


Abstract
We used an in silico approach to predict microRNAs (miRNAs) genome-wide in the brown alga Ectocarpus siliculosus. As brown algae are phylogenetically distant from both animals and land plants, our approach relied on features shared by all known organisms, excluding sequence conservation, genome localization and pattern of base-pairing with the target. We predicted between 500 and 1500 miRNAs candidates, depending on the values of the energetic parameters used to filter the potential precursors. Using quantitative polymerase chain reaction assays, we confirmed the existence of 22 miRNAs among 72 candidates tested, and of 8 predicted precursors. In addition, we compared the expression of miRNAs and their precursors in two life cycle states (sporophyte, gametophyte) and under salt stress. Several miRNA precursors, Argonaute and DICER messenger RNAs were differentially expressed in these conditions. Finally, we analyzed the gene organization and the target functions of the predicted candidates. This showed that E. siliculosus miRNA genes are, like plant miRNA genes, rarely clustered and, like animal miRNA genes, often located in introns. Among the predicted targets, several widely conserved functional domains are significantly overrepresented, like kinesin, nucleotide-binding/APAF-1, R proteins and CED-4 (NB-ARC) and tetratricopeptide repeats. The combination of computational and experimental approaches thus emphasizes the originality of molecular and cellular processes in brown algae.
INTRODUCTION
MicroRNAs (miRNAs) are short single-stranded RNA molecules, which are able to regulate gene expression by interfering with messenger RNAs (mRNAs). Since their discovery in the nematode Caenorhabditis elegans (1,2), their taxonomic coverage has been extended to other animals, plants, green algae and viruses [reviewed in (3)]. The miRNAs from different lineages belong to a common class of functional molecules, in which several ubiquitous families share extensive sequence similarity. However, their biogenesis, primary and secondary structures and mode of action are not exactly the same between plants and animals (4). Distinct from both the opisthokonts (animals, fungi) and the archaeplastida (plants, green and red algae), the heterokonts form a large eukaryotic phylum comprising unicellular (e.g. diatoms), syncytial (e.g. oomycetes) and multicellular organisms (e.g. brown algae). Phylogenetic analyses showed that heterokonts diverged from the ancestors of the opisthokont and the archaeplastida phyla more than one billion years ago (5), enabling a large field of alternative molecular strategies to adapt, develop and evolve. In this perspective, identifying and studying miRNAs in heterokonts is likely to uncover new features and mechanisms. Advanced genomic studies were carried out in several organisms of the heterokont phylum: oomycetes Phytophthora sp. (6), Hyaloperonospora sp. (7) and Pythium sp. (8); diatoms Thalassiosira sp. (9) and Phaeodactylum tricornutum (10). Yet, no miRNAs were reported in these organisms; only 13 precursors were found in the latter (11). The brown alga Ectocarpus siliculosus [Ectocarpales, Phaeophyceae; see (12)] genome has recently been published (13), and the identification of several specific proteins involved in the miRNA biogenesis (Argonaute: AGO1 and DICER: DCL1) provided a good presumption of the presence of miRNAs in this alga. Among the results obtained by the annotation consortium, a deep-sequencing approach followed by a computational filtering allowed the detection of nine different miRNA candidates for which targets could be predicted, and 14 other miRNA candidates without a predicted target. However, organisms with an miRNA machinery are expected to be able to produce and use hundreds of miRNAs (3).
Computational methods aiming at the ab initio identification of miRNAs in a newly sequenced genome [reviewed in (14–16)] are either based on sequence conservation, or inspired by the knowledge about their biogenesis and function. Sequence conservation can only be used when an extensive repertoire of miRNAs is available in a closely related species, which is not the case for E. siliculosus. Similarly, the phylogenetic position of heterokonts, apart from both animals and plants, does not permit to refer to animal- or plant-specific structural features or miRNA biogenesis processes. Thus, our identification and filtering criteria must rely on the common features of miRNAs shared by the organisms investigated to date. These features are as follows: (i) the primary transcript is processed into a precursor called the pre-miR. Alternatively, pre-miRs can derive from introns of protein-coding genes (17,18). In any case, the pre-miR is folded as a stable hairpin, which exhibits specific structural features (19); (ii) this structured RNA is recognized and processed by an enzyme called DICER (20), which excises a short (possibly imperfect) duplex made of the miRNA and its complementary strand: the miRNA*. The cutting points may vary by one or two nucleotides, thus allowing the precursor to generate alternative duplexes (21); (iii) the miRNA strand is specifically incorporated into a ribonucleoproteic complex called RNA-induced silencing complex (RISC, including the protein Argonaute), and serves as a guide to bind the complex to the target mRNA, using an imperfect sequence complementarity. It is possible that one single miRNA interacts with several mRNAs, and reciprocally, one mRNA can be targeted by more than one miRNA (22–24). The subsequent inhibition of target translation can be caused by the interference of the RISC complex with regulatory elements, the destabilization of the mRNA and/or its cleavage at the RISC binding site. In much less documented cases, the target expression is enhanced by an unknown process (25). The repression by cleavage requires a better complementarity between the miRNA and the target mRNA, and is mostly (but not exclusively) found in plants, while the other processes are typical of (but not exclusive to) animals (26).
Here, we apply a genome-wide approach to extend the set of miRNA candidates in E. siliculosus using a computational identification and filtering, followed by an experimental search of selected candidates under a variety of conditions. Relying on common features, we identified a comprehensive list of 568 miRNA candidates in the genome of E. siliculosus, from which 22 were experimentally validated. We also analyzed their specific features in terms of sequence, genomic organization and putative biological functions.
MATERIALS AND METHODS
Data collection and preparation
The genome sequences (super-contigs with length >2 kb), primary annotation and protein sequences (version June 2010) were retrieved from the E. siliculosus annotation Web site (https://bioinformatics.psb.ugent.be/gdb/ectocarpus/). These data were processed to obtain a suitable partition of the sequences (shown as ‘re-assignation’ in Figure 1). We identified all the ribosomal RNAs (rRNAs), including those that were not annotated, using BLAST (27) to search for sequences similar to the EMBL entries EF990201 [partial rRNA gene of E. siliculosus (28)] and D16558 [complete rRNA gene of the closely related species Scytosiphon lomentaria (29)]. We also identified the transfer RNAs (tRNAs) using the tRNAsan-SE 1.23 software (30) downloaded from the author's Web site (http://lowelab.ucsc.edu/tRNAscan-SE/). The rRNA and tRNA sequences were stored as separate data sets, and masked in the genomic sequence. In the remaining genome sequence, mRNA annotation was found to miss an important information: in 16 254 mRNAs, no 3′UTR was annotated for 8677, no 5′UTR was annotated for 12 598 mRNAs and 7602 mRNAs had no annotation for untranslated region (UTR) at all. In all these cases, we added putative UTRs to the annotated mRNA sequence, by extending the first exon in 5′ and/or the last exon in 3′. The length of the putative 5′UTRs was computed as the 95th percentile of the length distribution of the annotated 5′UTRs upstream of the first coding exon, i.e. 374 nt. Similarly, 3′UTR of 1734 nt was added downstream the last exons of genes without an annotated 3′UTR. As a summary of this preparation step, the genome was sorted into five sets: rRNAs, tRNAs, mRNAs, introns and intergenic sequences.
Figure 1.

Flowchart of the in silico analysis. Data are represented in boxes, processes in ovals. Colour code: black: initial data; red: mRNAs or parts of mRNAs; green: intergenic/non–protein-coding RNA; gray: preexisting software; purple: our software. The resulting in silico predictions were then tested for experimental validation.
