


Abstract
Infection by oncogenic viruses is a frequent cause for tumor formation as observed in cervical cancer. Viral oncoproteins cause inactivation of p53 function and false transcriptional regulation of central cell cycle genes. Here we analyze the regulation of Plk4, serving as an example of many cell cycle- and p53-regulated genes. Cell cycle genes are often repressed via CDE and CHR elements in their promoters and activated by NF-Y binding to CCAAT-boxes. In contrast, general activation of Plk4 depends on NRF1 and CRE sites. Bioinformatic analyses imply that NRF1 and CRE are central elements of the transcriptional network controlling cell cycle genes. We identify CDE and CHR sites in the Plk4 promoter, which are necessary for binding of the DREAM (DP, RB-like, E2F4 and MuvB) complex and for mediating repression in G0/G1. When cells progress to G2 and mitosis, DREAM is replaced by the MMB (Myb-MuvB) complex that only requires the CHR element for binding. Plk4 expression is downregulated by the p53-p21WAF1/CIP1-DREAM signaling pathway through the CDE and CHR sites. Cell cycle- and p53-dependent repression is abrogated by HPV E7 oncoprotein. Together with genome-wide analyses our results imply that many cell cycle genes upregulated in tumors by viral infection are bound by DREAM through CDE/CHR sites.
INTRODUCTION
Centrioles are essential for the formation of centrosomes, and alteration of centrosome numbers has been associated with genome instability and tumor formation. Thus, precise control of centriole biogenesis is required for proper chromosome segregation and genomic stability. Polo-like kinase 4 (Plk4, also known as Sak or Stk18) is a serine/threonine kinase, which controls correct centriole biogenesis (1–4). Therefore, regulation of Plk4 protein levels is crucial for proper centriole duplication (5,6). Plk4 is expressed only at low levels in G0 and G1. The protein increases in S phase and is maximally expressed in G2 and mitosis (7).
Synthesis of proteins displaying this expression pattern is often controlled on the transcriptional level through cell cycle-dependent elements (CDE) and cell cycle genes homology regions (CHR) in the promoter (8). Activation of most CDE/CHR-regulated genes is conferred by nuclear factor Y (NF-Y) bound to CCAAT-boxes (8–11).
Recently, the DREAM (DP, RB-like, E2F4 and MuvB) complex was identified as the protein complex, which binds to CHR elements (12). This complex is conserved in Drosophila, Caenorhabditis elegans and mammals (13–17). In mammalian cells, DREAM consists of E2F4, DP1, p130 and p107 in addition to RBBP4 and the MuvB-like Lin proteins LIN9, LIN37, LIN52 and LIN54 that form its MuvB-core (15–17). The DREAM complex forms in G0 and early G1. Formation of the complex is necessary for repression of its target genes. When a cell progresses through the cell cycle, E2F4/DP1 and p130/p107 are replaced by B-Myb forming a complex named MMB (Myb-MuvB) that can activate gene expression in S phase (12,15–21).
Transcriptional repression is a hallmark of p53-mediated DNA damage response (22). Recently, we have reported a mechanism that controls p53-dependent repression by the p53-p21WAF1/CIP1-DREAM-CHR pathway (23). After DNA damage and p53 induction, expression of p21WAF1/CIP1 is activated by p53, leading to formation of DREAM and its association with cell cycle promoters (23,24). DREAM binding to the CHR in the target promoter then leads to transcriptional repression (23). This mechanism does not involve p53 binding to the repressed target promoter (23).
p53 normally protects cells from centrosome duplication, mitotic spindle abnormality and failure in cytokinesis (25). Furthermore, it is well established that cells infected with human papilloma virus express the oncoproteins E6 and E7, which interfere with the p53 and retinoblastoma (RB) pocket protein-E2F pathways (26–29). Human papilloma virus (HPV) E7 expression interferes with centriole biogenesis and Plk4 protein level is limiting for aberrant centriole duplication (30–32). Interestingly, it has been shown that Plk4 transcriptional activation increases on HPV-16 E7 induction (33). However, the underlying mechanism remains unidentified.
In this report, we describe the regulation of the Plk4 promoter, which serves as an example for a number of cell cycle- and p53-regulated genes. We identify a CDE/CHR tandem element that binds the DREAM complex and mediates repression in G0 and G1. When cells progress to G2 and mitosis, DREAM is replaced by the MMB complex. General activation of Plk4 depends on the nuclear respiratory factor 1 (NRF1) and cAMP response element (CRE) sites instead of the established NF-Y/CCAAT system. Bioinformatic motif analyses suggest this to be a common mechanism for CDE/CHR-controlled genes as an alternative to the NF-Y/CCAAT system. Importantly, Plk4 mRNA levels are downregulated by the p53-p21WAF1/CIP1-DREAM signaling pathway requiring both the CDE and the CHR sites. Furthermore, cell cycle- and p53-dependent repression is abrogated by HPV E7 oncoproteins. Together with genome-wide analyses, our results imply that many cell cycle genes upregulated in tumors by viral infection are bound by DREAM through CDE/CHR sites.
MATERIALS AND METHODS
Plasmids, RNAi and DNA probes
The mouse Plk4 promoter with a size of 453 bp [nt −510 to −57, named mPlk4 wild type (wt)] was amplified from NIH3T3 cell DNA and ligated in the pGL4.10 vector (Promega, Madison, WI, USA). Mutations were introduced with the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA, as shown in Table 1). The plasmid used for Bmyb-knockdown (sh-Bmyb1) was a kind gift from Kenneth Boheler (34).
Table 1.
Mutations introduced into the CDE, CHR, NRF1 and CRE sites of the mouse Plk4 promoter
 |
The human p53 expression plasmids pcDNA-p53wt and pcDNA-p53mut were produced by amplifying the insert of pCMV-p53wt and pCMV-p53mut R175H (kindly provided by Bert Vogelstein) and ligating into pcDNA3.1HisC (Invitrogen, Carlsbad, CA, USA) (23). Expression plasmids for human p21WAF1/CIP1, pCEP-p21wt and pCEP-p21mut were generously provided by Bert Vogelstein (35). Expression plasmids (pCMV) for HPV-16 E7 wt and ΔDLYC-mutant were kindly provided by Karl Münger (36).
Nrf1 small interfering RNA (siRNA) has been previously published as Nrf1#1 (37) and was obtained as Stealth RNA interference (RNAi) (Invitrogen). Stealth RNAi negative control (Invitrogen) served as siControl.
DNA probes for affinity purification with the same sequence as mPlk4 were obtained by polymerase chain reaction (PCR) using a biotinylated primer for labeling the 3′-end (pGL4-Biotin-rev 5′-TGT TTT TGG CAT CTT CCA TG-3′, Invitrogen). As a negative control, a fragment of the pGL4.10 vector was used (220 bp from the multiple cloning site, amplified with pGL4-for 5′-CGA TAG TAC TAA CAT ACG CTC TCC A-3′).
Cell culture, synchronization and drug treatment
NIH3T3 and T98G cells were used to synchronize mouse and human cells, respectively, by serum starvation. NIH3T3 cells displayed better transient transfection efficiencies than T98G cells, while T98G cells yielded stronger signals in chromatin immunoprecipitation (ChIP) experiments. HCT116 cells were used for experiments on p53-dependent gene regulation as p53- and p21-knockout cells lines were available. HeLa cells were used to assess PLK4 regulation in HPV-infected cancer cells.
HCT116 wt, HCT116 p53−/− and HCT116 p21−/− cells, kindly provided by Bert Vogelstein (38), and NIH3T3, T98G and HeLa cells (DSMZ, Braunschweig, Germany) were grown in Dulbecco's modified Eagle's medium (DMEM; Lonza, Basel, Switzerland) supplemented with 10% fetal calf serum (FCS) (Biochrom, Berlin, Germany) and penicillin/streptomycin (PAA Laboratories, Pasching, Austria) and maintained at 37°C and 10% CO2. Cells transfected for RNAi were grown in medium without penicillin/streptomycin.
Stably transfected HCT116 cells were generated by transfection with pCMV-HPV16-E7 wt or pCMV-HPV16-E7 ΔDLYC and selection with G418/Geneticin (PAA Laboratories) at a concentration of 0.6 mg/ml.
NIH3T3 and T98G cells were synchronized by serum starvation (exchanging growth medium with 10% FCS for DMEM without FCS) for 60 and 72 h, respectively. Cells were stimulated for cell cycle reentry using DMEM with 20% FCS and collected at given time points. NIH3T3 cells were synchronized by double thymidine and thymidine nocodazole block (39). Doxorubicin was used at a final concentration of 0.2 µg/ml for 24 or 48 h. Roscovitine (Adipogen, San Diego, CA, USA) was dissolved as a 25 mM stock solution in dimethyl sulfoxide and used at a final concentration of 25 µM for 24 h.
Transfections and luciferase assays
Transfections and measurements of cell cycle-dependent promoter activity with luciferase reporter assays were carried out as described previously (12).
For Bmyb knockdown experiments, NIH3T3 cells were plated in 12-well plates (19 000 cells per well). After 8 h, cells were transfected by lipofection with 1.5 µl of Fugene 6 (Promega), 0.2 µg of promoter reporter plasmid (pGL4.10, Promega), 0.4 µg of pSuper construct and 0.025 µg of Renilla luciferase plasmid (pGL4.70, Promega) per well. After synchronization by double thymidine and thymidine nocodazole block, cells were collected (39).
For Nrf1 knockdown experiments, HCT116 cells were plated in 12-well plates (50 000 cells per well). After 8 h, cells were transfected by lipofection with 1.0 µl of DharmaFect DUO (Dharmacon, Lafayette, CO, USA), 0.15 µg of promoter reporter plasmids (pGL4.10), 0.020 µg of Renilla luciferase plasmid (pGL4.70) and 100 nM siRNA per well. Seventy-two hours after transfection, cells were collected.
For measuring p53- and p21-dependent promoter activity with luciferase reporter assays, HCT116 cells were plated in 24-well plates (75 000 cells per well) and transfected by lipofection with Fugene 6. Cells were cultured overnight before cotransfection of 0.25 µg of promoter reporter plasmids (pGL4.10) along with 0.025 µg of constructs expressing wt or mutant p53 or p21 proteins and 0.025 µg of Renilla luciferase plasmid (pGL4.70, Promega). After 24 h, cells were collected by adding 100 µl of passive lysis buffer (Promega).
For HPV-16 E7 expression experiments, HCT116 cells were plated in 12-well plates (120 000 cells per well) and transfected by lipofection with Fugene 6. Cells were cultured overnight before cotransfection of 0.2 µg of promoter reporter plasmids (pGL4.10) along with 0.3 µg of constructs expressing E7 wt or E7 ΔDLYC and 0.0025 µg of Renilla luciferase plasmid (pGL4.70).
