


Abstract
The hepatitis C virus (HCV) RNA genome contains multiple structurally conserved domains that make long-distance RNA–RNA contacts important in the establishment of viral infection. Microarray antisense oligonucelotide assays, improved dimethyl sulfate probing methods and 2′ acylation chemistry (selective 2’-hydroxyl acylation and primer extension, SHAPE) showed the folding of the genomic RNA 3′ end to be regulated by the internal ribosome entry site (IRES) element via direct RNA–RNA interactions. The essential cis-acting replicating element (CRE) and the 3′X-tail region adopted different 3D conformations in the presence and absence of the genomic RNA 5′ terminus. Further, the structural transition in the 3′X-tail from the replication-competent conformer (consisting of three stem-loops) to the dimerizable form (with two stem-loops), was found to depend on the presence of both the IRES and the CRE elements. Complex interplay between the IRES, the CRE and the 3′X-tail region would therefore appear to occur. The preservation of this RNA–RNA interacting network, and the maintenance of the proper balance between different contacts, may play a crucial role in the switch between different steps of the HCV cycle.
INTRODUCTION
The hepatitis C virus (HCV) genome is an ∼9.6-kb-long, positive, single-stranded RNA molecule that codes for a single open reading flanked by untranslatable regions (UTRs) (1,2). The viral genome plays important roles in temporally and spatially regulated viral processes. In early infection, HCV protein synthesis is initiated by an internal ribosome entry site (IRES)-dependent mechanism (3,4) different to the cap-dependent method used for the translation of most cellular mRNAs. The IRES element is mostly located at the 5′UTR and spans a short stretch of the core coding sequence (5,6) (Figure 1A). The minimal IRES contains a subset of regulatory elements organized into two major structural domains (7) (II and III), plus a short stem-loop (domain IV) bearing the initiation codon (8). These structural elements guide the direct recruitment of the 40S ribosomal subunit (9–12) and the further binding of eIF3 (13,14), which aids the incorporation of the ternary complex eIF2-tRNAiMet and the joining of the 60S subunit (15). This mechanism minimizes protein factor requirements and simplifies the pathway for the assembly of the translationally active 80S ribosome. Further, the presence of domains at the 3′ end of the genomic RNA influences translation efficiency, most likely by the acquisition of a closed-loop topology resembling the circular structure adopted by cellular mRNAs (16–21). This conformation would help the 3′UTR for retaining 40S ribosomal subunits during the translation termination step, thus ensuring efficient initiation of the next translation round (22).
Figure 1.

Secondary structure of the 5′ and 3′ ends of the HCV genome. (A) Diagram showing the secondary structure of the 5′ and 3′ regions of the HCV genome including functional RNA domains involved in long-range RNA–RNA contacts. The 3′ end of the viral genomic RNA is organized into two well-defined structural elements: the CRE region and the 3′X-tail, separated by a hypervariable sequence (HV) and a polyU/UC stretch. The 3′X-tail folds into two different conformers. Pseudoknot elements within the IRES element are marked as PK1 and PK2. Dimerization linkage sequence (DLS), start and stop translation codons are indicated by arrows. The gray solid lines identify long-distance RNA–RNA interactions involving the IRES, the CRE and the 3′X-tail regions. (B) Diagram of the six HCV transcripts used in the present work. fluc, Firefly luciferase coding sequence.
Once viral proteins levels have reached a certain threshold, the genomic RNA serves as a template for replication. This replication is initiated at the 3′UTR but is also dependent on the 5′ end of the HCV RNA (23,24). The 3′UTR (Figure 1A) is organized into three conformational modules that define (i) a hypervariable region (HV), (ii) a polyU/UC stretch of variable length and composition and (iii) the essential 3′X-tail (24–29). The 3′X-tail theoretically folds into one of two mutually exclusive conformations (30) (Figure 1A). Both predicted structures preserve the 3′SLI domain at the end of the 3′ terminus, which influences replication efficiency (31) and confers specificity upon the initiation site (32). The 55-nt-long segment upstream of the 3′SLI folds either as two stem-loops, named 3′SLII and 3′SLIII (26), or as a single stem-loop exposing a 16-nt-long palindromic sequence (the dimer linkage sequence [DLS]) (30) (Figure 1A). It has been proposed that these different conformers assume different functions during the HCV cycle (33,34), but the nature of the stimulus that promotes the acquisition of one isoform or the other remains unknown. During replication, the increase in the genome copy number unleashes the formation of homodimers by the interaction of two viral RNA molecules through the DLS motif, most likely in the presence of the core chaperone protein (30,33,35). Thus, packaged viral RNA molecules are finally enveloped and removed to the extracellular environment.
The proper balance between these processes must be maintained if viral adaptive fitness and persistence are to be achieved. The transitions between them must be also finely regulated, avoiding the potential steric hindrance of the translation and replication machineries. To achieve this, the virus establishes a dynamic complex network of contacts between the available functional genomic RNA domains, which may change depending on viral needs at different steps over the course of infection. In addition to the well-known protein-mediated 5′UTR-3′UTR bridges (17–19,36–38), other cis-acting elements participate in this interacting web to provide benefits such as the formation of direct RNA–RNA contacts that minimize protein requisites. Phylogenetically conserved RNA secondary structured domains have been reported within the 3′ end of the HCV coding sequence (39). Among them, the stem-loop 5BSL3.2 has been identified embedded in a cruciform structure that acts as a cis-essential element during viral RNA synthesis (the cis-acting replicating element [CRE]) (40,41) (Figure 1A). By the establishment of direct RNA–RNA interactions, the stem-loop 5BSL3.2 participates in viral translation and replication. Thus, the apical loop is complementary to the apical loop of the 3′SLII within the 3′X-tail (34,42–44), while the 8-nt bulge (see Figure 1A) establishes one of two possible contacts: one with the apical loop of the IIId subdomain of the IRES region (44,45), the other with the apical loop of the stem-loop SL9110 upstream of the CRE element (34,43,44,46) (Figure 1A). It has recently been reported that all these interactions are equally probable and show similar dissociation constants (Kd) (34,44). Choosing between different contacts may therefore depend on the presence of host or viral factors, which would help to bring the ends of the HCV genome into close proximity. The acquisition of a circular topology is advantageous with respect to the execution of different viral processes and switching between them.
The discovery of a network of long-range direct RNA–RNA interactions has prompted the study of the structural changes that take place in the involved regions. Analysis of the complexes SL9110-5BSL3.2-3′SLII and IRES-CRE by several techniques in replication-competent RNA transcripts (43,47) has recently provided solid evidences for the existence of these interactions, and demonstrated that such contacts promote conformational tuning events in functionally essential RNA elements. However, little is known about the role that the interaction IRES-CRE plays in the folding of the 3′X-tail. Because the CRE is a critical partner in the structural organization of the 3′X-tail, the interplay with the IRES element likely leads to conformational rearrangements in the genomic RNA 3′ terminus. The present work shows that the IRES and the CRE elements induce by themselves fine structural reorganizations within the 3′X-tail. Further, the combined presence of both regions exerts a potent effect on the architecture of the essential domains 3′SLII and 3′SLIII, which switch to a single stem-loop conformation that exposes the DLS motif in an apical loop. This suggests that the IRES-CRE interaction may modulate 3′ end folding in the absence of protein factors. The results agree with the previous findings that the 3′ end of the HCV RNA genome, which contains both the CRE and the 3′UTR elements, fine-tunes the 3D structure of the IRES region (47). As a consequence, the 3′X-tail suffers conformational rearrangements that might be related to the switch between viral replication and genomic dimerization.
