich and can react directly with Selectfluor.32 The resulting benzylic fluorides were also not stable. Converting the phenol group to an acetoxy group effectively suppressed the side-reactions but the conversion was moderate (6). The highly electron-deficient 4-ethylbenzonitrile could also be fluorinated to give 9, providing evidence against the electron-transfer mechanism wherein the photosensitized 9-fluorenone acted as an electron acceptor.33 Because visible instead of UV light was used to promote the photoexcitation, some aromatic halides could be tolerated. Fluorination of 4-fluoro and 2-chloro substituted ethylbenzene gave 10 and 11 in good yields without dehalogenation.34 However, reactions involving the 3- and 4-chloro substituted and 3-bromo substituted ethylbenzenes were less clean, giving 12–14 in moderate yields.
Figure 2.

Scope of the benzylic monofluorination. Conditions: 0.2 mmol substrate, 5 mol % 9-fluorenone, 2.0 equiv Selectfluor, 2.5 mL acetonitrile, CFL irradiation, 27 °C, isolated yield. aDetermined by 19F NMR using C6H5F as an external standard. bIsolated yield from 20 mmol of 1 and 20 h reaction time. cIsolated yield over two steps after reduction of the ketone.
Heteroatoms on the alkyl chain were also well-tolerated (15–23). There was no elimination of the β-heteroatom despite the intermediacy of a benzylic α-radical (15 and 17). While it is well-documented that photoexcited benzophenone can effectively abstract the hydrogen atom adjacent to an oxygen atom,22 we obtained 17 and 18 cleanly without complications. Substrates containing a tertiary alcohol, ketone, carboxylic acid, carboxylic ester, or phthalimide group could also be fluorinated smoothly (19–23). However, purification of ketone 20 led to partial elimination of the fluoride. We therefore isolated the benzylic fluoride as the corresponding alcohol after reduction of the ketone to avoid the formation of benzylideneacetone. Primary and secondary alcohols were not compatible. Selectfluor is known to directly oxidize them to acyl fluorides and ketones, respectively.35
This photoreaction can also be used to fluorinate the N-phthaloyl-protected phenylalanine methyl ester and amphetamine. Both 24 and 25 were obtained in moderate yield as a 1:1 mixture of diastereomers. The observed lack of diastereoselectivity is consistent with our proposed radical mechanism (Figure 1c). Deprotecting 25 gave β-fluoroamphetamine in three steps from a commercially available alcohol (see Supporting Information). Fluorinating the highly reactive diphenylmethane also gave 26 smoothly. However, we were not able to isolate 26 as a pure compound because of hydrolysis issues.
In addition to the methylene group, we found that methyl (27–29) and methine (30–32) groups can also be monofluorinated. Both electron-donating and withdrawing substituent groups on the aromatic ring can be tolerated. However, elimination of the fluoride from 30 and 31 to give stable tertiary benzylic cations occurred readily, preventing the isolation of these two products as pure compounds.
While aryl boronates can react with Selectfluor directly,36 amine-stabilized MIDA boronate37 did not react under our photolysis conditions. Given that boronates can undergo a variety of transformations, 29 is a valuable α-fluorotolyl building block. Selective monofluorination (>20:1) of a methyl or methylene group was achieved because introducing a fluorine atom increased the bond dissociation energy of the adjacent C–H.38
Difluorination
C–H difluorination is a challenging and underdeveloped area of research. Currently, there is no report of selective gem-difluorination using C–H activation methods. Traditional electron-transfer methods gave little control on the degree of fluorination.39 Therefore, gem-difluorides are usually synthesized by deoxodifluorination of ketones or dethiodifluorination of dithianes.40 Baran recently reported a C–H alkylation method that allows for the introduction of an α,α-difluoroalkyl group to heterocycles.3 Complimentary to this radical alkylation method, our ketone-catalyzed photolytic C–H fluorination reaction can be used to directly introduce two fluorine atoms to the benzylic position of aromatic compounds. This is the first report of selective C–H gem-difluorination.
