

Abstract
The bone marrow is an important site for the interrelated processes of hematopoiesis, granulopoiesis, erythropoiesis and lymphopoiesis. A wide variety of microbial challenges are associated with profound changes in this compartment that impact on hematopoietic differentiation and mobilization of a variety of cell types. This article reviews some of the key pathways that control BM homeostasis, the infectious and inflammatory processes that affect the BM, and how addressing the knowledge gaps in this area has the potential to widen our comprehension of immune homeostasis.
INTRODUCTION
The bone marrow (BM) is a critical site of immune cell development, erythropoiesis, and provides a niche for plasma cells and memory T cells. While the cell populations and structural elements in the BM are typically characterized by composition and morphology (1, 2), the application of novel imaging technologies, such as intravital imaging and laser scanning cytometry, has allowed the field to better define the microenvironments within this complex organ. Similarly, the use of conditional knock out technologies has helped to clarify the factors that maintain stem cell populations and support the development of hematopoietic precursors and immature B cells (3–6). Using these advances, recent sub-setting of stromal and precursor populations in the BM has provided insights into their behavior in the endosteal and perivascular compartments (3–6). In addition to the central role of the BM in maintaining immune homeostasis, the ability to generate and mobilize immune cells in response to infection is a key function of this system. Notably, emergency granulopoiesis and rapid mobilization of neutrophils from the BM is key for resistance to many pathogens. Similarly, increased erythropoiesis can be a physiological response to acute inflammation, but certain infections can lead to the depletion of erythroid precursors, and the development of anemia. The overarching goal of this review is to discuss the role of the BM niche in the host response to infection, illustrate the impact of infectious diseases on this compartment, and highlight some of the major questions in the field.
Hematopoiesis and the HSC niche
Hematopoiesis is the process by which hematopoietic stem cells (HSC) differentiate into immune cells through a series of lineage commitments. Lineageneg Sca-1+ ckit+ cells (LSKs, reviewed extensively in (7–9)), include the earliest hematopoietic precursors in the BM with the potential to develop into multiple lineage-specific progenitors, such as common lymphoid and myeloid progenitors and megakaryocyte or erythrocytic precursors (Figure 1). Of note, only a small percentage of LSKs are HSCs; the majority of the LSK population represents a variety of multipotent or lineage committed cells. At steady state, this differentiation is a complex but well-ordered process, leading to the development of lymphocytes, granulocytes, and myeloid cells.
Figure 1.
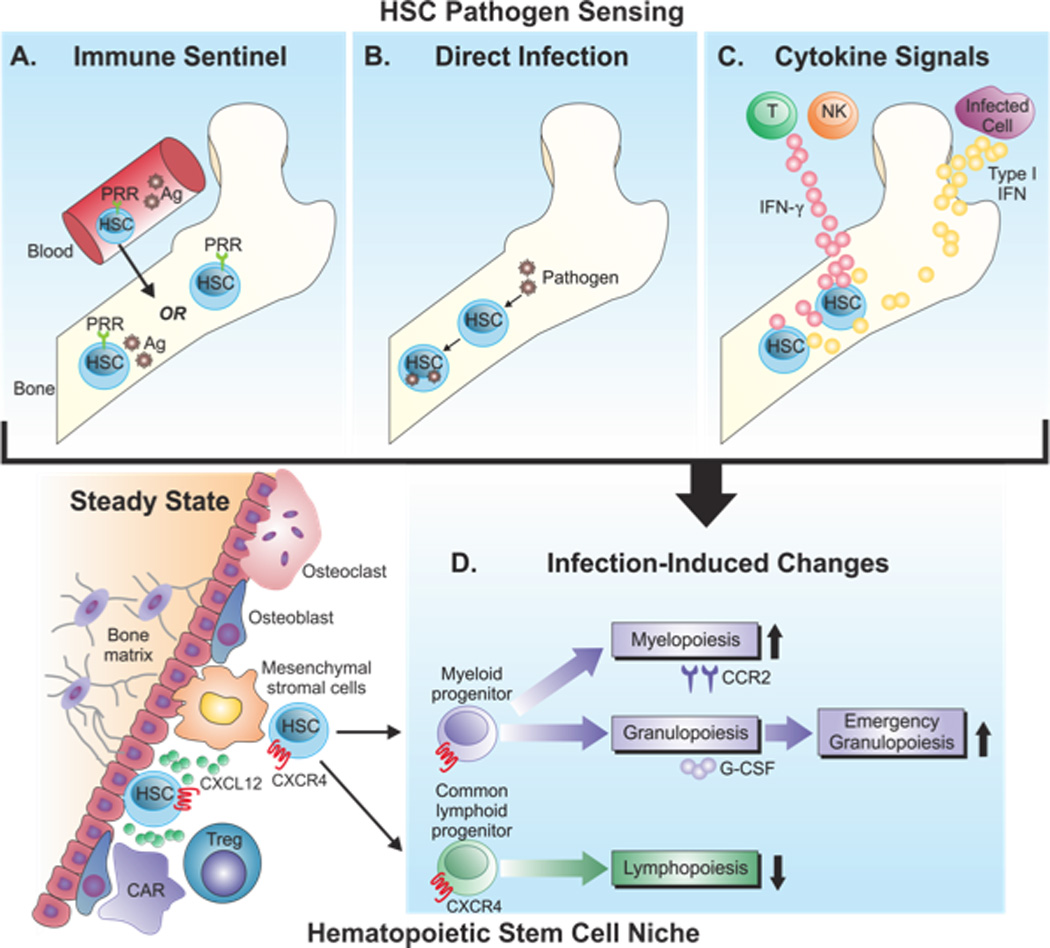
Hematopoietic stem cell responses to infection. (A–C). Potential routes of pathogen sensing by HSCs. (A) HSCs in the BM can express PRRs, such as TLRs, thus, when the BM is directly infected, these cells may recognize pathogen-derived antigen or immune products produced in response to infection within the BM. Alternatively, circulating HSCs in the blood expressing PRRs may recognize PAMPs and traffic back to the BM to relay these signals. (B) HSCs in the BM may be directly infected by pathogens, such as JCV and respond directly to this challenge. (C) Cytokines produced at distal sites, such as IFN-γ or type I interferons, may enter the BM and signal HSCs to initiate BM responses. Infection-induced changes in the BM (D). HSCs are maintained by stromal cells and CXCL12-CXCR4 interactions. Although HSCs are depicted here in endosteal regions, there is also evidence for their presence in perivascular niches. These cells can differentiate into myeloid or common lymphoid progenitors, which then undergo myelopoiesis, granulopoiesis, and/or lymphopoiesis. The impact of infection on these processes is shown, with increased myelopoiesis and emergency granulopoiesis correlating with decreased lymphopoiesis.
