


Abstract
We applied Illumina Human Methylation450K array to perform a genomic-scale single-site resolution DNA methylation analysis in neuronal and nonneuronal (primarily glial) nuclei separated from the orbitofrontal cortex of postmortem human brain. The findings were validated using enhanced reduced representation bisulfite sequencing. We identified thousands of sites differentially methylated (DM) between neuronal and nonneuronal cells. The DM sites were depleted within CpG-island–containing promoters but enriched in predicted enhancers. Classification of the DM sites into those undermethylated in neurons (neuronal type) and those undermethylated in nonneuronal cells (glial type), combined with findings of others that methylation within control elements typically negatively correlates with gene expression, yielded large sets of predicted neuron-specific and non–neuron-specific genes. These sets of predicted genes were in excellent agreement with the available direct measurements of gene expression in human and mouse. We also found a distinct set of DNA methylation patterns that were unique for neuronal cells. In particular, neuronal-type differential methylation was overrepresented in CpG island shores, enriched within gene bodies but not in intergenic regions, and preferentially harbored binding motifs for a distinct set of transcription factors, including neuron-specific activity-dependent factors. Finally, non-CpG methylation was substantially more prevalent in neurons than in nonneuronal cells.
INTRODUCTION
Epigenetic mechanisms, including DNA methylation and histone modification, are an integral part of a multitude of brain functions that range from basic cellular tasks to the development of the nervous system to higher order cognitive processes (1). Recently, a substantial body of evidence has surfaced, suggesting that several neurodevelopmental, neurodegenerative and neuropsychiatric disorders are in part caused by aberrant epigenetic modifications (2–4). Therefore, a thorough characterization of the epigenetic status of the brain is critical for understanding the molecular basis of its function in health and disease.
In mammals, DNA methylation plays a critical role in genomic imprinting, and X chromosome inactivation, as well as cellular differentiation and development, and is generally considered to be associated with transcriptional repression (5–7). It involves almost exclusively the formation of 5-methylcytosine (5-mC) in CpG dinucleotides. To a much lesser extent, cytosine methylation occurs also in non-CpG contexts. Although previously considered to be largely absent from adult somatic cells (8,9), non-CpG methylation has recently been detected in several human somatic tissues, and found to be particularly prevalent in the adult human and mouse brain (10,11). DNA methylation is extremely important both for the establishment of cell-type–specific identities in the nervous system (12) and in mediating environmentally induced changes in the adult brain, being a critical component of various processes and conditions including memory formation, stress responses, depression and drug addiction (13–16). Despite its importance, the DNA methylation profile of the brain, especially (owing to the obvious experimental difficulties) in humans, has not been sufficiently explored, and, when examined, was studied mostly using bulk brain tissues (11,17–22). These studies have shown that DNA methylation significantly varies between different brain regions as well as between white and gray matter of the same region (17,20,23,24). The brain, however, is characterized by multifaceted complexity, including heterogeneity of cell types, such as neurons and glia, as well as subpopulations within these cell types. These cell types are differentially distributed among brain regions that themselves are heterogeneous in cytoarchitecture, connectivity and function. Hence, to achieve meaningful insight into the epigenetic landscape of the brain (including DNA methylation profile), the epigenetic marks should be studied within individual cell types that are captured from specific brain regions. Indeed, recent reports have clearly demonstrated significant differences in DNA methylation patterns between neuronal and nonneuronal cells (25,26), and suggested that the previously reported epigenetic variation among brain regions could be largely owing to differences in neuron to glia ratios (26).
Because of our interest in genomic regulation of gene expression and its possible role in psychiatric disorders, we performed a genomic-scale single-site resolution analysis of DNA methylation in two subpopulations of brain cells, neurons and nonneuronal cells (primarily glial), both obtained from a specific area of the human prefrontal cortex (PFC), medial orbitofrontal cortex (mOFC), which is implicated in particular behavioral domains, including behavioral inhibition, impulsivity and aggression (27–29). We focused on two key questions: first, which genomic regions harbor DNA methylation differences that distinguish mature neurons from nonneuronal cells? Second, how do these methylation differences relate to cell-type–specific gene expression?
We found that sites that are differentially methylated (DM) between neurons and nonneuronal cells are mostly located distally from the transcription start sites (TSS) and are significantly enriched within predicted enhancers. Conversely, these sites are depleted from CpG islands and, consequently, from the high CpG density promoters. Using several independent approaches, we confirmed that DNA methylation across the entire gene locus is highly predictive of cell-type–specific gene expression. Finally, we report that non-CpG methylation is significantly more abundant in the neuronal compared with nonneuronal cells. Our results provide a resource for understanding the mechanisms of cell-type–specific gene expression in the adult mammalian brain.
MATERIALS AND METHODS
Nuclei Separation by fluorescence-activated cell sorting
Dissected mOFC tissue was ground on liquid nitrogen, resuspended in ice-cold lysis buffer (0.1% Triton, 0.32 M sucrose, 5 mM CaCl2, 3 mM MgCl2, 10 mM Tris–HCl), filtered through a cell strainer, and centrifuged for 5 min at 300g. The pellet was resuspended in blocking buffer (1% goat serum, 2 mM MgCl2, Tris-buffered saline) and incubated for 45 min with Alexa488-conjugated anti-NeuN antibodies (Millipore) (1:1000 dilution). Next, second centrifugation step (15 min, 2800g) through a layer of 1.1 M sucrose was performed, and the resulted pellet was resuspended in phosphate-buffered saline. The DNA dye 7-AAD (Sigma) was added to a final concentration of 2 µg/ml, and the sample was subjected to the fluorescence-activated cell sorting (FACS) procedure using FACS Vantage with DiVa (excitation wavelength 488 nm). Finally, the sorted nuclear fractions were precipitated by centrifugation at 4000 rpm for 20 min at 4°C and stored frozen at −80° C until DNA isolation.
DNA methylation analyses
DNA methylation profiling on Illumina’s Infinium Human Methylation450K array (HM450K) (Illumina Inc.) was performed as previously described (30). Enhanced reduced representation bisulfite sequencing assay (ERRBS) was performed by the Epigenomics Core of the Weill Cornell Medical College (WCWCM; New York) as described in (31). The procedures and quality control measures are detailed in Supplementary Methods.
Genome-wide genotyping and structural variant detection
Genome-wide genotyping and structural variant detection was performed with the Illumina Infinium HumanOmni1 Quad v1.0 Beadarray using standard procedures (Infinium® HD Assay Super Protocol Guide Part # 11 322 427 Rev. C). Genotyping was used for the analysis of the genetic background (see Supplementary Methods). Copy number variation (CNV) calls were made using the following parameters in the CNV partition v3.1.6 plug-in in Genome Studio: confidence threshold = 35; GC wave adjustment; minimum probe count = 10; minimum CNV size = 10 kb. Genomic analysis confirmed the absence of large-scale genomic structural defects such as aneuploidy.
Comparison of the DM regions with histone modification profiles
We used ChIP-seq data generated in (32) to obtain genome-wide maps of H3K4me3, H3K4me1 and H3K27ac profiles in the adult midfrontal and inferior temporal lobes. We also included two nonbrain tissues as negative controls (adult liver, and CD20 naive primary cells). The data were downloaded from http://www.broadinstitute.org/pubs/epigenomicsresource, and the UCSC liftOver tool was used to convert position data from hg18 to hg19 assembly (33). Because enhancer-associated H3K4me1 and H3K27ac histone modifications also partially overlap with promoter regions, we considered only H3K4me1 and H3K27ac peaks that are located further than 2.5 kb from the TSS.
Weighted gene co-expression network analysis
The BrainCloud gene expression microarray data (Gene Expression Omnibus: accession GSE30272) from postnatal subcohort (N = 220) were used for weighted gene co-expression network analysis (WGCNA). Gene expression data were preprocessed as described (34). Network construction was performed using the blockwiseModules function in the WGCNA package (35). The adjacency matrix was calculated by raising the correlation matrix to the power of 6, as determined using the scale-free topology criterion (36). For each pair of genes, a topological overlap measure was calculated and probes were organized into modules using hybrid dynamic tree-cutting (37). The minimum module size was set to 30 genes and the minimum height for merging modules was set at 0.15. The WGCNA userListEnrichment function for the ‘brain’ data sets was used for cell-type–specific annotation of the modules. Enrichment with genes that were predicted to be expressed in neurons or nonneuronal cells based on their DNA methylation profile was calculated using the cumulative hypergeometric probability implemented in the phyper function in R.
Analysis of transcription factor binding motifs in DM regions
We used the Hypergeometric Optimization of Motif Enrichment (HOMER) (v4.1, 11-2-2012, http://biowhat.ucsd.edu/homer/) for known motif discovery. HOMER screens the list of previously determined motifs against the target and background sequences. We used the sites with nonsignificantly changed (NS) methylation as the background. The DM regions were defined as sequences within 250 bp upstream and downstream from each DM site. We corrected for imbalances in the sequence content of target and background sequences by conducting GC-content and short oligo sequences normalization procedures.
Laser-microdissection
Laser-microdissection (LMD) was performed using Leica AS LMD system (Leica Microsystems Inc.) using tissue sections prepared from the PFC of four subjects. The preparation of sections and LMD procedures were performed as described in (38) (See Supplementary Figure S22 for details).
RNA extraction, cDNA synthesis and quantitative real-time polymerase chain reaction
RNA extraction from LMD-microdissected preparations was performed using the PicoPure RNA isolation Kit (Invitrogen). cDNA synthesis was performed using iScript cDNA synthesis kit (BioRad). Quantitative real-time polymerase chain reaction (qPCR) was performed using EagleTaq Master Mix (Roche), specific TaqMan probes (Applied Biosystems) and a touch-down cycle: 10 min at 95°C, 10 cycles of 15 s at 95°C, 60 s at 70–61°C (annealing temperature is decreased 1°C after each cycle), followed by 40 cycles of 15 s at 95°C and 60 s at 60°C. The expression of target genes was compared between neuronal and white matter preparations within each sample using DeltaCt procedure and normalization to the expression levels of 18S RNA.