As described in the previous section, introducing a fluorine atom impedes the second fluorination at the same site. Therefore, sequential fluorination is particularly challenging. Under our standard monofluorination conditions (5 mol % 9-fluorenone, 2.0 equiv Selectfluor), there was little or no difluorination products. For example, fluorination of 1 gave only 2% of 33 (Table 2, entry 1). Increasing the amount of Selectfluor had little effects (entry 2). Further extending the reaction time led to decomposition without significantly increasing the yield of gem-difluoride 33 (entry 3).
Table 2.
Effects of the catalyst and fluorine donor on the C–H difluorination of 1.a
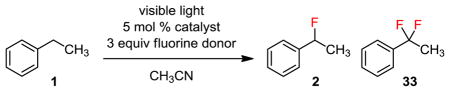
| ||||
|---|---|---|---|---|
| Entry | Catalyst | Fluorine source | Reaction time | Yieldb 2/33 |
| 1 | A | Selectfluorc | 24 h | 86%/2% |
| 2 | A | Selectfluor | 24 h | 81%/5% |
| 3 | A | Selectfluor | 96 h | 29%/9% |
| 4 | B | Selectfluor | 24 h | 11%/20% |
| 5 | C | Selectfluor | 24 h | 0%/31% |
| 6 | D | Selectfluor | 24 h | 7%/15% |
| 7 | C | NFSI | 24 h | 26%/0% |
| 8 | C | Selectfluor II | 24 h | 0%/83% |
| 9 | A | Selectfluor II | 24 h | 62%/2% |
Irradiated with an 11 W IKEA E26 CFL Bulb.
Determined by 19F NMR using C6H5F as an external standard.
2 equiv Selectfluor.
We envisioned that modulating the reactivity of the ketone catalyst and fluorine donor would lead to improved reactivity toward difluorination. Indeed, we obtained 20% yield of 33 when using the more electron-rich catalyst 4,4′-dimethoxybenzophenone (entry 4). Further improvement was achieved with xanthone, which led to the formation of 33 in 31% yield (entry 5). However, introducing two electron-donating methoxyl groups to xanthone resulted in decreased reactivity (entry 6).
Having identified xanthone as a better catalyst, we turned our attention to the fluorine source. Consistent with the monofluorination reactions, NFSI was a less effective fluorine donor (entry 7). However, we observed smooth difluorination when using xanthone with Selectfluor II (entry 8). Reaction of 1 with 5 mol % xanthone and 3 equiv Selectfluor II under visible light irradiation gave 83% yield of gem-difluoride 33 and no monofluoride 2. The major byproduct was acetophenone. We confirmed that the use of Selectfluor II alone was not sufficient to improve the reaction. Using 9-fluorenone as the catalyst and Selectfluor II as the fluorine donor gave only 2% yield of 33 (entry 9).
Under these new conditions, a broad range of substrates can be difluorinated cleanly to give no or less than 5% yield of the monofluorination products (Figure 3). Both methylene (33–40) and methyl groups (41–44) can be difluorinated well. A variety of substituent groups can be tolerated on the aromatic ring (34–36 and 41–44) and on the side-chain (37–40). Specifically, this includes alkyl (34 and 41), aromatic (35 and 42), and heteroatom (36, 43, and 44) substitution on the aromatic ring, and chloro (37), hydroxyl (38), and carbonyl (39) substitution on the side-chain. Again, MIDA boronate is compatible, giving 44 as a valuable α,α-difluorotolyl building block. However, 33, 36, and 43 were too volatile to isolate.
Figure 3.
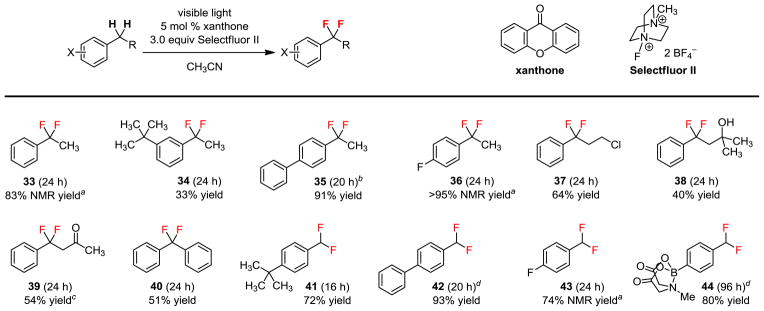
Scope of the benzylic difluorination. Conditions: 0.1 mmol substrate, 5 mol % xanthone, 3.0 equiv Selectfluor II, 2.5 mL acetonitrile, CFL irradiation, 27 °C, isolated yields. aDetermined by 19F NMR using C6H5F as an external standard. b5 mol % 9-fluorenone, 3.0 equiv Selectfluor. cIsolated yield over two steps after reduction of the ketone. d5 mol % 9-fluorenone, 4.0 equiv Selectfluor.
