


Abstract
We have used model substrates carrying modified nucleotides at the site immediately 5′ of the canonical RNase P cleavage site, the −1 position, to study Escherichia coli RNase P RNA-mediated cleavage. We show that the nucleobase at −1 is not essential but its presence and identity contribute to efficiency, fidelity of cleavage and stabilization of the transition state. When U or C is present at −1, the carbonyl oxygen at C2 on the nucleobase contributes to transition-state stabilization, and thus acts as a positive determinant. For substrates with purines at −1, an exocyclic amine at C2 on the nucleobase promotes cleavage at an alternative site and it has a negative impact on cleavage at the canonical site. We also provide new insights into the interaction between E. coli RNase P RNA and the −1 residue in the substrate. Our findings will be discussed using a model where bacterial RNase P cleavage proceeds through a conformational-assisted mechanism that positions the metal(II)-activated H2O for an in-line attack on the phosphorous atom that leads to breakage of the phosphodiester bond.
INTRODUCTION
Transfer RNA (tRNA) genes are transcribed as precursors with extra residues at their 5′- and 3′-ends that have to be removed to generate functional tRNAs. The endoribonuclease—RNase P—is responsible for removing the extra 5′ residues, i.e. the 5′ leader. In Bacteria, RNase P consists of one protein and one RNA subunit, referred to as the C5 protein and RNase P RNA (RPR), respectively; in Archaea and Eukarya, the number of proteins is expanded. Irrespective of origin, the catalytic activity resides in the RNA moiety and the RNA alone can mediate cleavage in the absence of the protein (1–3). However, recent data suggest the existence of RNase P-like activities based solely on protein (4,5). Several determinants in the tRNA precursor substrate influence binding and cleavage efficiency [for recent reviews see (6–8)]. For one of these, the residue at −1, genetic and biochemical data suggest that it interacts with a conserved A at position 248 in Escherichia coli (Eco) RPRwt. This interaction is referred to as the N−1/A248 interaction [(6,9); see Figure 1A]. The recently published structure of RNase P in complex with tRNA positions A248 close to the 5′ termini of the tRNA (11); however, it is still unclear if and how N−1 in the substrate interacts with A248.
Figure 1.
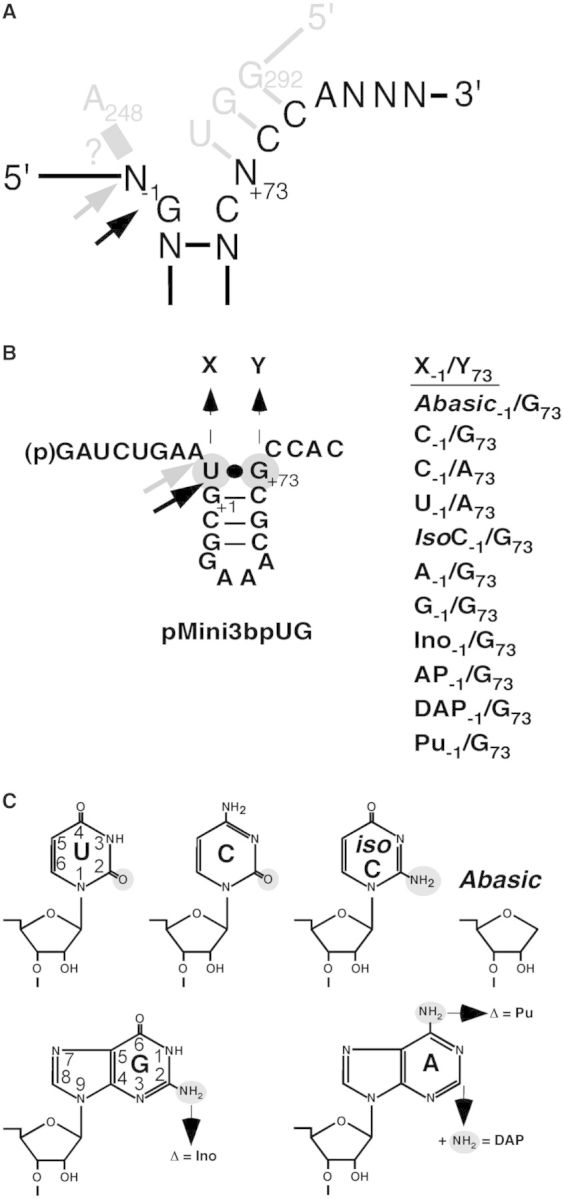
Structures of the model hairpin loop substrate pMini3bp and structure of uridine, cytidine, iso-cytidine, guanosine and adenosine. (A) Illustration of the N−1/248 - and the RCCA-RPR interactions [interacting residues underlined (6,9,10)]. Substrate residues are marked in black and residues marked in grey represent the RPR. Black and grey arrows mark the canonical and the alternative cleavage sites, respectively. ? indicate if and how residues −1 and 248 interact (see the main text). (B) Structure of pMini3bpUG and its derivatives. The black arrow marks the canonical cleavage site at +1 and the grey arrow marks the alternative cleavage site −1. X and Y indicate where base changes were introduced as indicated on the right side. Abasic, deletion of the base; IsoC, iso-cytidine; Ino, inosine; 2AP, 2-amino purine; DAP, 2;6-diamino purine; Pu, purine. (C) Structure of the bases uridine (U), cytidine (C), iso-cytidine (IsoC), Abasic, guanosine (G) and adenosine (A). Chemical groups marked in small grey circles refer to the groups that were substituted or deleted as indicated. The ring numbering for U and G is as indicated. For details see text.
The identity of the −1 residue varies in tRNA precursors. In E. coli, the majority (≈57%) carry a uridine at −1, whereas in others, e.g. Mycobacterium tuberculosis, a cytidine is more frequent (12,13). In a tRNA precursor context, in vitro data suggest that substrates with U at −1 bind to RPR with higher affinity compared with substrates with other −1 residues (9,14). Moreover, for substrates with C at −1, which can pair with a guanosine at position +73 [the discriminator base position; (15)], in vitro and in vivo studies show that cleavage occurs at the correct site between positions −1 and +1 (the canonical site or the +1 site), as well as at an alternative site between residues at −1 and −2, i.e. at −1 (6–8,16). Cleavage of substrates with C−1/G+73 and U−1/G+73 pairs has furthermore demonstrated that optimal cleavage of the former substrate requires higher Mg2+-concentrations (14). The rate of cleavage for Eco RPRwt as a function of the identity of the residue at −1, however, does not change to any significant extent in a tRNA precursor context (9,14). In contrast, for model hairpin loop substrates (see Figure 1B), the −1 identity influences the rate of cleavage (6,7,13,14,17). Hence, the effect of the −1 residue in a pre-tRNA context is likely to be obscured by other elements affecting RPR-mediated cleavage (14). Moreover, there is no information about the contribution of the chemical groups on the −1 base to catalysis. Therefore, we decided to reinvestigate the impact of the identity of the −1 residue on cleavage using a model substrate. Our specific objectives were to identify chemical groups of the −1 nucleobase that contribute to catalysis and to determine if the nucleobase at −1 is absolutely necessary for cleavage. As available data suggest that the protein subunit of RNase P does not interact with the residue at −1 (6–8), we studied cleavage mostly in the absence of the protein. Our results show that the nucleobase at the −1 position is not necessary for cleavage in the context of a model hairpin loop substrate. However, the presence, identity and specific chemical groups at −1 influence cleavage site selection and the kinetics of cleavage. The −1 position contributes to the stabilization of the transition state for cleavage at the canonical cleavage site. We will discuss our data in view of the recently solved structure of RNase P in complex with tRNA (11) and a model that emphasizes the importance of the positioning of Mg(II) at and near the cleavage site for ensuring cleavage at the correct site.
MATERIALS AND METHODS
Preparation of substrates, RPRs and C5 protein
The different pMini3bp derivatives except pMini3bpAbasicG and pMini3bpIsoCG were purchased from Dharmacon (Lafayette, CO, USA). The pMini3bpAbasicG and pMini3bpIsoCG were synthesized in-house essentially according to Wincott et al. (18). The rSpacer and isoC phosphoramidites were purchased from MedProbe AS, Norway (Glen Research, USA) and ChemGenes, USA, respectively. All the RNA substrates were purified on 15% denaturing polyacrylamide gels and extracted overnight using a Biotrap device following the manufacturer’s procedures (Schleicher and Schuell, GmbH, Germany; Elutrap in USA and Canada). This was followed by phenol-chloroform extraction according to standard procedures. The different substrates were labeled with 32P at the 5′-end with [γ-32P]ATP as described elsewhere (14).
The construction of the Eco RPRG248 encoding gene has been reported elsewhere (19). The Eco RPRwt and Eco RPRG248 were generated as T7 RNA polymerase run-off transcripts (20), whereas the C5 protein was purified from an E. coli BL21(DE3) strain harbouring the plasmid pET33b carrying the His6-C5 gene (N-terminal fusion). Briefly, the pET33b plasmid with the His6-C5 gene behind an isopropyl β-D-1-thiogalactopyranoside-inducible promoter was transformed into E. coli BL21(DE3). The cells were grown at 37°C in LB liquid medium supplemented with 50 µg/ml kanamycin. At OD600 = 0.6, isopropyl β-D-1-thiogalactopyranoside was added to a final concentration of 1.5 mM and the culture was incubated for another 4 h. The His6-C5 protein was purified essentially as described by Feltens et al. (21), and the protein concentration was determined using the standard Bradford protein assay.