Given the diverse functions of the BM, it is not surprising that this organ is comprised of distinct anatomical compartments. For example, within the BM, HSCs are distributed primarily in or near the endosteal region or the interface between medullary bone and stromal cells (Figure 1). This is a site with a distinctive micro-anatomic circulatory system, though recent evidence indicates that perivascular niches also support HSC populations (6). The retention of HSCs in this environment is thought to promote survival and/or maintain hematopoietic progenitors in the quiescent G0 phase of the cell cycle, allowing these cells to self renew and offering a ready pool of cells for rapid emergence (10–12). The direct interactions between vascular stromal cells and nestin-negative mesenchymal progenitors and between osteoblastic cells and HSCs themselves promote HSC survival and control niche size (5, 13).
Several chemokines and adhesion molecules, notably CXCL12 and VLA-4 (14), contribute to HSC localization and maintenance, and the local production of SCF by mesenchymal and perivascular stromal cells, as well as endothelial cells, promotes the generation and maintenance of HSCs (6). Perhaps the best-studied chemokine-receptor pair in this process is CXCL12-CXCR4 and disruption of this pathway leads to alterations in cellular retention in the BM, including mobilization of early lymphoid progenitors and HSCs (14, 15). However, these cell types occupy distinct niches, populated by discrete populations of CXCL12-producing cells (16). Thus, expression of CXCL12 by endothelial cells, perivascular stromal cells, and osteoblasts supports specific cell types within distinct niches. For example, the use of lineage-specific deletions established that nestin-negative mesenchymal progenitors, not CXCL12-abundant reticular cells (which make the majority of CXCL12 (12, 17, 18)), are the critical source of CXCL12 required to maintain HSC in the BM niche (11). This study also demonstrated that other stromal cells are key sources of CXCL12 required for survival of B cell progenitors. Of note, pre, pro, and mature B cells, as well as granulocytes, express high levels of CXCR4, though mature and immature B cell subsets are the least responsive to CXCL12. These differential sensitivities imply that later stage B cell populations are poised to traffic out of the BM, whereas early stage B cells are more receptive to signals that maintain their BM localization (19). Understanding these events has led to the utilization of blocking reagents for CXCL12 and CXCR4, which mobilize HSCs from the BM, resulting in improved harvesting of stem cells for transplantation (14, 17, 20).
Inflammation, infection, and hematopoiesis
It has long been recognized that systemic infection with a variety of bacterial, viral, and parasitic organisms can result in profound alterations in the BM, many of which appear to be part of a conserved ahost response to microbial challenge (Figure 1). For example, during malaria and toxoplasmosis (and other systemic challenges), there is an increase in granulocytes in the BM, but a transient decrease in the numbers of lymphocytes, erythrocytes, and megakaryocytes, despite low parasite burdens in this site (21–24). Increased populations of LSKs and/or HSCs in the BM are the hallmark of many experimental infections (25–31). Thus, challenge with Plasmodium chabaudi or Pneumocystis carinii, organisms not typically found in the BM, leads to increased LSK and HSC populations in the BM and circulation, followed by an increase in multipotent progenitor cells (25, 26). Similarly, murine ehrlichiosis results in increased numbers of LSK cells, though these cells appear to have a defect in their ability to differentiate (32, 33). For many of these examples, it is unclear whether these alterations are secondary consequences of systemic inflammation or part of a coordinated host response to limit infection.
It should be noted that some of the responses associated with diverse infections are context dependent, which likely reflects different host-pathogen interactions. Thus, whether a pathogen can establish infection in the BM and which cell types it infects are relevant factors. This has led to an interest in understanding whether HSCs are inherently resistant to infection (9), however, many of these studies have used pathogens that do not commonly infect the BM or pathogens that typically require phagocytosis, a process that HSCs cannot perform. For instance, Mycobacterial species can be isolated from the BM under a variety of circumstances, that range from asymptomatic patients to AIDs patients with overt clinical disease, and a recent report highlighted that M. tuberculosis resides latently in mesenchymal stem cells, which are phagocytic (34). The importance of understanding these specifics is exemplified by the disparate responses associated with different viruses. Thus, the presence of the non-cytolytic lymphocytic choriomeningitis virus (LCMV) in the BM contributes to the reduced ability of HSCs to engraft in this site (35, 36). JC virus, the cause of progressive multifocal leukoencephalopathy (PML) in immune-compromised patients, can infect HSCs and B cells and is known to persist within the BM. Notably, several antibody-mediated therapies in humans that target LFA-1, VLA-4 or CD20 lead to mobilization of pre-B and B cells and CD34+ HSC progenitors from the BM. It has been proposed that in the context of reduced immune surveillance, these events promote the dissemination of JCV to the brain (37, 38). Cytomegalovirus (CMV) can also infect stromal and mononuclear cells within the BM, which has been linked to a reduced ability to make progenitor colonies (39). Although the examples discussed above focus on organisms found in the BM during infection, as described earlier, reductions in precursor populations can also occur with infectious challenges that do not establish in this site. For example, marked decreases in CD34+ hematopoietic precursors have been reported during HIV and SIV infection without detection of local virus (40–43).
Whether HSCs have an active role in immune sensing remains an open question (Figure 1) and it has been proposed that the expansion of HSC populations may serve as a component of the primary response, as well as a mechanism to replenish depleted progenitor populations (9). In adults, small numbers of HSCs appear to traffic between the BM and circulation, perhaps acting as a form of immune surveillance that can relay distal signals to the BM (44, 45). Evidence in favor of surveillance activity includes HSC expression of Toll-like receptors (TLR) (Figure 1) (9). Furthermore, TLR signaling in LSKs and other hematopoietic progenitors results in myeloid differentiation (46), while TLR9 is required in a model of HSV-1 infection for HSCs to produce dendritic cells (47). There are studies in which MyD88, the adaptor molecule involved in TLR and IL-1 signaling, has been shown to be critical for infection-induced granulopoiesis, myelopoiesis, and mobilization of these populations. Thus, infection with vaccinia virus in vivo, or culture of LSKs with C. albicans in vitro led to elevated numbers of LSKs and differentiated myeloid cells in the BM in a MyD88-dependent manner (27, 28). However, the observation that the increase in LSKs present in a model of bacterial sepsis or infection with S. aureus is MyD88-independent (30) highlights the gaps in our understanding of the crosstalk between the peripheral immune response and the BM compartment. Nevertheless, this literature provides a direct link between pathogen recognition and mobilization of the appropriate innate populations required to control infection.