Mechanistic Studies
We confirmed that this C–H fluorination reaction requires both visible light and the ketone catalyst. First, a 10.5 W household light-emitting diode (LED) lamp can be used as the alternative light source (Table 1, entry 8). Second, there was no reaction when the reaction mixture was stirred in dark at 50 °C without a catalyst (Table 1, entry 9), suggesting that the reaction did not occurred through a thermal radical process.13m The reaction can also be switched on and off by turning on and off the light (Figure 4). Third, the visible light-active photocatalyst Ir(ppy)3, having the same triplet energy (ET = 55 kcal/mol)41 as 9-fluorenone, did not catalyze this reaction (Table 1, entry 10). It is therefore less likely that 9-fluorenone functions as an energy transferring photosensitizer. While it is possible that 9-fluorenone forms an exciplex with 1, we did not observe changes in UV-vis absorption when mixing 1 with 9-fluorenone in acetonitrile (see Supporting Information). We also confirmed that the 9-fluorenone-catalyzed fluorination of 1 was not promoted by the insignificant amount of UV light generated by CFL, because it also proceeded smoothly in the presence of a 400 nm longpass filter.
Figure 4.

9-Fluorenone-catalyzed benzylic fluorination of 1 is a light-dependent reaction.
To gain further insights into the reaction mechanism, we measured the intermolecular kinetic isotope effect (KIE) (Figure 5). A competition experiment using a 1:1 mixture of 1 and 1-d2 provided a 65:35 mixture of 2 and 2-d2, corresponding to a KIE value of 1.9. The modest primary KIE is consistent with the hypothesis that a non-metal radical C–H abstraction is involved in the rate-limiting step.42
Figure 5.
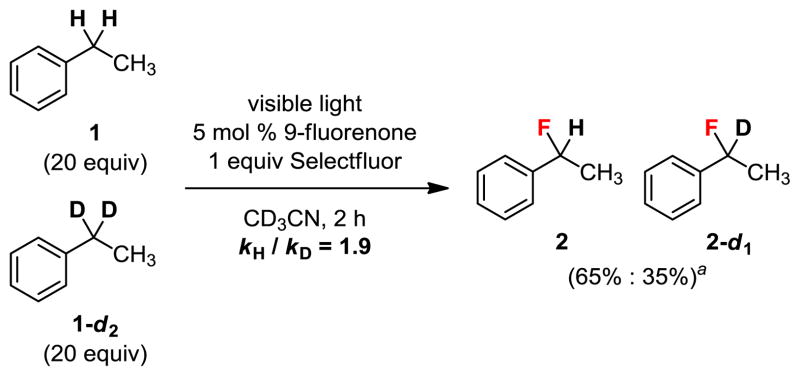
Intermolecular KIE of ethylbenzene (1). aProduct ratio determined by 19F NMR.
We also studied the electronic and steric effects on the site selectivity using intermolecular competition experiments with toluene (45), ethylbenzne (1), and cumene (47) (Figure 6a–c). The ratio of fluorination at the primary versus secondary benzylic site was 1:2.4, secondary versus tertiary benzylic site 3.5:1, and primary versus tertiary benzylic site 1.3:1. Therefore, the site preference for this reaction is 2° > 1° ≈ 3°. We have further confirmed that electron-rich substrates reacted faster than the electron-deficient ones when the steric factors are comparable. For example, fluorinating a 1:1 mixture of 48 and 49 gave 4 as the only product (figure 6d).
Figure 6.

Relative reactivity of different C–H groups. aProduct ratio determined by 19F NMR.
To demonstrate the practicality of this photoreaction, we carried out the monofluorination reaction of 1 on a gram scale (Figure 7). Reaction of 2.12 g (20 mmol) of ethylbenzene (1) with 1.5 equiv Selectfluor in the presence of 5 mol % 9-fluorenone in acetonitrile gave 2 (2.11 g) in 85% isolated yield. A household CFL and regular glassware were used. No specialized photochemical equipment was used for any of the experiments described herein.