Assay conditions—cleavage by RPR alone and in the presence of the C5 protein
The RPR alone reactions were conducted in buffer C [50 mM Mes (pH 6.1 at 37°C), 0.8 M NH4Cl] and indicated Mg(OAc)2 concentrations. Before adding the preheated (37°C) substrate, the RPRs were pre-incubated at 37°C in buffer C and Mg(OAc)2 for at least 10 min to allow proper folding. In the Mg2+ titration experiments, the concentrations of substrates were 0.02 µM while the concentration of RPRs varied between 0.8 and 5.2 µM [the concentration varied depending on substrates and RPR combination, see Figure legend 3; (22)].
All the reactions with the His6-C5 protein were carried out in buffer A [50 mM Tris–HCl (final pH 7.2), 5% (w/v) PEG 6000, 100 mM NH4Cl] supplemented with 10 mM MgCl2. The RPR was pre-incubated in buffer A at 37°C for 10 min. The His6-C5 was added, and incubation was continued for an additional 10 min followed by addition of preheated (37°C) substrate. The concentrations of RPR and His6-C5 were 0.004 µM and 0.21 µM [empirically determined; see also e.g. (23)], respectively, and the concentration of substrate was 0.02 µM.
To terminate the reactions, double volumes of stop solution were added (10 M urea, 100 mM EDTA).
Determination of the kinetic constants under single-turnover conditions
The kinetic constants kobs and kobs/Ksto (=kcat/Km) were determined under saturating single-turnover conditions in buffer C at pH 6.1 and 800 mM Mg(OAc)2. At this pH, the chemistry of cleavage is rate-limiting (24). The final concentration of the substrate was 0.02 µM, and the concentration of RPRs varied between 0.8 and 29 µM, depending on the substrate–RPR combination. Given that the substrates were labeled at the 5′-end, we used the 5′ cleavage fragments in our activity measurements. While calculating the rate, the incubation times for each substrate and RPR combination were adjusted to ensure that the velocity measurements were in the linear range, i.e. ≤40% of the substrate had been consumed.
The kobs and kobs/Ksto values were obtained by linear regression from Eadie–Hofstee plots as described elsewhere [(25); see also (26,27)].
RESULTS
To test the contribution of residue −1 to cleavage, we used the short model substrate, pMini3bp [Figure 1; (12)], in which cleavage relies on residues near the cleavage site, mainly the N−1/A248 and the RCCA-RPR interactions (interacting residues underlined, Figure 1A; 6,9,10). For this purpose, we generated pMini3bp variants carrying changes at −1 (Figure 1B). These substrates were studied with respect to (i) cleavage site recognition, where cleavage between −1 and +1 is referred to the canonical or correct site (Figure 1) and (ii) Mg2+ dependence. We also determined the kinetic constants kobs and kobs/Ksto (kobs/Ksto = kcat/Km; see later in the text) for cleavage under saturating-single turnover conditions at pH 6.1. At this pH, previous data have suggested that the cleavage of other model hairpin loop substrates is rate-limiting [see e.g. Ref. (24)]. In the simplified scheme the kinetic constant kobs reflects the rate of cleavage as indicated while the rate constant kobs/Ksto = k+1 (Scheme 1). Under these conditions, we argue that Ksto ≈ Kd because k−1 >> kobs in the cleavage of pMini3bpUG by Eco RPRwt, where k−1 corresponds to the rate of dissociation (Supplementary Figure S1; for experimental details see Supplementary data and Refs (25,28,29); we assume that this is the case for all the pMini3bp variants). Going from ES1 to ES2 involves breaking the −1/+73 base pair in the substrate (e.g. as when C is at −1 and G at +73; see Figure 1B) and positioning of the Mg2+ responsible for generating the nucleophile. Furthermore, the ES1 to ES2 route results in cleavage at the canonical cleavage site +1, whereas ES1 to ES1* describes the pathway (and e.g. does not involve breaking of the −1/+73 pair if present) that gives cleavage at the alternative site −1 [see Figure 1; see also (19)].
Scheme 1.

The nucleobase at −1 is not essential for cleavage
To understand if the nucleobase at −1 is necessary for cleavage, we prepared a substrate in which the base had been deleted, pMini3bpAbasicG (Figure 1B). Eco RPRwt cleaved this substrate at two positions, at the canonical site +1 and at −1, with low efficiency compared with cleavage of pMini3bpUG where the base is present (Figure 2A). Cleavage of pMini3bpAbasicG at +1 and at −1 occurred throughout the entire concentration range of Mg2+ tested (Figure 3A); however, we noted that there was less cleavage at −1 at lower concentrations of Mg2+ (Supplementary Figure S2). The Mg2+ requirement for optimal cleavage was found to be comparable with that for cleavage of pMini3bpUG (Figure 3B). However, compared with cleavage of precursor tRNAs and other longer model substrates [cf. Figures 5 and 6 in Ref. (14)], cleavage of the substrate lacking the nucleobase at −1 and the other pMini3bp variants (see later in the text) required a higher concentration of Mg2+ to reach optimal cleavage.
Figure 2.
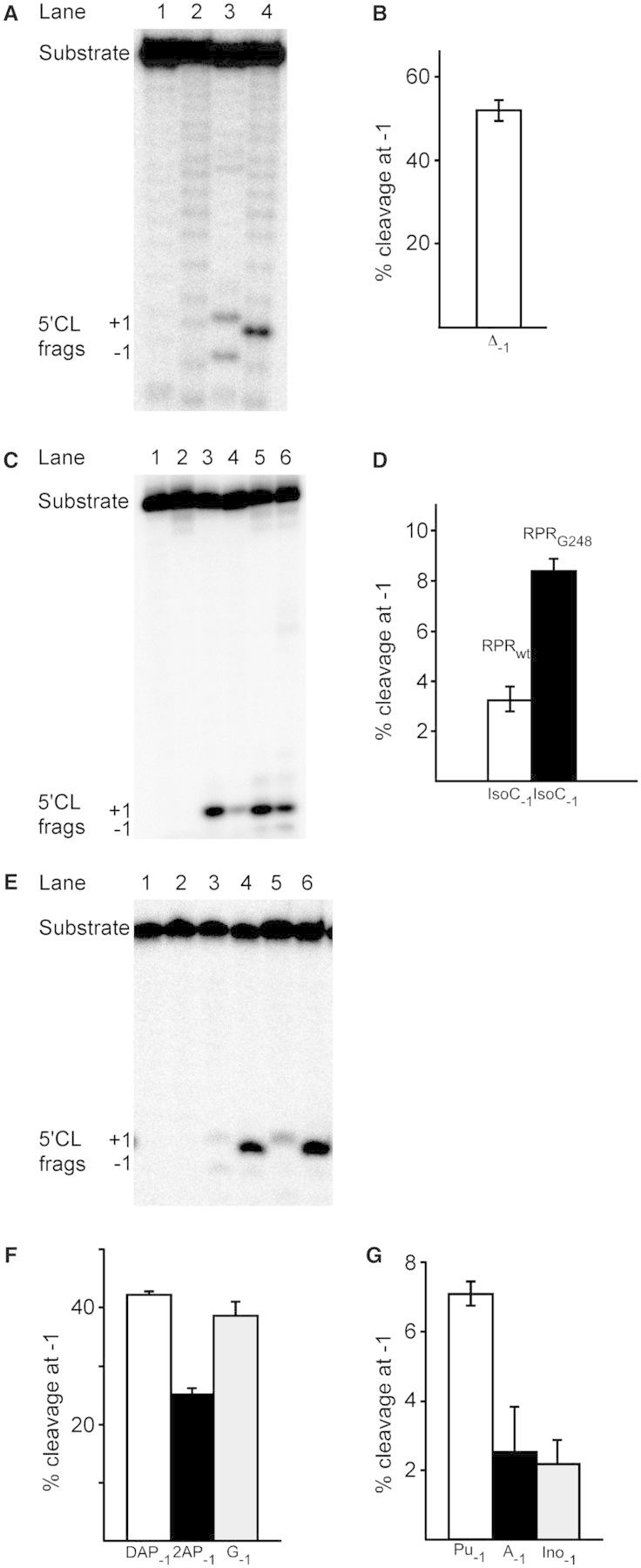
Cleavage of pMini3bp variants with Eco RPR. (A) pMini3bpUG and pMini3bpAbasicG; the reaction was performed at 37°C in buffer C containing 800 mM Mg2+ (see ‘Materials and Methods’). Lanes 1 (pMini3bpAbasicG) and 2 (pMini3bpUG), controls incubation without Eco RPRwt; lanes 3 (pMini3bpAbasicG) and 4 (pMini3bpUG) incubation with Eco RPRwt for 1179 min and 20 s, respectively. The concentrations of Eco RPRwt and substrate were ≈0.8 and 0.02 µM, respectively. The 5'CL frags indicate the 5′ cleavage fragments generated after cleavage at +1 and −1. The difference in migration of the +1 cleavage fragments in lanes 3 and 4 is likely because of the absence of the −1 nucleobase in the 5′ cleavage product after cleavage of pMini3bpAbasicG at +1. For experimental details see ‘Materials and Methods’. (B) Frequency of cleavage of pMini3bpAbasicG at the alternative site −1 with Eco RPRwt at 800 mM Mg2+. For the calculations of the percentage of cleavage, we used the 5′ cleavage fragments and the data are the mean and experimental errors of at least three independent experiments. The error bars indicate experimental errors and the frequencies of cleavage at −1 were calculated as described previously (30). For experimental details, see figure legend 2A. (C) Cleavage of pMini3bpUG and pMini3bpIsoCG with Eco RPRwt and Eco RPRG248; the reaction was performed at 37°C in buffer C containing 800 mM Mg2+ (see ‘Materials and Methods’). Lanes 1 (pMini3bpUG) and 2 (pMini3bpIsoCG), controls incubation without Eco RPR for 90 min; lanes 3 and 4, pMini3bpUG incubated with Eco RPRwt and Eco RPRG248, respectively, for 20 s; lanes 5 and 6, pMini3bpIsoCG incubated with Eco RPRwt (60 min) and Eco RPRG248 (90 min), respectively. The concentrations of Eco RPR and substrate were ≈0.8 and 0.02 µM, respectively. The 5'CL frags indicate the 5′ cleavage fragments generated after cleavage at +1 and −1. (D) Frequency of cleavage of pMini3bpIsoCG at the alternative site −1 (mean and experimental errors of at least three independent experiments) with Eco RPRwt and Eco RPRG248 at 800 mM Mg2+. For experimental details and calculations, see figure legends 2B and 2C. (E) Cleavage of pMini3bpGG and pMini3bpAG with Eco RPRwt in the absence and in the presence of C5. Lanes 1 (pMini3bpGG) and 2 (pMini3bpAG), controls incubation without Eco RPRwt and C5 for 180 min; lanes 3 (pMini3bpGG) and 4 (pMini3bpAG) cleavage without C5 for 180 min; lanes 5 (pMini3bpGG) and 6 (pMini3bpAG) cleavage with C5 for 1 min. 5′ CL Frags +1 and −1 mark the migrations of the 5′ cleavage fragments. The reactions were carried out at 37°C and 800 mM Mg2+ and 10 mM Mg2+ as described in ‘Materials and Methods’. In the absence of C5, the concentration of Eco RPRwt was 3.2 µM, whereas it was 0.002 µM in its presence. The substrate concentration was 0.02 µM. (F) Percentage of cleavage at −1 for substrates carrying 2;6-diaminopurine (DAP), 2-aminopurine (2AP) or guanosine (G) at −1 as indicated. The reactions were performed in the absence of C5 in buffer C at 800 mM Mg2+ as described earlier in text and in ‘Materials and Methods’. The calculations were done as described in figure legend 2B. (G) Percentage of cleavage at −1 for substrates carrying purine (Pu), adenosine (A) or inosine (Ino) at −1 as indicated. The reactions were performed in the absence of C5 in buffer C at 800 mM Mg2+ as described earlier in text and in ‘Materials and Methods’. The calculations were done as described in figure legend 2B.