RESULTS
Subjects and the analyzed brain region
To focus our analysis on the epigenetic features that specifically distinguish neuronal and nonneuronal cells in human brain, we made an effort to eliminate the common confounding factors that could affect the epigenetic patterns. To this end, we used human postmortem brain specimens from six Caucasian male individuals of similar age (22–29 years old at the time of death) (Supplementary Table S1) and similar genetic background (Supplementary Methods and Supplementary Figure S1), without any neurological or psychiatric conditions diagnosed at the time of death (Supplementary Methods). We focused on the ventral extent of the PFC that is commonly referred to as the OFC. The OFC is not functionally homogenous, and there are significant differences in projection fields for different subregions of the OFC (39). We targeted the more lateral subregion of the OFC, which projects to central striatal regions that are believed to be involved to a greater extent in modulating ‘impulsive action’ as opposed to ‘impulsive choice’ (40). Thus, in all six brain specimens, a distinct small area (specifically, a surface area of the mOFC just lateral to the gyrus rectus) was dissected and analyzed (Supplementary Figure S2).
Nuclei separation by FACS
Separation of neuronal and nonneuronal nuclei was performed by FACS using anti-NeuN antibodies (whose antigen is specific for neuronal cells) and a modified protocol that was based on previously published methods (41,42). We used the DNA-binding dye 7-AAD and the anti-NeuN antibodies directly conjugated with the fluorophore. As a result, we were able to routinely obtain well-separated NeuN(+) and NeuN(−) nuclear fractions, with the width of separation reaching up to an order of magnitude of the NeuN signal intensity (Figure 1). In addition, both fractions contained well-defined DNA content because aggregates, nuclei of dividing cells and debris were excluded in the process of sorting (Figure 1). We confirmed the purity of each fraction using a modified version of the protocol that included staining of sorted nuclei with antibodies against a nonneuronal, oligodendrocyte-specific nuclear marker OLIG2 (42) (Supplementary Figure S3). It should be noted that the nonneuronal NeuN(−) fraction consists mostly of the glial cells (such as oligodendrocytes, astrocytes and microglia) as well as a small population of endothelial cells.
Figure 1.
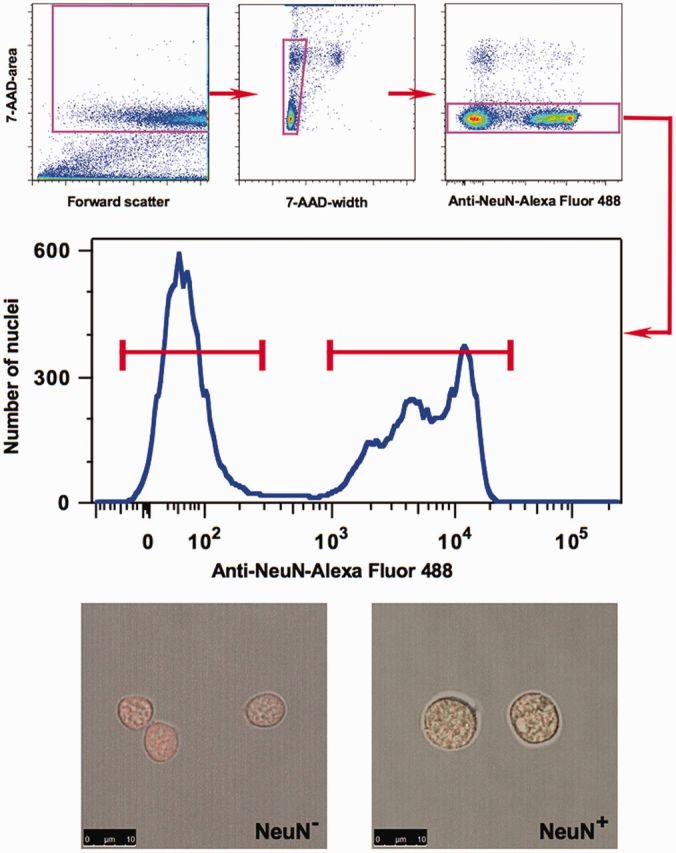
Separation of neuronal and nonneuronal nuclei from the human OFC by FACS. A representative experiment is illustrated. Upper panel: Sequential gating steps in the FACS protocol: exclusion of debris, nuclear fragments, doublets of nuclei and nuclei of dividing cells. Middle Panel: FACS separation of neuronal and nonneuronal nuclei based on NeuN-labeling intensity. Lower Panel: Representative microscopic view of nuclei from sorted neuronal (NeuN+) and nonneuronal (NeuN) fractions. Green color—NeuN staining, red color—DNA dye staining.
General characterization of the DNA methylation data
For each subject, DNA was extracted from the neuronal and nonneuronal fractions and submitted to the HM450K array protocol (30). Each sample was processed in two replicate experiments. The HM450K array examines the methylation status of ∼480 000 individual cytosine positions across the human genome. The content includes coverage of 99% of the RefSeq protein coding genes with multiple probes per gene and 96% of the CpG islands from the UCSC database (30). In addition, HM450K covers high and low CpG density promoters (http://fantom.gsc.riken.jp/4/) (43) and predicted enhancers (44–46). Although the array mostly targets CpG dinucleotide sites, ∼3000 array probes are reserved for non-CpG methylation sites. The methylation level at each CpG or non-CpG locus is described by ‘the beta value’ (β), which is defined as the ratio of the methylated probe intensity to the sum of methylated and unmethylated probe intensities. The resulting β can range from 0 (no methylation) to 1 (complete methylation).
We first filtered out several groups of probes that could potentially produce spurious results, including probes containing common (minor allele frequency > 1%) single-nucleotide polymorphisms (SNPs), probes within the identified CNVs or any probes displaying a missing value in more than one sample (Supplementary Figures S4 and S5 and Supplementary Table S2). In both neuronal and nonneuronal samples, the methylation frequency of individual sites showed the expected bimodal distribution (either hypo- or hypermethylated) (Supplementary Figure S6). However, because the HM450K array does not probe CpG sites in repetitive elements and transposons that are predominantly hypermethylated, the percentage of hypermethylated sites was lower in our samples compared with the published data obtained in several cell types by whole-genome bisulfite sequencing (8,47,48). Unsupervised hierarchical clustering of DNA methylation data from all 24 samples showed perfect separation into the neuronal and nonneuronal groups (Figure 2A). We then examined the mean (across all six subjects) methylation levels at each site between the pairs of experimental replicates. A nearly perfect correlation between the replicates was observed when the two experiments for the same cell-type were compared (R values = 0.99) (Figure 2B; upper panel). In contrast, the correlations were lower (both R values = 0.92) when neuronal versus nonneuronal methylation levels were compared in each of the two experiments (Figure 2B; lower panel). Collectively, in addition to demonstrating high reproducibility between the technical replicates, these results clearly indicate the existence of significant differences in DNA methylation between neuronal and nonneuronal cellular populations at the level of individual sites.
Figure 2.

Significant differences in DNA methylation between neuronal and nonneuronal cellular populations in the human OFC. (A) Unsupervised hierarchical clustering of the DNA methylation data obtained in the neuronal and nonneuronal samples from the OFC of six subjects in two independent replicate experiments, using 453 221 filtered sites that comprised the final data set. Cell type was the largest source of variability in these data compared with the effects of the experimental replicate or heterogeneity among the subjects. (B) Scatter plots comparing averaged (across all six subjects) DNA methylation levels in the neuronal versus nonneuronal samples obtained in two replicate experiments. Each point represents an average beta value for one site from the array. The data demonstrate high level of reproducibility between the technical replicates as well as significant differences in DNA methylation patterns between neuronal and nonneuronal cells.
In contrast to differences between cell types (Figure 2A), we observed low variability in DNA methylation among individuals (Supplementary Figure S7). When all HM450K sites were considered, pairwise Spearman correlation coefficients calculated among the six subjects within both neuronal and nonneuronal samples for each of the two replicate experiments ranged between 0.97 and 0.98 (Supplementary Table S3). Similar results were obtained when the analysis was restricted to sites with intermediate levels of methylation (average β between 0.2 and 0.8) (Supplementary Table S3). The low interindividual variability (compared with highly significant differences between the cell types) was also confirmed by analysis of variance (ANOVA) (Supplementary Figure S8 and Supplementary Table S4).
Differential methylation in neuronal versus nonneuronal cells
We applied a paired t-test to calculate the difference in methylation between neuronal and nonneuronal cellular populations. The analysis was performed separately for each of the two experimental replicates. Based on Illumina recommendations, to achieve 99% confidence of detection (30), we considered a site to be DM if it showed an absolute value of difference between β values in neuronal versus nonneuronal cells >0.2 [|delta(β)| > 0.2]. In addition, false discovery rate was applied at 1% level to correct for multiple testing. We defined the DM sites with higher DNA methylation in nonneuronal versus neuronal cells as ‘neuronal undermethylated’ (NUM) sites. For the sake of clarity and because the nonneuronal fraction consists mostly of the glial cells (see above), we referred to the DM sites with higher DNA methylation in neuronal versus nonneuronal cells as ‘glial undermethylated’ (GUM) sites. Given the well-established negative correlation between methylation and expression in CpG-rich promoters (49–51), we considered NUM and GUM sites as ‘neuronal-type’ (linked to neuron-specific gene expression) and ‘glial-type’ (linked to glia-specific gene expression), respectively. Applying these criteria, we identified 26 556 NUM and 30 672 GUM CpG sites in the first experimental data set, and 26 609 NUM and 31 205 GUM CpG sites in the replicate data set. Among these sites, 23 670 NUM and 27 742 GUM sites showed differential methylation in both experiments, and these ‘overlapping’ subsets of sites were used in all subsequent analyses (Supplementary Figure S4).
Differential methylation in CpG islands and related features
CpG islands represent an important class of regulatory regions in mammalian genomes that are characterized by high local concentration of CpG sites (52). The CpG islands usually have a low level of methylation, and are often found in the vicinity of promoters (53,54). In addition to CpG islands, two related genomic features known as ‘shores’ and ‘shelves’ are also abundantly represented in the HM450K array (92% and 86% coverage, respectively). The shores are defined as 2-kb regions flanking CpG islands, whereas shelves are located within 2 kb outside of the shores. According to a recent study, most tissue-specific differential DNA methylation is situated not in the CpG islands themselves but within the shores (18).