MATERIALS AND METHODS
DNA templates and RNA synthesis
DNA molecules coding for FCU and ICU transcripts (Figure 1B) used in microarray assays were obtained as previously described (21). Briefly, for the FCU molecule, T7pFCU and T7pICU were amplified from the plasmid pGL-ICU (21) using the primers 5′T7pFLuc and 3′HCV, while 5′T7pHCV and 3′HCV were used for the ICU construct (see Supplementary Table S1). The resulting RNA molecules therefore had the correct 3′ ends.
To monitor dimethyl sulfate (DMS) and N-methyl-isatoic anhydride (NMIA) reactivity at the 3′ end of the constructed transcripts U, CU, I + U and I + CU (Figure 1B), a 45-nt cassette was attached to their 3′ termini. This cassette has been previously described to fold autonomously into two short stable hairpins (48) that do not interfere with the predicted folding of the HCV 3′UTR (data not shown). It also contains a primer binding site for efficient cDNA synthesis. Briefly, molecules T7pU and T7pCU carrying this cassette at their 3′ termini were obtained from the plasmid pGL-ICU (21): amplification with the oligonucleotides T7pHCV-9380 and 3′HCV_cassette generated the template construct T7pU. Primers 5′T7pHCV-9181 and the 3′HCV_cassette were used for the amplification of the variant T7pCU (Supplementary Table S1). DNA molecules coding for constructs I + U and I + CU encompassing the cassette were generated by amplification from their respective vectors pGLI + U and pGLI + CU, previously obtained by site-directed mutagenesis (47). Subsequent amplification with the primer set T7p5′HCV and the 3′HCV_cassette (Supplementary Table S1) yielded DNA templates T7pI + U and T7pI + CU, from which the respective RNA variants (I + U and I + CU) were synthesized by in vitro transcription (see below).
RNA synthesis was performed using the RiboMAX™-T7 large-scale RNA production system (Promega), following the manufacturer’s instructions. Transcripts were purified as previously described (47). RNA yield was determined by ultraviolet spectrophotometry (A260) and the extent of protein and carbohydrate contaminations assessed by the A260/A280 and A260/A230 ratios, respectively. The integrity of the RNA molecules was examined by denaturing agarose-formaldehyde gel electrophoresis.
Microarray design, RNA hybridization and data analysis
Forty six antisense DNA oligonucleotides complementary to CRE and the 3′UTR of HCV (genotype 1b) were designed to specifically hybridize with sequences of 14 consecutive nucleotides of the viral genome, overlapping by seven nucleobases with the adjacent oligos. Each high pressure (or high performance) liquid chromatography-purified oligonucleotide (IBA GmbH) was composed of a C6-amino linker motif at its 5′ end, followed by either (TCC)5, (TTC)5, TTTTTTTTTTTTCCC or CCCCTTCCCCTTCCC tracks (designed to avoid self-annealing within the oligo) as spacer regions, and the specific antisense sequence at the 3′ end of the oligo (Supplementary Table S2). The names of these oligonucleotides correspond to the nucleotide on the HCV sequence complementary to the 3′ end of the oligo. One oligonucleotide with unrelated sequence (belonging to the 5′ genomic region of the foot-and-mouth disease virus) was included as negative hybridization control. The 47 oligonucleotides were diluted, spotted onto super-epoxy surfaces and fixed as described (47,49). The microarrays were hybridized with 300 ng of fluorescently labeled RNA transcripts (47) under non-denaturing conditions (using the same buffer selected for selective 2’-hydroxyl acylation and primer extension (SHAPE) analysis: 100 mM HEPES, pH 8.0, 100 mM NaCl, 1 mM MgCl2) for 2 h at 37°C. Washed and dried slides were immediately scanned using a Genepix 4100 scanner. Data were retrieved using Genepix Pro 6.0 software.
The hybridization of each RNA transcript to the microarrays was measured in three independent experiments. In all cases, the raw fluorescence signal was corrected by subtraction of the local background. To compare the signal detected in the three independent hybridization assays, the mean fluorescence signal yielded by the 20 µM spots was normalized against the mean fluorescence of either the spots corresponding to the CRE region (spanning nucleotides 9190–9372), or those corresponding to the 3′UTR region (oligonucleotides 9379–9592), after excluding outliers (oligonucleotides 9190 and 9383) (Supplementary Table S2). The interquartile range of the hybridization values was calculated to identify outliers. The outlier with the lowest value was established as the upper limit of the accessibility range. Quartile 3 defined the lower limit of the accessibility range.
DMS probing
DMS chemical probing was performed essentially as previously described (47). Briefly, 1 µg of purified HCV RNA was denatured for 3 min at 95°C before transferring to ice for 15 min. The RNA was then incubated for 5 min at 37°C in folding buffer (50 mM HEPES, pH 8.0, 100 mM NaCl, 1 mM MgCl2). Reactions were initiated by the addition of 1 µl of freshly diluted DMS in ethanol (1:5). Chemical probing proceeded for 1 min at 37°C in the presence of tRNA (2 µg) and was stopped by the addition of 300 mM sodium acetate, pH 5.2. Control DMS reactions were performed in parallel. The RNA was ethanol-precipitated, washed twice with 80% ethanol and subjected to primer extension for the detection of the modified A and C residues, as previously described (47). Primer Std (see Supplementary Table S1), which anneals within the structure cassette placed at the 3′ end of transcripts under study, was fluorescently labeled with NED (to detect untreated probes), VIC (for mapping positive reactions), FAM and PET (for sequencing reactions). A fraction of the resulting cDNA samples were purified and resolved as previously reported (47). Electropherograms were analyzed using ShapeFinder software (50). Normalized DMS reactivity values for each nucleotide position were obtained by dividing each value by the average intensity of the 10% most reactive residues, after excluding outliers calculated by box plot analysis (51).
SHAPE analysis
SHAPE analyses were performed by treatment with NMIA as previously reported (47). Briefly, 1 µg of RNA was denatured as described above under the same ionic conditions. 2′-adduct formation was achieved by the addition of 6 mM NMIA in dimethyl-sulfoxide for 5 min at 37°C. Reactions were stopped by ethanol precipitation. The RNA was subsequently washed twice with 80% ethanol and subjected to primer extension as described above. Normalized NMIA reactivity values for each nucleotide position were calculated as indicated for DMS probing.
Secondary structure prediction
RNA secondary structure models were generated using ShapeKnots software (52), including the structural constraints derived from NMIA relative reactivity data.
Statistical analysis
All accessibility and reactivity data are presented as means ± standard deviation. Data points were compared using the unpaired two-tailed Mann–Whitney test (53). Significance was set at P ≤ 0.05. P values for reactive positions are included in the corresponding Supplementary Tables.
RESULTS
Comparative structural analysis showed the HCV IRES region to influence the conformation of the genomic RNA 3′ end
The finding that the 3′ end of the HCV genome fine-tunes the folding of the IRES element (47) points to the existence of long-range RNA–RNA interactions between both termini of the viral genome. It therefore seemed likely that the conformation of the 3′ end relies on the presence of the IRES element. To test this hypothesis, the accessibility pattern of the genomic 3′ end was subjected to microarray antisense oligonucleotide analyses in the presence and absence of the IRES. This methodology has been successfully used in conformational studies of HCV RNA (47,49), foot-and-mouth disease virus genome (54,55) and human immunodeficiency virus RNA (56). Also, it has provided evidence suggesting the specific structural tuning of the HCV IRES to be mediated by the 3′ region of the viral genome (47).