Figure 3.
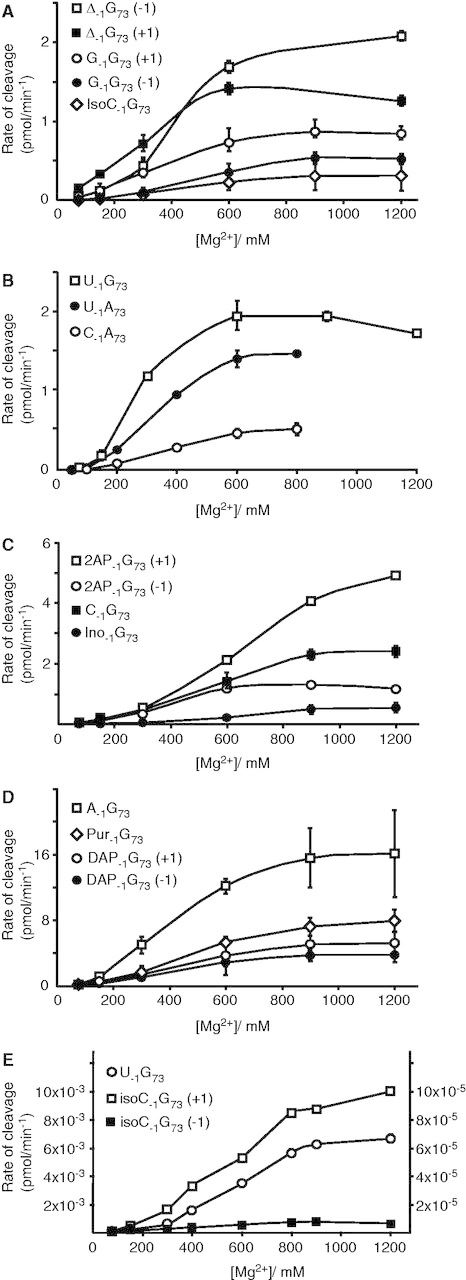
Cleavage of the pMini3bp variants by Eco RPR as a function of Mg2+ concentration. Panels A–E show the Mg2+ profiles for Eco RPRwt (A–D) and Eco RPRG248 (E). The experiment was performed under single-turnover conditions at 37°C as described in ‘Materials and Methods’ and the concentration of substrates was 0.02 µM, whereas the concentration of Eco RPR (in parenthesis) varied according to the following: Panel A pMini3bpAbasicG (Δ−1 G+73, 3.2 µM); pMini3bpGG (G−1 G+73, 5.2 µM); pMini3bpIsoCG (IsoC−1 G+73, 3.2 µM); Panel B pMini3bpUG (U−1 G+73, 0.8 µM); pMini3bpUA (U−1 A+73, 3.1 µM); pMini3bpCA (C−1 A+73, 3.1 µM); Panel C pMini3bp2APG (2AP−1 G+73, 5.2 µM); pMini3bpCG (C−1 G+73, 1.6 µM); pMini3bpInoG (Ino−1 G+73, 5.2 µM); Panel D pMini3bpAG (A−1 G+73, 5.2 µM); pMini3bpPuG (Pur−1 G+73, 5.2 µM); pMini3bpDAPG (DAP−1 G+73, 5.2 µM); Panel E (cleavage with Eco RPRG248) pMini3bpUG (U−1 G+73, 0.8 µM; left y-axis); pMini3bpIsoCG (IsoC−1 G+73, 0.8 µM; right y-axis). For the calculations, we used the 5′ cleavage fragments and the data are the mean of at least three independent experiments. The bars indicate the experimental errors.
Next, we determined the kinetic rate constants at saturating Mg2+ concentration (800 mM). As shown in Table 1, deleting the nucleobase at −1 (pMini3bpAbasicG) resulted in a 4200-fold reduction in both kobs and kobs/Ksto for cleavage at +1 compared with cleavage of pMini3bpUG at the same position. Given that pMini3bpAbasicG is cleaved at +1 and at −1, we compared the rate constants for cleavage at these positions. The kobs values for cleavage at +1 and −1 were essentially the same, whereas kobs/Ksto was slightly higher for cleavage at −1. Following the argument that Ksto ≈ Kd (see earlier in text), we calculated the Kd values using the data for cleavage at +1. This revealed that there was no significant effect on binding comparing pMini3bpAbasicG and pMini3bpUG.
Table 1.
The kinetic constants kobs and kobs/Ksto for cleavage of the various substrates by wild type Eco RPR
| Substrate/[Mg2+] | kobs (min−1) | kobs/Ksto (min−1 µM−1) | Kd (µM) |
|---|---|---|---|
| pMini3bpUG | |||
| 160 mM | 0.86 ± 0.12 | 0.31 ± 0.12 | 2.8 |
| 800 mM | 4.2 ± 0.48 | 1.6 ± 0.5 | 2.6 |
| pMini3bpAbasicG | |||
| 800 mM | |||
| +1 | 0.0010 ± 0.000006 | 0.00038 ± 0.000053 | 2.6 |
| −1 | 0.0012 ± 0.00004 | 0.0010 ± 0.00014 | 1.2 |
| pMini3bpCG | |||
| 160 mM | 0.0065 ± 0.0005 | 0.0006 ± 0.00012 | 11 |
| 800 mM | 0.6 ± 0.3 | 0.19 ± 0.12 | 3.2 |
| pMini3bpUA | |||
| 160 mM | 0.8 ± 0.06 | 0.11 ± 0.01 | 7.3 |
| 800 mM | 4.8 ± 0.95 | 3.7 ± 0.4 | 1.3 |
| pMini3bpCA | |||
| 160 mM | 0.29 ± 0.033 | 0.095 ± 0.015 | 3.1 |
| 800 mM | 1.6 ± 0.2 | 1.3 ± 0.2 | 1.2 |
| pMini3bpIsoCG | |||
| 160 mM | 0.0046 ± 0.0003 | 0.0013 ± 0.0003 | 3.5 |
| 800 mM | 0.062 ± 0.014 | 0.009 ± 0.001 | 6.9 |
| pMini3bpAG | |||
| 800 mM | 0.11 ± 0.061 | 0.034 ± 0.01 | 3.2 |
| pMini3bpGG | |||
| 800 mM | 0.0088 ± 0.0036 | 0.0012 ± 0.0002 | 7.3 |
| pMini3bpInoG | |||
| 800 mM | 0.22 ± 0.056 | 0.099 ± 0.026 | 2.2 |
| pMini3bp2APG | |||
| 800 mM | 0.016 ± 0.007 | 0.0088 ± 0.003 | 1.8 |
| pMini3bpDAPG | |||
| 800 mM | 0.025 ± 0.0048 | 0.013 ± 0.002 | 1.9 |
| pMini3bpPuG | |||
| 800 mM | 0.029 ± 0.0094 | 0.023 ± 0.007 | 1.3 |
The experiments were performed under single-turnover conditions at different Mg2+ concentrations at pH 6.1 as described in ‘Materials and Methods’. The final concentration of substrate was >10 nM. The concentration of Eco RPR was varied between 0.8 and 29 µM dependent on the substrate. For details regarding the calculation of Kd, see the main text. The data represent mean and experimental errors calculated from at least three independent experiments.