We found that the fraction of the sites located within CpG islands among the DM sites was significantly lower than the fraction of CpG island sites in the HM450K array as a whole, indicative of depletion of the DM sites in the CpG islands [odds ratio (OR) = 0.30, P < 1e−275, by Fisher’s exact test] (Supplementary Figure S9A). When the GUM and the NUM sites were considered separately, comparable depletion of the DM sites in CpG islands was observed for both cell types (ORs 0.25 and 0.29 for the NUM and GUM sites, respectively; P < 1e−275) (Supplementary Figure S9B and C). We also detected moderate enrichment of the NUM sites located in CpG island shores (OR = 1.5, P = 3.5e−166) and to a lesser extent in shelves (OR = 1.34, P = 8e−45) (Supplementary Figure S9B and C), whereas the GUM sites did not show notable enrichment or depletion in these regions.
Differential methylation within different genomic regions
Next we analyzed the distribution of the DM sites among various genomic regions as defined according to the UCSC browser annotation (55). In particular, ‘promoter’ is defined as the region within 1000 bp upstream and 100 bp downstream of TSS of a gene. We observed a significant depletion of the NUM and GUM sites in the promoters (OR = 0.37 and 0.61; P < 1e−252) (Supplementary Figure S10A). This was in line with our finding of the depletion of the DM sites in CpG islands (see Supplementary Figure S9) because CpG islands overlap with the majority of annotated mammalian promoters (53,54). Both the NUM and the GUM sites were overrepresented in introns (OR = 2.02 and 1.27, P < 1e−275 and 2.7e−74, respectively). However, only the GUM, but not the NUM sites, were enriched in intergenic regions (OR = 1.40, P = 3.2e−134, and OR = 0.94, P = 2.3e−5, respectively) (Supplementary Figure S10A).
Genes with CpG island-containing promoters (hereinafter CpG promoters) mostly encode housekeeping proteins that are expressed in all tissues (53), but also include a substantial number of master developmental regulators such as HOX genes (56). In contrast, non-CpG promoter genes tend to have more restricted cell-specific expression patterns and are expressed later in development during tissue differentiation. We calculated the relative abundances of DM sites in these two types of promoters. We defined a promoter as a CpG promoter if it overlapped with an annotated CpG island by at least 200 bp. The GUM but not the NUM sites were more likely to reside within non-CpG promoters (OR = 1.5, P = 1.2e−114). In contrast, both the GUM and the NUM sites were depleted to a similar extent in CpG promoters (ORs 0.23 and 0.24, respectively, P values < 1e−275) (Supplementary Figure S10B).
Distribution of DM sites by distance from TSS: enrichment of DM sites in predicted enhancers
We next examined the distribution of the DM sites as a function of the distance from the nearest TSS (Figure 3). In agreement with our findings that DM sites were underrepresented in CpG islands and/or in CpG promoters (Supplementary Figures S9 and S10), we observed depletion of both the NUM and the GUM sites in the vicinity of TSS (the density of the DM sites was <1 at a distance of <1000 bp from TSS in Figure 3). However, the GUM sites were more abundant than the NUM sites proximal to TSS, in line with our observation that the GUM (but not the NUM) sites are enriched within non-CpG promoters (Supplementary Figure S10B). There was also a conspicuous peak in the NUM data profile outside the promoter region (∼1000–4000 bp to TSS; Figure 3), which largely originated from the contribution of the NUM sites that are enriched within CpG island shores (Supplementary Figures S9B and S11).
Figure 3.

Density distribution of differential DNA methylation as function of distance from TSS. The distance is represented on a log scale. For each bin, the normalized density of sites was calculated as [(number of DM sites per bin)/(total number of DM sites)]/[(number of sites in the filtered HM450K array per bin)/(total number of sites in the filtered HM450K array)]. For both the NUM sites (shown as filled circles) and the GUM sites (shown as open circles), the normalized density of the DM sites was <1 at a distance of <1000 bp from TSS, and was >1 at a distance of >1000 bp from TSS, suggesting that the DM sites were depleted in the promoters but enriched within distal regulatory elements. More GUM than NUM sites resided proximal to TSS. Arrow indicates an enrichment of the NUM sites outside promoter regions (∼1000–4000 bp from TSS), which was largely explained by the overrepresentation of the NUM sites in CpG-island shores (Supplementary Figure S8). Sites with nonsignificantly changed methylation (NS) are shown as open squares.
Most importantly, the data clearly demonstrated that both the NUM and the GUM sites were significantly more likely to reside distal to TSS (density of DM sites > 1 at a distance of > 1000 bp to TSS in Figure 3). Because the HM450K array covers a significant number of ENCODE-predicted enhancers, the enrichment of the DM sites at positions distal to TSS could be, at least in part, explained by the high prevalence of the DM sites within enhancers. Indeed, we found that both the NUM and the GUM sites were significantly enriched within HM450K-annotated enhancers (OR = 1.7 and 3.0, respectively, P < 1.0e−275) (Figure 4A). As expected, the majority of the DM sites in HM450K-annotated enhancers were located distally from TSS (Supplementary Figure S12).
Figure 4.

Overrepresentation of differential DNA methylation in predicted enhancers. (A) The NUM and GUM sites were enriched in predicted enhancers, defined according to the HM450K array annotation. (B) Comparison of the DM sites (this study) with genome-wide mapping of distal H3K4me1, distal H3K27ac and H3K4me3 marks in different tissues and cells (32). Left panel: The NUM and GUM sites were highly overrepresented in distal H3K4me1 peaks in the adult frontal and temporal lobes; however, in control nonbrain specimens (liver or blood cells) the enrichment was less pronounced. Middle panel: The NUM and GUM sites were highly enriched in distal H3K27ac peaks in frontal and temporal lobe; in contrast, the NUM sites were only marginally enriched in nonbrain tissues. Right panel: the NUM and GUM sites were underrepresented in H3K4me3 peaks in all specimens.
The recent ChIP-Seq study by the NIH Roadmap Epigenomics Mapping Consortium (REMC) examined histone modifications in many human cell lines and tissues including several regions of the brain (32). The data encompass genome-wide measurements of histone modifications including histone 3 lysine 4 mono-methylation (H3K4me1), histone 3 lysine 27 lysine acetylation (H3K27ac) and histone 3 lysine 4 tri-methylation (H3K4me3). Enrichment of H3K4me3 is indicative of an active promoter, whereas H3K4me1 and H3K27ac outside promoter regions are generally considered to mark enhancers (46,57,58). We reasoned that promoters and enhancers predicted from these experiments (done on bulk human brain tissue rather than on isolated cell types) would be informative for assigning enhancer status to the DM sites. Because the H3K4me1 and H3K27ac marks are also known to overlap with promoter regions, we used only distal H3K4me1 and H3K27ac peaks, specifically those that are located >2.5 kb from TSS. We found that both the NUM and the GUM sites were more likely to reside within distal H3K27ac- and H3K4me1-enriched regions in the frontal and temporal lobes compared with the enhancer-associated regions identified by the same criteria in the control specimens (liver, or CD20 cells) (Figure 4B). The effect was more pronounced for the NUM sites, probably because of the significant heterogeneity of cells within our nonneuronal population (e. g the presence of microglia).
The REMC study also reported that in the human brain tissues the majority of the H3K4me1 marks (∼70%) reside within genes, whereas the marks of the Polycomb-repressed state are significantly enriched within intergenic regions, suggestive of a preferential utilization of regulatory elements within introns in highly specialized adult brain cells (32). Thus, we examined the distribution of the NUM and GUM sites within the distal H3K4me1 and H3K27ac marks. In line with REMC study, we observed significant enrichment of the intronal NUM sites but significant depletion of the intergenic NUM sites that reside within both enhancer-associated marks (Supplementary Figure S13). The GUM sites localized within H3K27ac regions showed a similar, albeit less pronounced pattern, but the GUM sites within H3K4me1 regions were not depleted in intergenic regions. These data suggest that the relatively higher density of enhancer-associated marks in introns versus intergenic regions is more pronounced in the neuronal than in the nonneuronal cells.
In contrast to the H3K4me1 and H3K27ac regions, both the NUM and the GUM sites were significantly depleted in the H3K4me3-enriched regions in frontal and temporal lobes (Figure 4B), in agreement with our findings that DM sites are underrepresented in promoters (Supplementary Figure S10). The NUM and GUM sites were also depleted in the H3K4me3-enriched regions in the control specimens. These data are consistent with recent findings in multiple human cell types demonstrating that the majority of promoters are active in multiple cell types, whereas the enhancers are significantly more cell-type–specific (46,57,59).
Evolutionary conservation of the NUM and GUM sites and regions that encopass these sites
We tested the evolutionary conservation of the NUM and GUM sites compared with sites with nonsignificantly changed methylation (NS sites). We used the phastCons46way track for vertebrates from UCSC, which provides a precalculated conservation score ranging from 0 to 1 for each position in the human genome based on multigenome alignment among vertebrates (60). A higher score for a specific position indicates that the site has a higher probability of being conserved. We compared the phastCons score values for all CpG sites within each category (NUM, GUM and NS) and found that the mean conservation for the NUM and GUM sites was significantly lower than the mean conservation among the NS sites (Supplementary Figure S14A). There was no significant difference between the NUM and the GUM conservation scores.
We also calculated the mean conservation in the regions encompassing 100, 250, 500 and 1000 bp in both directions from each NUM and GUM site (NUMR and GUMR, respectively). The conservation within the NUMR and GUMR was compared with the mean conservation of (i) regions of the same size that are adjacent to the NUMRs and GUMRs in both directions (for example, for a 100-bp NUMR or GUMR, the comparison was done with the 100-bp sequences located immediately upstream and immediately downstream of the respective region of interest); (ii) the regions of the same size that encompass RefSeq annotated TSS (i.e. promoter regions); (iii) RefSeq annotated coding exons. This analysis showed that NUMR and GUMR are significantly less conserved compared with the regions that encompass TSS and coding exons (Supplementary Figure S14B). This is in line with the preferential concentration of both the NUM and the GUM sites within enhancers in contrast to the depletion of the DM sites in promoters (see also Figure 4A). As reported previously, compared with promoters, enhancers tend to be more variable among species (61). Conversely, for all fragment sizes tested (200, 500, 1000, 2000 bp), the NUMR and GUMR are more conserved than the adjacent sequences of the same length (Supplementary Figure S14B), suggesting functional importance of the NUMR and GUMR.