Erythropoiesis
The suppression of erythropoiesis and development of anemia is characteristic of many infections (48). For organisms, such as Plasmodium and Babesia sp, that directly infect erythrocytes, there is a clear link to erythrocyte destruction that can eventually lead to a depletion of erythoid precursor cells in a chronic setting. For other pathogens, such as the African Trypanosomes, the presence of parasites in the blood stream is associated with damage of erythrocytes, elevated erythrophagocytosis and, ultimately, decreased erythropoiesis (49). In other settings, severe anemia associated with the loss of erythroid precursors also has an immune component (50, 51); Ehrlicia muris and Toxoplasma gondii do not infect erythrocytes, but these distinct challenges lead to a reduction in erthyroid precursors and severe anemia (32). While the cytokines IFN-γ, IL-6, and IL-15 are implicated in immune-mediated anemia (24, 52), it remains unclear whether this response simply reflects an interesting epi-phenomenon or is part of a conserved host response that limits availability of host cells for organisms that do infect erythrocytes.
Granulopoiesis and Myelopoiesis in the BM
Increased granulopoiesis within the BM is a hallmark of acute infection or inflammation in experimental and clinical settings that gives rise to short-lived neutrophils, basophils, and eosinophils (21, 23, 32, 53–56). It has long been recognized that increased circulating levels of basophils and eosinophils are characteristic of many helminth parasites, but how the immune system communicates with the BM to promote this process has been unclear. For Trichuris muris, a nematode parasite of mice that is restricted to the gut, this challenge leads to epithelial cell production of TSLP that induces basophil production in the BM (57). In contrast, elevated neutrophil numbers are characteristic of many bacterial infections and G-CSF promotes “emergency granulopoiesis” in the BM (58, 59). At the molecular level, steady state granulopoiesis is regulated through the transcription factor C/EBPα, whereas C/EBPβ and STAT3 mediate G-CSF-dependent granulopoiesis (60, 61). Naïve mice depleted of neutrophils or injected with the adjuvant alum (which induces granulocyte mobilization) exhibit emergency granulopoiesis and proliferation of HSCs in a G-CSF- and C/EBPβ-dependent manner (62). This body of work illustrates the feedback mechanisms that allow cells in the BM to respond rapidly to changes in the periphery and suggests the presence of a density sensing mechanism that regulates granulopoietic activity (62).
The BM is also an active site of myelopoiesis, leading to the production of monocytes and macrophages. BM macrophages have an important role in maintaining the HSC niche (63), yet the populations that are mobilized in response to infection have key roles in the development and resolution of inflammation, as well as acting as potent anti-microbial effectors (64). In mice challenged with T. gondii or Listeria monocytogenes, the CCR2-dependent mobilization of monocytes out of the BM is essential to control these organisms (64–67). During Ehrlichia infection, IFN-γ is required to activate macrophages to control this intracellular bacterium, but IFN-γ also contributes to the diminished hematopoietic progenitor population in the BM (33). Indeed, the ability of IFN-γ to induce SOCS3 in granulocyte-macrophage progenitors leads to reduced G-CSF signaling and a shift from neutrophil production to myeloid differentiation (68). In this context, it is tempting to speculate that systemic IFN-γ (or direct TLR signaling) provides a mechanism to tailor BM output to the class of pathogen. However, the identification of an IFN-γ-dependent atypical progenitor population of IL-7R+ckithi cells, with predominantly myeloid potential, that is involved in clearance of P. chabaudi (26), illustrates the broad effects of IFN-γ on myelopoiesis.
B cell lymphopoiesis and homeostasis
The BM is also a site of B cell development and many infections can profoundly impact this process (47, 69–74). Challenge with influenza or LCMV results in a transient decrease in pro, pre, and immature B cells in the BM, which is in part dependent on TNFα and lymphotoxin α (71, 72). In a model of bacterial sepsis, the early depletion of B cell progenitors is delayed in MyD88-deficient mice, indicating a role for TLR or IL-1 family members (30). Although the physiological significance of these events remains to be defined, the block in B cell development correlates with reduced humoral responses to irrelevant antigens (71, 72). Interestingly, under inflammatory conditions, there is an inverse correlation between the induction of granulopoiesis and decreased lymphopoiesis in the BM (75). In one experimental system, treatment of mice with incomplete Freund’s adjuvant results in an increase in granulocyte numbers, but a decline in the numbers of B (and T) lymphocytes in the BM (76). Similarly, during infection with the bacterium E. muris, a transient decrease in B220+ cells in the BM is accompanied by an increase in granulocytes (32). Insight into how these events may be coordinated is provided by the observation that, although lymphoid and granulocytic precursors express CXCR4, the disruption of the CXCL12/CXCR4 axis during inflammation leads to preferential loss of B cell precursors, potentially providing space to generate additional granulocytes required for resistance to infection (75). These studies highlight the coordinated changes that occur in hematopoietic processes in the BM during infection, that are presumably required to allow the development of appropriate responses to different classes of pathogen.
The BM as a niche for plasma cells and memory T cells
The BM also provides a niche for long-lived plasma cell populations that continually produce antibodies against previously encountered antigens. The ability of eosinophils, basophils, megakaryocytes, and stromal cells in this site to produce BLyS, April, IL-6, and CXCL12, is required for the survival and retention of plasma cells (3, 4, 77–83). As noted earlier, infection can lead to alterations in many of these cell populations, and there is evidence that without eosinophils, alternative plasma cell survival niches can be established in the spleen (78). Memory CD4+ and CD8+ T cells, also reside within the BM, maintained by stromal cells that produce the cytokines IL-7 and IL-15 that act as survival and proliferative factors for these populations (84–89). In humans, it has been shown that memory T cells in the BM are more highly activated and polyfunctional than those isolated from blood, though memory T cells from blood can develop a similar phenotype after culture with IL-15 (87, 90). Thus far, it is unclear whether the profound infection-induced changes in the BM impact on the (ill defined) memory T cell and plasma cell niches or on the function of these populations. Understanding how different infections influence the homeostasis of memory cell populations in the BM may provide opportunities to manipulate these niches, and so aid in the design of vaccines that induce long lasting immunity.