ND = not determined.
Taken together, these data show that the nucleobase at −1 is not essential for cleavage of the model substrate used here, but it does affect both the kinetics and accuracy of Eco RPR-mediated cleavage.
The identity of residue −1 contributes to catalysis
We previously reported that the kinetic constant kobs (see above Scheme 1) decreased 100-fold when U was replaced with C at the −1 position in the pMini3bp model substrate. These earlier experiments were done at 160 mM Mg2+, a non-optimal concentration [Table 1; cf. cleavage of pMini3bpUG and pMini3bpCG Figures 5 and 6 (14)]. Hence, to investigate the impact of the identity of the residue at −1, we now determined the optimal Mg2+ concentration for cleavage of these two substrates. A comparison of the Mg2+ profiles for cleavage of pMini3bpUG (Figure 3B) and pMini3bpCG (Figure 3C) revealed that cleavage of the latter required a higher Mg2+ concentration for optimal cleavage. This is consistent with our previous findings where we used pre-tRNA and longer model substrates [cf. Figures 5 and 6 in (14)].
Next, we determined kobs and kobs/Ksto for cleavage of pMini3bpUG and pMini3bpCG at 800 mM Mg2+, a concentration that resulted in optimal cleavage for both substrates (see earlier in text). As shown in Table 1, we detected an almost 10-fold reduction in both these constants due to substitution of U with C at −1. However, it was not possible to determine whether the reduced cleavage of the C−1 variant was due to the identity of residue −1 or due to pairing between C−1 and G+73 (Figure 1B). Hence, we generated two new variants, pMini3bpUA and pMini3bpCA, where pairing between residues −1 and +73 was expected to be weaker and absent, respectively. This would allow us to study if kobs and kobs/Ksto did differ for substrates with U or C at the −1 position.
An analysis of the cleavage as a function of Mg2+ concentration suggested that pMini3bpUA was cleaved more efficiently at position +1 (compared with pMini3bpCA) with no detectable cleavage at −1 (Figure 3B). The determination of kobs and kobs/Ksto at two Mg2+ concentrations (160 and 800 mM; Table 1) revealed a 3-fold higher kobs value for pMini3bpUA irrespective of Mg2+ concentrations. At saturating Mg2+ (800 mM), kobs/Ksto was also increased by a factor of three for pMini3bpUA suggesting that the Kd is not changed relative to pMini3bpCA. Rather the difference comparing U and C at −1 is related to a change in kobs (Scheme 1).
In conclusion, the identity of residue −1 in Eco RPR-mediated cleavage contributes to catalysis with model hairpin loop substrate (see also later in text). U carries oxygen at position 4 (O4), whereas C has an exocyclic amine (N4) at this position (Figure 1B). Thus, the O4 of U at −1 likely confers only a minor advantage in RPR-mediated catalysis. This is consistent with our previous finding that the kinetic rate constants for cleavage of other model hairpin substrates with U−1/A+73 and C−1/A+73 under multiple turnover conditions are similar (13). Moreover, the replacement of U with C at −1 (pMini3bpUA and pMini3bpCA) resulted in a 3-fold effect on the kinetic constants, whereas almost a 10-fold effect was observed comparing cleavage of pMini3bpUG and pMini3bpCG. This indicates that the possibility to form a strong −1/+73 pair affects the kinetics of cleavage.
The oxygen at C2 of U−1 and C−1 contributes to catalysis
Both U and C carry oxygen at C2 (O2) on the nucleobase (Figure 1C). To understand the influence of O2 on cleavage, we generated a pMini3bp derivative pMini3bpIsoCG in which O2 of U (or C) at position −1 is substituted with an exocyclic amine, 2NH2. Eco RPRwt cleaved pMini3bpIsoCG preferentially (≈97%; Figure 2D) at the canonical site +1 irrespective of Mg2+ concentration but at a reduced rate relative to cleavage of pMini3bpUG. Moreover, as observed for the other pMini3bp variants, cleavage of pMini3bpIsoCG required high Mg2+ concentration for optimal cleavage (Figure 3A).
Determination of the kinetic constants at 800 mM Mg2+ revealed an ≈70- and >150-fold reduction in kobs and kobs/Ksto, respectively, compared with when pMini3bpUG was used. Using these data, Kd was calculated to be just ≈3-fold higher relative to the Kd value for pMini3bpUG (Table 1). These data suggest that O2 of U (or C) at −1 contributes significantly to Eco RPR-mediated catalysis of a model substrate and can, therefore, be considered to be a positive determinant.
Presence of the exocyclic amine (2NH2) of G at −1 has a negative impact on cleavage at the canonical site +1
Bacteria also have pre-tRNAs with purines at −1; E. coli carry ≈13% and 8% A and G at −1, respectively (13). Processing studies with Eco RPRwt (with and without the C5 protein) and yeast pre-tRNASer derivatives show that cleavage between pyrimidines and purines is preferred to cleavage between two purines (31). Therefore, we decided to study the influence of a purine at −1 on substrate cleavage (all substrates have G at the +1 position; Figure 1). We were particularly interested to understand the impact of the exocyclic amine at C2 on the nucleobase (2NH2; Figure 1C) in RPR-mediated cleavage. We predicted that the 2NH2 at −1 (when present) would have a negative impact on cleavage by affecting the kinetic rate constants. Hence, six pMini3bp variants with different purines at −1 were generated (see Figure 1B) and subjected to cleavage by Eco RPRwt. As with the substrates discussed earlier in the text, we studied cleavage site recognition and Mg2+ dependence and determined the kinetic constants kobs and kobs/Ksto at 800 mM Mg2+.
As seen with the other pMini3bp variants, cleavage of the substrates with the different purine derivatives at −1 required high Mg2+ concentration for optimal cleavage (Figure 3A, C and D). Moreover, introduction of G at −1 resulted in substantial cleavage at the alternative site −1 (Figure 2E and F). However, the frequency of cleavage at −1 depended on the Mg2+ concentration; there was less cleavage at −1 at lower Mg2+ concentrations (Supplementary Figure S2). When A was present at −1, cleavage occurred preferentially at the correct site +1 (Figure 2E and G). The substrate carrying inosine at −1 was also cleaved mainly at +1 (≈2% cleavage at −1; Figure 2G). As pMini3bpAG, this substrate lacks the 2NH2 group (compared with G at −1; pMini3bpGG versus pMini3bpInoG and pMini3bpAG; see Figure 1C). In contrast, the pMini3bp2APG and pMini3bpDAPG (with 2-AminoPurine and 2;6-DiAminoPurine at −1, respectively, Figure 1B and C) substrates were both cleaved more frequently at the alternative site −1 (Figure 2F; For pMini3bp2APG and pMini3bpDAPG there was no variation in cleavage site selection as a function of Mg2+ with the possible exception in the cleavage of pMini3bp2APG at high Mg2+, Supplementary Figure S2). As in the substrate with G at −1, both pMini3bp2APG and pMini3bpDAPG carry the 2NH2 group on the nucleobase. Moreover, a slight increase in the frequency of cleavage at −1 was detected when comparing cleavage of pMini3bpAG and pMini3bpPuG (Figure 2G). Cumulatively these data indicate that deletion of 6NH2 at −1 in a model substrate context marginally affect cleavage site recognition, whereas an exocylic amine (2NH2) at C2 on the purine base has a negative impact on the selection of the cleavage site.
Determination of the kinetic constants, kobs and kobs/Ksto (Table 1) revealed that both were lower for substrates with A or G at −1 relative to those with U or C (cf. pMini3bpAG or pMini3bpGG versus pMini3bpUG or pMini3bpCG). This is particularly apparent when G is present at −1 (pMini3bpGG). In this case, there is an almost 500 - and >1000-fold reduction (for cleavage at +1) in kobs and kobs/Ksto, respectively, compared with cleavage of pMini3bpUG. For the substrate with A at −1 (pMini3bpAG versus pMini3bpUG), kobs was reduced ≈40-fold and kobs/Ksto ≈50-fold. When the 2NH2 of G−1 (pMini3bpInoG versus pMini3bpGG) was deleted both kobs and kobs/Ksto increased. By contrast, introduction of an exocyclic amine at C2 (pMini3bp2APG and pMini3bpDAPG) resulted in a decrease compared with cleavage of the substrate with A at −1 (pMini3bpAG; Table 1). Deletion of 6NH2 from the A at −1 (Figure 1C) decreased mainly the kobs (3 - to 4-fold), whereas kobs/Ksto changed only marginally (cf. cleavage of pMini3bpAG and pMini3bpPuG; Table 1). A comparison of Kd values revealed only a modest increase (2 - to 3-fold) for the substrate with G at −1 relative to the substrates with U or C at −1. These data emphasize that the presence of 2NH2 on the purine base at −1 has a negative impact on the kinetics of cleavage, in agreement with our prediction. The presence of the exocyclic amine at C6 (6NH2) of A at −1, however, appears to have only a modest influence on cleavage efficiency.
In conclusion, RPR-mediated cleavage between purines in a model substrate context is unfavourable. An exocyclic amine at C2 on the purine base has a negative impact on cleavage both with respect to accuracy and kinetics of cleavage at +1. From our data, it also appears that a chemical group at C6 contributes to catalysis.