For cell sorting, pEGFP (Clontech, Mountain View, CA, USA) was cotransfected with the promoter reporter plasmid at a 1:3 ratio.
Fluorescence-activated cell sorting analysis
Cells were fixed for at least 12 h at 4°C in one volume phosphate buffered saline/1 mM EDTA and three volumes of absolute ethanol. DNA was stained with propidium iodide (Sigma) at a final concentration of 10 µg/ml in presence of RNase A (10 µg/ml). Alternatively, cells were collected in phosphate buffered saline/1 mM EDTA, and DNA was stained with Hoechst 33342 (Invitrogen) at a final concentration of 10 µg/ml and incubated for 15 min at 37°C. DNA content per cell was measured by flow cytometry on an LSR II instrument (Becton Dickinson, Franklin Lakes, NJ, USA). Cell sorting was carried out on a FACSVantage SE (Becton Dickinson). Data analysis was carried out with WinMDI 2.9 software.
DNA affinity purification
DNA affinity purification of protein complexes with density-arrested, asynchronously growing and doxorubicin-treated cells was performed as described previously (12).
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis and immunoblot
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis and western blot were performed following standard protocols (40). If necessary, blots were stripped or probes were blotted again to detect multiple proteins. The following antibodies were used for the detection of DREAM/MMB complex components: E2F4 (C-20, Santa Cruz Biotechnology, Santa Cruz, CA, 1:750 dilution), p130 (C-20, Santa Cruz Biotech., 1:1000), p107 (C-18, Santa Cruz Biotech., 1:1000), DP1 (K-20, Santa Cruz Biotech., 1:500), B-Myb (N-19, Santa Cruz Biotech., 1:500) and LIN9 (ab62329, Abcam, Cambridge, UK, 1:1000 dilution). The B-myb LX015.1 monoclonal antibody for detection of mouse B-myb was a kind gift from Roger Watson (41), and the LIN37 (1:1000) and LIN54 (1:750) polyclonal antibodies were kind gifts from James DeCaprio (15). Nrf-1 was detected with Nrf-1 polyclonal antibody (1:5000), a kind gift from Kimitoshi Kohno (42). Creb-1 polyclonal antibody (C-21, Santa Cruz Biotech., 1:200) was used for detection of Creb-1. Band intensities were quantified by densitometric analyses using Multi Gauge v3.1 Quant (Fujifilm, Tokio, Japan).
Chromatin immunoprecipitations
T98G, HCT116 and HeLa wt cells were cross-linked with 1% formaldehyde for 10 min at room temperature. ChIPs were performed as described previously (12). The following antibodies were used for precipitation of DREAM complex components: E2F4 (C-20, Santa Cruz Biotech.), p130 (C-20, Santa Cruz Biotech.), B-Myb (N-19, Santa Cruz Biotech.), p53 (FL-393, Santa Cruz Biotech.), p53 (Ab-6, DO-1, Calbiochem), LIN9 (ab62329, Abcam, Cambridge, UK) and another LIN9 and LIN37 antibodies were kind gifts from James DeCaprio (15). A nontargeting rabbit antibody was used as a control for nonspecific signals. For all precipitations 1–2 µg of antibody and 20–35 µl of Protein G Dynabead suspension (Invitrogen) were used. Immunoprecipitated DNA was used as template for quantitative real-time PCR as described previously (12). The following primers were used for qPCR: hPLK4-for 5′-GGC CCC GAA GTC TAG AAC C-3′, hPLK4-rev 5′-CGG AAA GTT CTC CCT GAC AC-3′; hGAPDHS-for 5′-AGA CCA GCC TGA GCA AAA GA-3′, hGAPDHS-rev 5′-CTA GGC TGG AGT GCA GTG GT-3′; hCDKN1A-for 5′-CTG AGC CTC CCT CCA TCC-3′, hCDKN1A-rev 5′-GAG GTC TCC TGT CTC CTA CCA TC-3′.
RNA extraction, reverse transcription and quantitative real-time PCR
Total RNA was isolated from cell lines using TRIzol Reagent (Invitrogen) following the manufacturer’s protocol. One-step reverse transcription and quantitative real-time PCR were performed with an ABI 7300 Real-Time PCR System (Applied Biosystems, Forster City, CA, USA) using QuantiTect SYBRGreen PCR Kit (QIAGEN). U6 was served as endogenous control. The following primers were used for qPCR: U6-for 5′-AAC GCT TCA CGA ATT TGC GT-3′, U6-rev 5′-CTC GCT TCG GCA GCA CA-3′; hPLK4-for 5′-CCA CAG ACA ACA ATG CCA AC-3′, hPLK4-rev 5′-GCA GAT TCC CAA ACC ACT GT-3′; mPlk4-for 5′-GAA ACA CCC CTC TGT CTT GG-3′, mPlk4-rev 5′-GCA TGA AGT GCC TAG CTT CC-3′; mBmyb-for 5′-AGG GAC TGC AAG CCT GTC TA-3′, mBmyb-rev 5′-GCA GCT ATG GCA ATC TCC TC-3′; mCcnb2-for 5′-TGA AAC CAG TGC AGA TGG AG-3′, mCcnb2-rev 5′-CTG CAG AGC TGA GGG TTC TC-3′; hNRF1-for 5′-CTT ACA AGG TGG GGG ACA GA-3′, hNRF1-rev 5′-CAA TGT CAC CAC CTC CAC AG-3′.
Statistics
Data are presented as means of ± standard deviations. The significance of the difference between two groups was assessed using the Student’s t-test. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; n.s.: not significant; 2 ≤ n ≤ 4.
Bioinformatics
Promoter regions were compiled from coding transcripts in the UCSC known Gene table (43) by using the 200 nt upstream and downstream from each transcription start site. The GREAT tool (44) in its current version 2.02 was used to identify regions that overlap with functional genes (with at least one annotated gene ontology term). Promoter regions were separated into sets of overlapping and nonoverlapping regions while overlapping regions were merged using BEDtools (45) to avoid redundancy. Corresponding sequences from nonredundant regions were used to search for transcription factor binding sites.
ChIP-chip binding data for DREAM was taken from Litovchick et al. (15) and lifted to the current human genome version (GRCh37/hg19) using UCSC liftover (46). Promoter regions covered to at least 90% by a protein's binding region are considered to be bound by this protein. Following Litovchick et al., a promoter was considered to be bound by DREAM if it is bound by at least E2F4, LIN9 and p130.
PhastCons conservation scores (47) obtained from the multiz46 alignment of placental mammalia (48) were used to calculate average sequence conservation. Transcription factor binding sites (represented as a set of strings) were searched in all promoter sequences on plus and minus strands and considered as hits if their average conservation was at least 0.8. String representation for the transcription factor binding sites was chosen as follows: CHR (TTTGAA, TTTAAA, CTTGAA, TAGGAA) (8); CCAAT (CCAAT); CRE (TGACGT); NRF1 (GCATGCG, GCACGCG, GCAGGCG). For sequences from the set of overlapping promoter regions, the affiliation of binding sites to either one of the contained transcriptional start sites (TSS) was assigned manually.
The HPV microarray data sets were manually compared with each other, with the original data set by Litovchick et al. (15) and with the processed data set described above.
RESULTS
Cell cycle-dependent transcription of the Plk4 gene is controlled by repression through a CDE/CHR tandem promoter element
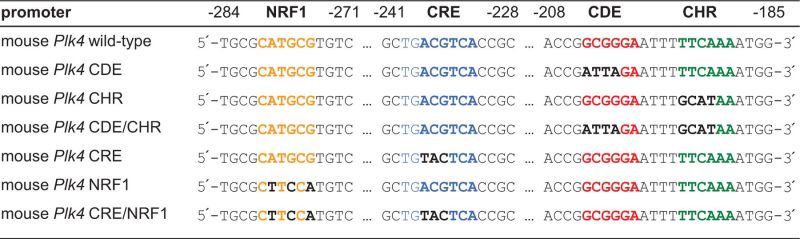
The Plk4 promoter region shows an unusual pattern of conserved elements (Figure 1A). We searched mammalian genomes from opossum to human for phylogenetically conserved sites and found putative CDE and CHR elements, separated by a standard 4 nt spacer. However, Plk4 promoters lack CCAAT-boxes, which are usually found in CDE/CHR-controlled promoters with two or three copies separated by 31–33 nt spacers (8). The only conserved and potentially activating transcription factor motifs in the promoter region are putative NRF1 and CRE sites (Figure 1A).
Figure 1.
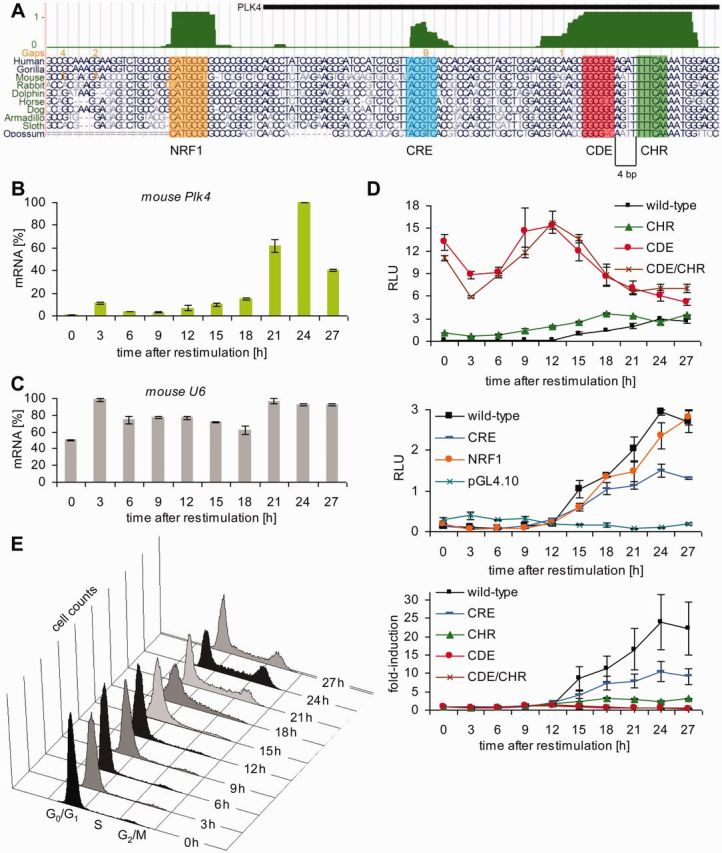
CDE and CHR sites, but not NRF1 and CRE elements, control repression of the Plk4 promoter in G0/G1. (A) UCSC genome browser graph displaying a part of the Plk4 promoter with the conserved NRF1, CRE, CDE and CHR sites. The vertebrate conservation track (PhastCons, dark green) highlights phylogenetically well conserved elements. Selected alignments from opossum to human are shown. (B) Expression of Plk4 mRNA during the cell cycle. NIH3T3 cells were arrested in G0 by serum starvation and stimulated to reenter the cell cycle by addition of FCS to the medium. Cells were collected every 3 h, RNA was extracted and the relative expression of Plk4 mRNA was quantified by real-time RT-PCR. (C) Expression of U6 mRNA served as control. (D) Luciferase reporter assays from NIH3T3 cells transfected with wt or mutant Plk4 promoter constructs. Mutants are named by the site that is mutated. NIH3T3 cells were arrested in G0 by serum starvation and stimulated to reenter the cell cycle by addition of FCS to the medium. Top and middle panel: RLU, relative light units. Both panels originate from one experiment, which was split in two graphs with different scaling. Wt Plk4 results are displayed in both panels. Bottom panel: fold-induction of RLUs relative to the 0 h time point. (E) Fluorescence-activated cell sorting analyses of cells used in D.