Constitution of a large set of potential miRNAs
The program findMiRNA (31) was downloaded from its author's Web site (http://sundarlab.ucdavis.edu/mirna/). We introduced a small modification to the code to predict the best secondary structure at a temperature of 13°C [the usual temperature for E. siliculosus cultures, see (32) instead of the default 37°C. This adapted version of findMiRNA was used to process (i) the ‘intergenic’ sequences (on both strands) and (ii) the introns (on the transcript strand only), both using the mRNAs as a reference (shown as ‘findMiRNA’ on Figure 1).
Reference RNA folding parameters
To build a set of reference values for the structural and topological properties of folded RNAs in E. siliculosus, we isolated local substructures from rRNAs and mRNAs. To do so, we predicted their complete optimal folding at 13°C (see above) using the RNAfold 1.8 software (33) downloaded from the Vienna RNA Package Web site (http://www.tbi.univie.ac.at/∼ivo/RNA/). Substructures made of 40–200 nt were extracted out of these complete structures. As tRNA sizes lie within this range, we used their full-length sequences.
We determined the structural and topological properties of the complete tRNAs and of the rRNA and mRNA substructures using the genRNAStats and RNAspectral programs (19) downloaded from their author's Web site (http://web.bii.a-star.edu.sg/∼stanley/Publications/Supp_materials/06-004-supp.html), and tuned to use a folding temperature of 13°C (see above). This step is shown as ‘RNAfold’ and ‘analyse’ in Figure 1. Its output is a list of five parameter values computed for each analyzed structured RNA sequence: normalized minimum free energy (Nmfe), normalized Shannon entropy (NQ), normalized base-pair distance (ND), degree of compactness (NF) and normalized base-pairing propensity (Nbp). Valid pre-miRs have a lower Nmfe, NQ, ND and NF, and a higher Nbp than other structured RNAs (19). Thus, we computed threshold values for each of these parameters as follows. As an example, for Nmfe, we computed three values: the Nmfe values above which were found 90% of the rRNA, mRNA and tRNA. The lowest among these three values was retained as Nmfe90. We computed NQ90, ND90 and NF90 by applying the same procedure. Nbp90 was computed the same way, only substituting the minimum among the three values having 90% of the distribution above them by the maximum among the three values having 90% of the distribution below them. For a more discriminant filter, we also computed Nmfe95, NQ95, ND95, NF95 and Nbp95 by the same method, using 95% of the corresponding distributions instead of 90%. This step is shown as ‘distribution analysis’ in Figure 1, and the resulting values are shown in Table 1. These statistical treatments were performed with R (34), obtained from the Comprehensive R Archive Network (http://cran.r-project.org/).
Table 1.
Secondary structure filter on pre-miRs
| Parameter | Short | Filter 90 | Number of passing 90 | Filter 95 | Number Of passing 95 |
|---|---|---|---|---|---|
| Normalized minimum free energy | Nmfe | <−0.750 | 86 485 | <−0.826 | 43 885 |
| Normalized Shanon entropy | NQ | <0.0565 | 73 518 | <0.0441 | 50 731 |
| Normalized base-pair distance | ND | <0.0244 | 88 088 | <0.0175 | 52 414 |
| Degree of compactness | NF | <0.382 | 350 532 | <0.325 | 332 241 |
| Normalized base-pairing propensity | Npb | >0.364 | 162 473 | >0.369 | 129 715 |
| All five filters | 1540 | 597 |
A valid pre-miR is expected to have a lower Nmfe, NQ, ND, NF and a higher Npb than other structured RNAs. For each parameter (rows 1–5), the filtering threshold ‘Filter n’ was defined as the lowest (except Npb: highest) nth percentile of the value distribution among the three RNA reference sets: tRNAs, rRNAs and mRNAs. The number of pre-miRNAs passing each individual filter is indicated on their respective row in the last column. The 6th row shows the number of pre-miRs passing all the filters of the two values of n presented: 95 and 90.
Selection of miRNA candidates
We computed the values of the five parameters described above on the predicted secondary structures of the 697 657 potential pre-miRs resulting from the search with findMiRNA. Each candidate was retained in the pre-miR90 set only if its values of Nmfe, NQ, ND and NF were, respectively, lower than Nmfe90, NQ90, ND90 and NF90, and its value of Nbp was higher than Nbp90. These thresholds are displayed as ‘filter 90’ in Table 1. Similarly, we used the values of Nmfe95, NQ95, ND95, NF95 and Nbp95 to design the ‘filter 95’ (Table 1) and to obtain the pre-miR95 subset of candidates. This step is shown as ‘filter’ on Figure 1.
A Support Vector Machine (SVM) approach was implemented using the R package e1071 (http://cran.r-project.org/web/packages/e1071) to filter the potential pre-miRs, using a discriminant kernel function adjusted for maximal separation between known pre-miRNAs (1000 animal and plant precursors from miRBase) and non-miRNA (1000 tRNAs, rRNA and mRNA hairpins). We used the same five parameters as those on which the filters were applied (see above). We did not succeed in obtaining a robust set of pre-miRNA candidates as an output: when several runs of the adjustment procedure were performed using random ‘positive’ and ‘negative’ sequences, the results of the prediction were different. This approach was not carried on further.
Expression level analysis
We sorted the sequences (tiles) of the E. siliculosus high-resolution transcription map (tiling array, Gene Expression Omnibus GSE19912) into seven sets: Exon, tRNA, Intergenic not candidate, Intergenic miR candidate, Intronic not candidate, Intronic miR candidate, Others (discarded from further analysis). The expression level for each tile was computed as the logarithm of its RNA expression normalized to its DNA expression signal. The statistical significance of the difference between ‘miR candidate’ and ‘not candidate’ sets was tested using the Student's t-test implemented in R (34).
Culture conditions and treatments
E. siliculosus uni-algal strain 32 (CCAP accession 1310/4, origin san Juna de Marcona, Peru) was cultivated in 10 L plastic flasks in a culture room at 13°C using filtered and autoclaved natural seawater enriched in Provasoli nutrients (32). Light was provided by daylight fluorescence tubes with a photon flux density of 40 µmol.m−2.s−1 for 14 h per day. Cultures were bubbled with filtered (0.22 µm) compressed air to avoid CO2 depletion. To conduct the chemical treatment experiments, sporophyte materials were transferred into Petri dishes containing artificial seawater enriched with Provasoli (ASW; see 32) for at least 18 h before treatments to acclimatize the cultures to the change of growth conditions. They were then treated with different chemicals for 6 h. To perform saline stresses, sporophytes were transferred for acclimatization to ASW for 1 week before applying the salt stresses. Hypo-saline stress corresponded to 56 mM and hyper-saline stress to 1470 mM NaCl in ASW (ASW contains 450 mM NaCl). Treatments were applied for 6 h before harvesting the tissues in liquid nitrogen for RNA extraction. Gametophytes were cultured in the same conditions as the sporophytes and collected before they reached maturity. For each treatment or condition, total RNAs were extracted from three independent biological replicates.
RNA extraction and cDNA synthesis
RNAs were extracted as described in Le Bail et al. (35). They were RNAse-free DNAse I-treated (Turbo DNAse, Ambion), cleaned up, diluted in RNAse-free water and quantified using a NanoDrop ND-1000 spectrophotometer. RNA integrity was verified on 1.5% agarose gel stained with ethidium bromide. From each RNA sample, 2 μg was polyadenylated by the poly(A) polymerase (PAP) of the Poly(A) Tailing Kit (Ambion) according to the manufacturer instructions, and reverse transcribed to cDNA using oligo(dT)12–18. Two types of reverse transcriptases of the ‘First Strand synthesis for RT-PCR’ kits (Invitrogen) were used: (i) the Superscript™ to detect and quantify the expression level of the 72 miRNAs in the sporophyte tissues; (ii) the Thermoscript™, which is particularly relevant to polymerize cDNAs from RNAs with stable secondary structures such as the pre-miRs. To allow comparison between pre-miR expression levels and miRNA, miRNA target genes, RNAse genes (AGO1: D7FQK3 and DCL1: D7FZW2) and reference genes EEF1A2 (D7FZS6), TUA (D8LPR8) and UBCE (D7G3Z7), the Thermoscript-amplified cDNAs were used to test the transcript level of these molecules in salt stress conditions and in gametophyte tissues.