Influence of the protein subunit of Eco RNase P
The RNase P protein, C5, binds to the 5′ leader of the substrate, and it affects binding and affinity of catalytic important Mg2+ (32–37). Also comparing cleavage with and without C5 of yeast pre-tRNASer with G at −1 indicates that the presence of the C5 protein affects cleavage site selection (31). Therefore, the C5 protein is a positive factor for cleavage and selection of the cleavage site. Hence, we inquired whether addition of C5 to the reaction also influenced the choice of cleavage site when cleaving the model hairpin loop substrate with G (or A) at −1. Figure 2E shows that the reconstituted Eco RNase P holoenzyme cleaved pMini3bpGG at +1 with no apparent cleavage at −1. No change in choice of cleavage site was observed for cleavage of the substrate having A at −1 with RPR or with the reconstituted Eco RNase P holoenzyme. However, the substrate with A at −1 was cleaved more efficiently compared with cleavage of the substrate with G at −1 also in the presence of the C5 protein (cf. lanes 5 and 6 Figure 2E).
These data show that the C5 protein suppressed cleavage at the alternative site −1 also in the case of model substrates carrying G at −1. This further indicates that C5 has a strong positive effect on the cleavage site recognition process.
Interaction between residue −1 and 248 in the RPR
The A at position 248 in the RPR (Figures 1A and 4) has been suggested to interact with the −1 residue in the substrate (9). In the crystal structure of RNase P in complex with tRNA, which represents a post cleavage state, A248 is positioned near the 5′ termini of the tRNA [(11); Figure 5A]. Nucleotide analogue-modification interference studies suggest that the Hoogsteen surface of A248 plays an important role in the interaction with the substrate (38). As shown here, substitution of O2 of U (or C) at −1 with NH2 affects the kinetics of cleavage (see earlier in text; pMini3bpIsoCG). Previous data reveal that the 2'OH of residue −1 contribute to catalysis [(7) and references therein]. The O2 and 2'OH of residue −1 are exposed on the same surface (Figure 5B; see later in text). Together this opens for the possibility that N7 and 6NH2 of A248 are hydrogen-bonded to the 2'OH and O2 of residue −1, respectively, in the transition state (Figure 4B). If this hypothesis is valid, changing of A248 to G in the RPR would restore the interaction between IsoC at −1 and 248 by forming hydrogen bonding between 2NH2 of IsoC and O6 of the G at 248. As a consequence, the rate of cleavage of pMini3bpIsoCG should increase using an RPR variant with G at 248 compared with cleavage with RPRwt. To test this prediction, we generated Eco RPRG248. Structural probing revealed no significant overall structural difference compared with Eco RPRwt [Supplementary Figure S3; see also (19). We note the appearance of additional cleavages, located between well-established lead(II)-induced sites IV and V, when Eco RPRG248 was subjected to RNase T1 cleavage. This might indicate a slight change in the structure in this region].
Figure 4.
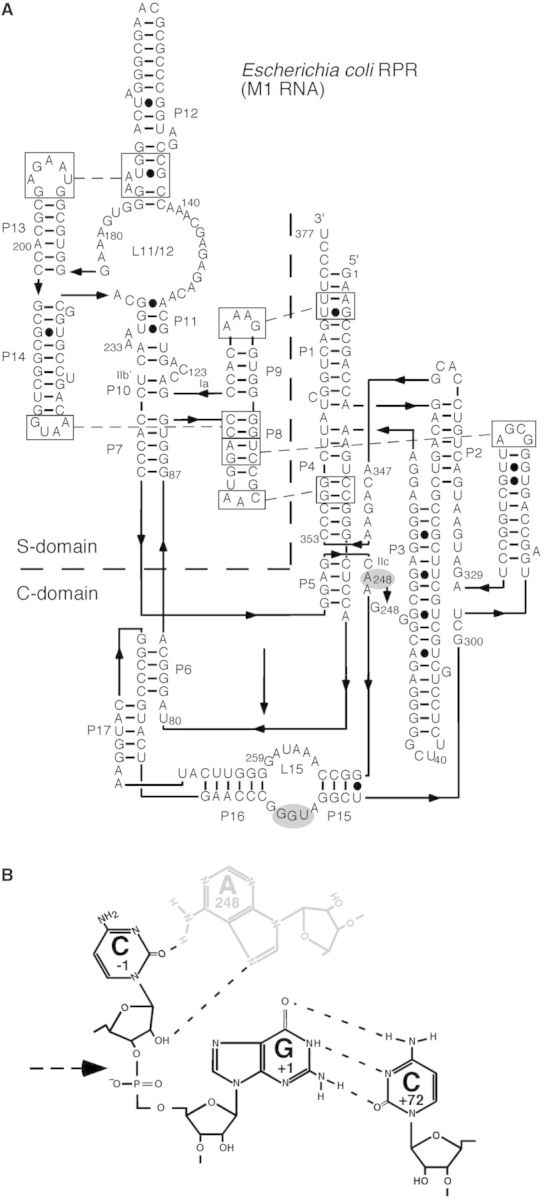
Secondary structure model of Eco RPR and a model of the N−1/A248 interaction. (A) Secondary structure of Eco RPRwt according to Massire et al. (39). Residue A248 that was changed to G is indicated in grey. (B) A putative model of the N−1/A248 interaction. Black and grey residues mark substrate and RPR residues, respectively, and the dashed arrow marks the scissile bond.
Figure 5.
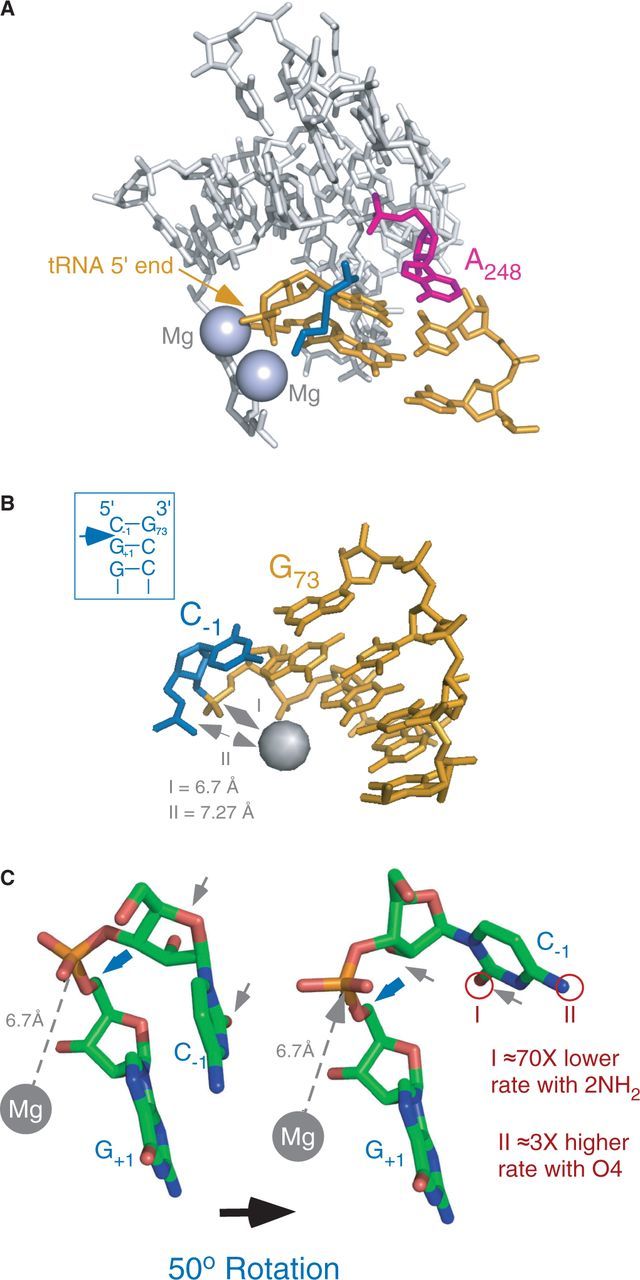
The structure of the RPR active site and model of the RNase P cleavage site. (A) The active site of RPR from the PDB structure 3OKB (11). Residues G+1 of the 5′ matured tRNA and A248 (E. coli numbering) are marked as indicated. The grey spheres corresponds to the Mg2+ ions observed in the structure, and the blue structure represents the 5′ leader that was soaked into the crystal. (B) Model of the RNase P cleavage site with C−1/G+73 and G+1/C+72. The model is part of the SRP RNA structure [PDB code 1LNT; (40)]. The phosphorous atom to be attacked is marked with the grey dashed double arrow. The light blue sphere represents Mg2+ bound in the minor groove and is positioned at a distance of 6.7 Å relative to phosphorous atom I, and 7.27 Å relative to phosphorous atom II; I and II are referred to as P(+1) and P(−1) in the main text. This distance allows for a nucleophilic attack on P(+1) by an activated H2O coordinated to a divalent metal ion (41,42). The boxed hairpin structure shows the sequence of SRP RNA that mimics the RNase P cleavage site. The blue arrow marks the cleavage site. (C) Model of the RNase P cleavage site showing the conformational change in the substrate that facilitates the nucleophilic attack on P(+1). The model was prepared using the structure shown in panel B and rotating the P-O5′ phosphoester bond (marked with a blue arrow) 50°. The rotation displaces the pro-Sp non-bridging oxygen and exposes the P(+1)-atom to an inline hypothetical nuclephilic attack by a Mg2+-activated H2O (grey dashed arrow). The grey arrows mark the 2′OH and O2 of C−1, and I and II refer to the changes in cleavage rates as a result of changing O2 to 2NH2 and O4 to 4NH2, respectively.