Microarray assays were performed with the previously described FCU construct (21), encompassing the fluc gene fused to the CRE region plus the 3′UTR (Figure 1B). The use of PCR-generated DNA templates ensured the presence of the precise 3′ ends. RNA transcripts were fluorescently labeled, renatured and hybridized under native conditions to a customized panel of 46 overlapping 14-mer antisense DNA oligonucleotides complementary to the whole 3′ end of the HCV RNA, as previously described (47,49,55). The fluorescence signal (mean ± SD for three independent assays) was determined for each oligonucleotide (Figure 2A). The resulting hybridization profile for the FCU molecule fitted perfectly with the previously mapped secondary structure: in general, little fluorescence was recorded in the helical regions while strong hybridization signals were recorded for the loops and their surrounding areas (Figure 2A). This map points to the existence of a preferred and stable secondary structure under the present assay conditions. Further tailoring of the IRES element to the 5′ end of the FCU construct (ICU molecule; Figure 1B) (21) promoted evident and significant alterations (P ≤ 0.05; see Supplementary Table S3 for a detailed list of P values associated with each oligonucleotide) in the accessibility profile throughout domain 5BSL3.2 (Figure 2B and C), with reductions in the fluorescence signal for the oligo 9267, mapping to the 5′ portion of the stem, and a concomitant increase in the hybridization capacity of the 3′ fragment of the stem and the internal loop (oligonucleotides 9288 and 9295, respectively). Additional significant increases (P ≤ 0.05) in relative fluorescence values were observed for those oligos covering domains 3′SLII and 3′SLI (Figure 2B and C), suggesting that the interplay 5BSL3.2-3′SLII may be affected by the presence of the 5′ end of the HCV genome. Together, these data support the idea that conformational reorganization events involving IRES-mediated RNA–RNA interactions occur throughout the whole 3′ end of the viral genome, being domains 5BSL3.2 and 3′SLII specially affected by this interplay (Figure 2C).
Figure 2.
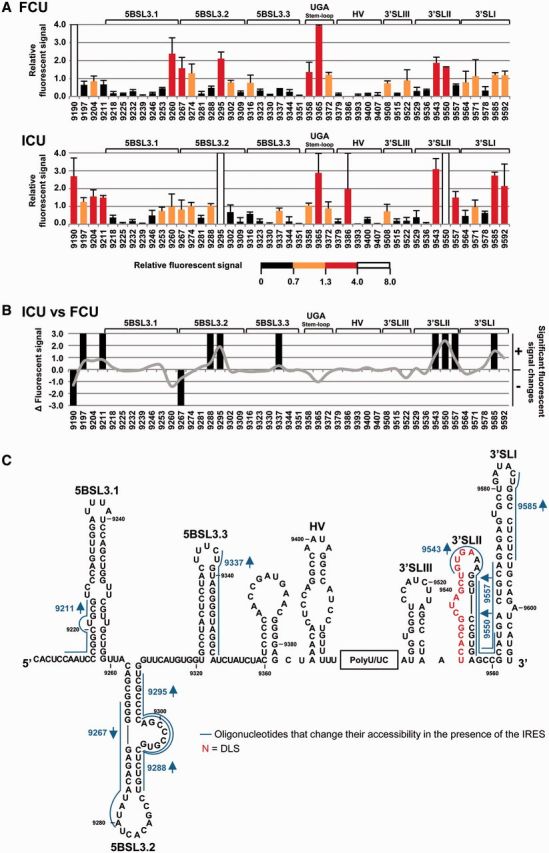
Structural analysis of the HCV genomic 3′ end by antisense DNA oligonucleotide microarrays in the presence of the IRES region. (A) Normalized hybridization signal of transcripts FCU and ICU plotted against each oligonucleotide (mean fluorescence hybridization signal ± standard deviation for three independent assays). The color code shows the four accessibility levels, as described in ‘Materials and Methods’ section. (B) Differences in fluorescence values for constructs ICU versus FCU with respect to different oligos are represented as a solid gray line. Significant variations (P ≤ 0.05) are qualitatively indicated as gain (+) or loss (−) of accessibility. Functional RNA domains are shown. (C) Figure shows the secondary structure of the CRE and the 3′UTR region and includes those antisense oligonucleotides that change their accessibility for the transcript ICU compared with FCU. Red nucleotides: dimerization linkage sequence (DLS).
To further map the specific residues affected by these rearrangements, both DMS chemical probing assays and SHAPE analyses with NMIA were performed. DMS reacts with susceptible groups of the nucleobases in unpaired A, C and G nucleotides to generate alkyl derivatives, while NMIA is an acylating agent that promotes the formation of 2′-O-adducts in flexible residues independent of their solvent accessibility (48,57). The subsequent use of both techniques provides data on the RNA secondary structure and the shape of its ribose-phosphate backbone, and thus offers complementary information on the 3D folding of target RNA molecules (47). The reaction conditions were set to minimize alternative suboptimal foldings and to reduce potential structural noise (see ‘Materials and Methods’ section). These reductions were considered of potential help in detecting local structural tuning processes probably missed in earlier studies. Reactive residues were detected by a high-throughput strategy involving primer extension with a set of fluorescent primers (see ‘Materials and Methods’ section). The reverse transcription elongation products were resolved by capillary electrophoresis and analyzed using ShapeFinder software (51).
Constructs CU and I + CU (Figure 1B) were subjected to structural mapping (45,47). These constructs, respectively, encompass the CRE region plus the 3′UTR, and both the latter plus the first 691 nt of the HCV genome (including the IRES element) at the 5′ end. These shortened transcripts are of better RNA quality with respect to ICU and FCU: they minimize 3′ heterogeneity but preserve the conformational fine-tuning effect that the HCV RNA 3′ end has on the IRES (47). Such RNA molecules were subjected to DMS and NMIA analysis and the reactivity pattern analyzed in four independent experiments to yield the mean relative reactivity values for each nucleotide, as previously described (47).
The DMS reactivity profile for the CU construct largely corresponded with the secondary structure data previously reported for the 3′ end of the HCV genome, with the most reactive residues in the apical and internal loops (26,41,42) (Figure 3A and Supplementary Figure S1). Similar analysis for the I + CU molecule allowed the potential conformational rearrangements within the 3′ end mediated by the 5′ terminus to be examined (Figure 3). The accessibility patterns of these constructs showed significant differences (P ≤ 0.05; see Supplementary Table S4), mainly involving an increase in DMS reactivity for the residues at the base of 3′SLIII (A9529 and A9530) and the apical loop of 3′SLII (A9547) (Figure 3). Significant reductions in solvent accessibility were also apparent within A9302 in the internal loop of domain 5BSL3.2, and residues A9278 and C9287 in the apical loop of 5BSL3.2. Together, these data support previous observations suggesting some interplay between the IRES, the CRE and the 3′X-tail region (43,44,47). Deviations in DMS sensitivity (both increases and reductions) were also detected throughout the HV region and the stem-loop containing the stop codon. Though none of these variations seem to substantially influence the secondary structure of the affected regions, they do reflect the subtle control of the IRES over the folding of the entire 3′ end.
Figure 3.

Differential DMS sensitivity of the HCV CRE and 3′UTR elements induced by the IRES region. (A) The histogram shows a detailed view of the DMS reactivity pattern for different regions of the CRE and the 3′UTR elements in the transcripts CU, I + CU, U and I + U. Data are the mean of three independent experiments ± standard deviation. Different accessibility values are color coded, as indicated. (B) Variations in the DMS reactivity profile throughout the different domains of the HCV 3′ end were quantified and represented as a solid gray line. Significant differences (P ≤ 0.05) are represented qualitatively as gain (+) or loss (−) of reactivity for construct I + CU with respect to CU RNA. Only reactive nucleotides (relative DMS reactivity ≥0.3) were taken into account in this analysis. Functional RNA domains are indicated. Numbers refer to the nucleotide positions of the HCV Con1 isolate (GenBank accession number AJ238799).