Real-time polymerase chain reaction
Oligonucleotide sequences were designed using Perl Primer (http://perlprimer.sourceforge.net) in the 3′UTR of the AGO1 and DCL1 mRNAs (Supplementary Table S1A), in the coding sequence of the predicted target mRNAs (Supplementary Table S1B) and for the miRNAs (Supplementary Table S1C) and the pre-miRNAs (Supplementary Table S1D). The Real-Time quantitative Polymerase Chain Reaction (RT-qPCR) reactions were performed in a 96-well thermocycler (Chromo4 System thermocycler; BioRad Laboratories) with SYBRgreen reaction mix from ABgene (AB-1162/B; ABgene France, Courtabœuf), for 15 min at 95°C, followed by 41 runs of 15 s at 95°C, 30 s at 60°C and 30 s at 72°C. Each sample was technically duplicated. The amplification efficiency was tested using a dilution series of either genomic DNA (for the AGO1, DCL1, miRNA target genes and the pre-miRs; see below) or of poly-adenylated cDNAs (for the miRNAs). The specificity of amplification was checked with a dissociation curve obtained by heating the samples from 65 to 95°C (measurement every 0.3°C). Experiments were carried out on three independent biological replicates. Pre-miRs were amplified from equivalent amount of cDNAs and RNAs, as well as on genomic DNA. As negative controls, non–reverse-transcribed RNAs were used in addition to water. The PCR products were loaded on a 4% agarose gel, stained with ethidium bromide and were sequenced using a Sanger-based method on a ABI 3130XL Genetic Analyser (Applied Biosystem, Life Technologies Corporation, USA).
Analysis of RT-qPCR data
The normalization of the PCR signals corresponding to mRNAs (AGO1, DCL1 and target genes) and pre-miRs was conducted following Hellemans et al. (36), except that instead of working on Cq values averaged over replicates, we normalized each measured amount to the amount of reference genes (EEF1A2, TUA and UBCE) in the same replicate. MiRNAs were normalized using the whole set of miRNAs as a reference set [as recommended in (37)]. The comparison between samples (gametophyte, hypo-, hyper-saline) and the control (sporophytes in normal culture conditions) was performed as a bidirectional t-test on the log-transformed normalized expression levels of the three replicates, using the Welch correction for inequality of variances. Samples with a P-value <0.05 were retained as significantly different from the control.
Genomic DNA extraction
Ectocarpus siliculosus genomic DNA was prepared as described in Le Bail et al. (35) and was used as a quantification reference for the RT-qPCR experiment and the calculation of PCR efficiency. A dilution series ranging from 47 to 60 730 copies (6 times dilution on 5 points) of the E. siliculosus genomic DNA was prepared and tested for the E. siliculosus AGO1, DCL1, EEF1A2, TUA and UBCE genes as well as for the pre-miR sequences.
Sequence conservation in the miRNA candidates
The known mature miRNA sequences were obtained from miRBase (http://microrna.sanger.ac.uk). We excluded the miRNA* sequences and extracted two subsets of miRNA sequences, one corresponding to Metazoa and the other to Viridiplantae. We computed the Levenshtein distances using our own Java implementation of the classical dynamic programming algorithm. These computations were performed for the predicted miRNAs from sets Mir95 and Mir90 versus the miRBase subsets, and for the miRBase subsets between them. We selected the lowest score for each predicted E. siliculosus miRNA against Metazoa or Viridiplantae, the lowest score for each plant miRNA against all Metazoa, and the lowest score for each metazoan miRNA against all Viridiplantae. To compute Hausdorff distances, we also selected the lowest scores for each Metazoa or Viridiplantae miRNA against the predicted E. siliculosus miRNA.
Protein domain analysis
The protein domains were searched using Interproscan (38), in the whole proteome of E. siliculosus. For each domain, we compared the number of proteins containing at least one instance of this domain among the whole proteome of E. siliculosus, and among the candidate targets. The P-value retained to estimate the overrepresentation of a motif occurring in m proteins in the whole proteome and in k proteins in the candidate targets was computed as PN,n,m(X ≥ k) where PN,n,m is the distribution function of the hypergeometric law, i.e. the probability to obtain at least k positive instances within a sample of n individuals drawn out of a population of N individuals containing m positive instances. Protein domains were considered overrepresented if the P-value was <0.05.
RESULTS
In silico identification of miRNAs, pre-miRs and target candidates
The global strategy for the in silico analysis is shown in Figure 1. We searched for miRNAs in two sets of non–protein-coding RNA sequences: (i) 12 798 ‘intergenic’ sequences, i.e. sequences that are neither in genes nor in rRNAs or tRNAs and (ii) 112 513 intron sequences. Each of these two sets of sequences was analyzed together with the 16 254 mRNAs of E. siliculosus, using the findMiRNA software (31). The intergenic sequences were searched on both strands (2 × 68 917 369 nt), while for introns, only the transcript strand was used (78 816 594 nt). As an output, we obtained 864 679 results from the intergenic sequences, and 403 761 from the introns. Each of these ∼1.27 million potential miRNA candidates is associated to one folded precursor and one target sequence located within an mRNA. The relationship between these three types of molecules is, however, not univocal: the results contain 516 147 unique miRNAs, 697 657 unique pre-miRs and 16 250 unique target mRNAs.
According to Ng Kwang Loong and Mishra (19), the pre-miRs exhibit values for several structural and topological parameters, which differ from those computed on other structured RNAs. We made use of this feature to filter the potential candidates. To do so, we determined on tRNAs, rRNAs and mRNAs, the distribution of values for five discriminant parameters: Nmfe, NQ, ND, NF and Nbp. As we had no prior idea about the number of miRNA genes in the E. siliculosus genome, we used the classical cutoff values of 95th and 90th percentiles to decide whether a potential pre-miR was sufficiently different from the ncRNAs. Likewise, the reference energetic values we used were not extracted from literature, but were computed on E. siliculosus RNAs. Thus, for each parameter, we computed two threshold values, one corresponding to the 95th percentile, the other the 90th percentile of its distribution (see Table 1). Noteworthy, only the combination of all of the filters allowed a drastic reduction in the number of candidates. Yet, the Nmfe appeared to be the most discriminant filter, as it allowed the smallest number of pre-miRs to pass through. Two sets of pre-miRs were derived from these values: (i) a pre-miR was retained in the set Pre95 when the values for the five parameters were beyond their respective threshold, corresponding to the 95th percentile; (ii) a second set, named Pre90, was similarly defined by reference to the 90th percentiles, but excluding those sequences already contained in Pre95. Pre95 contained 597 pre-miRs, which were able to generate 568 different miRNAs (set Mir95, see Supplementary Tables S2 and S3A and B), which in turn were predicted to interact with 498 target mRNAs (set Tg95, see Supplementary Table S4). Similarly, Pre90 contained 943 pre-miRs, Mir90 is made of 922 miRNAs (Supplementary Tables S2 and S3a and b), predicted to interact with 1153 target mRNAs (set Tg90, see Supplementary Table S4).