As shown in Figure 2C and D, Eco RPRG248 cleaved pMini3bpUG and pMini3bpIsoCG preferentially at the correct site, +1. For pMini3bpIsoCG, we also observed ≈9% cleavage at −1, which was higher at lower Mg2+ (Supplementary Figure S2). Moreover, compared with Eco RPRwt, the G248 variant showed similar Mg2+ profiles in cleavage of pMini3bpUG and pMini3bpIsoCG (Figure 3E). Determinations of the apparent rate constants (kapp) at 800 mM Mg2+ revealed that Eco RPRG248 cleaved these two substrates with slightly reduced rates relative to the wild type (Table 2). Importantly, the rate of cleavage did not improve as predicted. Rather, the rate was lower with Eco RPRG248 compared with Eco RPRwt. These findings, therefore, suggest that 6NH2 of A248 is not engaged in hydrogen bonding with O2 of U (or C) at −1 or at least not in the context of a model substrate (see also the ‘Discussion’).
Table 2.
Apparent rates, kapp, for cleavage of pMini3bpUG and pMini3bpIsoCG with Eco RPRwt and Eco RPRG248
| Substrate |
Eco RPR variant |
|
|---|---|---|
| Wt | G248 | |
| pMini3bpUG | ||
| +1 | 1.5 ± 0.03 | 0.98 ± 0.06 |
| pMini3bpIsoCG | ||
| +1 | 0.026 ± 0.002 | 0.011 ± 0.0004 |
| −1 | ND | 0.001 ± 0.00007 |
kapp = pmol cleaved per min. The experiments were performed in buffer C containing 800 mM Mg2+ at 37°C. The concentrations of RPRs and substrates were 4 and 0.02 µM, respectively. The data represent mean and experimental errors calculated from at least three independent experiments.
ND = not determined.
DISCUSSION
Here we provide data that the nucleobase, the identity and specific chemical groups of the nucleobase at position −1 in a model substrate contribute significantly to catalysis in the E. coli RPR alone reaction. In particular, our data suggest that the carbonyl oxygen (O2) at C2 on C and U at −1 is a positive factor, whereas a purine (A or G) at −1 and the presence of an exocyclic amine at C2 (2NH2) have a negative impact on the kinetics and accuracy of cleavage. The nucleobase at −1 is, however, not essential for cleavage although it affects the kinetic rate constants, kobs and kobs/Ksto. It contributes to catalysis by stabilizing the transition state, at least in the context of a model substrate, and this corresponds to ≈5.1 kcal [using kobs values for cleavage of pMini3bpAbasicG and pMini3bpUG at +1 and ΔΔG = −RTlnkobs(Δ−1)/kobs(U−1); (43)]. Of these 5.1 kcal, the contribution of the O2 (with U at −1) amounts to 2.6 kcal [ΔΔG = −RTlnkobs(IsoC−1)/kobs(U−1)], which corresponds roughly to one hydrogen bond. In accordance with this is that the presence of 2NH2 on a purine at −1 destabilizes the transition state with up to 2 kcal (using kobs values for cleavage of pMini3bpInoG and pMini3bpGG at +1). Interestingly, Baidya et al. (44) used modified pyrimidines at position 17 (5′ residue of the cleavage site) in the hammerhead ribozyme and showed that an abasic residue at position 17 almost abolished cleavage. They also suggested that O2 of C17 makes an important contribution to catalysis by stabilizing the transition-state structure. The recently solved structure of the Schistosoma mansoni hammerhead ribozyme suggests hydrogen bonding between O2 of C17 at the cleavage site and 6NH2 of A13 that presumably helps to stabilize the transition state (45). However, the recent crystal structure of bacterial RNase P in complex with tRNA (11) does not reveal any structural information about the nature of the interaction between residue −1 in the substrate and the RPR (see later in text). We conclude that the O2 (in the case of U or C) at position −1 in a model substrate acts as a positive determinant in Eco RPR-mediated catalysis whereas 2NH2 (in the cases of U, C or purines) at this position has a negative impact.
Comparing cleavage of pre-tRNA versus model hairpin loop substrates
Studies where pre-tRNAAsp was used (9,46) show modest changes in both kobs and Kd (except for the variant with C at −1 where Kd increased 230-fold, possibly owing to the C at −1 being paired with the G at +73 in the substrate). In contrast, cleavage of model substrates (this report) caused an almost 500-fold change in kobs (for G at −1) with only small changes in Kd. Moreover, model substrates with G at −1 (or purines with 2NH2 or where the nucleobase had been deleted at −1) resulted in significant cleavage at −1 whereas the other −1 variants were cleaved mainly at +1 (Figure 2 and Supplementary Figure S2). This is in contrast to the U at −1 substitution in pre-tRNAAsp, which did not change the cleavage site. However, cleavage of pre-tRNAAsp at the alternative site −1 was observed when residue −1 was changed together with replacement of the 2'OH with 2'H at −1, i.e. two cleavage site determinants had been changed (9,46). A comparison of the structures of pre-tRNAAsp and pMini3bpUG reveals striking similarities at and near their respective cleavage sites
[black circles correspond to residues that are not shown and the canonical cleavage site is between U−1 and G+1; Figure 1 and (46)]. The T-loop/stem, known to affect site selection (14,22), is absent in the pMini3bp model substrates. This makes them dependent on the remaining determinants, e.g. the −1 residue (Figure 1A). Together this argues that the impact of the −1 residue in a pre-tRNA (all ribo) context is obscured by the presence of several residues and regions that interact with the RPR on formation of the RPR-substrate complex. Nevertheless, for both model and pre-tRNA substrates, the base at −1 as well as its identity influences cleavage efficiency and site recognition but it is not essential for cleavage. However, as shown in this report using model substrates, specific chemical groups on the −1 nucleobase influence both transition-state stabilization and accuracy in Eco RPR-mediated cleavage.
We have reported elsewhere that the presence of an exocyclic amine (2NH2) on the nucleobase at +1 plays an important role for cleavage at the correct (+1) site (17). Our present data show that replacement of U (or C) at −1 with purines with 2NH2 in a model substrate had a negative impact on the kinetic constants with significant cleavage at the alternative site −1. On the basis of these data we suggest that 2NH2 (in the case of G) at −1 functions as a positive determinant for cleavage at −1, i.e. at the site immediately 5′ of a purine carrying 2NH2 (see Figure 1). At the same time, it acts as a negative factor for cleavage at +1. This would explain why the efficiency of cleavage between two G residues is low (31). Most bacterial tRNAHis precursors [and pre-tRNASeCys; Ref (47)] carry G at both −1 and +1, and this model also provides one reason to why bacterial RPR cleaves pre-tRNAHis and pre-tRNASeCys at −1 and not between two G-residues [reviewed in (6)]. However, we have to consider that addition of the C5 protein, which interacts with the substrate 5′ leader (see earlier in text), resulted in cleavage mainly at +1 even with G at −1 in the model substrate (Figure 2E). It is conceivable that this is due to G−1 in the model substrate is not being paired with a C at +73, in contrast to pre-tRNAHis and pre-tRNASeCys in which the G at −1 is paired to a C. The presence of a G−1/C+73 pair in different precursor substrates plays an important role for cleavage at −1 (6). Also, E. coli pre-tRNA(valU) and pre-tRNA(thrW) carry G at −1 (48) but in those two cases G−1 is unlikely to pair with the discriminator base (which is A in both cases) at position +73. Another factor to consider is that the difference in cleavage site selection of the G−1 model substrate with and without the C5 protein may be an effect of the higher Mg2+ concentration used in the ‘RPR alone reaction’ (Supplementary Figure S2). Evidently, the role of the C5 protein in the cleavage site recognition process in the processing of different precursors requires further studies. The present study together with our previous data (17), however, indicates the importance of the presence of G (and the 2NH2) 3′ of the scissile bond at least in the ‘Eco RPR alone reaction’. In this context, we note the importance of the exocyclic amine of a guanosine marking the cleavage site in the reaction catalyzed by the group I ribozyme (49), indicating similarities with group I RNA and RPR-mediated cleavage.
Interaction between the −1 residue in the substrate and Eco RPR and positioning of Mg2+ at the cleavage site
A248 has been suggested to play a key role in the interaction with residue −1; this interaction is referred to as the N−1/A248 interaction [(9,38,46,50); see also (6) and Figure 1A]. The crystal structure of the RNase P-tRNA complex, which represents the post-cleavage state, does not provide any information about the interaction between the RPR and the −1 residue (11). Although it cannot be excluded that the Watson–Crick surface of A248 is involved in pairing with residue −1 as suggested elsewhere (9,46), we consider this unlikely. The reasons are: (i) as mentioned earlier in the text, the identity of residue −1 varies and must be taken into account; (ii) N3-methyl-U at −1 in a model substrate did not affect the kinetic rate constants, kobs and kobs/Ksto, to any significant extent compared with a substrate with an unmodified U at −1 (51); and (iii) a model substrate with C−1/G+73 is cleaved with increased frequency at −1 (and not a decrease in cleavage at the alternative site −1, as would be predicted if it was base pairing between −1 and 248) when A248 in Eco RPRwt was substituted with a G (19). Alternatively, given that the Hoogsteen surface of A248 is facing the tRNA 5′-end in the crystal structure (Figure 5A) it is possible that N7 and 6NH2 interact with specific groups of the −1 residue. This would be consistent with nucleotide analogue-modification interference studies, which suggest that the Hoogsteen surface of A248 plays an important role on interaction with the substrate (38). Here a possibility is that 6NH2 and N7 of A248 interact with O2 (in the case of U and C) and the 2'OH (Figure 4B; see also below) of residue −1, respectively. The lack of rescue in cleavage of pMini3bpIsoCG with RPRG248 argues against this alternative; however, we cannot conclusively exclude this possibility (see later in the text).