To further investigate the relationship between the IRES, the CRE and the 3′X-tail region, SHAPE analyses were performed on the transcripts CU and I + CU. The NMIA reactivity profile for the 5BSL3.2 and 3′SLII domains in the CU transcript greatly resembled that previously shown in replication-competent RNA constructs derived from genotype 1b (43) (Figure 4A and Supplementary Figure S2), suggesting that these elements contain all the minimal requisites for autonomous folding to occur. Interestingly, comparisons with the profile for the I + CU transcript confirmed some of the observations previously derived from DMS analysis and also provided important new structural data (Figure 4). Major significant changes (P ≤ 0.05; see Supplementary Table S5) affected the flexibility of the ribose–phosphate backbone in the nucleotide positions at the base of 3′SLIII (Figure 4B). SHAPE analyses also revealed significant variations in the reactivity of the 3′SLII domain’s upper portion. Modifications involved significant reductions (P ≤ 0.05; Supplementary Table S5) in NMIA sensitivity for residues C9541, U9542 and G9543, and a concomitant increase for the nucleotides closing the apical loop, i.e. A9539, G9540, A9548 and U9551 (Figure 4). In addition, the presence of the IRES element also enhanced NMIA reactivity at positions G9295, U9296 and G9301 (P ≤ 0.05) in the internal loop of 5BSL3.2. Again, these data support the existence of a network of RNA–RNA interactions involving the IRES and different structural elements of the 3′ end. A summary of the results derived from the comparison of the data obtained by DMS probing and SHAPE analysis, in the absence and presence of the IRES, is provided in Figure 5.
Figure 4.

The SHAPE profile of the HCV 3′ end is influenced by the IRES region. (A) Histograms showing a detailed view of the SHAPE profile for different domains within the HCV RNA 3′ end in the molecules CU, I + CU, U and I + U. Values are the mean of at least three independent experiments ± standard deviation. The color code denotes differences in NMIA reactivity. (B) The line graph shows differences in NMIA sensitivity throughout the HCV RNA 3′ end for transcript I + CU with respect to molecule CU. Significant variations (P ≤ 0.05) in the SHAPE pattern for reactive positions (NMIA reactivity > 0.3) are also qualitatively indicated by (+) (increase) or (−) (decrease). Functional genomic domains are noted. Nucleotide numbering is indicated according to Figure 3.
Figure 5.

The CRE and the 3′UTR conformation is fine-tuned by the HCV IRES as detected by DMS chemical probing and SHAPE analysis. Comparison of the results obtained by DMS and NMIA probing regarding the conformation of the HCV 3′ end for the I + CU molecule with respect to construct CU. Filled circles: significant increase (P ≤ 0.05) in NMIA reactivity; open circles: significant reduction (P ≤ 0.05) in NMIA reactivity; filled triangles: significant increase (P ≤ 0.05) in DMS reactivity; open triangles: significant reduction (P ≤ 0.05) in DMS reactivity. Red nucleotides: dimerization linkage sequence (DLS). Gray residues: not analyzed.
Therefore, the results of microarray assays, DMS chemical probing and SHAPE analysis confirm the acquisition of a preferential secondary structure by the 3′ end of the HCV genome, which can be fine-tuned by the IRES element to achieve a dynamic 3D conformation involving a complex network of direct RNA–RNA interactions.
The CRE region influences the conformation of the 3′UTR
The HCV IRES element promoted major changes in the DMS and NMIA reactivity profiles of the 3′X-tail in the presence of the CRE element. Based on this observation, we further examined the contribution of these independent regions to the folding of the whole 3′UTR.
DMS chemical probing and SHAPE analyses of the U transcript, which carries the whole 3′UTR (Figure 1B), were performed. The relative reactivity (mean ± SD for at least three independent experiments) was calculated for each nucleotide position, allowing the construction of DMS and NMIA profiles (Figures 3A and 4A). Such data for the 3′X-tail were largely consistent with the acquisition of one major fold defined by three stem-loops (25,26) (Figures 3A and 4A). This reinforces the idea that, in the absence of additional genomic elements, the 3′X-tail adopts a preferential secondary and tertiary structure. Interestingly, while the HV region appeared highly sensitive to both reagents, the 3′X-tail showed reduced DMS and NMIA reactivity, suggesting that this element shows limited solvent accessibility and little flexibility of its constituting nucleotides. These results indicate the existence of two conformational modules within the 3′UTR, a potentially crucial feature for regulatory folding events.
The DMS reactivity profiles obtained for the 3′UTR alone (U variant) were then compared with those recorded in the presence of the CRE region (CU transcript) (Figures 3A and 6A). Small but significant secondary structure variations were observed within the 3′X-tail (P ≤ 0.05; Figure 6 and Supplementary Table S6). Importantly, reductions in DMS sensitivity were seen for positions A9346–A9347 in the apical loop of 3′SLII (Figure 6). This agrees well with previous observations of an interaction involving these residues and the apical loop of the 5BSL3.2 domain (34,42,43). Changes in the accessibility of the 3′SLII loop were accompanied by an increase in DMS reactivity for residues A9533 and C9534 at the base of the 3′SLII stem-loop. These observations imply the existence of subtle secondary structure rearrangements dependent on the presence of the CRE region in this crucial domain. Other significant deviations (both increases and reductions; P ≤ 0.05) in the DMS reactivity pattern observed for the CU molecule with respect to the U transcript were observed in the 5′ end of the HV region (Figures 3A, 6A and Supplementary Table S6). Because this domain participates in no known interaction with either the CRE or the 3′X-tail, and because the observed changes follow no clear variation pattern, these deviations most likely result from the existence of a structurally diverse pool of molecules for the HV region, even under the used stringent conditions. This observation also supports the idea of a 3D organization for the two possible modules (the HV region and the 3′X-tail) within the 3′ end, as proposed above.
Figure 6.

Comparison of the DMS pattern for the HCV 3′UTR with different 5′ tails. (A) Significant variations (P ≤ 0.05) in the DMS profile for those reactive positions (>0.3) in each construct under study (CU, I + U and I + CU) with respect to molecule U are highlighted in different colors: CU: blue; I + U: red; I + CU: green. Qualitative increases (+) and reductions (−) are represented. RNA domains are depicted. (B) Sequence and secondary structure of the 3′X-tail region summarizing the data presented in Figure 6A. Filled triangles: significant increase (P ≤ 0.05) in DMS reactivity; open triangles: significant reduction (P ≤ 0.05) in DMS reactivity. Red nucleotides: dimerization linkage sequence (DLS). Gray residues: not analyzed.
SHAPE analysis also revealed small but significant variations (P ≤ 0.05) within the 3′X-tail in the presence of the CRE (CU variant) compared with the U transcript (Figures 4A and 7A, and Supplementary Table S7). The changes affected single residues throughout the 3′SLIII domain as well as positions U9542 and A9547 in the apical loop of 3′SLII (Figure 7B). This suggests differences in local flexibility and conformation, not necessarily dependent on solvent accessibility or on the formation of further secondary foldings. As seen in DMS analysis, the HV region in the CU and U transcripts showed different NMIA reactivity (Figures 4A, 7A and Supplementary Table S7).
Figure 7.

SHAPE analysis shows rearrangement events in the HCV 3′X-tail influenced by the CRE and the IRES regions. (A) Graph shows significant (P ≤ 0.05) gains (+) or losses (−) in NMIA reactivity for the 3′UTR element in the presence of the CRE (CU), the IRES (I + U) or both elements (I + CU) with respect to transcript U. Color code is as noted in Figure 6A. Functional domains are indicated. (B) Summary of significant reactivity differences (P ≤ 0.05) observed in SHAPE profiles for the molecules CU, I + U and I + CU with respect to transcript U. Filled circles: significant increase (P ≤ 0.05) in NMIA reactivity; open circles: significant reduction (P ≤ 0.05) in NMIA reactivity. Red nucleotides: dimerization linkage sequence (DLS). Gray residues: not analyzed.
Taken together, these results show that the effect of the CRE on the structure of the 3′X-tail mainly involves the 3′SLII domain, and that the conformational rearrangement is likely triggered by the establishment of RNA–RNA interactions. Further, the integration of these contacts within the overall folding of the 3′X-tail may involve sporadic reorganization processes by which domains surrounding the 3′SLII may change their flexibility or solvent accessibility.