We measured Plk4 mRNA expression during the cell cycle in NIH3T3 cells. The typical expression pattern of Plk4, with low mRNA levels in G0/G1 and increasing expression in S phase, reaching a maximum in G2 and mitosis (Figure 1B), resembles the pattern observed from genes regulated by CDE and CHR cell cycle elements (8). Expression levels of U6 served as control, which are only influenced to a small extent by the cell cycle (Figure 1C). We created mutants in the putative transcription factor binding sites and tested them together with the wt promoter in reporter assays during the cell cycle (Table 1, Figure 1D and E). Transcription from the wt promoter essentially reflected the expression of the Plk4 mRNA, indicating that the relevant gene fragment was selected to be tested as a promoter (Figure 1B and D, middle panel).
Cell cycle-dependent stimulation of transcription from the Plk4 promoter in the luciferase reporter assay was >25-fold when comparing the 0-h time point to the highest expression in G2/M (Figure 1D, bottom panel). Mutation of the CDE or both the CDE and CHR resulted in a substantial loss of cell cycle regulation with a general increase in promoter activity in the early phases of the cell cycle. Alteration of the CHR alone also led to a clear deregulation, however, not as substantial as the CDE mutant (Figure 1D, top and bottom panel). The pattern of deregulation showed that CDE and CHR are bona fide functional cell cycle elements. Analyses of mutants in the putative CRE and NRF1 sites led to only a small or essentially no change in cell cycle regulation, respectively (Figure 1D, middle panel). The data indicate that cell cycle-dependent transcription of Plk4 is controlled by the CDE/CHR tandem element in which the CDE plays a more significant role in mediating transcriptional repression than the CHR.
DREAM complex components bind through the CDE and CHR sites to the Plk4 promoter
Recently, we have shown that the DREAM protein complex can bind to CHR elements (12). Thus, we tested whether the CDE/CHR site responsible for Plk4 promoter regulation can bind components representative for DREAM. Western analyses of DNA affinity purifications from G0 cell nuclear extracts showed in vitro binding of DREAM components p130, E2f4, Lin9 and Lin37 to a wt Plk4 promoter probe (Figure 2A). Binding of DREAM components to CDE or CHR promoter mutants appeared to be close to the negative control background level (Figure 2A). Thus, the CDE appears to be required for DREAM binding to the Plk4 promoter (Figure 2A). Combined mutation of CRE and NRF1 sites hardly affected DREAM binding (Figure 2A).
Figure 2.

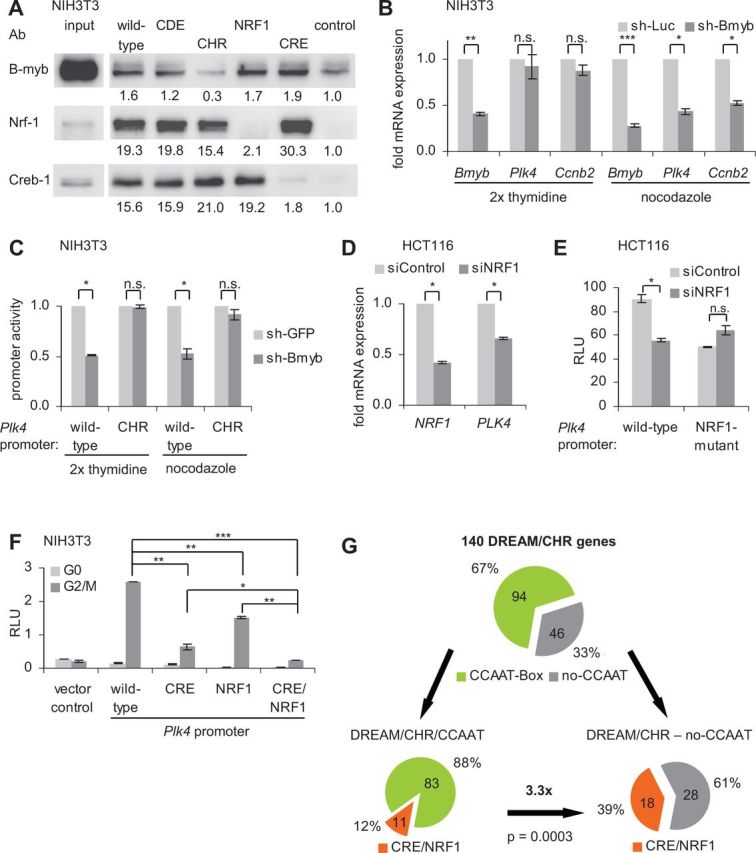
The DREAM complex components bind through the CDE and CHR and not through NRF1 or CRE sites to the Plk4 promoter. (A) Nuclear extracts from density-arrested NIH3T3 cells were analyzed by DNA affinity purification with wt, CRE/NRF1, CDE or CHR mutant Plk4 promoter probes followed by western blot. An irrelevant segment from the pGL4.10 vector served as negative control. Band intensities were quantified by densitometric analyses. The intensities relative to the control are given below the bands. (B) In vivo protein binding to the Plk4 promoter in serum-starved and restimulated T98G cells was tested by ChIP.
For ChIP, serum-starved or restimulated human T98G cells were used. Both DREAM components, E2F4 and LIN9, were bound to the Plk4 promoter in G0 cells, and association substantially decreased in cell populations with an enriched G2/M fraction. In contrast, binding of B-Myb to the Plk4 gene increased after the shift toward G2/M (Figure 2B, Supplementary Figure S1A).
Recently, the transcription factor FoxM1 was found to bind cell cycle genes via MMB (49–51) and that the resulting complex requires the CHR but not the CDE as binding site (51). Consequently, we also tested the Plk4 gene for binding of FoxM1. However, no FoxM1 association to Plk4 was detected by ChIP (data not shown). Furthermore, the Plk4 locus was not observed to bind FoxM1 in ChIP-Seq analyses (51).
The Plk4 promoter is activated through NRF1 and CRE sites. B-Myb contributes to maximal expression and is bound through the CHR in proliferating cells
Binding of B-myb to Plk4 in proliferating cells is also observed in vitro using DNA affinity purification (Figure 3A). Comparing binding to the different probes, it appears that B-myb is bound to the Plk4 promoter through the CHR but not the CDE. Furthermore, the other potentially activating transcription factors Nrf-1 and Creb-1 were bound independently from the CDE and CHR through their NRF1 and CRE recognition sites, respectively (Figure 3A).
Figure 3.
The Plk4 promoter is activated through NRF1 and CRE sites. B-Myb contributes to maximal expression and is bound through the CHR in proliferating cells. (A) Protein binding to the wt and mutant Plk4 promoter was analyzed with nuclear extracts from asynchronous growing NIH3T3 cells by DNA affinity purification and western blot. Band intensities were quantified by densitometric analyses. The intensities relative to the control are given below the bands. (B) Double thymidine- or nocodazole-blocked NIH3T3 cells were cotransfected with a vector expressing shRNA targeting Bmyb (sh-Bmyb) or an shRNA construct expressing a nontargeting luciferase shRNA (sh-Luc) as a negative control and with EGFP. Cells were sorted for green fluorescence before mRNA preparation. Relative expression of Bmyb, Plk4 and Ccnb2 mRNA was quantified by RT-PCR and normalized to U6 RNA levels. (C) Luciferase reporter assays were performed with wt and CHR mutant Plk4 promoter reporter constructs from double thymidine- or nocodazole-blocked NIH3T3 cells. Cells were cotransfected with sh-Bmyb or a shRNA construct expressing a nontargeting GFP-shRNA as a negative control. (D) Asynchronous growing HCT116 cells were transfected with siRNA targeting NRF1 or with siControl. Relative expression of NRF1 and PLK4 mRNA was quantified by RT-PCR and normalized to U6 RNA levels. (E) Luciferase reporter assays with wt or NRF1-mutant Plk4 promoter constructs. HCT116 cells were cotransfected with siRNA targeting NRF1 or with siControl. (F) Luciferase reporter assays from NIH3T3 cells transfected with wt or mutant Plk4 promoter constructs. NIH3T3 cells were arrested in G0 by serum starvation and stimulated to reenter the cell cycle by addition of FCS to the medium. After 24 h of serum addition, cells were enriched in G2/M phases. Twenty four hours after addition of serum, NIH3T3 cells were enriched in G2/M cell cycle phases. RLU, relative light units. (G) Bioinformatic identification of CCAAT, CRE and NRF1 elements in DREAM/CHR promoters.
The B-myb-containing MMB complex forms in S and G2 phases and is required for induction of gene expression in late S, G2 and M phases (12,15,16,19,20,49). However, mutation of the CHR did not lead to reduced activity of the Plk4 promoter in luciferase reporter assays during late cell cycle phases (Figure 1D, top panel). This contrasts prior findings for cyclin B2, but is in line with results obtained for the Ube2c promoter (12). Bmyb-knockdown by shRNA reduces Plk4 and Ccnb2 expression in nocodazole-blocked G2/M cells but not in double thymidine-arrested G1/S cells (Figure 3B, Supplementary Figure 1B). Since B-myb binds through the CHR (Figure 3A) (12), we compared wt and the CHR mutant promoter function in reporter assays (Figure 3C, Supplementary Figure S1C). Activity of the wt promoter was reduced while the CHR mutant yielded only small changes (Figure 3C). However, also in double thymidine-arrested cells the Plk4 promoter activity was reduced even though MMB was reported to be inactive during this phase of the cell cycle (49). There is no obvious explanation for the discrepancy between the two assays. Taken together, these results indicate that B-myb, as part of the MMB complex, not only binds to the CHR but is also necessary for full activity of the Plk4 wt promoter.