Validation of the HM450K methylation data by reduced representation bisulfite sequencing
To validate the results obtained by the HM450K array, we applied ERRBS to a subset of the study cohort (three of the six subjects). Similar to RRBS, ERRBS uses the MspI restriction, which leads to a bias for CpG-rich regions of the genome. However, ERRBS provides an extended coverage for regions lying outside of CpG islands (31). To ensure the reproducibility of measurements across the full range of values, the analysis was restricted to sites with an at least 10× coverage (11). On average, 3 192 726 ± 151 816 (mean ± SD) CpG sites were detected in the three neuronal and three nonneuronal samples that we assessed, of which 2 255 360 were present in all six samples and were used in subsequent analyses (see Supplementary Figures S15–S20).
The ERRBS data set provided a greater coverage of both CpG and non-CpG methylation sites than did the HM450K data set. Moreover, the sets of methylation sites obtained by these two methods complemented each other. For example, a more detailed assessment of CpG islands and shores was achieved by the ERRBS than by the HM450K method, whereas non-CpG island promoters were better represented in the HM450K array (see Supplementary Figures S9B, S10B, S15, S17–S18). There were 113 858 CpG sites that overlapped between the ERRBS and HM450K data sets. Similar to the analyses of the entire HM450K (Figure 2) or ERRBS data sets (Supplementary Figure S15A), unsupervised hierarchical clustering of DNA methylation data obtained for these overlapping sites showed perfect separation into the neuronal and nonneuronal groups (Supplementary Figure S16A). In addition, within each cell type group, a clear division was detected between the data obtained with the two methods (Supplementary Figure S16A), indicating a greater magnitude of differences between the platforms compared with differences among the individuals (also see Supplementary Table S5A for the analysis of correlation between the ERRBS and HM450K data sets in each individual). The mean methylation levels (across all subjects) at each of the overlapping sites showed a strong positive correlation between the ERRBS and HM450K experiments for samples of the same cell type; the correlations were lower when neuronal versus nonneuronal methylation levels were compared (Supplementary Figure S16B). This relationship was reproduced when only a subset of sites with an intermediate range of DNA methylation levels (beta values: 0.3–0.7) was analyzed although lower correlations were detected (Supplementary Figure S16C and D, Supplementary Table S5B), reflecting the inherent differences between the two methods.
Applying criteria comparable with those used for the HM450K analysis [difference in percentage of methylation > 20, Q-value < 0.01] to the 2 255 360 sites that were detected in all six samples by ERRBS, we found 91 336 CpG sites that were undermethylated in neurons compared with nonneuronal (mostly glial) cells (NUMERRBS sites) and 167 251 CpG sites that were undermethylated in nonneuronal compared with neuronal cells (GUMERRBS sites). Importantly, among the CpG sites that overlapped between the ERRBS and HM450K data sets, there were no sites with opposite types of differential methylation detected by the two methods (Supplementary Table S6). The analysis of the distribution of the ERRBS-detected DM sites within different genomic regions largely confirmed the findings obtained with the HM450K method (see details in Supplementary Figures S17–S20). Specifically, both NUMERRBS and GUMERRBS sites were significantly depleted in CpG islands and promoters, and the NUMERRBS sites were more likely to be found in intragenic regions (exons and introns) versus intergenic regions, whereas the GUMERRBS sites showed enrichment in both introns and intergenic regions. Both the NUMERRBS and GUMERRBS sites were significantly depleted in the vicinity of TSS, but were significantly more likely to reside distal to TSS. The distal NUMERRBS and GUMERRBS sites strongly overlapped with the distal H3K4me1 and H3K27ac marks from the REMC study (indicative of enhancers), whereas the NUMERRBS and GUMERRBS sites were depleted from the regions enriched in the H3K4me3 mark (indicative of promoters).
Correlation between methylation status and cell-type–specific gene expression
We labeled each HM450K array-annotated gene (see Supplementary Methods) as neuronal if it had more NUM than GUM sites, or as nonneuronal if it had more GUM than NUM sites, thus predicting 5098 neuron- and 6821 non–neuron-specific genes (Table 1 for the top 25 neuronal and nonneuronal genes; Supplementary Table S7 for the entire gene list). The majority of the genes with known patterns of expression showed the expected (neuronal or nonneuronal, respectively) methylation profile, thus demonstrating the negative correlation between cell-type–specific gene expression and differential DNA methylation measured across the entire gene locus (Table 1 and Figure 5A). We also independently validated these results using bisulfite sequencing (see Supplementary Methods) of six loci within genes that were known or predicted to be neuronal (DNMT3A, SHANK2, ZFYVE28) or nonneuronal (GFAP, MOG, TMEM140) based on their HM450K array DNA methylation profiles (Figure 5B and Supplementary Figure S21, Supplementary Table S8). However, a substantial minority of known cell-type–specific genes displayed no obvious methylation preference or even showed a preference opposite to the expected (Table 1). Some of these genes showed no consistent pattern of differential methylation across the gene locus, suggesting that their cell-specific gene expression is determined by regulatory elements localized only within a specific region (see Supplementary Figure S21 for examples). For example, although ABR had more NUM than GUM sites within the entire gene locus, and was, therefore, classified as neuronal, there was a strong nonneuronal-type signal in the promoter region of ABR, which correlates with its observed cell specificity (Supplementary Figure S22).
Table 1.
DNA methylation-based prediction of neuronal versus non–neuronal-cell–specific gene expression
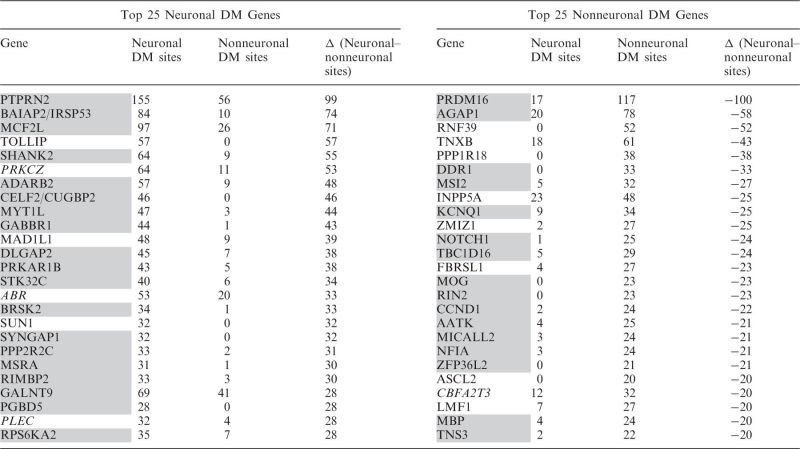 |
Predictions were made based on the number of the NUM or GUM sites assigned to the gene. Genes were sorted by the difference in the number of the NUM and GUM sites, and the top 25 protein-coding genes of each type are shown here. Color denotes comparison between predictions based on gene methylation status in humans (this study) and on differential expression status (neuronal or glial) in mouse brain (Cahoy et al., 2008) (61). Gray color—correct prediction, italic—contrary to the data by Cahoy et al., not marked—no information on neuronal versus glial cell specificity was available in Cahoy et al. based on the most relaxed criteria (2× enrichment) or in the available literature.
Figure 5.
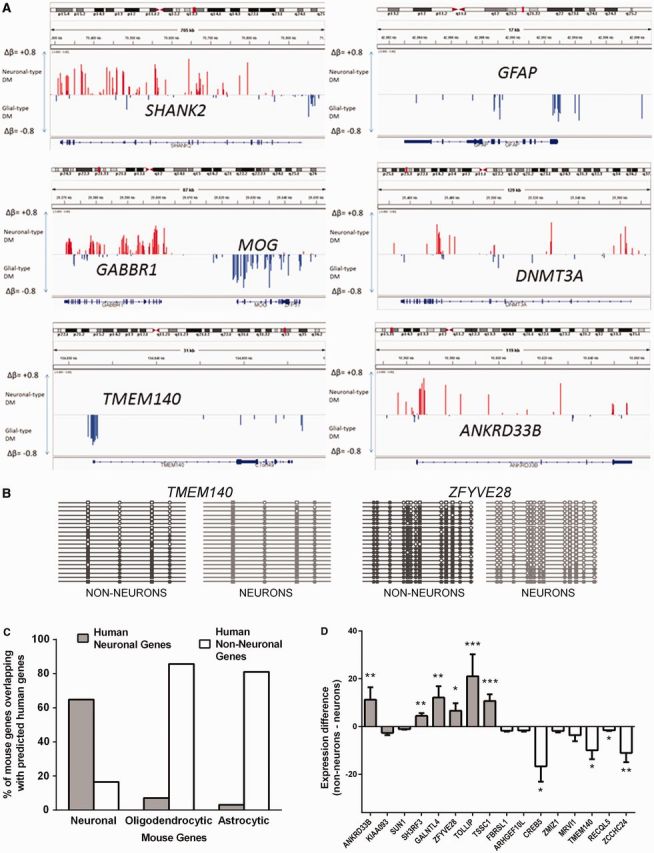
Differential DNA methylation profile predicts cell-specific gene expression. (A) Depiction of differential DNA methylation signatures at individual gene loci. Visualization is made using IGV (62). Shown are methylation profiles for several known neuronal (SHANK2, GABBR1, DNMT3A) or glial (MOG, GFAP) genes, as well as for putative non–neuron-specific (TMEM140) and neuron-specific (ANKRD33B) genes. Red and blue bars represent Δβ for sites that are undermethylated in neurons or nonneuronal cells (denoted here as ‘glial’), respectively. (B) Validation of the results obtained in HM450K array using bisulfite sequencing. Between 18 and 20 clones were analyzed for each fragment and cell type. Shown are results for TMEM140 (genomic location: 7: 134 833 108–134 833 366), predicted to be expressed mostly in nonneuronal cells, and ZFYVE28 (genomic location: 4: 2 303 549–2 303 774), predicted to be expressed mostly in neurons. CpG sites that are present in the HM450K array are denoted by squares; other CpG sites are denoted by circles. Methylated sites are filled. Methylation profiles obtained from neuronal and nonneuronal nuclei are shown in light gray color and dark gray color, respectively. (C) Cellular specificity of genes predicted by their DM in the human OFC highly correlates with cell-specific expression of these genes in the mouse brain. Shown is the percent of overlap between human neuron- and non–neuron-specific genes (predicted from their differential methylation in this study) and mouse neuron-, astrocyte- and oligodendrocyte-specific genes identified in flow cytometry–sorted cellular populations by (61). (D) We carried our qPCR in four individuals using RNA specimens obtained from the laser microdissected neurons (from deep layers of gray matter) and small areas of adjacent white matter in the PFC (see Methods and Supplementary Figure S19). Shown are fold differences that represent neuronal to white matter (positive values) or white matter to neuronal (negative values) gene expression of genes for which a cell-specific expression pattern was predicted from the methylation data. Gray bars, genes with predicted neuron-specific expression; white bars, genes with predicted non–neuron-specific expression. *P < 0.05; **P < 0.01; ***P < 0.001.