As an alternative procedure, we built a SVM, similar to an approach that has proven efficiency, for instance, to predict human pre-miRs (39). When we applied this method in E. siliculosus, the selected pre-miR candidates were not the same among repeats of the procedure adjusted on different sets of ‘positive’ and ‘negative’ sequences. A thorough analysis of the data, function parameters and results showed that this was probably due to the distribution of the discriminant factors, which contained a significant number of extreme values. Therefore, the approach based on cutoff filters set on quantiles [for a similar technique, see for instance (40)] appeared to be more suitable to these data.
Experimental validation of the predictions
The main drawback of the in silico approach is the large number of false-positive instances it produces (14). For this reason, we performed an experimental check, to evaluate the ratio of correct predictions. It is expected that miRNA genes located in so-called ‘intergenic’ regions are expressed at a detectable level, while the rest of these regions should only be detected as ‘noise’ in quantitative detection experiments. Similarly, miRNAs issued from introns should be retained, in contrast to regular introns, which are destroyed after excision. In both cases, the detectable amount of pre-miRs should be statistically distinguishable from the expression level of the regions from which they are issued. We used the high-resolution transcriptome map to isolate, among the intergenic or intronic sequences, those corresponding to the predicted pre-miR (both MiR90 and MiR95). As a comparison, we also included data from the exonic regions and tRNAs. The distribution of the expression levels of these various sets are shown on Figure 2. In both intergenic and intronic sequences, the expression level of the predicted pre-miRs is significantly higher than the expression level of the other sequences (Student t-test, α = 10−2). The intronic predicted miRNAs have an expression level similar to that of exons. In addition, we observed that in both the intergenic and the intronic sequences, the predicted pre-miRs in the set MiR95 are expressed at an even higher level than those in the set MiR90 (not shown). These results showed that the sets of predicted miRNAs were strongly biased toward highly expressed and stable RNA sequences, which confirmed the statistical enrichment of these sets in actual miRNA sequences.
Figure 2.

Expression level of various sequence sets extracted from a whole-genome tiling array experiment. The predicted pre-miRNA (‘miR cand.’, lines 2 and 4) are compared with the other sequences (‘Not cand.’, lines 1 and 3) of the same genomic origin (‘Intergenic’ or ‘Intronic’). Exons and tRNAs are also displayed. Each ‘miR cand.’ line is significantly higher than the corresponding ‘Not cand.’ line.
Among the 1488 miRNAs retained in Mir95 or Mir90, we extracted at random a subset of 36 sequences from each set, and quantified their expression by RT-qPCR (41) in E. siliculosus sporophyte filaments. We could detect a specific expression characterized by a unique dissociation curve with the expected half-dissociation temperature (Tm) for 22 different miRNAs (Table 2). Figure 3 shows the variable relative expression level (compared with tRNA-Leu) of the detected miRNAs. As a control, we attempted to amplify 13 randomly chosen non-retained potential miRNAs (i.e. instances filtered out from the structural filtering step). In agreement with the predictions, we could not detect any of them (not shown). Among the 22 validated miRNAs, 16 were from the set Mir95, and 6 from the set Mir90. Thus, these data show that increasing the threshold from the 90th to the 95th percentile enhances the ratio of experimental validation from ∼30 (22/72) to ∼44% (16/36). Extrapolating this result to the whole prediction of 568 miRNAs in Mir95 suggests that our most stringently filtered set contains ∼252 valid miRNAs.
Table 2.
Experimentally validated miRNAs
| miRNA name | miRNA sequence | Target(s) | ||||
|---|---|---|---|---|---|---|
| miR95_0055 | GUAGCCAAGAUCGGGUGCACCCGG | D7FJK8 | ||||
| miR95_0064 | UCCUGCAGCCGCUGCCGC | D8LF98 | D8LGC5 | |||
| miR95_0076 | ACUUCGCCGCCGGCGCAG | D7FT76 | ||||
| miR95_0168 | GUCCCAUGGUGUCCCAUGGGAC | D7FWG6 | D8LJ42 | |||
| miR95_0195 | CCAAGAUCGGGUGCACCCGGUGGAA | D7FWA3 | ||||
| miR95_0207 | CCGCUUGGUUCCGGCGCAGAUUUCG | D8LFK3 | D7FWF7 | D7FRZ9 | ||
| miR95_0213 | UCGUACAGUGGCUCAGCCUC | D7FWM6 | D7G720 | |||
| miR95_0257 | UCUCAUGGGCGCCCAUGGG | D7G179 | ||||
| miR95_0295 | GCCGCCGUUCCUGCAGCCGC | D8LS33 | ||||
| miR95_0324 | GUAUGGCCUGUGACCGCUCG | D7G722 | ||||
| miR95_0365 | AGUUGAUAGCUUCCGAGAAUCAGUG | D8LID2 | ||||
| miR95_0400 | AACCAAGAUGGCCUGGAUUUGCG | D8LNB1 | D8LNB2 | D8LNB6 | D7FM32 | D8LNC7 |
| miR95_0424 | UUGGUUCCGGCGUAGAUC | D7G7S8 | ||||
| miR95_0439 | GUCCGCAGCCGCCGCCGC | D8LBP6 | D8LBX5 | D7FMJ4 | D7FGT5 | D8LJR3 |
| miR95_0483 | AAGUUGAUAGCUUCCGAGAAUCAGU | D8LID2 | ||||
| miR95_0485 | CUGGCGCCCCAGGGCGG | D7FQ65 | D8LEB3 | D7FML8 | D7FI70 | |
| miR90_0079 | UAUAGAAGCCGAAAUCAAA | D7G3H8 | D7FRR2 | |||
| miR90_0193 | CAUGGAACUCCAUGGAACUCCAUG | D7FM32 | ||||
| miR90_0201 | CCACAGCAGUCACCACUAGCUUCAA | D7G014 | D8LGE5 | |||
| miR90_0682 | CGCCGACGGUUGCCGCUGC | D8LLD2 | ||||
| miR90_0796 | GUCUGGGGACUAUGUUAACC | D7FPI3 | ||||
| miR90_0805 | AGCAAAGUUGAUAGCUUCCGAG | D8LID2 | ||||
The first part of the name, ‘miR95’ or ‘miR90’, indicates the set from which the candidate was drawn (see Table 1). The mature sequence of each miRNA is shown, together with the list of predicted target proteins, for which the Uniprot accession numbers are indicated.
Figure 3.

Detection by reverse-transcriptase quantitative PCR of 22 candidate miRNAs in E. siliculosus sporophyte tissues grown in normal culture conditions. The expression level was normalized to the level of tRNA-Leu.
To reinforce the biological relevance of this experimental validation, we attempted to detect the precursors of the 22 detected miRNAs, using a PCR-based approach (Figure 4A; see Supplementary Table S1D for pre-miR sequences and position of the oligonucleotides). We could detect amplification of a PCR product for eight of them, the five most expressed ones being displayed in Figure 4B. Sequencing these PCR products showed that their primary structure was confirmed in all cases.
Figure 4.

Pre-miR detection by RT-qPCR amplification. (A) Example of primer design for PCR assay. Oligonucleotides used for the RT-qPCR are shown as arrows, and the predicted miRNA sequence is shaded in gray. To ensure a high specificity, one of the oligonucleotides was designed, when possible, in the terminal loop. (B) Visualization of assay results. For each predicted precursor, the PCR assay was conducted from H2O, gDNA (2.3 ng), total RNAs (40 ng) and cDNAs (40 ng equivalent of total RNAs) and run on a 4% agarose gel. Their predicted size in base pairs is indicated in the right side of the figure. Leftovers of primers or primer dimers are visible in the bottom part of each photography.