Impact of systemic cytokine responses on the bone marrow compartment
While some changes that occur in the BM during certain infections may be attributed to the local presence of pathogens, perhaps the most common scenario is that the production of cytokines at distal sites affects the BM (Figure 1) (91). Thus, type I interferons can shift HSCs out of cell cycle arrest and induce proliferation and differentiation, ultimately resulting in decreased numbers of HSCs (92). In murine models of influenza or Sendei virus, production of Type I interferons in the lung leads to upregulation of antiviral genes in hematopoietic cells in the BM (93). Whether these factors are produced at sufficiently high levels in the lungs to have systemic effects in the BM or there is a mechanism to relay these signals to the BM is unclear. As discussed earlier, IFN-γ can modify myelopoiesis (26, 33, 68), and other aspects of hematopoiesis (94) and the high systemic levels of IFN-γ characteristic of many infections suggest it would have a major impact on the BM. During chronic infection with Mycobacterium avium, this production of IFN-γ can activate HSCs from the quiescent state (95). Additionally, IFN-γ-mediated induction of SOCS1 (an inhibitor of cytokine signaling) inhibits the ability of the cytokine thrombopoietin to activate STAT5, which is required for HSC self renewal, leading to a reduction in the number of HSCs (94).
Systemic levels of IL-1 and TNF are also characteristic of many infectious challenges and have been linked to alterations in the BM. TNFα treatment results in a reduction in lymphocyte progenitor populations in the BM, while IL-1β elicits increased granulocyte precursors (76). Moreover, CXCR4-deficient mice or mice treated with pertussis toxin, which blocks chemokine signaling, given IL-1 or TNFα have increased B cell and myeloid progenitors in the circulation, indicating that CXCR4-CXCL12 interactions facilitate cytokine-mediated regulation of B cell and myeloid cell retention in the BM (76). Additionally, TNF-α and IL-1, (and RANKL and M-CSF,) can induce osteoclast differentiation from mononuclear precursor cells, and subsequent inflammatory osteolysis, a complication in many infectious, inflammatory, and neoplastic diseases (96–98). Additional studies are required to understand the physiological significance of these cytokine-mediated changes in the BM and whether they impact on the development of immune responses that are tailored to specific pathogens.
The bone marrow as an immune privileged site
As described above, the BM can be a site of infection, but is sensitive to the systemic effects of microbial challenge at distal locations and it is likely that these processes could be detrimental to essential stem cell populations. Consequently, mechanisms to temper the adverse effects of inflammation on different stem cell niches may be necessary. Although the BM lacks a physical barrier to exclude immune cells, there are elements of immune privilege in this compartment that may protect progenitors from immune-mediated damage or inflammatory signals that could lead to transformation of these long-lived pluripotent cells. Notably, as much as ~25% of BM CD4+ T cells are FoxP3+ Treg cells, a much higher frequency than the 5–10% typically present in other sites. Intravital imaging of the BM of FoxP3-GFP reporters revealed that Tregs are predominantly located in or near the endosteal region, with the majority of HSCs found in close proximity to or in contact with Tregs (99). The significance of these populations in infection has not been addressed, however, in an allogeneic transfer model, depletion of Tregs prevented engraftment of an allo-hematopoietic stem cell progenitor population (99). After BM transplantation, BM T cells express a unique profile of surface markers and cytokine production, with high expression of CD44, CD62L, and CD45RB, higher levels of IFN-γ, IL-4, and IL-10, and decreased IL-2 secretion, compared to those in other sites, and an increased ability to protect from Graft Versus Host Disease (GVHD) (100). Similar findings were reported in human patients, wherein increased Tregs positively correlated with lower incidence of GVHD (100, 101). More recently, in two models of arthritic disease, Tregs in the BM inhibited TNF-mediated bone damage (102), as well as plasma cell accumulation (103). Taken together, these studies suggest that the Treg population in the BM creates a suppressive environment, which establishes a specialized niche for HSCs. Although Treg populations at other sites can be significantly altered during infection (104–106), how those in the BM are influenced by inflammation or infection and whether they preserve different niches or are resident or transient populations represent distinct gaps in our understanding of BM dynamics.
CONCLUSIONS
There have been significant advances in characterizing the effects of infection and inflammation on the function of the BM, but major gaps remain in our understanding of whether these changes impact the ability of a host to control pathogens. In some situations, such as emergency granulopoiesis or monocyte mobilization, it is easy to link these processes with resistance to infection. Alternatively, it is possible that some of the global changes in BM cell populations reflect a shift in energetic resources from hematopoiesis to the support of effector populations required for pathogen control. Interestingly, diminished BM hematopoietic progenitor populations during infection are frequently accompanied by the development of extramedullary hematopoiesis in the spleen and liver (31, 33, 107), which may open niches and resources for the process of granulopoiesis and myelopoiesis. Additionally, the basis for and biological impact of the infection-induced blockade of B cell development in the BM remains unclear (108). In the context of infection, the presence of microbial antigens in the BM while B cells are undergoing selection could lead to the development of B cells that are tolerant to pathogen antigens or even to the deletion of pathogen-specific B cells. To the best of our knowledge, this idea has not been tested, but this phenomena may provide a mechanism to limit deleterious effects of infection on humoral immunity.
Regardless, despite dramatic changes in numerous resident BM populations in response to inflammation, the BM niche does appear to return to a normal steady-state. Whether this disruption impacts long-term hematopoiesis is not yet appreciated. Consequently, there are open questions about the processes that lead to restoration of this environment and whether they are different than those involved in the initial seeding of the BM. Understanding how restoration occurs may translate into more effective strategies to achieve BM reconstitution after irradiation, infection, or other processes that disrupt BM homeostasis. Indeed, since cancer stem cells can reside in the BM in a dormant state (109), incorrect reseeding of the BM or contamination of this immune privileged site could provide protected niches for these cells. Finally, while histological studies of the BM have provided the foundation to characterize this compartment (2), improved imaging technologies continue to improve our knowledge of the cell-cell interactions involved in the regulation of the BM niche. Combined with infectious models that have different effects on BM hematopoiesis, these technologies can be used to better understand the normal developmental processes that occur in the BM.
ACKNOWLEDGEMENTS
The authors would like to thank Deborah Argento for graphics assistance with figure design.