We then performed a functional annotation analysis of genes that we predicted to be neuronal or nonneuronal based on their DNA methylation profile using the MetaCore tool (www.genego.com). For the predicted neuronal gene set (5098 genes), we detected enrichment for neuron-related terms in all examined ontology databases (Supplementary Table S9). In contrast, we did not detect any neuron-associated terms in any of these ontologies for the predicted nonneuronal genes (6821 genes).
Next, we investigated the overlap between our DNA methylation-based predictions and the published data set of mouse neuron-, astrocyte- and oligodendrocyte-specific genes (61). In that study, a gene was defined as cell-type–specific if its expression within a particular cell type was at least 20 times higher compared with the other two cell types. We detected a highly significant overlap (i.e. a substantially greater number of orthologs than expected by chance) between the set of the predicted human neuron-specific genes and the set of neuron-specific genes in the mouse brain (Figure 5C and Supplementary Table S10). Similarly, both the astrocyte- or oligodendrocyte-specific mouse gene lists showed a nonrandom overlap with the nonneuronal gene list inferred in this work.
We also compared our DNA methylation-based predictions of cell-type–specific gene expression with the respective predictions derived from the analysis of the human transcriptome. We applied an in silico tissue dissection statistical approach that is based on WGCNA (36,63). WGCNA identifies biologically relevant patterns in high-dimensional gene expression data sets by grouping genes into modules with strongly covarying patterns across the sample set. WGCNA can distinguish gene expression patterns associated with specific cell types (e.g. neurons or glia) that are present in a heterogeneous sample (e.g. whole human cortex) thanks to the distinct transcriptional profiles of these cell types and variation in their relative proportions across samples (64). Modules generated by this unbiased approach are then examined for cell-type–specificity as determined by enrichment analysis using neural cell type-enriched gene sets found in previous studies (61,65–70). We applied WGCNA to the postnatal subcohort (N = 220) of the gene expression microarray data set from the BrainCloud study, which analyzed PFC samples obtained from a large collection of autopsy brain specimens (Gene Expression Omnibus; accession GSE30272) (34). WGCNA resulted in well-defined gene co-expression modules (N = 77) with specific anatomical distributions, consistent with previous studies in the brain tissues (64,68) (Table 2). The identified gene modules were frequently related to primary neural cell types or molecular functions. In particular, we detected numerous modules consisting of genes with enhanced expression of neurons or of different types of glia (microglia, astrocytes and oligodendrocytes) (Table 2). Next, we applied the hypergeometric distribution method (71) to compare the co-expression modules with the sets of genes that were identified as neuronal, nonneuronal or non–cell-type–specific (containing equal numbers of the NUM and GUM sites) on the basis of the DNA methylation profiles. This comparison revealed a significant enrichment for methylation-predicted neuronal genes in modules related to glutamatergic and GABAegric (e.g. parvalbumin-expressing) neurons, whereas the predicted nonneuronal genes were highly specific for oligodendrocyte-, microglia- and astrocyte-related modules (Table 2). In contrast, the set of non–cell-type–specific genes showed no enrichment in any of the BrainCloud co-expression modules.
Table 2.
Cell-type–specific genes (neuronal or nonneuronal) predicted by their differential DNA methylation are significantly enriched in the co-expression modules identified in BrainCloud transcription data set by WGCNA
| BrainCloud co-expression module | Num-ber of genes per mo-dule | Annotation of module with brain data set implanted in the UserListEnrichment WGCNA function | Module type | BrainCloud P value | Predicted neuronal genes (N = 3422) |
Predicted nonneuronal genes (N = 4925) |
No prediction (N = 11 661 genes) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Overlap | FER | Bonf. Corr. P value | Overlap | FER | Bonf. Corr. P value | Overlap | FER | Bonf. corr. P value | |||||
| M3 | 1227 | blue_M2_Oligodendrocytes__HumanMeta | OLIG | 2.3e−222 | 115 | 0.76 | 1 | 341 | 1.57 | 7.8e−17 | 497 | 0.96 | 1 |
| M10 | 404 | brown_M3_Astrocytes__HumanMeta | ASTRO | 1.3e−135 | 29 | 0.58 | 1 | 145 | 2.02 | 3.5e−16 | 155 | 0.91 | 1 |
| M13 | 318 | pink_M10_Microglia(Type1)__HumanMeta | MG | 1.3e−89 | 25 | 0.64 | 1 | 114 | 2.02 | 1.8e−12 | 101 | 0.76 | 1 |
| M30 | 155 | Neuron_probable_Cahoy/green_M10_ GlutamatergicSynapticFunction__CTX | NEU_ GLU | 7.0e-13/5.9e−11 | 56 | 2.93 | 4.6e-12 | 16 | 0.58 | 1 | 68 | 1.05 | 1 |
| M64 | 59 | orange_M5_Microglia(Type2)__CTX | MG | 2.0e−29 | 2 | 0.28 | 1 | 28 | 2.68 | 3.5e−05 | 11 | 0.44 | 1 |
| M2 | 1298 | black_M1_PvalbInterneuron_HumanMeta | PV+ | 4.0e−13 | 264 | 1.65 | 8.0e-15 | 227 | 0.99 | 1 | 623 | 1.14 | 0.001 |
| M36 | 150 | brown_M3_Astrocytes_HumanMeta | ASTRO | 2.5e−65 | 14 | 0.76 | 1 | 61 | 2.29 | 8.3e−09 | 54 | 0.86 | 1 |
| M5 | 625 | Neuron_probable__Cahoy | NEU | 8.2e−29 | 143 | 1.86 | 2.0e-11 | 85 | 0.77 | 1 | 311 | 1.19 | 0.01 |
| M60 | 68 | green_M10_GlutamatergicSynaptic Function__CTX | GLU | 1.3e−15 | 29 | 3.46 | 9.1e-08 | 8 | 0.66 | 1 | 28 | 0.98 | 1 |
| M23 | 197 | Autism_associated_module_M12Voineagu/ Neuronprobable_Cahoy | NEU_ AUTISM | 4.2e−11/1.1e−10 | 60 | 2.47 | 2.7e-09 | 22 | 0.63 | 1 | 103 | 1.25 | 0.5 |
BrainCloud microarray data were obtained from Gene Expression Omnibus (accession GSE30272). WGCNA was performed as described in ‘Materials and Methods’ section. Among 5098 predicted neuronal genes, 3422 genes were present in the BrainCloud cohort. Among 6821 predicted nonneuronal genes, 4925 genes were present in the BrainCloud cohort. Among genes that were not predicted to be expressed in a cell-type–specific manner, 11 661 genes were present in the BrainCloud Cohort. The enrichment was determined by hypergeometric distribution. Shown are P-values that were Bonferroni-corrected (Bonf.-corr.) for multiple testing (N = 231 tests; 77 modules × 3 gene lists). NEU, neuron; ASTRO, astrocyte; OLIG, oligodendrocyte; MG, microglia; PV+, parvalbumin-expressing neurons; GLU, glutamatergic neurotransmission; FER, fold enrichment ratio. The FER is calculated using a population size that was defined as the total number of genes with evidence of robust expression in the BrainCloud (BC) cohort (N genes BC = 27 777); 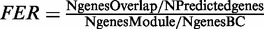
Ideally, experimental validation of the correlation between the DNA methylation status of a gene and its expression would require generating both sets of data from the same samples. However, in contrast to studies in cell lines or whole brain tissues, a direct comparison between DNA methylation and RNA expression within separate cell populations obtained from the human postmortem brain is not currently feasible because nuclei-sorting protocols are not compatible with preserving high-quality RNA that is required for gene expression profiling. Nevertheless, we performed LMD to isolate specimens comparable with those that were used in the present study for the analysis of DNA methylation after nuclei sorting. Specifically, we microdissected neuronal profiles from the gray matter as well as small areas of adjacent white matter; the latter represented the mixed population of cells that was mostly devoid of neurons (Supplementary Figure S23). Using qPCR, we first confirmed the enrichment of the obtained specimens for the known neuronal and glial markers, respectively (Supplementary Figure 24). We then tested if neuronal versus nonneuronal specificity of genes, for which the expression pattern had not been previously studied, could be predicted based solely on their patterns of differential DNA methylation. Because LMD provides limited amounts of material, we selected a small set of predicted neuronal and nonneuronal genes (10 in each group), which to our knowledge were not previously described as being predominantly expressed in the neuronal or glial cells. Among the 16 genes that showed sufficient level of expression in the microdissected samples, six predicted neuronal genes (ANKRD33B, SH3RF3, GALNTL4, ZFYVE28, TOLLIP, TSSC1), and four predicted nonneuronal genes [TMEM140 (see Figure 5A), CREB5, RECQL5, ZCCHC24] demonstrated significant enrichment (P < 0.05) in the respective cell population, whereas the remaining six genes were not significantly enriched in any of the two cell populations (Figure 5D).