RPR catalysis depends on divalent metal ions, preferentially Mg2+, and two Mg2+ are positioned close to the tRNA 5′-end in the RNase P-tRNA complex (Figure 5A). In our model, the Mg2+ that activates the nucleophilic water is positioned 6.7 Å from the phosphorous between −1 and +1 [P(+1) in Figure 5B] in the substrate (7,52,53). The distance between this Mg2+ and the phosphorous between −1 and −2 [P(−1) in Figure 5B] is just 0.57 Å longer. Hence, changing the identity of residues or chemical groups in the vicinity of the cleavage site can affect the charge distribution. This would then shift the positioning of Mg2+ that influences the rate of cleavage and site selection. This model is consistent with our data where substitutions of the nucleobase and chemical groups at −1 or at position 248 in the RPR affected the rate constants and site selection [see also (19); it also raises the possibility that the change in charge distribution owing to the presence of G at 248 is why we did not observe rescue of cleavage of pMini3bpIsoCG with Eco RPRG248]. In addition, deletion of the exocyclic amine (2NH2) of the G at +1 in a model substrate influenced the charge distribution at the cleavage site (17). Substitutions in the T-loop also influenced both the rate and site of cleavage in metal(II)-ion induced hydrolysis of yeast tRNAPhe (54,55).
A model for displacement of the −1 residue in the Eco RPR-substrate complex
For bacterial RPR, the 2'OH at −1 plays an important role for catalysis (6–8). It contributes 2.3 kcal to the stabilization of the transition state in cleavage of a hairpin loop model substrate using the same formula discussed earlier in text and data from Brännvall and Kirsebom (51). As reported here, the contribution of the carbonyl oxygen O2 of C (or U) at −1 in a model substrate to transition-state stabilization was 2.6 kcal (see earlier in text). The 2'OH and O2 of C at −1 in a model of the cleavage site were exposed on the same surface (marked with grey arrows in Figure 5C). We cannot exclude that one or both these groups form hydrogen bonds with the RPR in the transition state. However, available data suggest that the 2'OH at −1 acts as an outer (or inner) sphere ligand for Mg2+ [(24,56–59); but see also (46) for an alternative interpretation]. On the basis of this, it is therefore conceivable that also the carbonyl oxygen of C (or U) at −1 is involved in Mg2+ binding although the crystal structure does not confirm this. Nevertheless, on RPR-substrate complex formation, where residue +73 pairs with U294 in the RPR, the C−1/G+73 (when present) opens. This conformational change is likely accompanied with displacement of the C at −1 and positioning of A248 such that it stacks over the G+1/C+72 base pair (Figure 5A). We propose that displacement of the −1 residue is the result of a 50-degree rotation (Figure 5C) such that O2 (in the case of U or C at −1) and the 2'OH are facing away from the scissile phosphate. As a consequence, the Mg2+-activated H2O bound to the substrate in the vicinity of the phosphorus [P(+1)] to be attacked is positioned for an in-line attack [Figure 5; Ref (7)]. At the same time, the 2'OH at −1 is prevented from attacking the phosphorus, which would lead to cleavage products with faulty ends, i.e. a 5'OH and a 2′; 3′ cyclic phosphate (24). This model is consistent with a conformational-assisted mechanism of cleavage but needs to be confirmed by a structure of RNase P in complex with its precursor substrate.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Swedish Research Council; Uppsala RNA Research Center (Swedish Research Council Linneus support).
Conflict of interest statement. Leif A. Kirsebom is on the board of directors of Bioimics AB.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr R. K. Hartmann for the plasmid pET33b with the His6-C5 gene, our colleagues for discussions and Dr A. C. Forster and Ms T. Bergfors for critical reading of the article.
REFERENCES
- 1.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 2.Pannucci JA, Haas ES, Hall TA, Harris JK, Brown JW. RNase P RNAs from some archaea are catalytically active. Proc. Natl Acad. Sci. USA. 1999;96:7803–7808. doi: 10.1073/pnas.96.14.7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kikovska E, Svärd SG, Kirsebom LA. Eukaryotic RNase P RNA mediates cleavage in the absence of protein. Proc. Natl Acad. Sci. USA. 2007;104:2062–2067. doi: 10.1073/pnas.0607326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, Rossmanith W. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008;135:462–474. doi: 10.1016/j.cell.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 5.Gobert A, Gutmann B, Taschner A, Gössringer M, Holzmann J, Hartmann RK, Rossmanith W, Giegé P. A single Arabidopsis organeller protein has RNase P activity. Nat. Struct. Mol. Biol. 2010;17:740–744. doi: 10.1038/nsmb.1812. [DOI] [PubMed] [Google Scholar]
- 6.Kirsebom LA. RNase P RNA mediated cleavage: Substrate recognition and catalysis. Biochimie. 2007;89:1183–1194. doi: 10.1016/j.biochi.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Kirsebom LA, Trobro S. RNase P RNA-mediated cleavage. IUBMB Life. 2009;61:189–200. doi: 10.1002/iub.160. [DOI] [PubMed] [Google Scholar]
- 8.Lai LB, Vioque A, Kirsebom LA, Gopalan V. Unexpected diversity of RNase P, an ancient tRNA processing enzyme: challenges and prospects. FEBS Lett. 2010;584:287–296. doi: 10.1016/j.febslet.2009.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zahler NH, Christian EL, Harris ME. Recognition of the 5′ leader of pre-tRNA substrates by the active site of ribonuclease P. RNA. 2003;9:734–745. doi: 10.1261/rna.5220703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirsebom LA, Svärd SG. Base pairing between Escherichia coli RNase P RNA and its substrate. EMBO J. 1994;13:4870–4876. doi: 10.1002/j.1460-2075.1994.tb06814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reiter NJ, Osterman A, Torres-Larios A, Swinger KK, Pan T, Mondragón A. Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature. 2010;468:784–789. doi: 10.1038/nature09516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kufel J, Kirsebom LA. Different cleavage sites are aligned differently in the active site of M1 RNA, the catalytic subunit of Escherichia coli RNase P. Proc. Natl Acad. Sci. USA. 1996;93:6085–6090. doi: 10.1073/pnas.93.12.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brännvall M, Pettersson BMF, Kirsebom LA. Importance of the +73/294 interaction in Escherichia coli RNase P RNA substrate complexes for cleavage and metal ion coordination. J. Mol. Biol. 2003;325:697–709. doi: 10.1016/s0022-2836(02)01195-6. [DOI] [PubMed] [Google Scholar]
- 14.Brännvall M, Kikovska E, Wu S, Kirsebom LA. Evidence for induced fit in bacterial RNase P RNA-mediated cleavage. J. Mol. Biol. 2007;372:1149–1164. doi: 10.1016/j.jmb.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 15.Crothers DM, Seno T, Söll D. Is there a discriminator site in transfer RNA? Proc. Natl Acad. Sci. USA. 1973;69:3063–3067. doi: 10.1073/pnas.69.10.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pettersson BMF, Kirsebom LA. The presence of a C−1/G+73 pair in a tRNA precursor influences processing and expression in vivo. J. Mol. Biol. 2008;381:1089–1097. doi: 10.1016/j.jmb.2008.06.077. [DOI] [PubMed] [Google Scholar]
- 17.Kikovska E, Brännvall M, Kirsebom LA. The exocyclic amine at the RNase P cleavage site contributes to substrate binding and catalysis. J. Mol. Biol. 2006;359:572–584. doi: 10.1016/j.jmb.2006.03.040. [DOI] [PubMed] [Google Scholar]
- 18.Wincott F, DiRenzo A, Shaffer C, Grimm S, Tracz D, Workman C, Sweedler D, Gonzalez C, Scaringe S, Usman N. Synthesis, deprotection, analysis and purification of RNA and ribozymes. Nucleic Acids Res. 1995;23:2677–2684. doi: 10.1093/nar/23.14.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu S, Kikovska E, Lindell M, Kirsebom LA. Cleavage mediated by the catalytic domain of bacterial RNase P RNA. J. Mol. Biol. 2012;422:204–214. doi: 10.1016/j.jmb.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 20.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acid Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feltens R, Gossringer M, Willkomm DK, Urlaub H, Hartmann RK. An unusual mechanism of bacterial gene expression revealed for the RNase P protein of Thermus strains. Proc. Natl Acad. Sci. USA. 2003;100:5724–5729. doi: 10.1073/pnas.0931462100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu S, Chen Y, Lindell M, Mao G, Kirsebom LA. Functional coupling between a distal interaction and the cleavage site in bacterial RNase P RNA mediated cleavage. J. Mol. Biol. 2011;411:384–396. doi: 10.1016/j.jmb.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 23.Vioque A, Arnez J, Altman S. Protein-RNA interactions in the RNase P holoenzyme from Escherichia coli. J. Mol. Biol. 1988;202:835–848. doi: 10.1016/0022-2836(88)90562-1. [DOI] [PubMed] [Google Scholar]