Genomic organization and sequence conservation of the predicted miRNAs
The experimental validation of 44% of predicted miRNAs in the set Mir95 allowed the assumption that this pool of miRNAs and the corresponding pre-miRs were a relevant population to investigate the genomic organization and sequence conservation. Among the 597 predicted pre-miRs in the set pre95, 407 (68.2%) came from intergenic regions and 190 (31.8%) from introns. Similarly, in the set pre90, 313 precursors were intronic (33.2% of the 943). We also searched for miRNA gene clusters, which we defined as three or more pre-miRs in a row (not necessarily on the same strand), separated by no more than 5 kb. We identified three such clusters, each made of three genes (Figure 5A). In one case, two pre-miRs of the same cluster shared extensive similarity (Figure 5B). Altogether, these observations suggest that pre-miRs of E. siliculosus are not predominantly organized as clusters.
Figure 5.

MiRNA clusters in E. siliculosus genome. (A) Three clusters were predicted, on three different super-contigs (sctg_XX). (B) Two of the predicted miRNAs composing the second cluster share extensive sequence similarity.
To assess a putative sequence conservation between E. siliculosus miRNAs and those found in other organisms, we compared the miRNA sequences in Mir95 and Mir90 to the whole content of miRBase (17 341 miRNAs, with 10 099 different sequences). No identical sequence was found. To estimate the proximity between these sequences, we computed the smallest Levenshtein (edition) distance between each sequence in Mir95 or Mir90 and the miRBase entries issued from Metazoa and Viridiplantae. The results in Figure 6 show that the predicted miRNAs of E. siliculosus were found to be different from both animal (Figure 6A) and land plant (Figure 6B) miRNAs. The mean lowest distance from miRNAs in Mir95+90 to Metazoa (6.92) appeared to be lower than to Viridiplantae (7.74), but this difference could not be interpreted as a higher sequence similarity with Metazoa than with Viridiplantae. Instead, it was owing to the fact that the number of sequences was higher in the former (11, 411) than in the latter (3, 246), thus increasing the likelihood that any sequence finds a more similar closest relative in Metazoa than in Viridiplantae. To enable comparisons, we computed the mean lowest distance for Viridiplantae versus Metazoa (7.11) and Metazoa versus Viridiplantae (7.72). As expected, the same bias was observed, while the pairwise distances themselves were obviously the same. An other measure of the divergence between sequence sets is the maximum distance between each element in one set and the closest element in the other set, known as the ‘Hausdorff distance’. We computed that the Hausdorff distance between Mir95+90 and the miRBase entries issued from Metazoa and Viridiplantae were 12 and 11, respectively. The Hausdorff distance between Metazoa and Viridiplantae was 15. Again, the differences between these values corresponded to an expected bias, as bigger sets have a higher probability to contain at least one highly divergent sequence. Noticeably, the edition distances were high compared with the length of the sequences considered, showing that in each of these sets, there was at least one sequence displaying a high level of divergence with any miRNA in the other sets with which the comparison was being performed. To summarize, these data showed that the distance between the predicted E. siliculosus miRNAs and known miRNAs in Metazoa or Viridiplantae were similar to the distance between the miRNAs of Viridiplantae versus Metazoa.
Figure 6.

Comparison between miRNA candidates and miRBase. Each chart shows the distribution of the Leveshtein distance for each sequence to the closest entry in a subset of miRBase. (A) Distance for Viridiplantae and E. siliculosus to Metazoa; (B) Distance for Metazoa and E. siliculosus to Viridiplantae.
Biological function of miRNAs in E. siliculosus
To propose biological functions for E. siliculosus miRNAs, we first examined their target mRNAs. We searched for protein motifs, which were overrepresented in the set of predicted targets. The most significant results are shown in Table 3 (a complete set of results is provided as Supplementary Table S5). We grouped the 15 overrepresented patterns into seven classes, according to their cellular function. The most represented classes were related to kinesin molecular motors and to tetratricopeptide repeats involved in nuclear protein import and mitotic spindle, suggesting altogether a role in nucleus organization and dynamics (42,43). Interestingly, proteins displaying an LNR domain (44) were also overrepresented, suggesting that cell differentiation processes could be subject to a control by miRNAs.
Table 3.
Overrepresentation of functional motifs in the predicted targets
| Bank | Name | Proteome | Targets | Overrepresentation | P-value | Function |
|---|---|---|---|---|---|---|
| Panther | PTHR19959:SF16 | 59 | 22 | 4.00 | 5.62E-09 | Kinesin |
| Fprint | PR00381 | 38 | 17 | 4.80 | 1.20E-08 | Kinesin light chain |
| Panther | PTHR19959 | 80 | 23 | 3.08 | 6.27E-07 | Structural constituent of cytoskeleton |
| Pfam | PF00931 | 31 | 12 | 4.15 | 1.09E-05 | NB-ARC domain |
| superfamily | SSF48452 | 231 | 41 | 1.90 | 4.23E-05 | TPR-like superfamily |
| Pfam | PF07721 | 21 | 9 | 4.59 | 5.43E-05 | Tetratricopeptide repeat |
| Smart | SM00028 | 124 | 26 | 2.25 | 6.43E-05 | Tetratricopeptide repeats |
| Prosite | PS50293 | 166 | 31 | 2.00 | 1.35E-04 | TPR repeat region |
| Pfam | PF00515 | 61 | 14 | 2.46 | 1.21E-03 | Tetratricopeptide repeat |
| Gene3D | G3DSA:1.25.40.10 | 326 | 45 | 1.48 | 4.94E-03 | TPR-like_helical |
| Pfam | PF08241 | 49 | 12 | 2.63 | 1.46E-03 | Methyltransferase |
| Panther | PTHR10108 | 33 | 9 | 2.92 | 2.55E-03 | Methyltransferase |
| Pfam | PF00066 | 31 | 9 | 3.11 | 1.58E-03 | LNR domain |
| Prosite | PS00120 | 13 | 5 | 4.12 | 4.76E-03 | Lipases, serine active site |
| Gene3D | G3DSA:2.160.20.10 | 98 | 17 | 1.86 | 8.82E-03 | Pectin lyase-like |
Motifs were grouped by similar function, and groups were sorted by ascending best P-value. ‘Proteome’ and ‘Targets’ show the number of proteins containing at least one instance of the motif in the whole genome and in the set of targets that we predicted in silico, respectively. The ‘Overrepresentation’ is the ratio of the two previous columns, each normalized to its respective total number of proteins in the set. The P-value is computed using the hypergeometric probability law.
As a second step, we searched for conditions able to induce or repress the expression of the pre-miRs, as well as the corresponding miRNAs and their target genes. In parallel, we studied the expression of AGO1 and DCL1. Because E. siliculosus is a marine macro-alga, we tested two salt stress conditions on the sporophyte organism: hyper- and hypo-osmotic stresses. In addition, a morphologically different phase of the E. siliculosus life cycle, the gametophyte organism, was tested. The transcript level of AGO1 and DCL1 was significantly higher (Student t-test, α = 0.05) in response to hyper-salt stress conditions. In contrast, both the hypo-osmotic stress and the gametophytic stage did not modify their expression (Figure 7A). The expression profile of four pre-miRs was affected by growth conditions (Figure 7B). A differential expression level was statistically validated for two miRNAs: one for a hyper- and one for a hypo-saline stress. In contrast, the difference of expression between the gametophytes and the sporophytes appeared to be lower. The changes in miRNA expression were not statistically supported (not shown). We also quantified the transcript level of the target genes, by using oligonucleotides downstream from the predicted miRNA recognition site. In these experimental conditions, we noticed that variations in expression were not statistically higher than interindividual variations between the biological replicates (not shown).
Figure 7.