Figure 7.
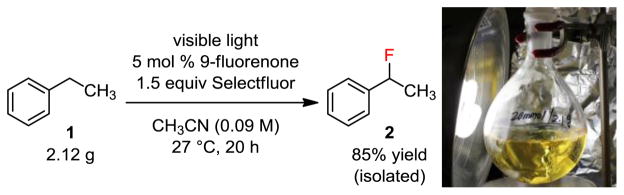
Demonstrating the practicality of the photolytic benzylic fluorination reaction.
SUMMARY
We have developed a photolytic method for direct benzylic fluorination. Cheap and readily available diarylketones were used as the catalysts. 9-Fluorenone catalyzed selective benzylic C–H monofluorination and xanthone gem-difluorination. Ketone-catalyzed photolytic C–H activation reactions were traditionally carried out with UV irradiation and a large amount of ketone. In contrast, we can use visible light to drive this C–H fluorination reaction. Only a low loading of the diarylketone catalysts is needed because of efficient catalyst turnover. Methyl, methylene, and methine groups can all be fluorinated, and a wide range of functional groups on the aromatic ring and side-chain can be tolerated. We are currently developing new catalyst systems for fluorinating the unactivated C–H groups.
Supplementary Material
Acknowledgments
Financial support was provided by NIH/NIGMS (R01-GM079554) and the Welch Foundation (I-1596). We thank Prof. Joseph Ready for helpful discussions.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information. Experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews, see: Böhm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Müller K, Obst-Sander U, Stahl M. ChemBioChem. 2004;5:637–643. doi: 10.1002/cbic.200301023.Müller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c.Hagmann WK. J Med Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f.Phelps ME. Proc Natl Acad Sci USA. 2000;97:9226–9233. doi: 10.1073/pnas.97.16.9226.Jeschke P. ChemBioChem. 2004;5:570–589. doi: 10.1002/cbic.200300833.Ameduri B, Boutevin B. Well-Architectured Fluoropolymers: Synthesis, Properties and Applications. Elsevier B.V; 2004. Okazoe T. Proc Jpn Acad, Ser B. 2009;85:276–289. doi: 10.2183/pjab.85.276.
- 2.For reviews, see: Rozen S. Eur J Org Chem. 2005:2433–2447.Sandford G. J Fluorine Chem. 2007;128:90–104.Kirk KL. Org Process Res Dev. 2008;12:305–321.Furuya T, Kuttruff CA, Ritter T. Curr Opin Drug Discov Devel. 2008;11:803–819.Furuya T, Klein JEMN, Ritter T. Synthesis. 2010:1804–1821. doi: 10.1055/s-0029-1218742.Tredwell M, Gouverneur V. Angew Chem Int Ed. 2012;51:11426–11437. doi: 10.1002/anie.201204687.Sibi MP, Landais Y. Angew Chem Int Ed. 2013;52:3570–3572. doi: 10.1002/anie.201209583.
- 3.For synthesis of benzylic difluorides by C–H alkylation, see: Fujiwara Y, Dixon JA, O’Hara F, Funder ED, Dixon DD, Rodriguez RA, Baxter RD, Herlé B, Sach N, Collins MR, Ishihara Y, Baran PS. Nature. 2012;492:95–99. doi: 10.1038/nature11680.Zhou Q, Ruffoni A, Gianatassio R, Fujiwara Y, Sella E, Shabat D, Baran PS. Angew Chem Int Ed. 2013;52:3949–3952. doi: 10.1002/anie.201300763.