Financial Support: NIH AI 042334, AAF, Commonwealth of Pennsylvania, T32 AI 055400
REFERENCES
- 1.Yang M, Busche G, Ganser A, Li Z. Morphology and quantitative composition of hematopoietic cells in murine bone marrow and spleen of healthy subjects. Ann Hematol. 2013;92:587–594. doi: 10.1007/s00277-012-1653-5. [DOI] [PubMed] [Google Scholar]
- 2.Travlos GS. Normal structure, function, and histology of the bone marrow. Toxicol Pathol. 2006;34:548–565. doi: 10.1080/01926230600939856. [DOI] [PubMed] [Google Scholar]
- 3.Tokoyoda K, Hauser A, Nakayama T, Radbruch A. Organization of immunological memory by bone marrow stroma. Nature reviews. Immunology. 2010;10:193–200. doi: 10.1038/nri2727. [DOI] [PubMed] [Google Scholar]
- 4.Tokoyoda K, Egawa T, Sugiyama T, Choi B-I, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20:707–718. doi: 10.1016/j.immuni.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, Park SY, Lu J, Protopopov A, Silberstein LE. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. 2013;15:533–543. doi: 10.1038/ncb2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–462. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krause DS, Scadden DT, Preffer FI. The hematopoietic stem cell niche--home for friend and foe? Cytometry B Clin Cytom. 2013;84:7–20. doi: 10.1002/cyto.b.21066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondo M. Lymphoid and myeloid lineage commitment in multipotent hematopoietic progenitors. Immunol Rev. 2010;238:37–46. doi: 10.1111/j.1600-065X.2010.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King KY, Goodell MA. Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nature reviews. Immunology. 2011;11:685–692. doi: 10.1038/nri3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma'ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–230. doi: 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, Harris S, Wiedemann LM, Mishina Y, Li L. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 14.Motabi IH, DiPersio JF. Advances in stem cell mobilization. Blood Rev. 2012;26:267–278. doi: 10.1016/j.blre.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 15.Ma Q, Jones D, Springer TA. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10:463–471. doi: 10.1016/s1074-7613(00)80046-1. [DOI] [PubMed] [Google Scholar]
- 16.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–235. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shiozawa Y, Taichman RS. Getting blood from bone: an emerging understanding of the role that osteoblasts play in regulating hematopoietic stem cells within their niche. Exp Hematol. 2012;40:685–694. doi: 10.1016/j.exphem.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugiyama T, Nagasawa T. Bone marrow niches for hematopoietic stem cells and immune cells. Inflamm Allergy Drug Targets. 2012;11:201–206. doi: 10.2174/187152812800392689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Honczarenko M, Douglas RS, Mathias C, Lee B, Ratajczak MZ, Silberstein LE. SDF-1 responsiveness does not correlate with CXCR4 expression levels of developing human bone marrow B cells. Blood. 1999;94:2990–2998. [PubMed] [Google Scholar]
- 20.Rettig MP, Ansstas G, DiPersio JF. Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia. 2012;26:34–53. doi: 10.1038/leu.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villeval JL, Gearing A, Metcalf D. Changes in hemopoietic and regulator levels in mice during fatal or nonfatal malarial infections. II. Nonerythroid populations. Exp Parasitol. 1990;71:375–385. doi: 10.1016/0014-4894(90)90063-i. [DOI] [PubMed] [Google Scholar]
- 22.Villeval JL, Lew A, Metcalf D. Changes in hemopoietic and regulator levels in mice during fatal or nonfatal malarial infections. I. Erythropoietic populations. Exp Parasitol. 1990;71:364–374. doi: 10.1016/0014-4894(90)90062-h. [DOI] [PubMed] [Google Scholar]
- 23.Petakov M, Stojanovic N, Jovcic G, Bugarski D, Todorovic V, Djurkovic-Djakovic O. Hematopoiesis during acute Toxoplasma gondii infection in mice. Haematologia (Budap) 2002;32:439–455. [PubMed] [Google Scholar]
- 24.Chou DB, Sworder B, Bouladoux N, Roy CN, Uchida AM, Grigg M, Robey PG, Belkaid Y. Stromal-derived IL-6 alters the balance of myeloerythroid progenitors during Toxoplasma gondii infection. J Leukoc Biol. 2012;92:123–131. doi: 10.1189/jlb.1011527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi X, Zhang P, Sempowski G, Shellito J. Thymopoietic and bone marrow response to murine Pneumocystis pneumonia. Infect Immun. 2011;79:2031–2042. doi: 10.1128/IAI.01213-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belyaev NN, Brown DE, Diaz AI, Rae A, Jarra W, Thompson J, Langhorne J, Potocnik AJ. Induction of an IL7-R(+)c-Kit(hi) myelolymphoid progenitor critically dependent on IFN-gamma signaling during acute malaria. Nat Immunol. 2010;11:477–485. doi: 10.1038/ni.1869. [DOI] [PubMed] [Google Scholar]
- 27.Singh P, Yao Y, Weliver A, Broxmeyer HE, Hong SC, Chang CH. Vaccinia virus infection modulates the hematopoietic cell compartments in the bone marrow. Stem Cells. 2008;26:1009–1016. doi: 10.1634/stemcells.2007-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yanez A, Murciano C, O'Connor JE, Gozalbo D, Gil ML. Candida albicans triggers proliferation and differentiation of hematopoietic stem and progenitor cells by a MyD88-dependent signaling. Microbes and infection / Institut Pasteur. 2009;11:531–535. doi: 10.1016/j.micinf.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 29.Zhang P, Nelson S, Bagby GJ, Siggins R, 2nd, Shellito JE, Welsh DA. The lineage-c-Kit+Sca-1+ cell response to Escherichia coli bacteremia in Balb/c mice. Stem Cells. 2008;26:1778–1786. doi: 10.1634/stemcells.2007-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scumpia P, Kelly-Scumpia K, Delano M, Weinstein J, Cuenca A, Al-Quran S, Bovio I, Akira S, Kumagai Y, Moldawer L. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. Journal of immunology (Baltimore, Md. : 1950) 2010;184:2247–2251. doi: 10.4049/jimmunol.0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glatman Zaretsky A, Silver JS, Siwicki M, Durham A, Ware CF, Hunter CA. Infection with Toxoplasma gondii alters lymphotoxin expression associated with changes in splenic architecture. Infect Immun. 