Furthermore, we addressed the role of Nrf-1 for activation of Plk4. siRNA-mediated downregulation of Nrf-1 mRNA resulted in a reduced Plk4 mRNA level (Figure 3D, Supplementary Figure S1D). The Plk4 wt promoter also displays a reduced activation after siNRF1 treatment. Conversely, the NFR1 mutant promoter displays not only lower activity than the wt promoter but also shows no further reduction in activity on siNRF1 treatment (Figure 3E). We tried to test the role of Creb-1 for activation. However, induction and inhibition of the cAMP-PKA pathway by forskolin and H89, respectively, resulted in such a significant shift of cell cycle distribution that the cell cycle-dependent change of promoter activity overshadowed a possible alteration by Creb-1 (data not shown). Similar shifts have been described for Creb-1-knockdown experiments (52,53). Thus, we were unable to discriminate whether Creb-1 instead of any other CRE-binding proteins activates the Plk4 promoter via the CRE site. Mutation of the CRE and NRF1 sites showed a reduction of promoter activity below vector control in promoter reporter assays and thus suggests that a substantial part of the Plk4 promoter activity is conferred through the two elements (Figure 3F).
Since activation of CDE/CHR-controlled promoters is usually achieved through NF-Y proteins binding to CCAAT-boxes (8,9), we investigated with bioinformatic tools how common CRE and NRF1 elements are in the group of CDE/CHR genes. Of 792 DREAM-binding genes (12,15), we found 140 promoter regions with conserved CHRs close to the TSS (±200 nt) (Figure 3G, Supplementary Table S1). In this group, 67% of the promoters harbor conserved CCAAT-boxes. Only 12% of this group of genes shows additional conserved CRE and/or NRF1 binding elements. However, 39% of the promoters not containing CCAAT-boxes have CRE and/or NRF1 binding sites in the DREAM/CHR fraction, which resembles an enrichment of 3.3-fold over the promoters with CCAAT-boxes (Figure 3G). In three genome-wide microarray data sets, we find all 18 genes of this fraction to be cell cycle-dependently regulated in at least one of the studies (39,49,54). Additionally, we find 12 (75%) of these genes to be p53-dependendly downregulated in at least one of three genome-wide microarray studies (55–57). However, a common promoter structure in which the CRE and NRF1 elements are placed relative to a CHR or the TSS could not be identified.
Taken together, these data show that most of the transcriptional activity of the Plk4 promoter is conferred through CHR, NRF1 and CRE sites and that CRE and NRF1 sites may serve as an alternative to transcriptional activation by NF-Y/CCAAT-boxes in DREAM/CHR-controlled genes.
p53-controlled downregulation of Plk4 expression depends on p21WAF1/CIP1 and binding of DREAM to the CDE and CHR sites
Several reports suggested that Plk4 expression can be downregulated by p53 (56,58,59). Experiments in NIH3T3 cells indeed showed that Plk4 expression is downregulated on doxorubicin-induced DNA damage, while CDKN1A (p21) expression increased (Figure 4A, Supplementary Figure S2A). To elucidate the mechanism of this regulation, we tested whether p21WAF1/CIP1 is required. HCT116 wt, HCT116 p53−/− and HCT116 p21−/− cells were treated with doxorubicin, and changes in Plk4 mRNA levels were assayed. In wt cells, Plk4 mRNA is downregulated about 4-fold after induction of DNA damage by doxorubicin. In cells that lack functional p53 or p21WAF1/CIP1, this effect is essentially lost, indicating that both tumor suppressors are required for p53-dependent downregulation of Plk4 on DNA damage (Figure 4B, Supplementary Figure S2A). Interestingly, the cyclin-dependent kinase (CDK) inhibitor roscovitine leads to reduced Plk4 expression not only in NIH3T3 and wt HCT116 cells but, in contrast to doxorubicin, also in HCT116 p53−/− or HCT116 p21−/− cells (Figure 4A and B). To further test for transcription factor binding sites potentially responsible for promoter repression, we performed reporter assays with wt and mutant Plk4 promoters after transfection of p53 and p21WAF1/CIP1 plasmids (Figure 4C, Supplementary Figure S2B and C). The cloned Plk4 promoter is similarly downregulated by p53 as the Plk4 mRNA. Importantly, p21WAF1/CIP1 appeared to be required since p53 was not able to substantially repress the Plk4 promoter in HCT116 p21−/− cells. In contrast, p21WAF1/CIP1 was sufficient for downregulation of Plk4 in the absence of p53, suggesting that p21WAF1/CIP1 is required downstream from p53 for repression. When looking at the transcription factor binding sites necessary for repression, we observed that both the CDE and the CHR elements are required for p53- and p21WAF1/CIP1-dependent repression. The NRF1 site did not seem to be involved in the transcriptional repression, whereas mutation of the CRE site resulted in a roughly 2-fold loss in transcriptional repression compared with the wt reporter when p53 or p21WAF1/CIP1 were overexpressed (Figure 4C, Supplementary Figure S2C). The p53 R175H mutant (p53mut) has been reported to be a structural and gain-of-function mutant (60,61). However, this mutant appears to have no gain-of-function effect on the transcriptional regulation of Plk4 when comparing the promoter reporter activities after coexpression with p53 and p21 constructs (Supplementary Figure S2C).
Figure 4.

p53-controlled downregulation of Plk4 expression depends on p21WAF1/CIP1 and binding of DREAM to the CDE and CHR sites. (A) Expression of Plk4 mRNA in NIH3T3 and in (B) HCT116 wt, p53−/− or p21−/− cells treated with doxorubicin and roscovitine for 24 h. Cells without treatment served as control. Relative expression of Plk4 mRNA was quantified by RT-PCR and normalized to U6 RNA levels. (C) Luciferase reporter assays with wt or mutant Plk4 promoter constructs were cotransfected in HCT116 p53−/− or HCT116 p21−/− cells with p53mut, p53wt, p21mut or p21wt expression vectors. Ratios of values from wt/mut transfections with p53 or p21 are shown as fold-repression relative to the vector control.
Since p53/p21-dependent repression was mediated through the CDE and CHR sites, we investigated DREAM and MMB binding to the Plk4 promoter in vivo. Chromatin from HCT116 cells before or after induction of DNA damage by doxorubicin was assayed by ChIP. Binding of p130 and E2F4, two representatives of the DREAM complex, to the Plk4 promoter increased after doxorubicin-induced DNA damage, although cell cycle distribution was shifted toward G2/M (Figure 5, top panel, Supplementary Figure S3B). Also LIN9 binding increased, whereas B-Myb binding did not change significantly. With p53 binding to the CDKN1A (p21WAF1/CIP1) promoter as a positive control, no direct binding of p53 to the Plk4 promoter was observed (Figure 5, top panel, Supplementary Figure S3). Conversely, in HCT116 p53−/− cells binding of the DREAM components p130 and E2F4 as well as LIN9 is reduced and binding of the MMB component B-Myb is increased after DNA damage-induced G2/M arrest (Figure 5, middle panel). This is consistent with the cell cycle-dependent shift from DREAM toward MMB binding (Figure 2B). Additionally, in HCT116 p21−/− cells we found low levels of DREAM binding to Plk4 and no induction after doxorubicin-induced DNA damage (Figure 5, bottom panel).
Figure 5.

In vivo protein binding to the Plk4 promoter in untreated or 48-h doxorubicin-treated HCT116 wt, p53−/− or p21−/− cells was tested by ChIP. Protein binding to the GAPDHS promoter served as a negative control.
These results suggest that p53-dependent downregulation of Plk4 on doxorubicin-induced DNA damage requires p21WAF1/CIP1 and the CDE/CHR tandem site. Although cell cycle distribution shifts toward G2/M after doxorubicin-induced DNA damage, binding of the DREAM complex increases in cells that have an intact p53-p21 pathway.
Deregulation of Plk4 expression after HPV-16 E7 induction depends on loss of DREAM binding to the CDE and CHR elements
An earlier study had described a deregulation of Plk4 expression on coexpression of HPV-16 E7 protein, however, without specifying the details of the mechanism involved (33). Disruption of the DREAM complex by HPV E7 has been reported previously (28). Thus, we addressed the question whether HPV E7 increases Plk4 expression by disrupting binding of the DREAM complex to the CDE/CHR site. First, we generated HCT116 cells stably expressing HPV-16 E7 wt and observed that Plk4 mRNA levels were increased compared with levels in HCT116 wt cells. Expression of the E7 ΔDLYC mutant, with a deletion in the pocket-protein binding-domain, served as a negative control (Figure 6A, Supplementary Figure S4A). To examine a possible influence of the CDE and CHR sites in the promoter, we cotransfected HCT116 wt, p53−/− and p21−/− cells with Plk4 promoter reporter and HPV-16 E7 constructs (Figure 6B, Supplementary Figure S4B). The Plk4 promoter is activated after expression of wt E7 compared with E7 ΔDLYC mutant. Activation of the Plk4 promoter by E7 is less prominent in cells lacking p53 and p21. Importantly, when the CDE and CHR sites in the promoter were mutated, deregulation after expression of HPV-16 E7 was lost (Figure 6B).
Figure 6.
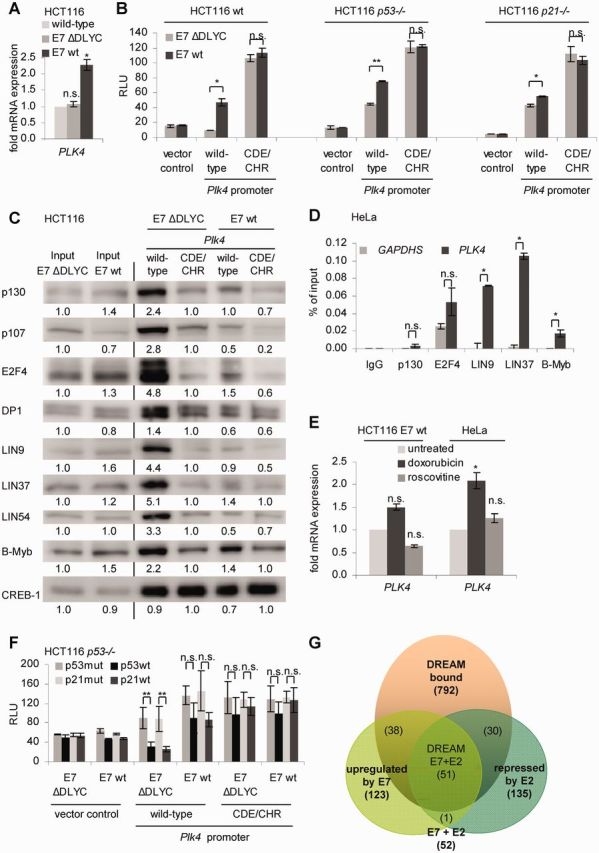
Deregulation of PLK4 expression after expression of HPV-16 E7 depends on loss of DREAM binding to the CDE and CHR elements. (A) HCT116 wt and HCT116 stably transfected with a plasmid encoding HPV-16 E7 wt or its ΔDLYC mutant. Relative expression of PLK4 mRNA was quantified by RT-PCR and normalized to U6 RNA levels. (B) Luciferase reporter assays from lysates of HCT116 wt; p53−/− or p21−/− cells transfected with plasmids expressing wt or CDE/CHR mutant Plk4 promoter constructs. Plasmids expressing HPV-16 E7 wt or its ΔDLYC mutant were cotransfected. Results are given as relative light units. (C) Nuclear extracts from HCT116 E7 wt and E7 ΔDLYC cells were analyzed by DNA affinity purification with wt and CDE/CHR mutant Plk4 promoter probes followed by western blot. Band intensities were quantified by densitometric analyses. Relative intensities are given below the bands. Intensities of input bands were normalized to E7 ΔDLYC. Binding intensities to Plk4 promoters were normalized to CDE/CHR mutant probes from E7 ΔDLYC extracts. (D) Protein binding to the PLK4 promoter in HeLa cells was assessed by ChIP. Protein binding to the GAPDHS promoter served as a negative control. (E) HCT116 E7 wt and HeLa cells were treated with doxorubicin and roscovitine for 24 h. Cells without treatment served as control. Relative expression of PLK4 mRNA was quantified by RT-PCR and normalized to U6 RNA levels. (F) Luciferase reporter assays with wt and CDE/CHR mutant Plk4 promoter reporter constructs in HCT116 p53−/− cells. Cells were cotransfected with p53mut, p53wt, p21mut or p21wt expression vectors and HPV-16 E7 wt or ΔDLYC mutant. Results are given as relative light units. (G) Venn diagram from DREAM-bound genes (12,15), genes upregulated by HPV E7 (64) and genes downregulated by HPV E2 proteins repressing E7 (65,66).