- 4.(a) Watson DA, Su M, Teverovskiy G, Zhang Y, García-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:1661–1664. doi: 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maimone TJ, Milner PJ, Kinzel T, Zhang Y, Takase MK, Buchwald SL. J Am Chem Soc. 2011;133:18106–18109. doi: 10.1021/ja208461k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Furuya T, Ritter T. Org Lett. 2009;11:2860–2863. doi: 10.1021/ol901113t. [DOI] [PubMed] [Google Scholar]; (b) Lee E, Kamlet AS, Powers DC, Neumann CN, Boursalian GB, Furuya T, Choi DC, Hooker JM, Ritter T. Science. 2011;334:639–642. doi: 10.1126/science.1212625. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lee E, Hooker JM, Ritter T. J Am Chem Soc. 2012;134:17456–17458. doi: 10.1021/ja3084797. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sladojevich F, Arlow SI, Tang P, Ritter T. J Am Chem Soc. 2013;135:2470–2473. doi: 10.1021/ja3125405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Kalow JA, Doyle AG. J Am Chem Soc. 2010;132:3268–3269. doi: 10.1021/ja100161d. [DOI] [PubMed] [Google Scholar]; (b) Katcher MH, Doyle AG. J Am Chem Soc. 2010;132:17402–17404. doi: 10.1021/ja109120n. [DOI] [PubMed] [Google Scholar]; (c) Katcher MH, Sha A, Doyle AG. J Am Chem Soc. 2011;133:15902–15905. doi: 10.1021/ja206960k. [DOI] [PubMed] [Google Scholar]
- 7.(a) Rauniyar V, Lackner AD, Hamilton GL, Toste FD. Science. 2011;334:1681–1684. doi: 10.1126/science.1213918. [DOI] [PubMed] [Google Scholar]; (b) Phipps RJ, Hiramatsu K, Toste FD. J Am Chem Soc. 2012;134:8376–8379. doi: 10.1021/ja303959p. [DOI] [PubMed] [Google Scholar]; (c) Mankad NP, Toste FD. Chem Sci. 2012;3:72–76. doi: 10.1039/C1SC00515D. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Phipps RJ, Toste FD. J Am Chem Soc. 2013;135:1268–1271. doi: 10.1021/ja311798q. [DOI] [PubMed] [Google Scholar]
- 8.(a) Hollingworth C, Hazari A, Hopkinson MN, Tredwell M, Benedetto E, Huiban M, Gee AD, Brown JM, Gouverneur V. Angew Chem Int Ed. 2011;50:2613–2617. doi: 10.1002/anie.201007307. [DOI] [PubMed] [Google Scholar]; (b) Gao Z, Lim YH, Tredwell M, Li L, Verhoog S, Hopkinson M, Kaluza W, Collier TL, Passchier J, Huiban M, Gouverneur V. Angew Chem Int Ed. 2012;51:6733–6737. doi: 10.1002/anie.201201502. [DOI] [PubMed] [Google Scholar]
- 9.(a) Fier PS, Hartwig JF. J Am Chem Soc. 2012;134:10795–10798. doi: 10.1021/ja304410x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fier PS, Luo J, Hartwig JF. J Am Chem Soc. 2013;135:2552–2559. doi: 10.1021/ja310909q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Yin F, Wang Z, Li Z, Li C. J Am Chem Soc. 2012;134:10401–10404. doi: 10.1021/ja3048255. [DOI] [PubMed] [Google Scholar]; (b) Li Z, Song L, Li C. J Am Chem Soc. 2013;135:4640–4643. doi: 10.1021/ja400124t. [DOI] [PubMed] [Google Scholar]
- 11.(a) Rueda-Becerril M, Sazepin CC, Leung JCT, Okbinoglu T, Kennepohl P, Paquin JF, Sammis GM. J Am Chem Soc. 2012;134:4026–4029. doi: 10.1021/ja211679v. [DOI] [PubMed] [Google Scholar]; (b) Leung JCT, Chatalova-Sazepin C, West JG, Rueda-Becerril M, Paquin JF, Sammis GM. Angew Chem Int Ed. 2012;51:10804–10807. doi: 10.1002/anie.201206352. [DOI] [PubMed] [Google Scholar]