The HCV IRES element promotes structural rearrangements within the 3′UTR
The influence of the IRES element on 3′X-tail conformation was examined by DMS chemical probing and SHAPE analysis of the I + U variant (47). This transcript encompasses the IRES element fused to the 3′UTR (Figure 1B). The results were compared with those obtained for transcript U.
In the presence of the IRES, the HV region and the 3′X-tail showed different behaviors (Figures 3A and 4A). While the HV region showed an overall reduction in sensitivity to NMIA in the I + U transcript, the 3′X-tail DMS and NMIA reactivity patterns were hardly affected by the presence of the IRES. Subsequent statistical analysis identified the specific and significant variations in these profiles (P ≤ 0.05; Supplementary Tables S6 and S7).
A comparison of the DMS patterns for the 3′X-tail in the presence and absence of the IRES (i.e. transcripts I + U and U) showed the existence of four IRES-dependent single-nucleotide reactivity deviations (Figures 3A, 6A and Supplementary Table S6) likely representative of minor local structural variations related to the creation or interruption of specific base pairs within the stems of domains 3′SLIII, 3′SLII and 3′SLI (Figure 6B). Interestingly one of these changes, affecting position C9534 in the stem of the 3′SLII, was completely reversed in the presence of the CRE region (CU construct; Figure 6A and B). This demonstrates the opposing effects of the IRES and CRE regions on certain positions in the 3′X-tail, and suggests that these elements compete for common interacting areas within the viral genomic 3′ end. Neither can we discard that this is the consequence of further conformational rearrangements in structurally related regions. The resulting effect might also extend to nucleotides surrounding the affected regions.
The NMIA probing profiles for the U and I + U transcripts confirmed that, as predicted from the DMS studies, the IRES element modulates the conformation of the 3′X-tail to some extent, promoting increases and reductions in NMIA reactivity at certain positions in the I + U molecule with respect to variant U (Figures 4A and 7A). Importantly, these changes did not affect the stems of the different domains—an effect that was seen in DMS analysis—but instead mainly occurred within the apical loops of 3′SLIII, 3′SLII and 3′SLI (Figure 7B).
Comparison of the DMS and NMIA profiles for the I + U and U transcripts showed the effect of the IRES element in minimizing the structural noise associated with the HV region (Figures 3A, 4A, 6A and 7A). To our knowledge, this is the first evidence to suggest an interplay between the IRES and the HV region that potentially results in the viral translation-enhancing effect exerted by the 3′UTR.
Interactions between the IRES and the CRE regions fine-tune the folding of the 3′UTR
The structural analyses described above show that both the CRE and the IRES regions may, by themselves, ultimately promote conformational reorganization processes within the 3′UTR of the HCV genome. However, it seems likely that the existence of long-range IRES-CRE contacts interferes with these structural tuning events and has an effect on the final shape of the 3′ end of the genomic RNA. We therefore compared the DMS and NMIA reactivity profiles for the transcripts I + CU and U.
Comparisons of the DMS reactivity data for the I + CU and U transcripts showed the presence of both the CRE and the IRES in the I + CU molecule to be a triggering factor for the structural rearrangement of the 3′X-tail (Figures 3A and 6A). The majority of the reactivity variations occurred within 3′SLII and were coincident with those previously detected in the CU transcript (A9533, C9534 and A9546), thus demonstrating that conformational tuning in these positions is primarily mediated by the CRE region (Figures 6A and 6B, and Supplementary Table S6). Interestingly, other changes previously detected in transcript CU with respect to U were not seen in the presence of the IRES. Single-nucleotide deviations in the reactivity pattern emerged for the I + CU construct (A9507 and A9530) (Figure 6). These results suggest that, besides the effect exerted by the CRE and IRES regions on the secondary structure of the 3′X-tail, further long-range RNA–RNA contacts between both ends of the viral genome may modulate, to some degree, the final conformation of the genomic RNA 3′ terminus.
This hypothesis was further supported by comparisons of the SHAPE patterns obtained for the construct I + CU with respect to U (Figures 4A and 7A). Numerous significant variations in the NMIA reactivity profile (P ≤ 0.05; see Supplementary Table S7) (mostly increases) were detected throughout the 3′X-tail region, with nucleotide flexibility generally greater when the IRES and the CRE were present in cis (Figure 7A). One of the most affected regions was the upper portion of domain 3′SLII (residues U9538–U9551) and the base of the stem in domain 3′SLIII (nucleotides A9507–U9511 and U9528–A9530; Figure 7 and Supplementary Table S7). It is remarkable that most of the observed differences were specific and strictly dependent on the simultaneous presence of both the CRE and the IRES regions. Only two of these changes were preserved both in the CU and I + CU molecules, affecting residues U9522 and A9529. Similarly, the increase in NMIA sensitivity for nucleotide A9547 was common to constructs I + U and I + CU with respect to U (Figure 7B). These observations point to a certain conservation of the structural effects triggered by CRE and IRES on the 3′X-tail. Moreover, they confirm the existence of substantial interdependence in the 3D folding for the 3′ and the 5′ ends of the HCV RNA and point to the IRES-CRE interaction as a critical factor for the structural tuning of the 3′X-tail.
DISCUSSION
Many processes of the viral cycles are commonly controlled by long-range interactions that involve essential RNA elements of the viral gemomes. These contacts frequently promote the direct recruitment of specific protein factors or the acquisition of preferential conformations by the affected regions. The HCV genome harbors a complex network of distant RNA–RNA interactions that exist in dynamic equilibrium. Together they modulate the initiation of essential viral events and control the switch between the different steps of the HCV cycle (21,33,42,44–47). We recently reported that HCV IRES folding is fine-tuned by the presence of the CRE and the 3′UTR elements, independent of protein factors (47). In the latter work, conformational rearrangements were found to affect key functional domains of the IRES in translation- and replication-competent genomes. These findings supported previous observations, confirming a functional role for both CRE and the 3′UTR in viral protein synthesis (16–21,42).
The results of the present work show that the IRES element exerts a structural tuning effect on the 3′ end of the viral genome, especially in the 3′X-tail. The present findings also suggest the IRES and the CRE regions to regulate the 3D reorganization of the 3′X-tail. This conclusion can be drawn from the fact that changes in 3′X-tail conformation cannot be explained by the presence of these elements on their own: rather, IRES-CRE interactions must be involved in displacing the CRE-3′X-tail contact to favor different folding. The present data support the existence of a complex RNA–RNA interaction network that operates between the IRES, the CRE and the 3′UTR. To our knowledge, this is the first report describing the existence of structural reorganizations within the 3′X-tail mediated by direct long-distance RNA–RNA contacts involving the 5′ end of the HCV genome.
The structural study of the HCV 3′ end via three different methodologies provided a complete overview of its 3D architecture. Further, DMS and NMIA probing assays were performed under stringent conditions with low magnesium concentrations and short treatment times to help minimize the existence of suboptimal foldings that might impede the detection of slight, but conformationally significant, structural variations.