2012;80:3602–3610. doi: 10.1128/IAI.00333-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacNamara KC, Racine R, Chatterjee M, Borjesson D, Winslow GM. Diminished hematopoietic activity associated with alterations in innate and adaptive immunity in a mouse model of human monocytic ehrlichiosis. Infect Immun. 2009;77:4061–4069. doi: 10.1128/IAI.01550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacNamara KC, Jones M, Martin O, Winslow GM. Transient activation of hematopoietic stem and progenitor cells by IFNgamma during acute bacterial infection. PLoS One. 2011;6:e28669. doi: 10.1371/journal.pone.0028669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Das B, Kashino SS, Pulu I, Kalita D, Swami V, Yeger H, Felsher DW, Campos-Neto A. CD271(+) bone marrow mesenchymal stem cells may provide a niche for dormant Mycobacterium tuberculosis. Sci Transl Med. 2013;5:170ra113. doi: 10.1126/scitranslmed.3004912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bro-Jorgensen K, Volkert M. Haemopoietic defects in mice infected with lymphocytic choriomeningitis virus. 1. The enhanced x-ray sensitivity of virus infected mice. Acta Pathol Microbiol Scand B Microbiol Immunol. 1972;80:845–852. doi: 10.1111/j.0365-5563.1973.tb00010.x. [DOI] [PubMed] [Google Scholar]
- 36.Petursson SR, Chervenick PA, Wu B. Megakaryocytopoiesis and granulopoiesis after murine cytomegalovirus infection. J Lab Clin Med. 1984;104:381–390. [PubMed] [Google Scholar]
- 37.Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, Major EO. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 2012;25:471–506. doi: 10.1128/CMR.05031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jing D, Oelschlaegel U, Ordemann R, Holig K, Ehninger G, Reichmann H, Ziemssen T, Bornhauser M. CD49d blockade by natalizumab in patients with multiple sclerosis affects steady-state hematopoiesis and mobilizes progenitors with a distinct phenotype and function. Bone Marrow Transplant. 2010;45:1489–1496. doi: 10.1038/bmt.2009.381. [DOI] [PubMed] [Google Scholar]
- 39.Apperley JF, Dowding C, Hibbin J, Buiter J, Matutes E, Sissons PJ, Gordon M, Goldman JM. The effect of cytomegalovirus on hemopoiesis: in vitro evidence for selective infection of marrow stromal cells. Exp Hematol. 1989;17:38–45. [PubMed] [Google Scholar]
- 40.Ganser A, Ottmann OG, von Briesen H, Volkers B, Rubsamen-Waigmann H, Hoelzer D. Changes in the haematopoietic progenitor cell compartment in the acquired immunodeficiency syndrome. Res Virol. 1990;141:185–193. doi: 10.1016/0923-2516(90)90020-j. [DOI] [PubMed] [Google Scholar]
- 41.Isgro A, Mezzaroma I, Aiuti A, De Vita L, Franchi F, Pandolfi F, Alario C, Ficara F, Riva E, Antonelli G, Aiuti F. Recovery of hematopoietic activity in bone marrow from human immunodeficiency virus type 1-infected patients during highly active antiretroviral therapy. AIDS Res Hum Retroviruses. 2000;16:1471–1479. doi: 10.1089/088922200750005994. [DOI] [PubMed] [Google Scholar]
- 42.Thiebot H, Vaslin B, Derdouch S, Bertho JM, Mouthon F, Prost S, Gras G, Ducouret P, Dormont D, Le Grand R. Impact of bone marrow hematopoiesis failure on T-cell generation during pathogenic simian immunodeficiency virus infection in macaques. Blood. 2005;105:2403–2409. doi: 10.1182/blood-2004-01-0025. [DOI] [PubMed] [Google Scholar]
- 43.Thomsen AR, Pisa P, Bro-Jorgensen K, Kiessling R. Mechanisms of lymphocytic choriomeningitis virus-induced hemopoietic dysfunction. J Virol. 1986;59:428–433. doi: 10.1128/jvi.59.2.428-433.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boiko JR, Borghesi L. Hematopoiesis sculpted by pathogens: Toll-like receptors and inflammatory mediators directly activate stem cells. Cytokine. 2012;57:1–8. doi: 10.1016/j.cyto.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ciriza J, Thompson H, Petrosian R, Manilay JO, Garcia-Ojeda ME. The migration of hematopoietic progenitors from the fetal liver to the fetal bone marrow: lessons learned and possible clinical applications. Exp Hematol. 2013;41:411–423. doi: 10.1016/j.exphem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 46.Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, Takatsu K, Kincade PW. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–812. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Welner RS, Pelayo R, Nagai Y, Garrett KP, Wuest TR, Carr DJ, Borghesi LA, Farrar MA, Kincade PW. Lymphoid precursors are directed to produce dendritic cells as a result of TLR9 ligation during herpes infection. Blood. 2008;112:3753–3761. doi: 10.1182/blood-2008-04-151506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stein BL. The anemia of inflammation. J Clin Rheumatol. 2012;18:437–442. doi: 10.1097/RHU.0b013e318278f553. [DOI] [PubMed] [Google Scholar]
- 49.Nishimura K, Nakaya H, Nakagawa H, Matsuo S, Ohnishi Y, Yamasaki S. Effect of Trypanosoma brucei brucei on erythropoiesis in infected rats. J Parasitol. 2011;97:88–93. doi: 10.1645/GE-2522.1. [DOI] [PubMed] [Google Scholar]
- 50.Hunfeld KP, Hildebrandt A, Gray JS. Babesiosis: recent insights into an ancient disease. Int J Parasitol. 2008;38:1219–1237. doi: 10.1016/j.ijpara.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Akinosoglou KS, Solomou EE, Gogos CA. Malaria: a haematological disease. Hematology. 2012;17:106–114. doi: 10.1179/102453312X13221316477336. [DOI] [PubMed] [Google Scholar]
- 52.Mullarky IK, Szaba FM, Kummer LW, Wilhelm LB, Parent MA, Johnson LL, Smiley ST. Gamma interferon suppresses erythropoiesis via interleukin-15. Infect Immun. 2007;75:2630–2633. doi: 10.1128/IAI.01836-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hartmann DW, Entringer MA, Robinson WA, Vasil ML, Drebing CJ, Morton NJ, True L. Regulation of granulopoiesis and distribution of granulocytes in early phase of bacterial infection. J Cell Physiol. 1981;109:17–24. doi: 10.1002/jcp.1041090103. [DOI] [PubMed] [Google Scholar]