Following the experiments on transcriptional regulation, we tested differential protein binding to the Plk4 promoter using extracts from HCT116 cells stably transfected with E7 wt- or E7 mutant-expressing constructs. Binding of the DREAM-specific components p130, p107, E2F4 and DP1 as well as the MuvB core components LIN9, LIN37 and LIN54 to the Plk4 wt probe was reduced when comparing E7 wt to E7 mutant extracts, whereas the MMB-specific component B-Myb appeared hardly to be affected. CREB-1 binding to the Plk4 promoter probe was essentially not affected by E7 wt expression or mutation of the CDE/CHR site (Figure 6C).
Binding of DREAM and MMB proteins was also tested by ChIP in HPV-18 E7-expressing HeLa cells. Comparing binding with the GAPDHS promoter as a negative control, no significant enrichment of DREAM-specific components p130 and E2F4 at the Plk4 promoter was observed. Still, the MuvB core proteins, LIN9 and LIN37, as well as the MMB-specific component B-Myb are bound to the Plk4 promoter (Figure 6D).
Furthermore, we tested whether wt E7 also impaired the p53-p21-DREAM-CDE/CHR pathway. HCT116 E7 wt cells and HeLa cells were treated with doxorubicin and roscovitine yielding no significant repression of Plk4 mRNA (Figure 6E, Supplementary Figure S4C). In contrast, HCT116 wt cells, without E7 expression, showed strong repression of Plk4 (Figure 4B). As described earlier, DNA damage-induced G2/M arrest in E7-expressing HCT116 E7 wt and HeLa cells is disrupted (Supplementary Figure S4C) (62). We also observed that E7-mediated deregulation of the p53-p21 pathway is mimicked by mutation of CDE and CHR sites (Figure 6F, Supplementary Figure S4D). However, the repression effect caused by p53 and p21 is slightly reduced in E7 ΔDLYC-expressing cells (Figure 6F) compared with cells not expressing E7 ΔDLYC (Figure 4C). Consistently, it had been published that E7 can interfere with p21 also in the absence of its pocket protein binding domain (63).
Next, we addressed the question whether HPV E7 protein-dependent deregulation of the DREAM-CDE/CHR system is a general mechanism. Two previous studies have analyzed deregulation of cellular genes after expression of HPV proteins in genome-wide approaches. In one study, Rosty et al. used microarray hybridizations with HPV-infected cervical cancer samples and correlated E7 expression with upregulation of cellular genes. They identified a cervical cancer proliferation cluster of 123 genes whose expression correlates with E7 mRNA levels and viral DNA load (64). Interestingly, when comparing these results with the study by Litovchick et al. (15), we found that 89 (72.4%) of these genes are also described to be bound by DREAM (Supplementary Table S2, Figure 6G). In a second study, a different approach used the expression of HPV E2, a transcriptional repressor of E6 and E7, in HPV-18-associated HeLa cells (65). After expressing E2, Thierry and coworkers identified 63 downregulated genes in a microarray analysis. Later, the same group reported the identification of additional 72 genes downregulated by E2 in a microarray experiment with higher coverage (66). Importantly, we observed that of the 135 genes combined from the two studies, 81 (60.0%) are also found to bind DREAM proteins. The overlap of the E2 and E7 studies consists of 51 genes. Strikingly, nearly all of the overlapping genes (50 or 98%) are also reported to bind DREAM (Supplementary Table S2, Figure 6G).
Taken together, these observations show that HPV E7 deregulates Plk4 expression by interfering with DREAM binding to the CDE/CHR tandem site. More importantly, HPV E7 appears to upregulate many important cell cycle genes by disturbing the DREAM-CDE/CHR pathyway.
DISCUSSION
In the present study, we show that Plk4 expression is activated through NRF1 and CRE promoter elements instead of the established NF-Y/CCAAT system. The cell cycle- and p53-depedent regulation of Plk4 is controlled by DREAM and MMB complex binding to a CDE/CHR tandem element and deregulated by the viral oncoprotein HPV E7.
Nrf-1 and Creb-1 can substitute for NF-Y in activating CDE/CHR promoters and link metabolism and cell cycle control
Most CDE/CHR-controlled genes described to date are activated through NF-Y bound CCAAT-boxes, with cyclin B2 as the most prominent example (8,9) (Supplementary Figure S5). Only Bub1b was shown to be activated differently, with ZNF143 serving as basal activator (67). Here we show that the Plk4 promoter is mainly activated through binding sites for Nrf-1 and Creb-1 (Supplementary Figure S5). Nrf-1 was initially described as a transcription factor binding to and activating metabolic genes, e. g. the cytochrome C promoter. The palindrome GCGCATGCGC and related sequences differing by few nucleotide exchanges were identified as Nrf-1 binding sites (68,69). Indeed, we find that Nrf-1 and Creb-1 bind to the phylogenetically conserved NRF1 and CRE sites in the Plk4 promoter, respectively (Figure 3A). Consistently, Nrf-1 and Creb-1 were identified to bind the Plk4 promoter locus in ENCODE ChIP-Seq experiments (70). The finding that Nrf-1 binds many genes also bound by p130 and E2F4 led to the hypothesis that Nrf-1 may provide a link between metabolism and the cell cycle (71). In that study, also Plk4 appeared in a ChIP-on-chip screening as bound by both p130/E2F4 and Nrf-1.
Nrf-1 and Creb-1 transcription factors were both described to be activated on serum addition (72). We could not confirm this observation for the NRF1 site in the Plk4 promoter. Nevertheless, we found an ∼2-fold contribution of the CRE site to cell cycle- and p53-dependent transcriptional regulation of Plk4 (Figures 1D and 4C). Consistently, a CRE site was earlier identified to contribute to p53-dependent transcriptional repression of cyclin A2 (73). These data are in agreement with the model that, among other intensively studied signaling pathways (74,75), Creb-1 is phosphorylated by GSK3-β during G0 and G1, leading to low expression of its target genes (76–78).
Considering these observations and the notion that most CDE/CHR-regulated genes were described to be activated by NF-Y/CCAAT, we used a motif search for phylogenetically conserved CCAAT-boxes, NRF1 and CRE sites in promoters reported to bind DREAM (Supplementary Table S1). About one-third of the genes harboring a conserved CHR and binding DREAM do not contain a conserved CCAAT-box (Figure 3G and Supplementary Table S1). Out of this group of genes, 18 (39%) hold conserved CRE and NRF1 sites. Significantly, this resembles a 3.3-fold enrichment of CRE/NRF1-site abundance over the group of CCAAT-box-containing DREAM/CHR genes (Figure 3G). We found all 18 genes in at least one of three genome-wide microarray studies to be regulated dependent on the cell cycle (39,49,54) and 12 (75%) to be p53-dependently downregulated in at least one of three genome-wide microarray data sets (55–57). In addition to Plk4, other genes coding for proteins that carry out important functions in the cell cycle are found in this fraction, namely Anln, Cdc7, Kif2c, Mad2l1, Ncapg, Ncaph, Ndc80, Rtkn2, Ska1, Smc4 and Top1. These observations are in agreement with a previous study that found NF-Y, Creb-1 and Nrf-1 binding sites to be enriched in genes that bind E2F4. This study also observed the presence of Nrf-1 sites to be significantly enriched with Creb-1 and E2F but not with NF-Y binding sites (79). Thus, basal activation through NRF1 and CRE sites might act as an alternative mechanism to NF-Y/CCAAT-dependent activation of DREAM/CHR-controlled genes (Supplementary Figure S5). Consistently, a study by the FANTOM consortium found Creb-1, Nrf-1, NF-Y, E2F1-5 and Myb to be part of 30 core nodes controlling the transcriptional regulation of growth arrest (80). Furthermore, the large fraction of cell cycle genes possibly activated by the metabolic transcription factors Nrf-1 and Creb-1 support a model by which the regulation of these genes links metabolism to cell cycle control.
The CDE site in addition to the CHR element is required for Plk4 regulation
Recently, we have reported that the cell cycle-dependent regulation of cyclin B2 and Ube2c genes essentially only requires the CHR, with the CDE either not being essential or nonexistent, respectively (12). In contrast, in case of Plk4, we observe that the CDE is functionally at least as important as the CHR for repression (Figure 1). In this promoter both the CHR and the CDE are required for binding of DREAM components (Figure 2A). Assessing the list of known CDE/CHR-regulated genes and combining it with recent data, it appears that CDEs can have a varying impact on promoter control (8,12) (Figures 1D, 2A and 7).
Figure 7.
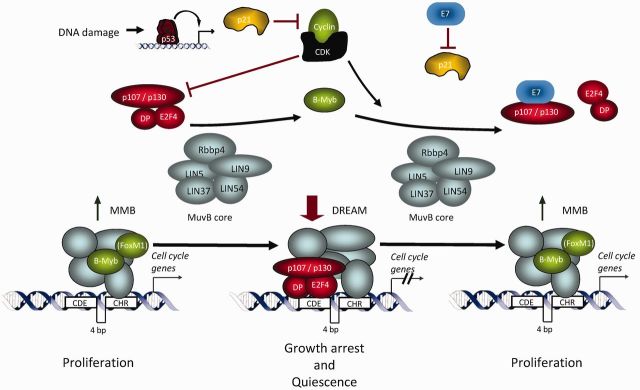
p53 controls transcriptional repression of target genes by an indirect mechanism through CDE and CHR elements. High levels of p53 lead to transcriptional activation of its target p21WAF1/CIP1. Inhibition of cyclin/CDK complex activity by p21WAF1/CIP1 leads to hypophosphorylation of p130. Hypophosphorylated p130, together with E2F4/DP1, can then form a repressive complex with the MuvB core replacing B-Myb in the MMB complex. MMB only requires the CHR element. The complex of p107/p130 and E2F4/DP1 with the MuvB core is named DREAM. DREAM requires both elements, the CDE and the CHR, for binding and repression. This resembles the protein complex binding to promoters during quiescence. Expression of the viral oncoprotein HPV E7 impairs p21WAF1/CIP1 function and blocks the pocket proteins p107/p130. Disruption of the DREAM complex then leads to derepression of cell cycle genes.