The microarray technique showed domains 5BSL3.2 and 3′SLII to efficiently hybridize with complementary oligonucleotides (Figure 2A). This reflects the ease with which they bind to other RNA targets, and is in good agreement with their roles as interacting partners in the complex network of contacts existing within the HCV genome. DMS probing and SHAPE analysis showed the existence of two well-differentiated structural modules corresponding to the functional regions CRE and 3′UTR (Figures 3A and 4A). Thus, while the CRE appeared as a packed rigid region with a low reactivity pattern, the 3′UTR element showed high reactivity values, with the exception of the highly compact 3′SLI. This feature reflects the structural plasticity of the 3′SLII and 3′SLIII domains. The presence of the IRES element (I + CU molecule) revealed statistically significant differences (detected by all three methodologies) in the structural map of the 3′X-tail (Figures 2–5). These results suggest that profound reorganization events occur in the 3D structure of the 3′UTR, which are dependent on the 5′ end of the HCV RNA. It should be noted that single nucleotide deviations within the 5BSL3.2 domain were detected by the DMS and NMIA probing assays, which correlated with the differential ability of 5BSL3.2 to interact with antisense oligonucleotides in the presence or absence of the IRES, as detected in microarray analysis (Figures 2C and 5). The variations seen in reactivity do not seem to involve long sequence stretches, nor do they entail great changes in relative reactivity. Nonetheless, they are reproducible and suggest subtle changes in the structure of the 5BSL3.2 domain mediated by the IRES element (Figures 3B, 4B and 5). Previous SHAPE analyses (43) returned an NMIA reactivity profile for the 5BSL3.2 domain indicative of a little reactive internal loop and a highly sensitive apical loop. This pattern was attributed to the formation of a double-pseudoknot motif involving the simultaneous interaction of 5BSL3.2-3′SLII-SL9110, excluding any possible interaction with the IRES. However, this 5BSL3.2 profile remained reproducible under the present conditions in the absence of SL9110, and was slightly tunable in the presence of the HCV IRES. This apparent discordance might be explained by the use of different experimental conditions (e.g., concentration of divalent cations, treatment time) and by different quantification pipelines, which may lead to substantial variations in the scoring of reactivity values (58). It is also noteworthy that the 5BSL3.2 domain is involved in multiple RNA–RNA contacts. Therefore, it seems likely that exchanges between different interacting partners cannot be detected by steady-state conformational mapping. Importantly, the fine-tuning conformational effect of the CRE and the 3′UTR on the IRES has already been confirmed by our group in translation and replication-competent RNA molecules (47), reinforcing the idea that complex interplay mediated by long-distant RNA–RNA interactions occur within the 5′ and the 3′ termini of the HCV genome. Finally, it has recently been reported that both 5BSL3.2-IRES and 5BSL3.2-9110 interactions are equally feasible; indeed, they have similar Kd values (44). This supports the hypothesis of a dynamic equilibrium between multiple RNA–RNA contacts, which may finally direct the acquisition of different and mutually exclusive structures within the 3′ end of the HCV RNA.
While the CRE and IRES regions on their own moderately affected the architecture of the 3′ terminus, when combined in cis they increased the magnitude and the length of the tuned region (Figures 3A, 4A, 6 and 7). Experimental constraints obtained through SHAPE analyses were then used to predict the secondary structure of the 3′X-tail by means of ShapeKnots software (52) (Figure 8A). The resulting models proposed the acquisition of the typical conformation comprising three stem-loops (3′SLIII, 3′SLII and 3′SLI) for the variants U, CU and I + U. Importantly, the presence of both the CRE and the IRES regions (transcript I + CU) induced the alternative, two stem-loop dimerizable conformation, in which the 3′SLIII and 3′SLII domains disappear to generate a single extended stem-loop with the DLS motif exposed in the apical loop (Figure 8A). These results provide, for the first time, structural evidence of the conformational switch that occurs within the 3′X-tail, and suggest the existence of interplay between the IRES and the CRE, as previously reported (45,47). It is noteworthy that, in a recently proposed model (44), transitions between both 3′X-tail isoforms are mainly dependent on the differential recruitment of protein factors. However, the present results suggest that both alternative foldings are not equally preferred within the genomic RNA. Indeed, their presence could be preferentially favored by a complex web of long-range RNA–RNA interactions established between RNA functional domains in the absence of protein factors. Proteins must play critical roles in the stabilization and functionality of this interaction network, but it should be emphasized that RNA–RNA contacts are alone sufficient to promote the shift between different 3′X-tail conformers.
Figure 8.

Impact of the CRE and the IRES regions on the secondary structure of the 3′X-tail and functional implications for the HCV cycle. (A) RNA secondary structure was predicted by means of the ShapeKnots software (52) using the SHAPE-obtained experimental constraints for the constructs U, CU, I + U and I + CU. The color code highlights differences in NMIA reactivity for each nucleotide position. Dimerization linkage sequence (DLS) is shadowed in transcript I + CU. Gray residues: not analyzed. (B) Model proposing the structural switch occurring in the 3′X-tail during the HCV cycle: (i) After virion entry, HCV translation is initiated in the cytosol by the direct recruitment of the 40S ribosomal subunit to the IIId subdomain of the IRES element and the subsequent binding of eIF3 to the apical portion of domain III. Under these conditions, the acquisition of a double pseudoknot motif involving the contacts SL9110-5BSL3.2-3′SLII (43) would be preferred. (ii) On translation, the accumulation of viral proteins promotes the migration of the viral RNA to the endoplasmic reticulum (ER) where the replication complex is assembled at the 3′ end of the HCV genome. Steric hindrance within the 3′X-tail would displace the conformational equilibrium toward the establishment of a long-distance interaction involving the IRES subdomain IIId and the stem-loop 5BSL3.2, thus impeding efficient translation (21). In this context, it is feasible that a pool of molecules could alternatively promote the contact SL9110-5BSL3.2, which is essential for viral replication (43,46). The newly synthesized, naked HCV transcripts would be gathered (iii), thus favoring the stabilization of the contact IIId-5BSL3.2, which would promote the structural switch of the 3′X-tail to the dimerizable two stem-loop conformation, with the dimerization linkage sequence (DLS) exposed in an apical loop. (iv) The presence of the core chaperone protein contributes to the formation of dimeric viral genomes (35), which would be finally encapsidated and enveloped before protrusion to the extracellular medium.
The present results, together with previously reported data (43,44,47), allow a working hypothesis to be envisioned in which, during early viral infection, the HCV IRES is occupied by the translational machinery, thus avoiding any contact with the 5BSL3.2 domain of the CRE region (Figure 8B). This would favor the establishment of the interaction 5BSL3.2-3′SLII, thus occluding the DLS motif. After viral translation, the whole 3′ end of the HCV genome (containing the CRE and the 3′X-tail) would be dedicated to the recruitment of the viral polymerase and other replication factors (both RNA and proteins) (29,36,46,59–63). In this context, both the IIId-5BSL3.2 and SL9110-5BSL3.2 interactions would be equally feasible. Swapping between them would contribute to the creation of a translationally repressed state (21) and to enhance the replicative process (46). Further accumulation of viral RNA molecules might then increase the proportion of naked HCV genomes containing both RNA conformations, thus favoring intermolecular contacts between those transcripts able to dimerize (44). Under these conditions, it seems likely that the structural equilibrium between the IIId-5BSL3.2 and SL9110-5BSL3.2 interactions would be preferentially displaced toward long-range IRES-CRE contact. This would increase the proportion of RNA genomes exposing the DLS motif in the apical loop of the dimerizable conformation, leading to the formation of dimeric genomic particles in the presence of the core chaperone protein (35). Importantly, it should be noted that during all this process the viral RNA is preferentially folded into a closed loop topology, which enables the formation of these long-distant RNA–RNA contacts. Therefore, the CRE region could swap between different partners over the viral cycle to control the course of infection.
In summary, the present results show that direct long-distance RNA–RNA contacts may mediate the conformational tuning of the essential 3′X-tail region of the HCV genome, and that these interactions occur in the absence of proteins. Such structural rearrangements are directly related to the acquisition of a dimerizable isoform by the 3′X-tail, and thus play an important role in the regulation of the switching between the different steps of the HCV cycle.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministerio de Ciencia e Innovación [BFU2009-08137 to A.B.-H. and BIO2010-20696 to C.B.]; Spanish Ministerio de Economía y Competitividad [BFU2012-31213 to A.B.-H.]; Junta de Andalucía [CVI-7430 to A.B.-H.]; Spanish Council for Scientific Research [201120E004 to A.B.-H.]; FEDER funds from the EU (to A.B.-H.) CIBERehd is funded by the Instituto de Salud Carlos III. Funding for open access charge: Spanish Council for Scientific Research.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Vicente Augustin, Beatriz Berzal-Herranz and María Fernández-Algar for excellent technical assistance.