- 12.(a) Topczewski JJ, Tewson TJ, Nguyen HM. J Am Chem Soc. 2011;133:19318–19321. doi: 10.1021/ja2087213. [DOI] [PubMed] [Google Scholar]; (b) Barker TJ, Boger DL. J Am Chem Soc. 2012;134:13588–13591. doi: 10.1021/ja3063716. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Suzuki S, Kitamura Y, Lectard S, Hamashima Y, Sodeoka M. Angew Chem Int Ed. 2012;51:4581–4585. doi: 10.1002/anie.201201303. [DOI] [PubMed] [Google Scholar]; (d) Cochrane NA, Nguyen H, Gagne MR. J Am Chem Soc. 2013;135:628–631. doi: 10.1021/ja3116795. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ye Y, Sanford MS. J Am Chem Soc. 2013;135:4648–4651. doi: 10.1021/ja400300g. [DOI] [PubMed] [Google Scholar]; (f) Tian T, Zhong WH, Meng S, Meng XB, Li ZJ. J Org Chem. 2013;78:728–732. doi: 10.1021/jo302099d. [DOI] [PubMed] [Google Scholar]
- 13.For early examples of C–H fluorination, see: Barton DHR, Hesse RH, Markwell RE, Pechet MM, Toh HT. J Am Chem Soc. 1976;98:3034–3035. doi: 10.1021/ja00426a071.Feiring AE. J Fluorine Chem. 1977;10:375–386.Sket B, Zupan M. J Org Chem. 1978;43:835–837.Stavber S, Zupan M. J Org Chem. 1983;48:2223–2226.Stavber S, Zupan M. J Org Chem. 1991;56:7347–7350.Stavber G, Zupan M, Stavber S. Tetrahedron Lett. 2007;48:2671–2673.Feiring AE. J Org Chem. 1979;44:1252–1254.Misaki S. J Fluorine Chem. 1981;17:159–171.Visser GWM, Bakker CNM, Halteren BWV, Herscheid JDM, Brinkman GA, Hoekstra A. J Org Chem. 1986;51:1886–1889.Meurs JHH, Sopher DW, Eilenberg W. Angew Chem Int Ed. 1989;28:927–928.Wang CM, Mallouk TE. J Am Chem Soc. 1990;112:2016–2018.Banks RE, Besheesh MK, Mohialdin-Khaffaf SN, Sharif I. J Fluorine Chem. 1997;81:157–161.Chambers RD, Kenwright AM, Parsons M, Sandford G, Moilliet JS. J Chem Soc Perkin Trans. 2002;1:2190–2197.
- 14.(a) Hull KL, Anani WQ, Sanford MS. J Am Chem Soc. 2006;128:7134–7135. doi: 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]; (b) McMurtrey KB, Racowski JM, Sanford MS. Org Lett. 2012;14:4094–4097. doi: 10.1021/ol301739f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Racowski JM, Gary JB, Sanford MS. Angew Chem Int Ed. 2012;51:3414–3417. doi: 10.1002/anie.201107816. [DOI] [PubMed] [Google Scholar]
- 15.(a) Wang X, Mei TS, Yu JQ. J Am Chem Soc. 2009;131:7520–7521. doi: 10.1021/ja901352k. [DOI] [PubMed] [Google Scholar]; (b) Chan KSL, Wasa M, Wang X, Yu JQ. Angew Chem Int Ed. 2011;50:9081–9084. doi: 10.1002/anie.201102985. [DOI] [PubMed] [Google Scholar]
- 16.(a) Liu W, Huang X, Cheng M-J, Nielsen RJ, Goddard WA, III, Groves JT. Science. 2012;337:1322–1325. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]; (b) Liu W, Groves JT. Angew Chem Int Ed. 2013;52:6024–6027. doi: 10.1002/anie.201301097. [DOI] [PubMed] [Google Scholar]
- 17.(a) Bloom S, Pitts CR, Miller DC, Haselton N, Holl MG, Urheim E, Lectka T. Angew Chem Int Ed. 2012;51:10580–10583. doi: 10.1002/anie.201203642. [DOI] [PubMed] [Google Scholar]; (b) Bloom S, Pitts CR, Woltornist R, Griswold A, Holl MG, Lectka T. Org Lett. 2013;15:1722–1724. doi: 10.1021/ol400424s. [DOI] [PubMed] [Google Scholar]
- 18.Amaoka Y, Nagatomo M, Inoue M. Org Lett. 2013;15:2160–2163. doi: 10.1021/ol4006757. [DOI] [PubMed] [Google Scholar]