Transcription factor-binding motifs in DM regions
We further asked whether the regions that encompass the DM sites (within 250 bp from each site) could contribute to cell-type–specific transcription via differential regulation by distinct sets of transcription factors (TFs). To this end, we compared the sets of predicted binding sites for known TFs between the NUM- and GUM-containing regions (NUMR and GUMR) using HOMER software for known motif discovery. Compared with the NS sites, our analysis identified a significant enrichment of TF motifs within both the NUMRs and the GUMRs; moreover, the majority of the identified motifs differ between the NUMRs and the GUMRs (see Supplementary Table S11 for the lists of the top 20 hits in each category). Both the NUMR- and the GUMR-enriched TF motif lists included many TFs (i.e. members of the EGR, MEF2 and SOX families) that play important roles in brain development and function (72–74), as well as TFs that are mostly expressed in the brain (e.g. members of RFX family) (75). The distribution of the TFs between the NUMR- and the GUMR-enriched lists also correlated well with the published data on their cell-type–specific expression in the mouse cortex (61). Specifically, EGR2, MEF2c, MEF2a and NEUROD1, whose consensus motifs were found enriched in the NUMRs but not in the GUMRs, are mostly expressed in neurons, whereas GUMR-enriched TFs (SOX2, SOX6, FOXO1 and TCF1) are mostly expressed in glia, with the sole exception of OLIG2.
A classification of predicted enhancers in 17 mouse tissues (including three adult brain tissues: cerebellum, cortex and olfactory bulb) has been recently developed using H3K4me1 marks, and TF binding motifs that were enriched in tissue-specific enhancers were identified (76). Among the 11 unique brain-specific motifs [Supplementary Table S12 in (76)], nine were present in the NUMR or GUMR lists (Supplementary Table S11). Moreover, all five motifs (RFX, X-BOX, MEF2A, ATOH1 and NP1) that were found to be enriched in the mouse cortex-specific enhancers were also present in our NUMR list. Likewise, using H3K4me1 profiling, the aforementioned REMC study assessed six different brain regions from the adult human brain and found two brain-specific clusters of predicted enhancers and 15 unique TF motifs enriched within these clusters [Clusters 18 and 19; Figure 2 and Supplementary Table S3 in (32)]. Eight of these 15 motifs (ZNF263, RFX, NF1, X-BOX, TAL1, SOX2, TLX and MYOD) were present among the top NUMR or GUMR TF motifs obtained in our analysis. Collectively, these findings agree with our observation that the NUM and GUM sites are enriched within predicted enhancers and accordingly are localized within regions that contain binding sites for cell-type–specific TFs.
The most interesting discovery in our motif-enrichment analysis is that the NUMRs seem to be enriched in TF motifs (i.e. MEF2C, NEUROD, AP-1 and EGRs) that mediate neuronal activity-dependent gene expression (77). Specifically, following neuronal stimulation, it has been found that the preexisting TFs (MEF2C and NEUROD) are activated through direct posttranscriptional modifications and facilitate the expression of promoter IV variant of Bdnf or the induction of dendritogenesis, respectively (73,78). AP1 and EGR are the products of immediate early genes (IEGs). The IEGs are activated in a rapid, transient and protein-synthesis–independent manner on neuronal stimulation, and their products in turn promote the transcription of additional activity-regulated genes. For example, EGR family members are rapidly induced in neurons by membrane depolarization and are implicated in regulation of memory (72), whereas c-Fos (a component of the dimeric AP-1 complex with Jun) has often been used as a marker of neuronal activation in different experimental contexts (77). The majority of the previous studies of the TFs mediating neuronal activity-dependent gene expression mostly focused on the promoters of activity-regulated genes (77). In line with a recent study (79), this work implicates enhancers as key players in cell-type–specific regulation of gene expression, by showing that the NUMRs are depleted in promoters but enriched in distal regulatory elements.
Non-CpG methylation in neuronal and nonneuronal cells
Although the HM450K array focuses on the ‘classical’ CpG-type methylation, it also probes 3091 non-CpG positions. In both replicate experiments, we found that the majority of these non-CpG sites were significantly more methylated in the neuronal compared with nonneuronal cells (Figure 6A). Although the number of non-CpG sites probed by the array is small, the neuron-specific methylation was observed in all six subjects in both replicate experiments (Figure 6B). To validate these findings, we used the ERRBS assay, which provided data for much larger population of non-CpG sites (∼10 000 000) with the coverage of at least 10 reads per site in each sample. At all methylation level intervals except for the lowest, neurons showed a greater number of methylated sites compared with nonneuronal cells (Figure 6C). In particular, we detected 20-fold more neuronal than nonneuronal non-CpG sites with methylation level > 20%, and 25-fold more neuronal than nonneuronal sites with methylation level > 40% (Figure 6C insert). Similar results were obtained when non-CpG sites located within the CHG or CHH sequence contexts were analyzed separately (Supplementary Figure S25). The mean level of methylation across all probed non-CpG sites was 2.6% in neurons and 0.36% in nonneuronal cells (a 7.2-fold difference). We also analyzed sequence context of the non-CpG methylation in the ERRBS assay, which was visualized using the WebLogo software (80). In agreement with recent findings (11), the neuronal non-CpG loci preferentially occur in a CACC sequence context (Figure 6D). We observed high pairwise correlation of non-CpG methylation among neuronal samples from different subjects (R values range from 0.86 to 0.88) (Supplementary Table S12), suggesting consistent patterns of non-CpG methylation in neurons. These correlations were lower (R = 0.66–0.74) for the nonneuronal samples. These data strongly suggest that non-CpG methylation does not occur in a random fashion, but rather constitutes a highly controlled process, which may play an important role in cell-specific epigenetic regulation.
Figure 6.
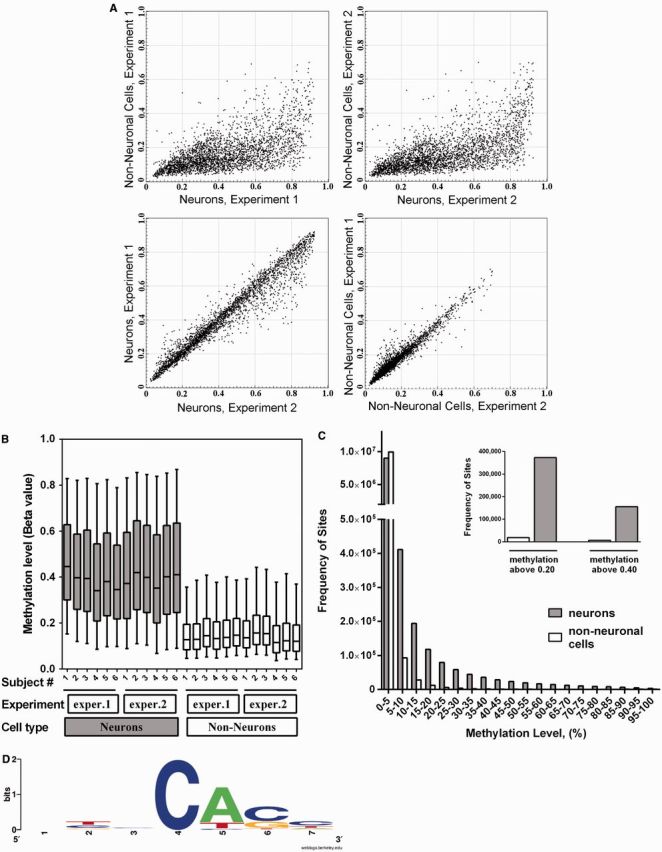
Non-CpG methylation in the adult human brain is restricted to neuronal cells. Data obtained with the Illumina HM450K array (A, B), and with the ERRBS methylation assay (C, D). (A) Scatter-plot comparison of non-CpG methylation in neuronal and nonneuronal cells at 3091 individual cytosine positions probed by the HM450K array. In both replicate experiments the majority of non-CpG methylation was detected in neuronal cells. (B) Box-and-whiskers plot comparing non-CpG methylation in individual samples. Boxes denote the median and the 25th and 75th percentiles of methylation level; whiskers denote the 5th and 95th percentiles. In both replicate experiments, the neuronal specificity of non-CpG methylation was observed in all six subjects. (C) Frequency histogram of non-CpG methylation levels in neurons versus nonneuronal cells. Data were obtained with ERRBS. Each bar in the figure represents a 5% bin interval of methylation level. Figure includes data for 10 086 642 non-CpG cytosine positions, which were present in both neuronal and nonneuronal data sets in all three subjects with coverage of at least 10 reads per site. Inset: Number of non-CpG cytosine sites with methylation level of >0.20 or 0.40. (D) Sequence context of non-CpG methylation at sites above 20% of methylation level in neuronal cells (N = 373 212 sites; based on ERRBS).
DISCUSSION
Recent findings of the ENCODE and REMC projects reveal exquisite cell-type specificity of noncoding regulatory elements (32,81). A major challenge for genome research is to characterize the epigenetic signatures that mark these functional regions, especially in tissues containing heterogeneous cell populations. This task is particularly daunting in the case of the brain, where various cell types are differentially distributed among anatomically and functionally diverse brain regions. As an initial attempt to address this challenge, in the present study we focused on DNA methylation profiling of the neuronal and nonneuronal cells, which were obtained from a discrete area of the human PFC and separated by FACS.
To the best of our knowledge, our findings represent the first demonstration that the in vivo DNA methylation differences that distinguish the two major types of cells in the brain (neuronal and nonneuronal) are mostly located distally from TSS and are probably positioned within cell-type–specific enhancers. In contrast, these cell-type–specific DNA methylation differences are depleted in CpG islands and consequently in CpG promoters. These observations are in agreement with the recent whole methylome study of the mouse embryonic stem cells (ESC) and ESC-derived neuronal progenitor cells (48) as well as with the previous studies that mapped the chromatin state in human cell lines (45,46,57), which indicate that enhancers constitute the most variable class of transcriptional regulatory elements among cell types and are probably of primary importance in driving cell-type–specific patterns of gene expression. Detailed mapping and understanding of enhancers is therefore critical for elucidating the mechanisms that control cell-type–specific gene expression, yet the incompleteness of the data on enhancers in the human genome has confined the majority of previous studies of gene regulatory networks to promoters.
We also found that the regions that encompass the NUM and GUM sites (NUMR and GUMR) are significantly enriched in known TF binding motifs, and that the majority of the identified motifs differ between the NUMRs and GUMRs. Most interestingly, we found that the NUMRs are enriched in the predicted binding sites of the TFs known to mediate the transcriptional control of activity-regulated genes in neuronal cells. In addition to emphasizing the importance of distal regulatory elements in the regulation of cell-type–specific transcriptional programs, these findings are also in line with the proposed role of DNA methylation in activity-dependent regulation of gene expression in the adult brain (82). However, it has also been suggested that DNA methylation may be a consequence of gene regulation rather than its cause (48,83). Our results do not allow us to distinguish between these possibilities.