REFERENCES
- 1.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 2.Takamizawa A, Mori C, Fuke I, Manabe S, Murakami S, Fujita J, Onishi E, Andoh T, Yoshida I, Okayama H. Structure and organization of the hepatitis C virus genome isolated from human carriers. J. Virol. 1991;65:1105–1113. doi: 10.1128/jvi.65.3.1105-1113.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsukiyama-Kohara K, Iizuka N, Kohara M, Nomoto A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992;66:1476–1483. doi: 10.1128/jvi.66.3.1476-1483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C, Sarnow P, Siddiqui A. Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. J. Virol. 1993;67:3338–3344. doi: 10.1128/jvi.67.6.3338-3344.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reynolds JE, Kaminski A, Kettinen HJ, Grace K, Clarke BE, Carroll AR, Rowlands DJ, Jackson RJ. Unique features of internal initiation of hepatitis C virus RNA translation. EMBO J. 1995;14:6010–6020. doi: 10.1002/j.1460-2075.1995.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang TH, Rijnbrand RC, Lemon SM. Core protein-coding sequence, but not core protein, modulates the efficiency of cap-independent translation directed by the internal ribosome entry site of hepatitis C virus. J. Virol. 2000;74:11347–11358. doi: 10.1128/jvi.74.23.11347-11358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kieft JS, Zhou K, Jubin R, Murray MG, Lau JY, Doudna JA. The hepatitis C virus internal ribosome entry site adopts an ion-dependent tertiary fold. J. Mol. Biol. 1999;292:513–529. doi: 10.1006/jmbi.1999.3095. [DOI] [PubMed] [Google Scholar]
- 8.Honda M, Ping LH, Rijnbrand RC, Amphlett E, Clarke B, Rowlands D, Lemon SM. Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology. 1996;222:31–42. doi: 10.1006/viro.1996.0395. [DOI] [PubMed] [Google Scholar]
- 9.Pestova TV, Shatsky IN, Fletcher SP, Jackson RJ, Hellen CU. A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev. 1998;12:67–83. doi: 10.1101/gad.12.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lytle JR, Wu L, Robertson HD. Domains on the hepatitis C virus internal ribosome entry site for 40s subunit binding. RNA. 2002;8:1045–1055. doi: 10.1017/s1355838202029965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berry KE, Waghray S, Doudna JA. The HCV IRES pseudoknot positions the initiation codon on the 40S ribosomal subunit. RNA. 2010;16:1559–1569. doi: 10.1261/rna.2197210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berry KE, Waghray S, Mortimer SA, Bai Y, Doudna JA. Crystal structure of the HCV IRES central domain reveals strategy for start-codon positioning. Structure. 2011;19:1456–1466. doi: 10.1016/j.str.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sizova DV, Kolupaeva VG, Pestova TV, Shatsky IN, Hellen CU. Specific interaction of eukaryotic translation initiation factor 3 with the 5′ nontranslated regions of hepatitis C virus and classical swine fever virus RNAs. J. Virol. 1998;72:4775–4782. doi: 10.1128/jvi.72.6.4775-4782.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Otto GA, Puglisi JD. The pathway of HCV IRES-mediated translation initiation. Cell. 2004;119:369–380. doi: 10.1016/j.cell.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 15.Fraser CS, Doudna JA. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat. Rev. Microbiol. 2007;5:29–38. doi: 10.1038/nrmicro1558. [DOI] [PubMed] [Google Scholar]
- 16.Ito T, Tahara SM, Lai MM. The 3′-untranslated region of hepatitis C virus RNA enhances translation from an internal ribosomal entry site. J. Virol. 1998;72:8789–8796. doi: 10.1128/jvi.72.11.8789-8796.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCaffrey AP, Ohashi K, Meuse L, Shen S, Lancaster AM, Lukavsky PJ, Sarnow P, Kay MA. Determinants of hepatitis C translational initiation in vitro, in cultured cells and mice. Mol. Ther. 2002;5:676–684. doi: 10.1006/mthe.2002.0600. [DOI] [PubMed] [Google Scholar]
- 18.Bradrick SS, Walters RW, Gromeier M. The hepatitis C virus 3′-untranslated region or a poly(A) tract promote efficient translation subsequent to the initiation phase. Nucleic Acids Res. 2006;34:1293–1303. doi: 10.1093/nar/gkl019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song Y, Friebe P, Tzima E, Junemann C, Bartenschlager R, Niepmann M. The hepatitis C virus RNA 3′-untranslated region strongly enhances translation directed by the internal ribosome entry site. J. Virol. 2006;80:11579–11588. doi: 10.1128/JVI.00675-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bung C, Bochkaeva Z, Terenin I, Zinovkin R, Shatsky IN, Niepmann M. Influence of the hepatitis C virus 3′-untranslated region on IRES-dependent and cap-dependent translation initiation. FEBS Lett. 2010;584:837–842. doi: 10.1016/j.febslet.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 21.Romero-López C, Berzal-Herranz A. The functional RNA domain 5BSL3.2 within the NS5B coding sequence influences hepatitis C virus IRES-mediated translation. Cell. Mol. Life Sci. 2012;69:103–113. doi: 10.1007/s00018-011-0729-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai Y, Zhou K, Doudna JA. Hepatitis C virus 3′UTR regulates viral translation through direct interactions with the host translation machinery. Nucleic Acids Res. 2013;41:7861–7874. doi: 10.1093/nar/gkt543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friebe P, Lohmann V, Krieger N, Bartenschlager R. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J. Virol. 2001;75:12047–12057. doi: 10.1128/JVI.75.24.12047-12057.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friebe P, Bartenschlager R. Genetic analysis of sequences in the 3′ nontranslated region of hepatitis C virus that are important for RNA replication. J. Virol. 2002;76:5326–5338. doi: 10.1128/JVI.76.11.5326-5338.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolykhalov AA, Feinstone SM, Rice CM. Identification of a highly conserved sequence element at the 3′ terminus of hepatitis C virus genome RNA. J. Virol. 1996;70:3363–3371. doi: 10.1128/jvi.70.6.3363-3371.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blight KJ, Rice CM. Secondary structure determination of the conserved 98-base sequence at the 3′ terminus of hepatitis C virus genome RNA. J. Virol. 1997;71:7345–7352. doi: 10.1128/jvi.71.10.7345-7352.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolykhalov AA, Mihalik K, Feinstone SM, Rice CM. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 2000;74:2046–2051. doi: 10.1128/jvi.74.4.2046-2051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yi M, Lemon SM. 3′ nontranslated RNA signals required for replication of hepatitis C virus RNA. J. Virol. 2003;77:3557–3568. doi: 10.1128/JVI.77.6.3557-3568.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yi M, Lemon SM. Structure-function analysis of the 3′ stem-loop of hepatitis C virus genomic RNA and its role in viral RNA replication. RNA. 2003;9:331–345. doi: 10.1261/rna.2144203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ivanyi-Nagy R, Kanevsky I, Gabus C, Lavergne JP, Ficheux D, Penin F, Fosse P, Darlix JL. Analysis of hepatitis C virus RNA dimerization and core-RNA interactions. Nucleic Acids Res. 2006;34:2618–2633. doi: 10.1093/nar/gkl240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim M, Kim H, Cho SP, Min MK. Template requirements for de novo RNA synthesis by hepatitis C virus nonstructural protein 5B polymerase on the viral X RNA. J. Virol. 2002;76:6944–6956. doi: 10.1128/JVI.76.14.6944-6956.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shim JH, Larson G, Wu JZ, Hong Z. Selection of 3′-template bases and initiating nucleotides by hepatitis C virus NS5B RNA-dependent RNA polymerase. J. Virol. 2002;76:7030–7039. doi: 10.1128/JVI.76.14.7030-7039.