- 19.(a) Tan X, Chen C. Angew Chem Int Ed. 2006;45:4345–4348. doi: 10.1002/anie.200601208. [DOI] [PubMed] [Google Scholar]; (b) Wang Q, Chen C. Org Lett. 2008;10:1223–1226. doi: 10.1021/ol800111j. [DOI] [PubMed] [Google Scholar]; (c) Gao S, Wang Q, Chen C. J Am Chem Soc. 2009;131:1410–1412. doi: 10.1021/ja808110d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gao S, Wang Q, Huang LJS, Lum L, Chen C. J Am Chem Soc. 2010;132:371–383. doi: 10.1021/ja908626k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wang X, Lu ZMJ, Tan X, Chen C. J Am Chem Soc. 2011;133:15350–15353. doi: 10.1021/ja207386q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang X, Wang XL, Tan X, Lu J, Cormier KW, Ma Z, Chen C. J Am Chem Soc. 2012;134:18834–18842. doi: 10.1021/ja309172t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Xia JB, Cormier KW, Chen C. Chem Sci. 2012;3:2240–2245. doi: 10.1039/C2SC20178J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Chen C. Vanadium-Catalyzed C–H Oxygenation and Fluorination. Gordon Research Conference on Organic Reactions & Processes; July 16, 2012. [Google Scholar]
- 20.Yang NC, Yang DDH. J Am Chem Soc. 1958;80:2913–2914. [Google Scholar]
- 21.(a) Breslow R. Chem Rec. 2001;1:3–11. doi: 10.1002/1528-0691(2001)1:1<3::AID-TCR3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]; (b) Breslow R, Winnik MA. J Am Chem Soc. 1969;91:3083–3084. [Google Scholar]; (c) Breslow R, Baldwin S, Flechtner T, Kalicky P, Liu S, Washburn W. J Am Chem Soc. 1973;95:3251–3262. doi: 10.1021/ja00791a031. [DOI] [PubMed] [Google Scholar]
- 22.For reviews, see: Fagnoni M, Dondi D, Ravelli D, Albini A. Chem Rev. 2007;107:2725–2756. doi: 10.1021/cr068352x.Hoffmann N. Chem Rev. 2008;108:1052–1103. doi: 10.1021/cr0680336.Bach T, Hehn JP. Angew Chem Int Ed. 2011;50:1000–1045. doi: 10.1002/anie.201002845.
- 23.An exception is the benzophenone-catalyzed photolytic radical conjugate addition reaction. However, the role of benzophenone is debatable because benzophenone is not required for this reaction. Fraser-Reid B, Holder NL, Yunker MB. J Chem Soc, Chem Commun. 1972:1286–1287.Sakakibara T, Nakagawa T. Carbohydr Res. 1987;163:239–246.Gómez AM, Mantecón S, Valverde S, López JC. J Org Chem. 1997;62:6612–6614.
- 24.Walling found in a radical reactivity study that cyclohexyl chloride can be formed from irradiating a mixture of cyclohexane-CCl4 with UV light in the presence of benzophenone. Walling C, Gibian MJ. J Am Chem Soc. 1965;87:3361–3364.
- 25.(a) Nicewicz DA, MacMillan DWC. Science. 2008;322:77–80. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nagib DA, MacMillan DWC. Nature. 2011;480:224–228. doi: 10.1038/nature10647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]; (b) Yoon TP, Ischay MA, Du J. Nat Chem. 2010;2:527–532. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]; (c) Yoon TP. ACS Catal. 2013;3:895–902. doi: 10.1021/cs400088e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; (b) Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617–1622. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]
- 28.For additional reviews, see: Zeitler K. Angew Chem Int Ed. 2009;48:9785–9789. doi: 10.1002/anie.200904056.Xuan J, Xiao WJ. Angew Chem Int Ed. 2012;51:6828–6838. doi: 10.1002/anie.201200223.
- 29.(a) Ref. 11. Nyffeler PT, Durón SG, Burkart MD, Vincent SP, Wong CH. Angew Chem Int Ed. 2005;44:192–212. doi: 10.1002/anie.200400648.
- 30.The photo-excitation of 9-fluorenone has been studied extensively. Unlike most other ketones, the first absorption band corresponds to a π→π* instead of n→π* excitation: Estrada LA, Yarnell JE, Neckers DC. J Phys Chem A. 2011;115:6366–6375. doi: 10.1021/jp200507j.