Interestingly, binding of MMB contrasts association of DREAM to the Plk4 promoter. MMB is only bound and contributes to transcriptional regulation through the CHR (Figures 3A-C and 7). These data are in line with earlier reports describing MMB binding and function through CHR sites (12,51). It had been suggested that the MMB complex activates gene expression only in late S, G2 and M phases (49). Consistent with this model, we find Plk4 mRNA levels only to be affected by Bmyb knockdown in G2/M but not in G1/S cells (Figure 3B). Luciferase reporter assays suggest that MMB-dependent regulation of Plk4 is conferred through the CHR (Figure 3C). However, mutation of the CHR does not lead to a reduced expression of Plk4 in late cell cycle phases as would be expected when MMB activates transcription through this site (Figure 1D, top panel). This contrasts the results obtained for cyclin B2, but is in line with results obtained for Ube2c (12). Thus, while MMB clearly contributes to Plk4 and Ube2c promoter regulation, it remains unclear why mutation of the MMB-binding CHR does not confer a loss of activity in luciferase reporter assays.
Additionally, binding of MMB to Plk4 appears to be weak relative to binding of DREAM. This becomes evident when comparing binding of LIN9 in G0 cells when DREAM is formed to binding in S/G2/M cells when MMB is formed (Figure 2B). This may explain why Plk4 was not found to bind B-Myb and LIN9 in a genome-wide screen (49). Despite its low affinity to Plk4, our results demonstrate that MMB is required for maximal Plk4 expression (Figure 3B and C).
The p53-p21WAF1/CIP1-DREAM pathway can depend on CDE sites
Recently, we reported that the p53-p21WAF1/CIP1 pathway controls transcriptional repression of cyclin B2 through DREAM binding solely to the CHR (23). In this report the CDE was shown not to be essential for DREAM binding or transcriptional repression since the human cyclin B2 promoter does not possess a functional CDE, and the CDE in the mouse cyclin B2 promoter only contributes slightly to regulation and binding (23). In contrast, in case of the Plk4 promoter, we observe the CDE to be required for regulation in addition to the CHR (Figures 4C and 7).
These observations correspond to the data for cell cycle-dependent regulation and binding of DREAM, where the CDE is required (Figure 2). From data on human cyclin B2, mouse Ube2C and human Kif23 promoter regulation and DREAM binding to the genes, an isolated CHR can be sufficient for DREAM function (12,23,81). In the mouse cyclin B2 and the mouse Plk4 promoters, a functional CDE 4-nt upstream of the CHR contributes to a small degree or is essential for regulation by DREAM, respectively (12,23) (Figures 1D, 2A and 4C). However, the criteria remain unclear why DREAM in some cases uses a CDE in addition to CHRs and in other examples solely relies on CHR sites (8).
We and others demonstrated a transcriptional downregulation of Plk4 by p53 (56,58,59). It was suggested that an Sp1/CRE-dependent (58) or a p130/p107-mediated pathway is responsible for p53-dependent regulation (59). Several reports suggested direct repression of G2/M genes by p53 binding to the target promoter (82,83). However, we do not observe p53 binding to the Plk4 promoter (Figure 5), which is consistent with genome-wide binding studies for p53 (84–86) and the mechanism identified for the regulation of cyclin B2 (23,87). Earlier, indirect mechanisms without p53 DNA binding to its downregulated target promoter were reported (22). The most detailed reports suggested p21WAF1/CIP1 as a link (23,24,81,88–92). Consistently, the CDK inhibitor p21WAF1/CIP1 operates upstream of the RB pocket protein-E2F pathway, and p21WAF1/CIP1/RB pocket protein-E2F signaling is important for chromosomal stability (93,94) (Figure 7). Here we show that Plk4 is downregulated p53- and p21WAF1/CIP1-dependently on doxorubicin-induced DNA damage (Figure 4A and B). Also, the CDK inhibitor roscovitine is able to mimic p21 function repressing Plk4 expression (Figure 4A and B). Following induction of p21WAF1/CIP1, DREAM forms and replaces MMB in binding to the Plk4 promoter (Figure 5, top panel). Conversely, doxorubicin-induced DNA damage does not induce DREAM binding to the Plk4 promoter in a p53−/− or p21−/− cells (Figure 5, middle and bottom panels). Consistent results were reported for cyclin B1, cyclin B2, Kif23, Mad2l1, PCNA and survivin promoters (23,24,81,92).
HPV E7 oncoprotein disrupts Plk4 transcriptional regulation through the CDE/CHR
Chromosomal instability induced by an impaired RB pocket protein-E2F pathway through HPV-16 E6 and E7 oncoproteins is a major cause for tumorigenic cell transformation (27,94). Tumor formation is often accompanied by aberrant centriole duplication and abnormal centrosome synthesis (95). Elevated Plk4 expression is required for aberrant centriole duplication in cancer cells (31,32). Consistently, we and others showed that HPV-16 E7 upregulates Plk4 expression (33) (Figure 6A and B). Our results demonstrate that this upregulation is caused by a loss of DREAM binding to the CDE/CHR tandem element (Figure 6C). This is consistent with the report that HPV-16 E7 causes disruption of the DREAM complex and upregulation of the cell cycle genes Bmyb and cyclin A (28). Additionally, expression of the E7 viral oncoprotein abrogates the p53-p21-DREAM-CDE/CHR pathway (Figure 6C–F). Other studies had shown a possible influence of HPV-16 E7 on the p53-p21 pathway (96–98). Roscovitine causes repression of Plk4 also in p53−/− and p21−/− cells but not in E7-expressing cells (Figures 4B and 6E). This indicates that E7 blocks the pathway downstream of CDK inhibition, namely on the level of DREAM.
Microarray studies that identified sets of genes related to cervical cancer reveal a strong overlap with genes also binding DREAM (12,15,64–66). These studies yielded an overlap of 52 genes, 51 of which are identified as DREAM targets (Figure 6G, Supplementary Table S2). Of these 51 genes, 32 (65.3%) do not only bind DREAM but also possess phylogenetically conserved CHR sites in their promoter regions close to the TSS (Supplementary Tables S1 and S2). Plk4 was identified only in one of the studies, indicating that selection criteria for the studies were stringent, excluding also positive hits. Thus, we looked at the data sets individually and found that 60 (42.9%) of 140 genes displaying DREAM binding and containing a conserved CHR close to their TSS were identified in at least one of the cervical cancer microarray studies (Supplementary Tables S1 and S2). We identified 18 putative DREAM/CHR-controlled genes to possess CRE/NRF1 sites as an alternative to CCAAT-boxes. Eight (44.4%) genes out of this group, namely Plk4, Anln, Cenpe, Kif2c, Mad2l1, Ncapg, Ndc80 and Smc4, were identified in at least one of the cervical cancer microarray studies (Figure 3G, Supplementary Table S1, Supplementary Table S2).
These observations lead to the conclusion that many genes relevant for cervical cancer progression are controlled by DREAM through CHR promoter elements (Figures 6G and 7, Supplementary Table S2).
Furthermore, this mechanism is also implicated in transformation by other tumor viruses. The RB pocket protein-E2F pathway is a target for several oncoproteins originating from DNA viruses with HPV E7, adenovirus early-region 1A (E1A) and polyomavirus large T-antigens as prominent examples (29,99). Consistent with this notion, also SV40 large T was recently reported to impair DREAM function (100,101).
We have described the regulation of the Plk4 promoter, which serves as an example for a number of cell cycle- and p53-regulated genes dependent on CDE and CHR sites. Our data suggest that NRF1 and CRE sites can function as an alternative to the NF-Y/CCAAT system in activating CDE/CHR-controlled cell cycle genes. Therefore, Nrf-1 and Creb-1 seem to be central components of the transcriptional network controlling cell cycle genes and in that role provide a potential link between metabolism and cell cycle control. The central mechanism in the regulation of Plk4 is a switch between DREAM and MMB complex binding to CDE and CHR sites. DREAM function and binding is disrupted by viral oncoproteins such as HPV E7. Together with genome-wide analyses, this implies that many cell cycle genes upregulated in HPV-induced tumors are bound by DREAM through CDE/CHR sites. These results imply that CDE/CHR-dependent regulation is central to viral oncogenesis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online, including [12,15,64–66].
FUNDING
Bundesministerium für Bildung und Forschung (BMBF) through grants by the Interdisciplinary Center for Clinical Research (IZKF) at the University of Leipzig (to K.E.); graduate fellowships provided by the Freistaat Sachsen and by the European Social Fund (to M.Q. and A.W., respectively). Funding for open access charge: Freistaat Sachsen.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Andrea Rothe and Carola Koschke for expert technical assistance, Kathrin Jäger and Andreas Lösche at the IZKF Leipzig core unit for performing FACS analyses, Karl Münger for HPV-16 E7 wt- and mutant-expressing plasmids, Bert Vogelstein for the kind gift of different HCT116 cell lines, p53- and p21WAF1/CIP1-expressing plasmids, Kenneth Boheler for Bmyb short hairpin construct, Roger Watson for B-myb monoclonal antibody, Larisa Litovchick and James DeCaprio for LIN9, LIN37 and LIN54 antibodies, Kimitoshi Kohno for Nrf-1 antibody and C. E. Engeland for critical reading of the manuscript.