Our data indicate that the DM sites are overrepresented in CpG island shores, and this effect is more pronounced for NUM than GUM sites. Differential methylation in CpG island shores was previously found to distinguish among tissue types (specifically, brain, liver, spleen and colon), as well as between normal and cancerous colon specimens (18). The importance of CpG island shore methylation for gene expression regulation was further corroborated in studies on individual genes (84–86). Although a large fraction of more distal NUM sites were localized within predicted enhancers (up to 50%; Supplementary Figure S12A), this fraction was much smaller (∼20%) among the proximal NUM sites. Thus, most of the neuronal-type DM within CpG island shores could not be assigned to the predicted enhancers and remains to be functionally characterized.
We also investigated how DM between neuronal and nonneuronal cells is related to cell-type–specific gene expression. An inverse correlation between DNA methylation and gene expression had previously been experimentally validated mostly for CpG island-containing promoters (49–51). The CpG islands make up only ∼1% of the human genome; for the remaining 99% that has a lower CpG content, the consequences of DNA methylation for gene regulation remain largely unclear, and have been reported to vary depending on the location of the CpG site, the specific gene or the specific cell type (11,17,87–89). Moreover, a positive correlation between gene body methylation and gene activity was reported in several highly proliferating human cells (8,88). This pattern, however, was not detected in slowly dividing tissues (e.g. kidney or lung) and was reversed in the brain (89). In agreement with that report, several lines of evidence presented in our work consistently demonstrate a highly predictive inverse correlation between cell-type–specific DNA methylation across the entire gene locus (including gene bodies and promoters) and cell-type–specific gene expression in the human brain.
The discovery of the striking difference in non-CpG methylation between neuronal and nonneuronal cells is intriguing. Previous studies have shown that non-CpG methylation levels are high in pluripotent cells, whereas somatic cell types exhibit low levels of non-CpG methylation (8,47). In addition, abundant non-CpG methylation has recently been discovered in adult mouse and human brain but not in the brain-derived cell lines (9,11). Here we report significantly higher levels of non-CpG methylation in neuronal compared with nonneuronal cells. Non-CpG methylation is associated with the DNMT3A activity (9). In the brain, DNMT3A is expressed mostly in neurons (90). This is compatible with the significantly greater number of NUM versus GUM sites within the DNMT3A gene itself, as detected in the present work (see Figure 5A). Thus, it appears likely that non-CpG methylation preferentially accumulates in nondividing neurons that express high levels of DNMT3A. Indeed, as suggested by a recent study of non-CpG methylation during germ-cell development, the presence and level of non-CpG methylation may be determined by the balance between de novo methylation activity and the rate of cell proliferation (91).
Neither the HM450K array nor the ERRBS distinguish between 5-mC and 5-hydroxymethylcytosine (5-hmC) modifications. It has been shown that 5-mC can be oxidized to 5-hmC (92), and 5-hmC can be an intermediate in DNA demethylation or constitute a distinct epigenetic mark (93). Interestingly, the highest levels of 5-hmC among all analyzed tissues have been reported for the adult brain (5-hmC% = 0.67) (94–96). Methods to analyze 5-hmC are in active development, and two single base-resolution protocols on a genomic scale have been recently published (97,98). A recent comprehensive study, which used bulk brain specimens, reported that 5-hmC is enriched within active genome regions in the fetal and adult mouse brain (99). Further studies will be required to determine the contribution and the role of 5-hmC in differential methylation among different cellular populations in the brain.
While this manuscript was under review, a new comparative analysis of DNA methylation in neuronal versus nonneuronal cells using whole-genome bisulfite sequencing has been published (99). This work has a substantially wider scope, but was mostly focused on global epigenome restructuring during mammalian development, to which end multiple specimens from the bulk human and mouse brain preparations of different ages were analyzed. The results reported for cell-type–specific DNA methylation in the adult brain samples—including the enrichment of the neuronal and nonneuronal DM regions in the intragenic regions and enhancers, the depletion of these regions within promoters, the global enrichment for non-CpG methylation (mainly in the CA context) in the neuronal compared with nonneuronal cells and the high interindividual correlation of non-CpG methylated sites in neurons—are completely in line with the findings that are described in the present work. However, in addition to the confirmation of these findings, our study, that was focused specifically on the differences between neuronal and nonneuronal DNA methylation in the human adult brain and was performed using an improved FACS protocol that provided more accurate separation of different cell types, allowed us to capture additional features. In particular, we observed the overrepresentation of cell-type–specific DM regions in CpG island shores, the enrichment of the binding motifs for neuron-specific TFs (including activity-dependent factors) within neuron-specific DM regions, and the strong association between DNA methylation within regulatory elements (including predicted enhancers) and cell-type–specific gene expression. These findings are expected to be important for future research of the role of DNA methylation in shaping specific cellular identities.
CONCLUSIONS
Methods such as HM450K array and ERRBS open up qualitatively new directions in functional genomics by allowing one to examine a given feature, in this case the methylation status of individual sites, on the whole-genome level. We took advantage of these technologies to perform a comprehensive comparative analysis of cytosine methylation in neuronal versus nonneuronal cells. Although there was no significant overall difference in the methylation levels, site-by-site analysis led to the identification of thousands of DM sites. The DM sites fell into two categories, namely those that are significantly undermethylated in neurons or in the nonneuronal cells, respectively. The DM sites showed a strongly nonuniform distribution among gene control elements. The predicted CpG-island promoters are depleted for DM sites, whereas the predicted enhancers are strongly enriched for DM sites. Given that methylation within control elements is typically negatively correlated with gene expression, these findings imply that the expression of neuron-specific and non–neuron-specific genes is associated with differential methylation of enhancers. The sets of neuron-specific and non–neuron-specific genes derived from differential methylation data indeed show excellent agreement with the available direct measurements of gene expression in human and mouse, suggesting that differential methylation is a robust predictor of cell-type–specific gene expression. Importantly, distinct neuron-specific DNA methylation trends were identified. In particular, neuronal-type differential methylation was significantly overrepresented in CpG island shores and was enriched within gene bodies but not in intergenic regions. In addition, we identified a significant excess of predicted binding motifs for neuron-specific TFs (including activity-dependent factors) in the vicinity of neuronal-type DM sites. Most strikingly, non-CpG methylation was found to be much more common in neurons than it is in nonneuronal cells. The high coverage brain-cell–specific DNA methylation information obtained in this work is expected to comprise a valuable resource for further study of cell-type–specific epigenetic marks and to enable new discoveries about the role of brain epigenetics in health and disease.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [100–107].
FUNDING
National Institute on Drug Abuse [R21DA031557 to S.D.]; National Institute of Mental Health [R21MH090352 to S.D.]; Hope for Depression Research Foundation grants (to S.D.); VISN 3 Mental Illness Research, Education and Clinical Center (MIRECC) (to S.D.). The work was also supported with resources and the use of facilities at the James J Peters VA Medical Center, Bronx, NY; Supported by intramural funds of the US Department of Health and Human Services (to National Library of Medicine) (to E.V.K.). Funding for open access charge: Authors’ funding.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Graff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol. Rev. 2011;91:603–649. doi: 10.1152/physrev.00012.2010. [DOI] [PubMed] [Google Scholar]
- 2.Urdinguio RG, Sanchez–Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 2009;8:1056–1072. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 3.Peter CJ, Akbarian S. Balancing histone methylation activities in psychiatric disorders. Trends Mol. Med. 2011;17:372–379. doi: 10.1016/j.molmed.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat. Med. 2012;18:1194–1204. doi: 10.1038/nm.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 6.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 7.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 8.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ziller MJ, Muller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS. Genet. 2011;7:e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, Dempster EL, Ren B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816–831. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23:555–567. doi: 10.1101/gr.147942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, Nakao M, Taga T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev. Cell. 2001;1:749–758. doi: 10.1016/s1534-5807(01)00101-0. [DOI] [PubMed] [Google Scholar]