- 31.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165–195. [Google Scholar]
- 32.Zupan M, Iskra J, Stavber S. Bull Chem Soc Jpn. 1995;68:1655–1660.Banks RE. J Fluorine Chem. 1998;87:1–17.(c) Ref. 13f. (d) Ref. 13l.
- 33.Mella M, Fagnoni M, Albini A. Eur J Org Chem. 1999:2137–2142.For a review of photosensitization, see: Kavarnos GJ, Turro NJ. Chem Rev. 1986;86:401–449.
- 34.For reviews of photolytic dehalogenation of aryl halides, see: Schutt L, Bunce NJ. In: In CRC Handbookof Organic Photochemistry and Photobiology. 2. Lenci F, Horspool W, editors. 1 & 2. CRC Press; 2003. Rossi RA, Pierini AB, Peñéñory AB. Chem Rev. 2003;103:71–168. doi: 10.1021/cr960134o.Han KL, He GZ. J Photochem Photobiol, C. 2007;8:55–66.
- 35.Banks RE, Lawrence NJ, Popplewell AL. Synlett. 1994:831–832. [Google Scholar]
- 36.Cazorla C, Métay E, Andrioletti B, Lemaire M. Tetrahedron Lett. 2009;50:3936–3938.(b) Ref. 5a. (c) Ref. 9b. (d) Ref. 12e.
- 37.(a) Gillis EP, Burke MD. J Am Chem Soc. 2007;129:6716–6717. doi: 10.1021/ja0716204. [DOI] [PubMed] [Google Scholar]; (b) Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.For example, the C–H bond dissociation energy for fluorinated methanes is 101.3, 103.2, 107.4 kcal/mol for H–CH2F, H–CHF2, H–CF3, respectively (CRC Handbook of Chemistry and Physics).
- 39.(a) Ref. 13b. (b) Ref. 13g. Laurent E, Marquet B, Tardivel R, Thiebault H. Tetrahedron Lett. 1987;28:2359–2362.Tajima T, Kurihara H, Nakajima A, Fuchigami T. J Electroanal Chem. 2005;580:155–160.
- 40.(a) Rozen S, Mishani E, Bar-Haim A. J Org Chem. 1994;59:2918–2918. [Google Scholar]; (b) Sasson R, Hagooly A, Rozen S. Org Lett. 2003;5:769–771. doi: 10.1021/ol034051n. [DOI] [PubMed] [Google Scholar]; (c) Hagooly A, Sasson R, Rozen S. J Org Chem. 2003;68:8287–8289. doi: 10.1021/jo034829i. [DOI] [PubMed] [Google Scholar]; (d) York C, Prakash GKS, Olah GA. Tetrahedron. 1996;52:9–14. [Google Scholar]; (e) Lal GS, Pez GP, Pesaresi RJ, Prozonic FM, Cheng H. J Org Chem. 1999;64:7048–7054. [Google Scholar]; (f) Singh RP, Chakraborty D, Shreeve JM. J Fluorine Chem. 2001;111:153–160. [Google Scholar]; (g) Reddy VP, Alleti R, Perambuduru MK, Welz-Biermann U, Buchholz H, Prakash GKS. Chem Commun. 2005:654–656. doi: 10.1039/b414254c. [DOI] [PubMed] [Google Scholar]; (h) Beaulieu F, Beauregard LP, Courchesne G, Couturier M, LaFlamme F, L’Heureux A. Org Lett. 2009;11:5050–5053. doi: 10.1021/ol902039q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Fukuhara T, Hara S. Synlett. 2009:198–200. [Google Scholar]; (j) Bloom S, Scerba MT, Erb J, Lectka T. Org Lett. 2011;13:5068–5071. doi: 10.1021/ol2019295. [DOI] [PubMed] [Google Scholar]
- 41.(a) Valentine D, Jr, Hammond GS. J Am Chem Soc. 1972;94:3449–3454. [Google Scholar]; (b) Chen FC, Chang SC, He G, Pyo S, Yang Y, Kurotaki M, Kido J. J Polym Sci, Part B: Polym Phys. 2003;21:2681–2690. [Google Scholar]
- 42.Buxton GV, Greenstock CL, Helman WP, Ross AB. J Phys Chem Ref Data. 1988;17:513–886. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.