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shetty S, Kim S, Shimakami T, Lemon SM, Mihailescu MR. Hepatitis C virus genomic RNA dimerization is mediated via a kissing complex intermediate. RNA. 2010;16:913–925. doi: 10.1261/rna.1960410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palau W, Masante C, Ventura M, Di Primo C. Direct evidence for RNA-RNA interactions at the 3′ end of the Hepatitis C virus genome using surface plasmon resonance. RNA. 2013;19:982–991. doi: 10.1261/rna.037606.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cristofari G, Ivanyi-Nagy R, Gabus C, Boulant S, Lavergne JP, Penin F, Darlix JL. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 2004;32:2623–2631. doi: 10.1093/nar/gkh579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ito T, Lai MM. Determination of the secondary structure of and cellular protein binding to the 3′-untranslated region of the hepatitis C virus RNA genome. J. Virol. 1997;71:8698–8706. doi: 10.1128/jvi.71.11.8698-8706.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang JW, Moyer RW. The effects of the conserved extreme 3′ end sequence of hepatitis C virus (HCV) RNA on the in vitro stabilization and translation of the HCV RNA genome. J. Hepatol. 2000;33:632–639. doi: 10.1034/j.1600-0641.2000.033004632.x. [DOI] [PubMed] [Google Scholar]
- 38.Kong LK, Sarnow P. Cytoplasmic expression of mRNAs containing the internal ribosome entry site and 3′ noncoding region of hepatitis C virus: effects of the 3′ leader on mRNA translation and mRNA stability. J. Virol. 2002;76:12457–12462. doi: 10.1128/JVI.76.24.12457-12462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tuplin A, Wood J, Evans DJ, Patel AH, Simmonds P. Thermodynamic and phylogenetic prediction of RNA secondary structures in the coding region of hepatitis C virus. RNA. 2002;8:824–841. doi: 10.1017/s1355838202554066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee H, Shin H, Wimmer E, Paul AV. Cis-acting RNA signals in the NS5B C-terminal coding sequence of the hepatitis C virus genome. J. Virol. 2004;78:10865–10877. doi: 10.1128/JVI.78.20.10865-10877.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.You S, Stump DD, Branch AD, Rice CM. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J. Virol. 2004;78:1352–1366. doi: 10.1128/JVI.78.3.1352-1366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friebe P, Boudet J, Simorre JP, Bartenschlager R. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 2005;79:380–392. doi: 10.1128/JVI.79.1.380-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tuplin A, Struthers M, Simmonds P, Evans DJ. A twist in the tail: SHAPE mapping of long-range interactions and structural rearrangements of RNA elements involved in HCV replication. Nucleic Acids Res. 2012;40:6908–6921. doi: 10.1093/nar/gks370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shetty S, Stefanovic S, Mihailescu MR. Hepatitis C virus RNA: molecular switches mediated by long-range RNA-RNA interactions? Nucleic Acids Res. 2013;41:2526–2540. doi: 10.1093/nar/gks1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Romero-López C, Berzal-Herranz A. A long-range RNA-RNA interaction between the 5′ and 3′ ends of the HCV genome. RNA. 2009;15:1740–1752. doi: 10.1261/rna.1680809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diviney S, Tuplin A, Struthers M, Armstrong V, Elliott RM, Simmonds P, Evans DJ. A hepatitis C virus cis-acting replication element forms a long-range RNA-RNA interaction with upstream RNA sequences in NS5B. J. Virol. 2008;82:9008–9022. doi: 10.1128/JVI.02326-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Romero-López C, Barroso-delJesus A, García-Sacristán A, Briones C, Berzal-Herranz A. The folding of the hepatitis C virus internal ribosome entry site depends on the 3′-end of the viral genome. Nucleic Acids Res. 2012;40:11697–11713. doi: 10.1093/nar/gks927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J. Am. Chem. Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 49.Martell M, Briones C, de Vicente A, Piron M, Esteban JI, Esteban R, Guardia J, Gomez J. Structural analysis of hepatitis C RNA genome using DNA microarrays. Nucleic Acids Res. 2004;32:e90. doi: 10.1093/nar/gnh088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vasa SM, Guex N, Wilkinson KA, Weeks KM, Giddings MC. ShapeFinder: a software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA. 2008;14:1979–1990. doi: 10.1261/rna.1166808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. Proc. Natl Acad. Sci. USA. 2009;106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hajdin CE, Bellaousov S, Huggins W, Leonard CW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc. Natl Acad. Sci. USA. 2013;110:5498–5503. doi: 10.1073/pnas.1219988110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lehmann EL, D′Abrera HJM. Nonparametrics: Statistical Methods Based On Ranks. 3rd edn. New York: Springer-Verlag; 2006. [Google Scholar]
- 54.Fernández N, Fernández-Miragall O, Ramajo J, García-Sacristán A, Bellora N, Eyras E, Briones C, Martínez-Salas E. Structural basis for the biological relevance of the invariant apical stem in IRES-mediated translation. Nucleic Acids Res. 2011;39:8572–8585. doi: 10.1093/nar/gkr560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernández N, García-Sacristán A, Ramajo J, Briones C, Martínez-Salas E. Structural analysis provides insights into the modular organization of picornavirus IRES. Virology. 2011;409:251–261. doi: 10.1016/j.virol.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 56.Ooms M, Verhoef K, Southern E, Huthoff H, Berkhout B. Probing alternative foldings of the HIV-1 leader RNA by antisense oligonucleotide scanning arrays. Nucleic Acids Res. 2004;32:819–827. doi: 10.1093/nar/gkh206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chamberlin SI, Weeks KM. Differential helix stabilities and sites pre-organized for tertiary interactions revealed by monitoring local nucleotide flexibility in the bI5 group I intron RNA. Biochemistry. 2003;42:901–909. doi: 10.1021/bi026817h. [DOI] [PubMed] [Google Scholar]
- 58.Leonard CW, Hajdin CE, Karabiber F, Mathews DH, Favorov OV, Dokholyan NV, Weeks KM. Principles for understanding the accuracy of SHAPE-directed RNA structure modeling. Biochemistry. 2013;52:588–595. doi: 10.1021/bi300755u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Banerjee R, Dasgupta A. Specific interaction of hepatitis C virus protease/helicase NS3 with the 3′-terminal sequences of viral positive- and negative-strand RNA. J Virology. 2001;75:1708–1721. doi: 10.1128/JVI.75.4.1708-1721.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tingting P, Caiyun F, Zhigang Y, Pengyuan Y, Zhenghong Y. Subproteomic analysis of the cellular proteins associated with the 3′ untranslated region of the hepatitis C virus genome in human liver cells. Biochem. Biophys. Res. Commun. 2006;347:683–691. doi: 10.1016/j.bbrc.2006.06.144. [DOI] [PubMed] [Google Scholar]
- 61.Foster TL, Belyaeva T, Stonehouse NJ, Pearson AR, Harris M. All three domains of the hepatitis C virus nonstructural NS5A protein contribute to RNA binding. J. Virol. 2010;84:9267–9277. doi: 10.1128/JVI.00616-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanamori H, Yuhashi K, Ohnishi S, Koike K, Kodama T. RNA-dependent RNA polymerase of hepatitis C virus binds to its coding region RNA stem-loop structure, 5BSL3.2, and its negative strand. J. Gen. Virol. 2010;91:1207–1212. doi: 10.1099/vir.0.016907-0. [DOI] [PubMed] [Google Scholar]
- 63.Oakland TE, Haselton KJ, Randall G. EWSR1 binds the hepatitis C virus cis-acting replication element and is required for efficient viral replication. J. Virol. 2013;87:6625–6634. doi: 10.1128/JVI.01006-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.