- 54.Mungyer G, Poels LG, Jerusalem C, Jerusalem R. Plasmodium berghei: influence on granulopoiesis and macrophage production in BALB/c mice. Exp Parasitol. 1983;56:266–276. doi: 10.1016/0014-4894(83)90072-3. [DOI] [PubMed] [Google Scholar]
- 55.Ojok L, Kaeufer-Weiss I, Weiss E. Bone marrow response to acute and chronic Trypanosoma congolense infection in multimammate rats (Mastomys coucha) J Comp Pathol. 2001;124:149–158. doi: 10.1053/jcpa.2000.0445. [DOI] [PubMed] [Google Scholar]
- 56.Van Ginderachter JA, Beschin A, De Baetselier P, Raes G. Myeloid-derived suppressor cells in parasitic infections. Eur J Immunol. 2010;40:2976–2985. doi: 10.1002/eji.201040911. [DOI] [PubMed] [Google Scholar]
- 57.Siracusa MC, Saenz SA, Hill DA, Kim BS, Headley MB, Doering TA, Wherry EJ, Jessup HK, Siegel LA, Kambayashi T, Dudek EC, Kubo M, Cianferoni A, Spergel JM, Ziegler SF, Comeau MR, Artis D. TSLP promotes interleukin-3-independent basophil haematopoiesis and type 2 inflammation. Nature. 2011;477:229–233. doi: 10.1038/nature10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood. 1991;78:2791–2808. [PubMed] [Google Scholar]
- 59.Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84:1737–1746. [PubMed] [Google Scholar]
- 60.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, Akashi K, Tenen DG. C/EBPbeta is required for 'emergency' granulopoiesis. Nat Immunol. 2006;7:732–739. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 61.Panopoulos AD, Zhang L, Snow JW, Jones DM, Smith AM, El Kasmi KC, Liu F, Goldsmith MA, Link DC, Murray PJ, Watowich SS. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood. 2006;108:3682–3690. doi: 10.1182/blood-2006-02-003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cain DW, Snowden PB, Sempowski GD, Kelsoe G. Inflammation triggers emergency granulopoiesis through a density-dependent feedback mechanism. PLoS One. 2011;6:e19957. doi: 10.1371/journal.pone.0019957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, Levesque JP. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116:4815–4828. doi: 10.1182/blood-2009-11-253534. [DOI] [PubMed] [Google Scholar]
- 64.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robben PM, LaRegina M, Kuziel WA, Sibley LD. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J Exp Med. 2005;201:1761–1769. doi: 10.1084/jem.20050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 68.de Bruin AM, Libregts SF, Valkhof M, Boon L, Touw IP, Nolte MA. IFNgamma induces monopoiesis and inhibits neutrophil development during inflammation. Blood. 2012;119:1543–1554. doi: 10.1182/blood-2011-07-367706. [DOI] [PubMed] [Google Scholar]
- 69.Bockstal V, Guirnalda P, Caljon G, Goenka R, Telfer JC, Frenkel D, Radwanska M, Magez S, Black SJ. T. brucei infection reduces B lymphopoiesis in bone marrow and truncates compensatory splenic lymphopoiesis through transitional B-cell apoptosis. PLoS Pathog. 2011;7:e1002089. doi: 10.1371/journal.ppat.1002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Castro-Eguiluz D, Pelayo R, Rosales-Garcia V, Rosales-Reyes R, Alpuche-Aranda C, Ortiz-Navarrete V. B cell precursors are targets for Salmonella infection. Microb Pathog. 2009;47:52–56. doi: 10.1016/j.micpath.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 71.Borrow P, Hou S, Gloster S, Ashton M, Hyland L. Virus infection-associated bone marrow B cell depletion and impairment of humoral immunity to heterologous infection mediated by TNF-alpha/LTalpha. Eur J Immunol. 2005;35:524–532. doi: 10.1002/eji.200425597. [DOI] [PubMed] [Google Scholar]
- 72.Sedger LM, Hou S, Osvath SR, Glaccum MB, Peschon JJ, van Rooijen N, Hyland L. Bone marrow B cell apoptosis during in vivo influenza virus infection requires TNF-alpha and lymphotoxin-alpha. J Immunol. 2002;169:6193–6201. doi: 10.4049/jimmunol.169.11.6193. [DOI] [PubMed] [Google Scholar]
- 73.Jyonouchi H, Kincade PW, Good RA. Immunosuppression of marrow B lymphocytes by administration of Corynebacterium parvum in mice. J Immunol. 1981;127:2502–2507. [PubMed] [Google Scholar]
- 74.Nagaoka H, Gonzalez-Aseguinolaza G, Tsuji M, Nussenzweig MC. Immunization and infection change the number of recombination activating gene (RAG)-expressing B cells in the periphery by altering immature lymphocyte production. J Exp Med. 2000;191:2113–2120. doi: 10.1084/jem.191.12.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med. 2005;201:1771–1780. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ueda Y, Yang K, Foster SJ, Kondo M, Kelsoe G. Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J Exp Med. 2004;199:47–58. doi: 10.1084/jem.20031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.O'Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, Lin LL, Mantchev GT, Bram RJ, Noelle RJ. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. 2004;199:91–98. doi: 10.1084/jem.20031330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chu VT, Berek C. The establishment of the plasma cell survival niche in the bone marrow. Immunol Rev. 2013;251:177–188. doi: 10.1111/imr.12011. [DOI] [PubMed] [Google Scholar]
- 79.Chu VT, Frohlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, Lee JJ, Lohning M, Berek C. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol. 2011;12:151–159. doi: 10.1038/ni.1981. [DOI] [PubMed] [Google Scholar]
- 80.Rodriguez Gomez M, Talke Y, Goebel N, Hermann F, Reich B, Mack M. Basophils support the survival of plasma cells in mice. J Immunol. 2010;185:7180–7185. doi: 10.4049/jimmunol.1002319. [DOI] [PubMed] [Google Scholar]
- 81.Winter O, Moser K, Mohr E, Zotos D, Kaminski H, Szyska M, Roth K, Wong DM, Dame C, Tarlinton DM, Schulze H, MacLennan IC, Manz RA. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood. 2010;116:1867–1875. doi: 10.1182/blood-2009-12-259457. [DOI] [PubMed] [Google Scholar]