- 24.Brännvall M, Kikovska E, Kirsebom LA. Cross talk in RNase P RNA mediated cleavage. Nucleic Acids Res. 2004;32:5418–5429. doi: 10.1093/nar/gkh883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sinapah S, Wu S, Chen Y, Pettersson BMF, Gopalan V, Kirsebom LA. Cleavage of model substrates by archaeal RNase P: role of protein cofactors in cleavage-site selection. Nucleic Acids Res. 2011;39:1105–1116. doi: 10.1093/nar/gkq732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hofstee BHJ. On the evaluation of the constants Vm and KM in enzyme reactions. Science. 1952;116:329–331. doi: 10.1126/science.116.3013.329. [DOI] [PubMed] [Google Scholar]
- 27.Dowd JE, Riggs DS. A comparison of estimates of Michaelis-Menten Kinetic constants from various linear transformations. J. Biol. Chem. 1965;240:863–869. [PubMed] [Google Scholar]
- 28.Stage-Zimmermann TK, Uhlenbeck OC. Hammerhead ribozyme kinetics. RNA. 1998;4:875–889. doi: 10.1017/s1355838298980876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen W-Y, Pulukkunat DK, Cho I-M, Tsai H-Y, Gopalan V. Dissecting functional cooperation among protein subunits in archaeal RNase P, a catalytic ribonucleoprotein complex. Nucleic Acids Res. 2010;38:8316–8327. doi: 10.1093/nar/gkq668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brännvall M, Kirsebom LA. Manganese ions induce miscleavage in the Escherichia coli RNase P RNA-catalyzed reaction. J. Mol. Biol. 1999;292:53–63. doi: 10.1006/jmbi.1999.3048. [DOI] [PubMed] [Google Scholar]
- 31.Krupp G, Kahle D, Vogt T, Char S. Sequence changes in both flanking sequences of a pre-tRNA influence the cleavage specificity of RNase P. J. Mol. Biol. 1991;217:637–648. doi: 10.1016/0022-2836(91)90522-8. [DOI] [PubMed] [Google Scholar]
- 32.Crary SM, Niranjanakumari S, Fierke CA. The protein component of Bacillus subtilis ribonuclease P increases catalytic efficiency by enhancing interactions with the 5′ leader sequence of pre-tRNAAsp. Biochemistry. 1998;37:9409–9416. doi: 10.1021/bi980613c. [DOI] [PubMed] [Google Scholar]
- 33.Loria A, Niranjanakumari S, Fierke CA, Pan T. Recognition of a pre-tRNA substrate by the Bacillus subtilis RNase P holoenzyme. Biochemistry. 1998;37:15466–15473. doi: 10.1021/bi9816507. [DOI] [PubMed] [Google Scholar]
- 34.Niranjanakumari S, Stams T, Crary SM, Christianson DW, Fierke CA. Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc. Natl Acad. Sci. USA. 1998;95:15212–15217. doi: 10.1073/pnas.95.26.15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jovanovic M, Sanchez R, Altman S, Gopalan V. Elucidation of structure-function relationships in the protein subunit of bacterial RNase P using a genetic complementation approach. Nucleic Acids Res. 2002;30:5065–5073. doi: 10.1093/nar/gkf670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurz JC, Fierke CA. The affinity of magnesium binding sites in the Bacillus subtilis RNase P-pre-tRNA complex is enhanced by the protein subunit. Biochemistry. 2002;41:9545–9558. doi: 10.1021/bi025553w. [DOI] [PubMed] [Google Scholar]
- 37.Day-Storms JJ, Niranjanakumari S, Fierke CA. Ionic interactionsbetween P RNA and P protein in Bacillus subtilis RNase P characterized using a magnetocapture-based assay. RNA. 2004;10:1595–1608. doi: 10.1261/rna.7550104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siew D, Zahler NH, Cassano AG, Strobel SA, Harris ME. Identification of adenosine functional groups involved in substrate binding by the ribonuclease P ribozyme. Biochemistry. 1999;38:1873–1883. doi: 10.1021/bi982329r. [DOI] [PubMed] [Google Scholar]
- 39.Massire C, Jaeger L, Westhof E. Derivation of the three-dimensional architecture of bacterial ribonuclease P RNAs from comparative sequence analysis. J. Mol. Biol. 1998;279:773–793. doi: 10.1006/jmbi.1998.1797. [DOI] [PubMed] [Google Scholar]
- 40.Deng J, Xiong Y, Pan B, Sundaralingam M. Structure of an RNA dodecamer containing a fragment from SRP domain IV of Escherichia coli. Acta Crystallogr. D Biol. Crysatallogr. 2003;59:1004–1011. doi: 10.1107/s0907444903006747. [DOI] [PubMed] [Google Scholar]
- 41.Brown RS, Dewan JC, Klug A. Crystallographic and biochemical investigation of the lead(II)-catalyzed hydrolysis of yeast phenylalanine tRNA. Biochemistry. 1985;24:4785–4801. doi: 10.1021/bi00339a012. [DOI] [PubMed] [Google Scholar]
- 42.Jovine L, Djordjevic S, Rhodes D. The crystal structure of yeast phenylalanine tRNA at 2.0 Å resolution: cleavage by Mg(2+) in 15-year old crystals. J. Mol. Biol. 2000;301:401–414. doi: 10.1006/jmbi.2000.3950. [DOI] [PubMed] [Google Scholar]
- 43.Wells JA. Additivity of mutational effects in proteins. Biochemistry. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 44.Baidya N, Ammons GE, Matulic-Adamic J, Karpeisky AM, Beigelman L, Uhlenbeck OC. Functional groups on the cleavage site pyrimidine nucleotide are required for stabilization of the hammerhead transition state. RNA. 1997;3:1135–1142. [PMC free article] [PubMed] [Google Scholar]
- 45.Martick M, Scott WG. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell. 2006;126:309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zahler NH, Sun L, Christian EL, Harris ME. The pre-tRNA nucleotide base and 2′-hydroxyl at N(−1) contribute to fidelity in tRNA processing by RNase P. J. Mol. Biol. 2005;345:969–985. doi: 10.1016/j.jmb.2004.10.080. [DOI] [PubMed] [Google Scholar]
- 47.Burkard U, Söll D. The unusually long amino acid acceptor stem of Escherichia coli selenocysteine tRNA results from abnormal cleavage by RNase P. Nucleic Acids Res. 1988;18:11617–11623. doi: 10.1093/nar/16.24.11617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pettersson BMF, Ardell DH, Kirsebom LA. The length of the 5′ leader of Escherichia coli tRNA precursors influences bacterial growth. J. Mol. Biol. 2005;351:9–15. doi: 10.1016/j.jmb.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 49.Strobel SA, Cech TR. Exocyclic amine of the conserved GU pair at the cleavage site of the Tetrahymena ribozyme contributes to 5′-splice site selection and transition state stabilization. Biochemistry. 1996;35:1201–1211. doi: 10.1021/bi952244f. [DOI] [PubMed] [Google Scholar]
- 50.Brännvall M, Pettersson BMF, Kirsebom LA. The residue immediately upstream of the RNase P cleavage site is a positive determinant. Biochimie. 2002;84:693–703. doi: 10.1016/s0300-9084(02)01462-1. [DOI] [PubMed] [Google Scholar]
- 51.Brännvall M, Kirsebom LA. Complexity in orchestration of chemical groups near different cleavage sites in RNase P RNA mediated cleavage. J. Mol. Biol. 2005;351:251–257. doi: 10.1016/j.jmb.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 52.Kikovska E, Mikkelsen NE, Kirsebom LA. Substrate discrimination in RNase P RNA-mediated cleavage: importance of the structural environment of the RNase P cleavage site. Nucleic Acids Res. 2005;33:6920–6930. doi: 10.1093/nar/gki344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kirsebom LA. Roles of metal ions in RNase P catalysis. In: Liu F, Altman S, editors. Protein Reviews Vol 10: Ribonuclease P. New York, NY: Springer Science+Business Media, LLC, 233 Springer Street; 2010. 10013, USA pp 113–134. [Google Scholar]
- 54.Behlen LS, Sampson JR, DiRenzo AB, Uhlenbeck OC. Role of the tertiary nucleotides in the interaction of yeast phenylalanine tRNA with its cognate synthetase. Biochemistry. 1990;29:2515–2523. doi: 10.1021/bi00462a014. [DOI] [PubMed] [Google Scholar]
- 55.Michalowski D, Wrzesinski J, Krzyzosiak W. Cleavage induced by different metal ions in yeast tRNA(Phe) U59C60 mutants. Biochemistry. 1996;35:10727–10734. doi: 10.1021/bi9530393. [DOI] [PubMed] [Google Scholar]
- 56.Perreault J-P, Altman S. Important 2′-hydroxyl groups in model substrates for M1 RNA, the catalytic RNA subunit of RNase P from Escherichia coli. J. Mol. Biol. 1992;226:399–409. doi: 10.1016/0022-2836(92)90955-j. [DOI] [PubMed] [Google Scholar]
- 57.Perreault J-P, Altman S. Pathway of activation by magnesium ions of substrates for the catalytic subunit of RNase P from Escherichia coli. J. Mol. Biol. 1993;230:750–756. doi: 10.1006/jmbi.1993.1197. [DOI] [PubMed] [Google Scholar]
- 58.Smith D, Pace NR. Multiple magnesium ions in the ribonuclease P reaction mechanism. Biochemistry. 1993;32:5273–5281. doi: 10.1021/bi00071a001. [DOI] [PubMed] [Google Scholar]
- 59.Persson T, Cuzic S, Hartmann RK. Catalysis by RNase P RNA: unique features and unprecedented active site plasticity. J. Biol. Chem. 2003;278:43394–43401. doi: 10.1074/jbc.M305939200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.