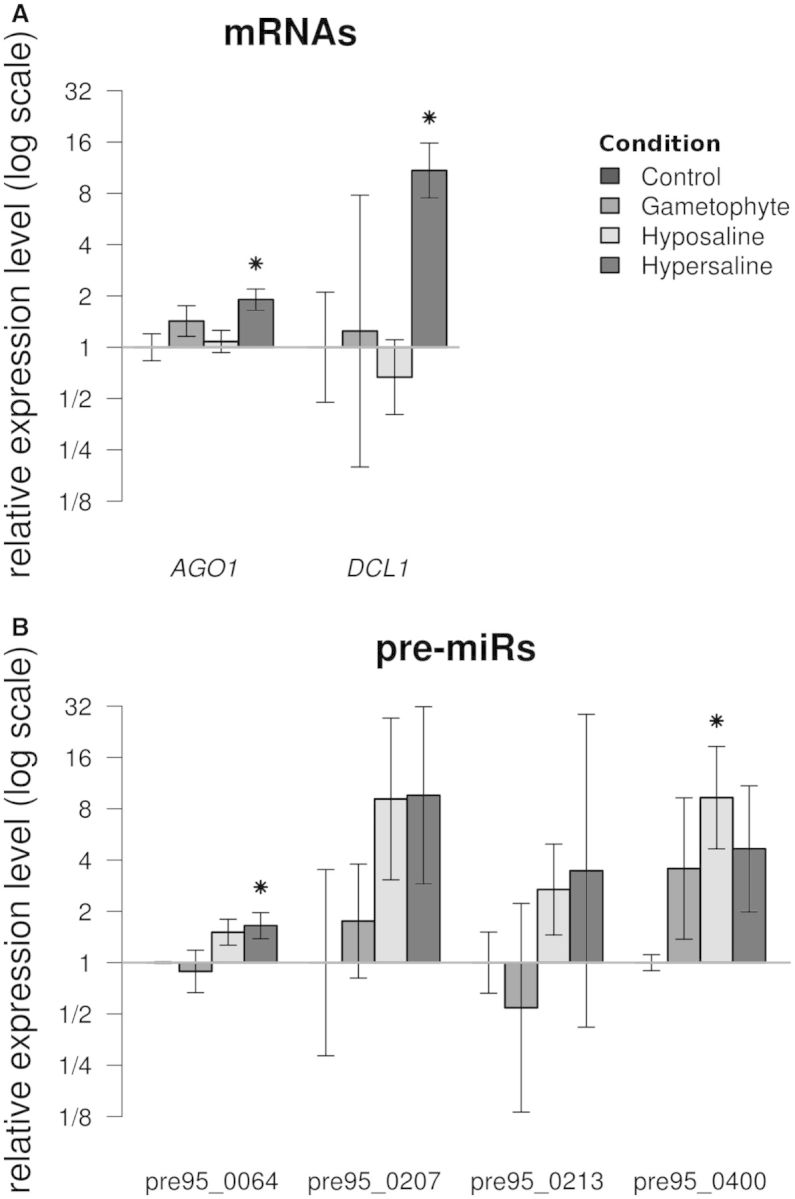
Transcript levels of miRNA processing proteins, pre-miRs, miRNAs and target genes measured by RT-qPCR in different algal materials. (A) AGO1 and DCL1 cDNAs. (B) Pre-miRs detected in Figure 4. The data are expressed as fold changes relative to the control. ‘Control’: sporophytes grown in normal culture conditions, used to set the reference value at 1; ‘Gametophyte’: gametophytes grown in normal culture conditions; ‘Hypo-saline’ and ‘Hyper-saline’: sporophytes subjected to corresponding osmotic stress conditions. Conditions for which the distribution of replicates values significantly differs from the control distribution (Student t-test, α = 0.05) are denoted by an asterisk.
DISCUSSION
This study presents the first genome-wide scale list of candidate miRNAs for an organism of the heterokont phylum. Our in silico search for new miRNA candidates in E. siliculosus was based on structural considerations, without any a priori on the sequence conservation or on the expression level of mature and/or precursor RNAs. Many features that are usually used to identify miRNA precursors in plants or animals had to be discarded because they were specific to one of these two kingdoms (45). The initial search step, however, was performed using findmiRNA, a software designed to detect a nearly perfect complementarity between the miRNA candidates and their target(s), as it is usual in plants. The rationale behind this choice is that a less-constrained search would have allowed to detect one or more target(s) for nearly any oligonucleotide with a length in the range expected for an miRNA (data not shown). Nevertheless, the higher expression level of the candidates, compared with sequences of the same origin (intergenic or intronic), constituted an emerging property of the predicted set of miRNAs. Our experimental confirmation suggested that the selection based on structural features of the precursors was efficient in reducing the ratio of false positives. This work also illustrated the fact that a large-scale prediction of miRNAs requires the combined use of computational and experimental analyses, whatever the order in which they are used (46). In any case, the relevance of the predictions relies on contextual information. For instance, any identification of miRNAs requires a clear distinction between coding (mRNAs) and noncoding (‘intergenic’ and intronic) sequences. Its accuracy is therefore strongly dependent on the initial assignment of nucleotides to these two sets. In particular, the annotation of UTRs can be critical, as miRNA target sites are expected to be found in UTRs (47,48). This is, however, one of the most difficult tasks in the primary annotation of a newly sequenced genome, and its output is not fully reliable. We tried to overcome these impediments by re-assigning the regions flanking the first and/or last exon to the mRNA sequences, in the case where the mRNA was devoid of a 5′UTR and/or a 3′UTR. Although this procedure lies on the reasonable hypothesis that the structure of the unknown UTRs is similar to that of the experimentally observed ones, it might nevertheless add some errors (both false positive and false negative) to the analysis. In these conditions, and after experimentally validating by RT-qPCR a subset of 72 miRNA candidates, we could extrapolate our prediction to a conservative number of 252 valid miRNAs. This number is likely under-estimated, as, in contrast to the in silico approach, the experimental detection of miRNAs is highly specific but suffers from a lack of sensitivity. Hence, many undetected candidates might be false negatives (15). In any case, the validation of each candidate and its implication in a given process would require a complex combination of ad hoc experiments.
The miRNAs identified in E. siliculosus display several specificities. First, they do not share significant sequence similarities with miRNAs already known in other species. Indeed, many miRNAs are species- or lineage-specific, and E. siliculosus is the first heterokont in which miRNAs are known. More precisely, the predicted miRNAs of E. siliculosus are as different from their closest animal miRNAs as plant miRNAs are, and as different from their closest plant miRNAs as animal miRNAs are. This is supported by the position of brown algae in the tree of life, distant from both the ‘opisthokonta’ and the ‘archaeplastida’. Secondly, their position within the genome was peculiar. While in the metazoan species studied to date pre-miR clustering is frequent (49), and examples are also known in plants [see for instance (50,51)], in E. siliculosus we found only three miRNA gene clusters, each comprising three genes. Interestingly, this low prevalence of miRNA gene clusters can be related to the low frequency of tandem repeats in the genome of E. siliculosus (13). In addition, only one of these clusters contained two pre-miRs sharing extensive similarity, whereas metazoan miRNA clusters are often made up of genes of the same family. From this criterion, the E. siliculosus miRNAs seem to be closer to plant miRNAs than to animal miRNAs. Conversely, about one-third of the predicted miRNA precursors were located in introns of protein-coding genes, a feature shared with human miRNAs, which are intronic in 25–40% of the cases (52), in contrast to plant miRNAs (53). Finally, several pre-miRs were detected, suggesting that these molecules have a sufficiently long lifetime, like most animal pre-miRs have, but usually not plant pre-miRs (54). Therefore, the genomic organization and biogenesis of E. siliculosus miRNAs share features with either animals or plants, again illustrating its original evolutionary history.