REFERENCES
- 1.Fode C, Motro B, Yousefi S, Heffernan M, Dennis JW. Sak, a murine protein-serine/threonine kinase that is related to the Drosophila polo kinase and involved in cell proliferation. Proc. Natl Acad. Sci. USA. 1994;91:6388–6392. doi: 10.1073/pnas.91.14.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005;7:1140–1146. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- 3.Sillibourne JE, Bornens M. Polo–like kinase 4: the odd one out of the family. Cell Div. 2010;5:25. doi: 10.1186/1747-1028-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Carcer G, Manning G, Malumbres M. From Plk1 to Plk5: functional evolution of polo-like kinases. Cell Cycle. 2011;10:2255–2262. doi: 10.4161/cc.10.14.16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cunha-Ferreira I, Rodrigues-Martins A, Bento I, Riparbelli M, Zhang W, Laue E, Callaini G, Glover DM, Bettencourt-Dias M. The SCF/Slimb ubiquitin ligase limits centrosome amplification through degradation of SAK/PLK4. Curr. Biol. 2009;19:43–49. doi: 10.1016/j.cub.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 6.Rogers GC, Rusan NM, Roberts DM, Peifer M, Rogers SL. The SCF Slimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. J. Cell Biol. 2009;184:225–239. doi: 10.1083/jcb.200808049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fode C, Binkert C, Dennis JW. Constitutive expression of murine Sak-a suppresses cell growth and induces multinucleation. Mol. Cell Biol. 1996;16:4665–4672. doi: 10.1128/mcb.16.9.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Müller GA, Engeland K. The central role of CDE/CHR promoter elements in the regulation of cell cycle-dependent gene transcription. FEBS J. 2010;277:877–893. doi: 10.1111/j.1742-4658.2009.07508.x. [DOI] [PubMed] [Google Scholar]
- 9.Müller GA, Heissig F, Engeland K. Chimpanzee, orangutan, mouse, and human cell cycle promoters exempt CCAAT boxes and CHR elements from interspecies differences. Mol. Biol. Evol. 2007;24:814–826. doi: 10.1093/molbev/msl210. [DOI] [PubMed] [Google Scholar]
- 10.Dolfini D, Gatta R, Mantovani R. NF-Y and the transcriptional activation of CCAAT promoters. Crit Rev. Biochem. Mol. Biol. 2012;47:29–49. doi: 10.3109/10409238.2011.628970. [DOI] [PubMed] [Google Scholar]
- 11.Salsi V, Caretti G, Wasner M, Reinhard W, Haugwitz U, Engeland K, Mantovani R. Interactions between p300 and multiple NF-Y trimers govern Cyclin B2 promoter function. J. Biol. Chem. 2003;278:6642–6650. doi: 10.1074/jbc.M210065200. [DOI] [PubMed] [Google Scholar]
- 12.Müller GA, Quaas M, Schümann M, Krause E, Padi M, Fischer M, Litovchick L, DeCaprio JA, Engeland K. The CHR promoter element controls cell cycle-dependent gene transcription and binds the DREAM and MMB complexes. Nucleic Acids Res. 2012;40:1561–1578. doi: 10.1093/nar/gkr793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korenjak M, Taylor-Harding B, Binne UK, Satterlee JS, Stevaux O, Aasland R, White-Cooper H, Dyson N, Brehm A. Native E2F/RBF complexes contain Myb-interacting proteins and repress transcription of developmentally controlled E2F target genes. Cell. 2004;119:181–193. doi: 10.1016/j.cell.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 14.Harrison MM, Ceol CJ, Lu X, Horvitz HR. Some C. elegans class B synthetic multivulva proteins encode a conserved LIN-35 Rb-containing complex distinct from a NuRD-like complex. Proc. Natl Acad. Sci. USA. 2006;103:16782–16787. doi: 10.1073/pnas.0608461103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S, Chen R, Washburn MP, Liu XS, DeCaprio JA. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell. 2007;26:539–551. doi: 10.1016/j.molcel.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 16.Schmit F, Korenjak M, Mannefeld M, Schmitt K, Franke C, von Eyss B, Gagrica S, Hanel F, Brehm A, Gaubatz S. LINC, a human complex that is related to pRB-containing complexes in invertebrates regulates the expression of G2/M genes. Cell Cycle. 2007;6:1903–1913. doi: 10.4161/cc.6.15.4512. [DOI] [PubMed] [Google Scholar]
- 17.Sadasivam S, DeCaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer. 2013;13:585–595. doi: 10.1038/nrc3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pilkinton M, Sandoval R, Song J, Ness SA, Colamonici OR. Mip/LIN-9 regulates the expression of B-Myb and the induction of cyclin A, cyclin B, and CDK1. J. Biol. Chem. 2007;282:168–175. doi: 10.1074/jbc.M609924200. [DOI] [PubMed] [Google Scholar]
- 19.Knight AS, Notaridou M, Watson RJ. A Lin-9 complex is recruited by B-Myb to activate transcription of G2/M genes in undifferentiated embryonal carcinoma cells. Oncogene. 2009;28:1737–1747. doi: 10.1038/onc.2009.22. [DOI] [PubMed] [Google Scholar]
- 20.Osterloh L, von Eyss B, Schmit F, Rein L, Hubner D, Samans B, Hauser S, Gaubatz S. The human synMuv-like protein LIN-9 is required for transcription of G2/M genes and for entry into mitosis. EMBO J. 2007;26:144–157. doi: 10.1038/sj.emboj.7601478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011;25:801–813. doi: 10.1101/gad.2034211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Böhlig L, Rother K. One function—multiple mechanisms: the manifold activities of p53 as a transcriptional repressor. J. Biomed. Biotechnol. 2011;2011:464916. doi: 10.1155/2011/464916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quaas M, Müller GA, Engeland K. p53 can repress transcription of cell cycle genes through a p21(WAF1/CIP1)-dependent switch from MMB to DREAM protein complex binding at CHR promoter elements. Cell Cycle. 2012;11:4661–4672. doi: 10.4161/cc.22917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mannefeld M, Klassen E, Gaubatz S. B-MYB is required for recovery from the DNA damage-induced G2 checkpoint in p53 mutant cells. Cancer Res. 2009;69:4073–4080. doi: 10.1158/0008-5472.CAN-08-4156. [DOI] [PubMed] [Google Scholar]
- 25.Fukasawa K. P53, cyclin-dependent kinase and abnormal amplification of centrosomes. Biochim. Biophys. Acta. 2008;1786:15–23. doi: 10.1016/j.bbcan.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 27.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 28.Nor Rashid N, Yusof R, Watson RJ. Disruption of repressive p130-DREAM complexes by human papillomavirus 16 E6/E7 oncoproteins is required for cell-cycle progression in cervical cancer cells. J. Gen. Virol. 2011;92:2620–2627. doi: 10.1099/vir.0.035352-0. [DOI] [PubMed] [Google Scholar]
- 29.Rozenblatt-Rosen O, Deo RC, Padi M, Adelmant G, Calderwood MA, Rolland T, Grace M, Dricot A, Askenazi M, Tavares M, et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature. 2012;487:491–495. doi: 10.1038/nature11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duensing S, Duensing A, Crum CP, Munger K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001;61:2356–2360. [PubMed] [Google Scholar]
- 31.Korzeniewski N, Zheng L, Cuevas R, Parry J, Chatterjee P, Anderton B, Duensing A, Munger K, Duensing S. Cullin 1 functions as a centrosomal suppressor of centriole multiplication by regulating polo-like kinase 4 protein levels. Cancer Res. 2009;69:6668–6675. doi: 10.1158/0008-5472.CAN-09-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holland AJ, Fachinetti D, Zhu Q, Bauer M, Verma IM, Nigg EA, Cleveland DW. The autoregulated instability of Polo-like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev. 2012;26:2684–2689. doi: 10.1101/gad.207027.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korzeniewski N, Treat B, Duensing S. The HPV-16 E7 oncoprotein induces centriole multiplication through deregulation of Polo-like kinase 4 expression. Mol. Cancer. 2011;10:61. doi: 10.1186/1476-4598-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tarasov KV, Tarasova YS, Tam WL, Riordon DR, Elliott ST, Kania G, Li J, Yamanaka S, Crider DG, Testa G, et al. B-MYB is essential for normal cell cycle progression and chromosomal stability of embryonic stem cells. PLoS One. 2008;3:e2478. doi: 10.1371/journal.pone.0002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 36.Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J. Virol. 2001;75:7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyamoto N, Izumi H, Miyamoto R, Kondo H, Tawara A, Sasaguri Y, Kohno K. Quercetin induces the expression of peroxiredoxins 3 and 5 via the Nrf2/NRF1 transcription pathway. Invest Ophthalmol. Vis. Sci. 2011;52:1055–1063. doi: 10.1167/iovs.10-5777. [DOI] [PubMed] [Google Scholar]
- 38.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 39.Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirschner RD, Sänger K, Müller GA, Engeland K. Transcriptional activation of the tumor suppressor and differentiation gene S100A2 by a novel p63-binding site. Nucleic Acids Res. 2008;36:2969–2980. doi: 10.1093/nar/gkn132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tavner F, Frampton J, Watson RJ. Targeting an E2F site in the mouse genome prevents promoter silencing in quiescent and post-mitotic cells. Oncogene. 2007;26:2727–2735. doi: 10.1038/sj.onc.1210087. [DOI] [PubMed] [Google Scholar]
- 42.Izumi H, Ohta R, Nagatani G, Ise T, Nakayama Y, Nomoto M, Kohno K. p300/CBP-associated factor (P/CAF) interacts with nuclear respiratory factor-1 to regulate the UDP-N-acetyl-alpha-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase-3 gene. Biochem. J. 2003;373:713–722. doi: 10.1042/BJ20021902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. The UCSC Known Genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 44.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 2006;34:D590–D598. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. Aligning multiple genomic sequences with the threaded blockset al.gner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012;26:474–489. doi: 10.1101/gad.181933.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Down CF, Millour J, Lam EW, Watson RJ. Binding of FoxM1 to G2/M gene promoters is dependent upon B-Myb. Biochim. Biophys. Acta. 2012;1819:855–862. doi: 10.1016/j.bbagrm.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 51.Chen X, Müller GA, Quaas M, Fischer M, Han N, Stutchbury B, Sharrocks AD, Engeland K. The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol. Cell Biol. 2013;33:227–236. doi: 10.1128/MCB.00881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aggarwal S, Kim SW, Ryu SH, Chung WC, Koo JS. Growth suppression of lung cancer cells by targeting cyclic AMP response element-binding protein. Cancer Res. 2008;68:981–988. doi: 10.1158/0008-5472.CAN-06-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan X, Wang S, Zhu L, Wu C, Yin B, Zhao J, Yuan J, Qiang B, Peng X. cAMP response element-binding protein promotes gliomagenesis by modulating the expression of oncogenic microRNA-23a. Proc. Natl Acad. Sci. USA. 2012;109:15805–15810. doi: 10.1073/pnas.1207787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bar-Joseph Z, Siegfried Z, Brandeis M, Brors B, Lu Y, Eils R, Dynlacht BD, Simon I. Genome-wide transcriptional analysis of the human cell cycle identifies genes differentially regulated in normal and cancer cells. Proc. Natl Acad. Sci. USA. 2008;105:955–960. doi: 10.1073/pnas.0704723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spurgers KB, Gold DL, Coombes KR, Bohnenstiehl NL, Mullins B, Meyn RE, Logothetis CJ, McDonnell TJ. Identification of cell cycle regulatory genes as principal targets of p53-mediated transcriptional repression. J. Biol. Chem. 2006;281:25134–25142. doi: 10.1074/jbc.M513901200. [DOI] [PubMed] [Google Scholar]