- 82.Slifka M, Antia R, Whitmire J, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 83.Manz R, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. 1997;388:133–134. doi: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- 84.Tokoyoda K, Radbruch A. Signals controlling rest and reactivation of T helper memory lymphocytes in bone marrow. Cell Mol Life Sci. 2012;69:1609–1613. doi: 10.1007/s00018-012-0969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tokoyoda K, Zehentmeier S, Hegazy A, Albrecht I, Grun J, Lohning M, Radbruch A. Professional memory CD4+ T lymphocytes preferentially reside and rest in the bone marrow. Immunity. 2009;30:721–730. doi: 10.1016/j.immuni.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 86.Kondrack R, Harbertson J, Tan J, McBreen M, Surh C, Bradley L. Interleukin 7 regulates the survival and generation of memory CD4 cells. J Exp Med. 2003;198:1797–1806. doi: 10.1084/jem.20030735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herndler-Brandstetter D, Landgraf K, Jenewein B, Tzankov A, Brunauer R, Brunner S, Parson W, Kloss F, Gassner R, Lepperdinger G, Grubeck-Loebenstein B. Human bone marrow hosts polyfunctional memory CD4+ and CD8+ T cells with close contact to IL-15-producing cells. J Immunol. 2011;186:6965–6971. doi: 10.4049/jimmunol.1100243. [DOI] [PubMed] [Google Scholar]
- 88.Becker T, Coley S, Wherry E, Ahmed R. Bone marrow is a preferred site for homeostatic proliferation of memory CD8 T cells. Journal of immunology (Baltimore, Md. : 1950) 2005;174:1269–1273. doi: 10.4049/jimmunol.174.3.1269. [DOI] [PubMed] [Google Scholar]
- 89.Parretta E, Cassese G, Barba P, Santoni A, Guardiola J, Di Rosa F. CD8 cell division maintaining cytotoxic memory occurs predominantly in the bone marrow. Journal of immunology (Baltimore, Md. : 1950) 2005;174:7654–7664. doi: 10.4049/jimmunol.174.12.7654. [DOI] [PubMed] [Google Scholar]
- 90.Palendira U, Chinn R, Raza W, Piper K, Pratt G, Machado L, Bell A, Khan N, Hislop A, Steyn R, Rickinson A, Buckley C, Moss P. Selective accumulation of virus-specific CD8+ T cells with unique homing phenotype within the human bone marrow. Blood. 2008;112:3293–3302. doi: 10.1182/blood-2008-02-138040. [DOI] [PubMed] [Google Scholar]
- 91.Lopez CB, Hermesh T. Systemic responses during local viral infections: type I IFNs sound the alarm. Curr Opin Immunol. 2011;23:495–499. doi: 10.1016/j.coi.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med. 2009;15:696–700. doi: 10.1038/nm.1973. [DOI] [PubMed] [Google Scholar]
- 93.Hermesh T, Moltedo B, Moran TM, Lopez CB. Antiviral instruction of bone marrow leukocytes during respiratory viral infections. Cell Host Microbe. 2010;7:343–353. doi: 10.1016/j.chom.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.de Bruin AM, Demirel O, Hooibrink B, Brandts CH, Nolte MA. Interferon-gamma impairs proliferation of hematopoietic stem cells in mice. Blood. 2013;121:3578–3585. doi: 10.1182/blood-2012-05-432906. [DOI] [PubMed] [Google Scholar]
- 95.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Teitelbaum SL. Osteoclasts; culprits in inflammatory osteolysis. Arthritis Res Ther. 2006;8:201. doi: 10.1186/ar1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chakravarti A, Raquil MA, Tessier P, Poubelle PE. Surface RANKL of Toll-like receptor 4-stimulated human neutrophils activates osteoclastic bone resorption. Blood. 2009;114:1633–1644. doi: 10.1182/blood-2008-09-178301. [DOI] [PubMed] [Google Scholar]
- 98.Bussard KM, Gay CV, Mastro AM. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008;27:41–55. doi: 10.1007/s10555-007-9109-4. [DOI] [PubMed] [Google Scholar]
- 99.Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, Gao W, Saito TI, Lo Celso C, Tsuyuzaki H, Sato T, Cote D, Sykes M, Strom TB, Scadden DT, Lin CP. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 2011;474:216–219. doi: 10.1038/nature10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zeng D, Hoffmann P, Lan F, Huie P, Higgins J, Strober S. Unique patterns of surface receptors, cytokine secretion, and immune functions distinguish T cells in the bone marrow from those in the periphery: impact on allogeneic bone marrow transplantation. Blood. 2002;99:1449–1457. doi: 10.1182/blood.v99.4.1449. [DOI] [PubMed] [Google Scholar]
- 101.Nguyen VH, Zeiser R, Negrin RS. Role of naturally arising regulatory T cells in hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2006;12:995–1009. doi: 10.1016/j.bbmt.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 102.Zaiss MM, Frey B, Hess A, Zwerina J, Luther J, Nimmerjahn F, Engelke K, Kollias G, Hunig T, Schett G, David JP. Regulatory T cells protect from local and systemic bone destruction in arthritis. J Immunol. 2010;184:7238–7246. doi: 10.4049/jimmunol.0903841. [DOI] [PubMed] [Google Scholar]
- 103.Jang E, Cho WS, Cho ML, Park HJ, Oh HJ, Kang SM, Paik DJ, Youn J. Foxp3+ regulatory T cells control humoral autoimmunity by suppressing the development of long-lived plasma cells. J Immunol. 2011;186:1546–1553. doi: 10.4049/jimmunol.1002942. [DOI] [PubMed] [Google Scholar]
- 104.Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, Pritchard GH, Silver JS, Bouladoux N, Stumhofer JS, Harris TH, Grainger J, Wojno ED, Wagage S, Roos DS, Scott P, Turka LA, Cherry S, Reiner SL, Cua D, Belkaid Y, Elloso MM, Hunter CA. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity. 2012;37:511–523. doi: 10.1016/j.immuni.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O'Brien S, Blank R, Lamb E, Natarajan S, Kastenmayer R, Hunter C, Grigg ME, Belkaid Y. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rossi MI, Dutra HS, El-Cheikh MC, Bonomo A, Borojevic R. Extramedullar B lymphopoiesis in liver schistosomal granulomas: presence of the early stages and inhibition of the full B cell differentiation. International immunology. 1999;11:509–518. doi: 10.1093/intimm/11.4.509. [DOI] [PubMed] [Google Scholar]
- 108.Rowland SL, Tuttle K, Torres RM, Pelanda R. Antigen and cytokine receptor signals guide the development of the naive mature B cell repertoire. Immunol Res. 2013;55:231–240. doi: 10.1007/s12026-012-8366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stefanovic S, Schuetz F, Sohn C, Beckhove P, Domschke C. Bone marrow microenvironment in cancer patients: immunological aspects and clinical implications. Cancer Metastasis Rev. 2013;32:163–178. doi: 10.1007/s10555-012-9397-1. [DOI] [PubMed] [Google Scholar]