In previously studied cases, the mechanisms by which miRNAs inhibit their target mRNA can be divided into two main classes: mRNA cleavage or translation repression. The AGO protein in the RISC complex is able to conduct mRNA cleavage if it contains a nucleolytic triad made of three conserved residues: Asp(760), Asp(846) and Asp/His(986) (numbering of Arabidopsis thaliana AGO1) (55). The AGO protein of E. siliculosus does contain these three residues, namely Asp (703), Asp (775) and His (912). It is thus expected to perform the endonucleolytic cleavage of the target mRNA. Despite numerous attempts (RACE-PCR on the target mRNA candidates), cleavage of the predicted target genes by miRNAs could not be demonstrated (data not shown). These negative results do not allow to rule out the possibility that miRNAs in E. siliculosus direct the cleavage of their target. However, future work should consider the hypothesis that, although the required residues are present in AGO, the mechanism by which miRNAs regulate their targets in E. siliculosus might rely on translation inhibition rather than on mRNA cleavage. A similar situation has been shown to occur in human (56). The actual mechanism of this effect remains to be demonstrated.
Expression studies performed by RT-qPCR revealed a possible involvement of the miRNA machinery in physiological processes. The four operating levels of this machinery corresponding to (i) the RISC RNAse Argonaute and the RNAse DICER, (ii) the pre-miR, (iii) the miRNA and (iv) the miRNA targets, were investigated using this approach. Both Argonaute (AGO1) and DICER (DCL1) genes and some of the pre-miRs tested were induced in response to a modification in salt concentration. These changes in AGO1 and DCL1 expression on stress distinguish E. siliculosus from other organisms. In animals, various stress conditions result in a decrease in DCL1 expression (57). In plants, the expression pattern of miRNA-related proteins is often complex, as it involves multi-copy genes, with divergent expression patterns within each family (58). For instance, in A. thaliana, all four DCL transcript levels are depleted on a salt stress, but each gene exhibits a distinct time course and intensity for this regulation (59). In Oryza sativa, only one AGO gene among 19 is induced in stress conditions, while none of the eight DCL genes is affected (60). Although a systematic study in a broad range of animals and plants remains to be conducted, the induction of DCL by a salt stress seems to be an exception, and might be a distinctive feature of this marine brown alga, along with the existence of one single instance of AGO1 and DCL1 genes. This can be related to the particular environmental conditions macro-algae have to face. These organisms are living attached to rocks or to other algae, and hence are subject to salt concentration variations during the day depending on tides and evaporation. We could not validate any differential expression of the corresponding target mRNAs. Therefore, if an miRNA-regulated process is involved in response to salt stress, we propose that its mechanism should rely on translational repression rather than cleavage and degradation of the target. In addition to a role in salt stress, miRNA-mediated gene expression regulation could also be involved in developmental processes. The target prediction by findmiRNA allows for substantial mismatches in miRNA-mRNA base pairing. This is in agreement with the addressing of the RISC complex to its target. However, this loose constraint might generate many false-positive targets. For this reason, we cannot analyze individual targets in the whole set of predictions, most of which have not been experimentally validated. However, unless there are reasons to suspect a coincidental specific bias toward a given class of proteins, the overrepresentation of a process among the predicted target can point to possible roles for miRNAs in cell processes. The predicted targets of E. siliculosus miRNAs represent a wide variety of functions and protein families, notably involved in nucleus dynamics, cell polarity and differentiation. These molecular functions, extensively conserved through the tree of life have been shown to be regulated by miRNAs in other organisms: kinesins are regulated by miRNAs in human (61), like the NB-ARC containing protein APAF1 (62). Similarly, expressed proteins containing kinesin and NB-ARC domains have been predicted to be regulated by miRNAs in A. thaliana and O. sativa (63). MiRNAs also regulate methyl-transferases (64) and Notch-related proteins (65). Interestingly, the recent characterization of the E. siliculosus morphogenetic mutant ‘étoile’ supported a role of Notch-related proteins in cell differentiation, with a mechanism, which remains to be identified (66). In addition, we predicted that miRNAs could be able to regulate some of the functional families identified as stress responsive by a transcriptomic study in E. siliculosus (67). Finally, in contrast to the already known miRNAs in E. siliculosus (13), we did not identify any bias toward the proteins containing Leucine-rich repeats.
In summary, the list of E. siliculosus miRNAs proposed in this study is a solid starting point for further investigations aiming at deciphering in detail their biological roles, as well as the molecular mechanisms by which they operate. In this perspective, brown algae represent a source of novelty because of their extraneous phylogenetic position.
ACCESSION NUMBERS
The validated pre-miRs were deposited in miRBase under the following accession numbers (see also Supplementary Table S1D): pre95_0213a = esi-MIR8618b; pre95_0055a = esi-MIR8619; pre95_0064a = esi-MIR8620; pre95_0207a = esi-MIR8622b; pre90_0829a = esi-MIR8623b; pre90_0257a = esi-MIR8623d; pre95_0365a = esi-MIR8624a; pre95_0400a = esi-MIR8625.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: French Centre National de la Recherche Scientifique and Université Pierre et Marie Curie.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The Interproscan analysis was run on the ABIMS platform (Station Biologique de Roscoff). The authors wish to thank Marie-Hélène Mucchielli-Giorgi for her expertise in SVM analysis, Martine Boccara (ENS, Paris), Martin Crespi, Christine Lelandais-Brière and Florian Frugier (ISV, CNRS Gif-sur-Yvette) for fruitful discussion and critical reading of the manuscript.