- 13.Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LaPlant Q, Vialou V, Covington HE, III, Dumitriu D, Feng J, Warren BL, Maze I, Dietz DM, Watts EL, Iniguez SD, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Labonte B, Suderman M, Maussion G, Navaro L, Yerko V, Mahar I, Bureau A, Mechawar N, Szyf M, Meaney MJ, et al. Genome-wide epigenetic regulation by early-life trauma. Arch. Gen. Psychiatry. 2012;69:722–731. doi: 10.1001/archgenpsychiatry.2011.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suderman M, McGowan PO, Sasaki A, Huang TC, Hallett MT, Meaney MJ, Turecki G, Szyf M. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc. Natl Acad. Sci. USA. 2012;109(Suppl 2):17266–17272. doi: 10.1073/pnas.1121260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ladd-Acosta C, Pevsner J, Sabunciyan S, Yolken RH, Webster MJ, Dinkins T, Callinan PA, Fan JB, Potash JB, Feinberg AP. DNA methylation signatures within the human brain. Am. J Hum. Genet. 2007;81:1304–1315. doi: 10.1086/524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, Wininger M, Colantuoni C, Weinberger DR, Kleinman JE, Lipska BK. DNA methylation signatures in development and aging of the human prefrontal cortex. Am. J. Hum. Genet. 2012;90:260–272. doi: 10.1016/j.ajhg.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, Coarfa C, Harris RA, Milosavljevic A, Troakes C, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13:R43. doi: 10.1186/gb-2012-13-6-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang D, Cheng L, Badner JA, Chen C, Chen Q, Luo W, Craig DW, Redman M, Gershon ES, Liu C. Genetic control of individual differences in gene-specific methylation in human brain. Am. J. Hum. Genet. 2010;86:411–419. doi: 10.1016/j.ajhg.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xin Y, O'Donnell AH, Ge Y, Chanrion B, Milekic M, Rosoklija G, Stankov A, Arango V, Dwork AJ, Gingrich JA, et al. Role of CpG context and content in evolutionary signatures of brain DNA methylation. Epigenetics. 2011;6:1308–1318. doi: 10.4161/epi.6.11.17876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS. Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh S, Yates AJ, Fruhwald MC, Miecznikowski JC, Plass C, Smiraglia D. Tissue specific DNA methylation of CpG islands in normal human adult somatic tissues distinguishes neural from non-neural tissues. Epigenetics. 2010;5:527–538. doi: 10.4161/epi.5.6.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwamoto K, Bundo M, Ueda J, Oldham MC, Ukai W, Hashimoto E, Saito T, Geschwind DH, Kato T. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 2011;21:688–696. doi: 10.1101/gr.112755.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013;8:290–302. doi: 10.4161/epi.23924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antonucci AS, Gansler DA, Tan S, Bhadelia R, Patz S, Fulwiler C. Orbitofrontal correlates of aggression and impulsivity in psychiatric patients. Psychiatry Res. 2006;147:213–220. doi: 10.1016/j.pscychresns.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Chambers CD, Bellgrove MA, Gould IC, English T, Garavan H, McNaught E, Kamke M, Mattingley JB. Dissociable mechanisms of cognitive control in prefrontal and premotor cortex. J. Neurophysiol. 2007;98:3638–3647. doi: 10.1152/jn.00685.2007. [DOI] [PubMed] [Google Scholar]
- 29.Zald DH, Andreotti C. Neuropsychological assessment of the orbital and ventromedial prefrontal cortex. Neuropsychologia. 2010;48:3377–3391. doi: 10.1016/j.neuropsychologia.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 30.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D, Hess JL, et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 2012;8:e1002781. doi: 10.1371/journal.pgen.1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, Durham T, Miri M, Deshpande V, De Jager PL, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 2006;34:D590–D598. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- 37.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the dynamic tree cut package for R. Bioinformatics. 2008;24:719–720. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 38.Dracheva S, Lyddon R, Barley K, Marcus SM, Hurd YL, Byne WM. Editing of serotonin 2C receptor mRNA in the prefrontal cortex characterizes high-novelty locomotor response behavioral trait. Neuropsychopharmacology. 2009;34:2237–2251. doi: 10.1038/npp.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Price JL. Definition of the orbital cortex in relation to specific connections with limbic and visceral structures and other cortical regions. Ann. N. Y. Acad. Sci. 2007;1121:54–71. doi: 10.1196/annals.1401.008. [DOI] [PubMed] [Google Scholar]
- 40.Dalley JW, Mar AC, Economidou D, Robbins TW. Neurobehavioral mechanisms of impulsivity: fronto-striatal systems and functional neurochemistry. Pharmacol. Biochem. Behav. 2008;90:250–260. doi: 10.1016/j.pbb.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 41.Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okada S, Saiwai H, Kumamaru H, Kubota K, Harada A, Yamaguchi M, Iwamoto Y, Ohkawa Y. Flow cytometric sorting of neuronal and glial nuclei from central nervous system tissue. J. Cell Physiol. 2011;226:552–558. doi: 10.1002/jcp.22365. [DOI] [PubMed] [Google Scholar]
- 43.Severin J, Waterhouse AM, Kawaji H, Lassmann T, van NE, Balwierz PJ, de Hoon MJ, Hume DA, Carninci P, Hayashizaki Y, et al. FANTOM4 EdgeExpressDB: an integrated database of promoters, genes, microRNAs, expression dynamics and regulatory interactions. Genome Biol. 2009;10:R39. doi: 10.1186/gb-2009-10-4-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van CS, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 46.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, van NE, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 49.Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, Baglivo I, Pedone PV, Grimaldi G, Riccio A, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell. 2011;44:361–372. doi: 10.1016/j.molcel.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Velasco G, Hube F, Rollin J, Neuillet D, Philippe C, Bouzinba-Segard H, Galvani A, Viegas-Pequignot E, Francastel C. Dnmt3b recruitment through E2F6 transcriptional repressor mediates germ-line gene silencing in murine somatic tissues. Proc. Natl Acad. Sci. USA. 2010;107:9281–9286. doi: 10.1073/pnas.1000473107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borgel J, Guibert S, Li Y, Chiba H, Schubeler D, Sasaki H, Forne T, Weber M. Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 2010;42:1093–1100. doi: 10.1038/ng.708. [DOI] [PubMed] [Google Scholar]
- 52.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 53.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl Acad. Sci. USA. 2006;103:1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Illingworth RS, Bird AP. CpG islands—'a rough guide'. FEBS Lett. 2009;583:1713–1720. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 55.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, et al. The UCSC genome browser database: update 2011. Nucleic Acids Res. 2011;39:D876–D882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanay A, O'Donnell AH, Damelin M, Bestor TH. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc. Natl Acad. Sci. USA. 2007;104:5521–5526. doi: 10.1073/pnas.0609746104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koch CM, Andrews RM, Flicek P, Dillon SC, Karaoz U, Clelland GK, Wilcox S, Beare DM, Fowler JC, Couttet P, et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 2007;17:691–707. doi: 10.1101/gr.5704207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, Laurance MF, Zhao W, Qi S, Chen Z, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc. Natl Acad. Sci. USA. 2006;103:17402–17407. doi: 10.1073/pnas.0608396103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat. Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- 66.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 67.Winden KD, Oldham MC, Mirnics K, Ebert PJ, Swan CH, Levitt P, Rubenstein JL, Horvath S, Geschwind DH. The organization of the transcriptional network in specific neuronal classes. Mol. Syst. Biol. 2009;5:291. doi: 10.1038/msb.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miller JA, Horvath S, Geschwind DH. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc. Natl Acad. Sci. USA. 2010;107:12698–12703. doi: 10.1073/pnas.0914257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fury W, Batliwalla F, Gregersen PK, Li W. Overlapping probabilities of top ranking gene lists, hypergeometric distribution, and stringency of gene selection criterion. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2006;1:5531–5534. doi: 10.1109/IEMBS.2006.260828. [DOI] [PubMed] [Google Scholar]
- 72.Poirier R, Cheval H, Mailhes C, Garel S, Charnay P, Davis S, Laroche S. Distinct functions of egr gene family members in cognitive processes. Front. Neurosci. 2008;2:47–55. doi: 10.3389/neuro.01.002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lyons MR, Schwarz CM, West AE. Members of the myocyte enhancer factor 2 transcription factor family differentially regulate Bdnf transcription in response to neuronal depolarization. J. Neurosci. 2012;32:12780–12785. doi: 10.1523/JNEUROSCI.0534-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stolt CC, Schlierf A, Lommes P, Hillgartner S, Werner T, Kosian T, Sock E, Kessaris N, Richardson WD, Lefebvre V, et al. SoxD proteins influence multiple stages of oligodendrocyte development and modulate SoxE protein function. Dev. Cell. 2006;11:697–709. doi: 10.1016/j.devcel.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 75.Aftab S, Semenec L, Chu JS, Chen N. Identification and characterization of novel human tissue-specific RFX transcription factors. BMC. Evol. Biol. 2008;8:226. doi: 10.1186/1471-2148-8-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu. Rev. Neurosci. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gaudilliere B, Konishi Y, de la Iglesia N, Yao G, Bonni A. A CaMKII-NeuroD signaling pathway specifies dendritic morphogenesis. Neuron. 2004;41:229–241. doi: 10.1016/s0896-6273(03)00841-9. [DOI] [PubMed] [Google Scholar]
- 79.Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011;14:1345–1351. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schubeler D. Molecular biology. Epigenetic islands in a genetic ocean. Science. 2012;338:756–757. doi: 10.1126/science.1227243. [DOI] [PubMed] [Google Scholar]
- 84.Perisic T, Holsboer F, Rein T, Zschocke J. The CpG island shore of the GLT-1 gene acts as a methylation-sensitive enhancer. Glia. 2012;60:1345–1355. doi: 10.1002/glia.22353. [DOI] [PubMed] [Google Scholar]
- 85.Nishioka M, Shimada T, Bundo M, Ukai W, Hashimoto E, Saito T, Kano Y, Sasaki T, Kasai K, Kato T, et al. Neuronal cell-type specific DNA methylation patterns of the Cacna1c gene. Int. J. Dev. Neurosci. 2013;31:89–95. doi: 10.1016/j.ijdevneu.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 86.Rao X, Evans J, Chae H, Pilrose J, Kim S, Yan P, Huang RL, Lai HC, Lin H, Liu Y, et al. CpG island shore methylation regulates caveolin-1 expression in breast cancer. Oncogene. 2012;32:4519–4528. doi: 10.1038/onc.2012.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 88.Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aran D, Toperoff G, Rosenberg M, Hellman A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011;20:670–680. doi: 10.1093/hmg/ddq513. [DOI] [PubMed] [Google Scholar]
- 90.Feng J, Chang H, Li E, Fan G. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J. Neurosci. Res. 2005;79:734–746. doi: 10.1002/jnr.20404. [DOI] [PubMed] [Google Scholar]
- 91.Ichiyanagi T, Ichiyanagi K, Miyake M, Sasaki H. Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Res. 2013;41:738–745. doi: 10.1093/nar/gks1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 2012;13:7–13. doi: 10.1038/nrg3080. [DOI] [PubMed] [Google Scholar]
- 94.Li W, Liu M. Distribution of 5-hydroxymethylcytosine in different human tissues. J. Nucleic Acids. 2011;2011:870726. doi: 10.4061/2011/870726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, Irier H, Upadhyay AK, Gearing M, Levey AI, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011;14:1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yu M, Hon GC, Szulwach KE, Song CX, Jin P, Ren B, He C. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat. Protoc. 2012;7:2159–2170. doi: 10.1038/nprot.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 99.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Drakenberg K, Nikoshkov A, Horvath MC, Fagergren P, Gharibyan A, Saarelainen K, Rahman S, Nylander I, Bakalkin G, Rajs J, et al. Mu opioid receptor A118G polymorphism in association with striatal opioid neuropeptide gene expression in heroin abusers. Proc. Natl Acad. Sci. USA. 2006;103:7883–7888. doi: 10.1073/pnas.0600871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nikoshkov A, Drakenberg K, Wang X, Horvath MC, Keller E, Hurd YL. Opioid neuropeptide genotypes in relation to heroin abuse: dopamine tone contributes to reversed mesolimbic proenkephalin expression. Proc. Natl Acad. Sci. USA. 2008;105:786–791. doi: 10.1073/pnas.0710902105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 105.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.