- 56.Böhlig L, Friedrich M, Engeland K. p53 activates the PANK1/miRNA-107 gene leading to downregulation of CDK6 and p130 cell cycle proteins. Nucleic Acids Res. 2011;39:440–453. doi: 10.1093/nar/gkq796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nikulenkov F, Spinnler C, Li H, Tonelli C, Shi Y, Turunen M, Kivioja T, Ignatiev I, Kel A, Taipale J, et al. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ. 2012;19:1992–2002. doi: 10.1038/cdd.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Tan M, Li L, Pamarthy D, Lawrence TS, Sun Y. SAK, a new polo-like kinase, is transcriptionally repressed by p53 and induces apoptosis upon RNAi silencing. Neoplasia. 2005;7:312–323. doi: 10.1593/neo.04325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jackson MW, Agarwal MK, Yang J, Bruss P, Uchiumi T, Agarwal ML, Stark GR, Taylor WR. p130/p107/p105Rb-dependent transcriptional repression during DNA-damage-induced cell-cycle exit at G2. J. Cell Sci. 2005;118:1821–1832. doi: 10.1242/jcs.02307. [DOI] [PubMed] [Google Scholar]
- 60.O'Farrell TJ, Ghosh P, Dobashi N, Sasaki CY, Longo DL. Comparison of the effect of mutant and wild-type p53 on global gene expression. Cancer Res. 2004;64:8199–8207. doi: 10.1158/0008-5472.CAN-03-3639. [DOI] [PubMed] [Google Scholar]
- 61.Di AS, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, Blandino G, Piaggio G. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10:191–202. doi: 10.1016/j.ccr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 62.Spardy N, Covella K, Cha E, Hoskins EE, Wells SI, Duensing A, Duensing S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009;69:7022–7029. doi: 10.1158/0008-5472.CAN-09-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shin MK, Balsitis S, Brake T, Lambert PF. Human papillomavirus E7 oncoprotein overrides the tumor suppressor activity of p21Cip1 in cervical carcinogenesis. Cancer Res. 2009;69:5656–5663. doi: 10.1158/0008-5472.CAN-08-3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosty C, Sheffer M, Tsafrir D, Stransky N, Tsafrir I, Peter M, de Crémoux P, de La RA, Salmon R, Dorval T, et al. Identification of a proliferation gene cluster associated with HPV E6/E7 expression level and viral DNA load in invasive cervical carcinoma. Oncogene. 2005;24:7094–7104. doi: 10.1038/sj.onc.1208854. [DOI] [PubMed] [Google Scholar]
- 65.Thierry F, Benotmane MA, Demeret C, Mori M, Teissier S, Desaintes C. A genomic approach reveals a novel mitotic pathway in papillomavirus carcinogenesis. Cancer Res. 2004;64:895–903. doi: 10.1158/0008-5472.can-03-2349. [DOI] [PubMed] [Google Scholar]
- 66.Teissier S, Ben KY, Mori M, Pautier P, Desaintes C, Thierry F. A new E6/P63 pathway, together with a strong E7/E2F mitotic pathway, modulates the transcriptome in cervical cancer cells. J. Virol. 2007;81:9368–9376. doi: 10.1128/JVI.00427-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Myslinski E, Gerard MA, Krol A, Carbon P. Transcription of the human cell cycle regulated BUB1B gene requires hStaf/ZNF143. Nucleic Acids Res. 2007;35:3453–3464. doi: 10.1093/nar/gkm239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Evans MJ, Scarpulla RC. NRF-1: a trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 1990;4:1023–1034. doi: 10.1101/gad.4.6.1023. [DOI] [PubMed] [Google Scholar]
- 69.Evans MJ, Scarpulla RC. Interaction of nuclear factors with multiple sites in the somatic cytochrome c promoter. Characterization of upstream NRF-1, ATF, and intron Sp1 recognition sequences. J. Biol. Chem. 1989;264:14361–14368. [PubMed] [Google Scholar]
- 70.ENCODE Project Consortium. A user's guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cam H, Balciunaite E, Blais A, Spektor A, Scarpulla RC, Young R, Kluger Y, Dynlacht BD. A common set of gene regulatory networks links metabolism and growth inhibition. Mol. Cell. 2004;16:399–411. doi: 10.1016/j.molcel.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 72.Herzig RP, Scacco S, Scarpulla RC. Sequential serum-dependent activation of CREB and NRF-1 leads to enhanced mitochondrial respiration through the induction of cytochrome c. J. Biol. Chem. 2000;275:13134–13141. doi: 10.1074/jbc.275.17.13134. [DOI] [PubMed] [Google Scholar]
- 73.Desdouets C, Ory C, Matesic G, Soussi T, Brechot C, Sobczak-Thepot J. ATF/CREB site mediated transcriptional activation and p53 dependent repression of the cyclin A promoter. FEBS Lett. 1996;385:34–38. doi: 10.1016/0014-5793(96)00330-4. [DOI] [PubMed] [Google Scholar]
- 74.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 75.Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal. 2004;16:1211–1227. doi: 10.1016/j.cellsig.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 76.Liang MH, Chuang DM. Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. J. Biol. Chem. 2006;281:30479–30484. doi: 10.1074/jbc.M607468200. [DOI] [PubMed] [Google Scholar]
- 77.Tullai JW, Chen J, Schaffer ME, Kamenetsky E, Kasif S, Cooper GM. Glycogen synthase kinase-3 represses cyclic AMP response element-binding protein (CREB)-targeted immediate early genes in quiescent cells. J. Biol. Chem. 2007;282:9482–9491. doi: 10.1074/jbc.M700067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tullai JW, Graham JR, Cooper GM. A GSK-3-mediated transcriptional network maintains repression of immediate early genes in quiescent cells. Cell Cycle. 2011;10:3072–3077. doi: 10.4161/cc.10.18.17321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elkon R, Linhart C, Sharan R, Shamir R, Shiloh Y. Genome-wide in silico identification of transcriptional regulators controlling the cell cycle in human cells. Genome Res. 2003;13:773–780. doi: 10.1101/gr.947203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Suzuki H, Forrest AR, van Nimwegen E, Daub CO, Balwierz PJ, Irvine KM, Lassmann T, Ravasi T, Hasegawa Y, de Hoon MJ, et al. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat. Genet. 2009;41:553–562. doi: 10.1038/ng.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fischer M, Grundke I, Sohr S, Quaas M, Hoffmann S, Knörck A, Gumhold C, Rother K. p53 and cell cycle dependent transcription of kinesin family member 23 (KIF23) is controlled via a CHR promoter element bound by DREAM and MMB complexes. PLoS One. 2013;8:e63187. doi: 10.1371/journal.pone.0063187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 83.Imbriano C, Gnesutta N, Mantovani R. The NF-Y/p53 liaison: well beyond repression. Biochim. Biophys. Acta. 2012;1825:131–139. doi: 10.1016/j.bbcan.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 84.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 85.Botcheva K, McCorkle SR, McCombie WR, Dunn JJ, Anderson CW. Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle. 2011;10:4237–4249. doi: 10.4161/cc.10.24.18383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, Denissov S, Stunnenberg HG, Lohrum M. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 2008;36:3639–3654. doi: 10.1093/nar/gkn232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dobbelstein M. Interchanging heads: p53 re-composes the DREAM/MMB complex to repress transcription. Cell Cycle. 2013;12:11. doi: 10.4161/cc.23169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Toledo SM, Azzam EI, Keng P, Laffrenier S, Little JB. Regulation by ionizing radiation of CDC2, cyclin A, cyclin B, thymidine kinase, topoisomerase IIalpha, and RAD51 expression in normal human diploid fibroblasts is dependent on p53/p21Waf1. Cell Growth Differ. 1998;9:887–896. [PubMed] [Google Scholar]
- 89.Gottifredi V, Karni-Schmidt O, Shieh SS, Prives C. p53 down-regulates CHK1 through p21 and the retinoblastoma protein. Mol. Cell. Biol. 2001;21:1066–1076. doi: 10.1128/MCB.21.4.1066-1076.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Löhr K, Möritz C, Contente A, Dobbelstein M. p21/CDKN1A mediates negative regulation of transcription by p53. J. Biol. Chem. 2003;278:32507–32516. doi: 10.1074/jbc.M212517200. [DOI] [PubMed] [Google Scholar]
- 91.Shats I, Milyavsky M, Tang X, Stambolsky P, Erez N, Brosh R, Kogan I, Braunstein I, Tzukerman M, Ginsberg D, et al. p53-dependent down-regulation of telomerase is mediated by p21waf1. J. Biol. Chem. 2004;279:50976–50985. doi: 10.1074/jbc.M402502200. [DOI] [PubMed] [Google Scholar]
- 92.Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell. 2011;19:701–714. doi: 10.1016/j.ccr.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barboza JA, Liu G, Ju Z, El-Naggar AK, Lozano G. p21 delays tumor onset by preservation of chromosomal stability. Proc. Natl Acad. Sci. USA. 2006;103:19842–19847. doi: 10.1073/pnas.0606343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Duensing A, Chin A, Wang L, Kuan SF, Duensing S. Analysis of centrosome overduplication in correlation to cell division errors in high-risk human papillomavirus (HPV)-associated anal neoplasms. Virology. 2008;372:157–164. doi: 10.1016/j.virol.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hickman ES, Bates S, Vousden KH. Perturbation of the p53 response by human papillomavirus type 16 E7. J. Virol. 1997;71:3710–3718. doi: 10.1128/jvi.71.5.3710-3718.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morozov A, Shiyanov P, Barr E, Leiden JM, Raychaudhuri P. Accumulation of human papillomavirus type 16 E7 protein bypasses G1 arrest induced by serum deprivation and by the cell cycle inhibitor p21. J. Virol. 1997;71:3451–3457. doi: 10.1128/jvi.71.5.3451-3457.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wells SI, Francis DA, Karpova AY, Dowhanick JJ, Benson JD, Howley PM. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21(CIP)-dependent pathways. EMBO J. 2000;19:5762–5771. doi: 10.1093/emboj/19.21.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Felsani A, Mileo AM, Paggi MG. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene. 2006;25:5277–5285. doi: 10.1038/sj.onc.1209621. [DOI] [PubMed] [Google Scholar]
- 100.Hauser S, Ulrich T, Wurster S, Schmitt K, Reichert N, Gaubatz S. Loss of LIN9, a member of the DREAM complex, cooperates with SV40 large T antigen to induce genomic instability and anchorage-independent growth. Oncogene. 2012;31:1859–1868. doi: 10.1038/onc.2011.364. [DOI] [PubMed] [Google Scholar]
- 101.Fine DA, Rozenblatt-Rosen O, Padi M, Korkhin A, James RL, Adelmant G, Yoon R, Guo L, Berrios C, Zhang Y, et al. Identification of FAM111A as an SV40 host range restriction and adenovirus helper factor. PLoS Pathog. 2012;8:e1002949. doi: 10.1371/journal.ppat.1002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.