REFERENCES
- 1.Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 2.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 3.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Axtell MJ, Westholm JO, Lai EC. Vive la différence: biogenesis and evolution of microRNAs in plants and animals. Genome Biol. 2011;12:221. doi: 10.1186/gb-2011-12-4-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon HS, Grant J, Tekle YI, Wu M, Chaon BC, Cole JC, Logsdon JMJ, Patterson DJ, Bhattacharya D, Katz LA. Broadly sampled multigene trees of eukaryotes. BMC Evol. Biol. 2008;8:14. doi: 10.1186/1471-2148-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haas BJ, Kamoun S, Zody MC, Jiang RHY, Handsaker RE, Cano LM, Grabherr M, Kodira CD, Raffaele S, Torto-Alalibo T, et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009;461:393–398. doi: 10.1038/nature08358. [DOI] [PubMed] [Google Scholar]
- 7.Baxter L, Tripathy S, Ishaque N, Boot N, Cabral A, Kemen E, Thines M, Ah-Fong A, Anderson R, Badejoko W, et al. Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science. 2010;330:1549–1551. doi: 10.1126/science.1195203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lévesque CA, Brouwer H, Cano L, Hamilton JP, Holt C, Huitema E, Raffaele S, Robideau GP, Thines M, Win J, et al. Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol. 2010;11:R73. doi: 10.1186/gb-2010-11-7-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Armbrust EV, Berges JA, Bowler C, Green BR, Martinez D, Putnam NH, Zhou S, Allen AE, Apt KE, Bechner M, et al. The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science. 2004;306:79–86. doi: 10.1126/science.1101156. [DOI] [PubMed] [Google Scholar]
- 10.Bowler C, Allen AE, Badger JH, Grimwood J, Jabbari K, Kuo A, Maheswari U, Martens C, Maumus F, Otillar RP, et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature. 2008;456:239–244. doi: 10.1038/nature07410. [DOI] [PubMed] [Google Scholar]
- 11.Huang A, He L, Wang G. Identification and characterization of microRNAs from Phaeodactylum tricornutum by high-throughput sequencing and bioinformatics analysis. BMC Genomics. 2011;12:337. doi: 10.1186/1471-2164-12-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charrier B, Coelho SM, Le Bail A, Tonon T, Michel G, Potin P, Kloareg B, Boyen C, Peters AF, Cock JM. Development and physiology of the brown alga Ectocarpus siliculosus: two centuries of research. New Phytol. 2008;177:319–332. doi: 10.1111/j.1469-8137.2007.02304.x. [DOI] [PubMed] [Google Scholar]
- 13.Cock JM, Sterck L, Rouzé P, Scornet D, Allen AE, Amoutzias G, Anthouard V, Artiguenave F, Aury J, Badger JH, et al. The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature. 2010;465:617–621. doi: 10.1038/nature09016. [DOI] [PubMed] [Google Scholar]
- 14.Lindow M, Gorodkin J. Principles and limitations of computational microRNA gene and target finding. DNA Cell Biol. 2007;26:339–351. doi: 10.1089/dna.2006.0551. [DOI] [PubMed] [Google Scholar]
- 15.Mendes ND, Freitas AT, Sagot M. Current tools for the identification of miRNA genes and their targets. Nucleic Acids Res. 2009;37:2419–2433. doi: 10.1093/nar/gkp145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allmer J, Yousef M. Computational methods for ab initio detection of microRNAs. Front. Genet. 2012;3:209. doi: 10.3389/fgene.2012.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, Rhoades MW, Burge CB, Bartel DP. The microRNAs of Caenorhabditis elegans. Genes Dev. 2003;17:991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai EC, Tomancak P, Williams RW, Rubin GM. Computational identification of Drosophila microRNA genes. Genome Biol. 2003;4:R42. doi: 10.1186/gb-2003-4-7-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng Kwang Loong S, Mishra SK. Unique folding of precursor microRNAs: quantitative evidence and implications for de novo identification. RNA. 2007;13:170–187. doi: 10.1261/rna.223807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hutvágner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002;297:2056–2060. doi: 10.1126/science.1073827. [DOI] [PubMed] [Google Scholar]
- 21.Vazquez F, Blevins T, Ailhas J, Boller T, Meins FJ. Evolution of Arabidopsis MIR genes generates novel microRNA classes. Nucleic Acids Res. 2008;36:6429–6438. doi: 10.1093/nar/gkn670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stark A, Brennecke J, Russell RB, Cohen SM. Identification of Drosophila MicroRNA targets. PLoS Biol. 2003;1:E60. doi: 10.1371/journal.pbio.0000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe Y, Yachie N, Numata K, Saito R, Kanai A, Tomita M. Computational analysis of microRNA targets in Caenorhabditis elegans. Gene. 2006;365:2–10. doi: 10.1016/j.gene.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 25.Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318:1931–1934. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 26.Gu S, Kay MA. How do miRNAs mediate translational repression? Silence. 2010;1:11. doi: 10.1186/1758-907X-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips N, Burrowes R, Rousseau F, de Reviers B, Saunders GW. Resolving evolutionary relationships among the brown algae using chloroplast and nuclear genes. J. Phycol. 2008;44:394–405. doi: 10.1111/j.1529-8817.2008.00473.x. [DOI] [PubMed] [Google Scholar]
- 29.Kawai H, Muto H, Fujii T, Kato A. A linked 5S rRNA gene in Scytosiphon lomentaria (scytosiphonales, phaeophyceae) J. Phycol. 1995;31:306–311. [Google Scholar]
- 30.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adai A, Johnson C, Mlotshwa S, Archer-Evans S, Manocha V, Vance V, Sundaresan V. Computational prediction of miRNAs in Arabidopsis thaliana. Genome Res. 2005;15:78–91. doi: 10.1101/gr.2908205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Bail A, Billoud B, Maisonneuve C, Peters A, Cock M, Charrier B. Early development pattern of the brown alga Ectocarpus siliculosus (ectocarpales, phaeophyceae) sporophyte. J. Phycol. 2008;44:1269–1281. doi: 10.1111/j.1529-8817.2008.00582.x. [DOI] [PubMed] [Google Scholar]
- 33.Hofacker I, Fontana W, Stadler P, Bonhoeffer L, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994;125:167–188. [Google Scholar]
- 34.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria; 2010. R Foundation for Statistical Computing. [Google Scholar]
- 35.Le Bail A, Dittami SM, de Franco P, Rousvoal S, Cock MJ, Tonon T, Charrier B. Normalisation genes for expression analyses in the brown alga model Ectocarpus siliculosus. BMC Mol. Biol. 2008;9:75. doi: 10.1186/1471-2199-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real–time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mestdagh P, Van Vlierberghe P, De Weer A, Muth D, Westermann F, Speleman F, Vandesompele J. A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol. 2009;10:R64. doi: 10.1186/gb-2009-10-6-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zdobnov EM, Apweiler R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 39.Ng Kwang Loong S, Mishra SK. De novo SVM classification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bioinformatics. 2007;23:1321–1330. doi: 10.1093/bioinformatics/btm026. [DOI] [PubMed] [Google Scholar]
- 40.Çakir M, Allmer J. Systematic computational analysis of potential RNAi regulation. In: Nazife B, Osman S, Mesude IC, Tolga C, editors. Toxoplasma gondii. 5th International Symposium on Health Informatics and Bioinformatics (HIBIT) Ankara, Turkey: Middle East Technical University; 2010. Vol. 5, pp. 31–38. [Google Scholar]
- 41.Shi R, Chiang VL. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques. 2005;39:519–525. doi: 10.2144/000112010. [DOI] [PubMed] [Google Scholar]
- 42.Paddy MR. The Tpr protein: linking structure and function in the nuclear interior? Am. J. Hum. Genet. 1998;63:305–310. doi: 10.1086/301989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tikhonenko I, Nag DK, Robinson DN, Koonce MP. Microtubule-nucleus interactions in Dictyostelium discoideum mediated by central motor kinesins. Eukaryot. Cell. 2009;8:723–731. doi: 10.1128/EC.00018-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fiúza U, Arias AM. Cell and molecular biology of Notch. J. Endocrinol. 2007;194:459–474. doi: 10.1677/JOE-07-0242. [DOI] [PubMed] [Google Scholar]
- 45.Bologna NG, Schapire AL, Palatnik JF. Processing of plant microRNA precursors. Brief. Funct. Genomics. 2013;12:37–45. doi: 10.1093/bfgp/els050. [DOI] [PubMed] [Google Scholar]
- 46.Mathelier A, Carbone A. MIReNA: finding microRNAs with high accuracy and no learning at genome scale and from deep sequencing data. Bioinformatics. 2010;26:2226–2234. doi: 10.1093/bioinformatics/btq329. [DOI] [PubMed] [Google Scholar]
- 47.Zhou X, Duan X, Qian J, Li F. Abundant conserved microRNA target sites in the 5'-untranslated region and coding sequence. Genetica. 2009;137:159–164. doi: 10.1007/s10709-009-9378-7. [DOI] [PubMed] [Google Scholar]
- 48.Brodersen P, Voinnet O. Revisiting the principles of microRNA target recognition and mode of action. Nat. Rev. Mol. Cell Biol. 2009;10:141–148. doi: 10.1038/nrm2619. [DOI] [PubMed] [Google Scholar]
- 49.Shomron N, Golan D, Hornstein E. An evolutionary perspective of animal microRNAs and their targets. J. Biomed. Biotechnol. 2009 doi: 10.1155/2009/594738. 2009, 594738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cui X, Xu SM, Mu DS, Yang ZM. Genomic analysis of rice microRNA promoters and clusters. Gene. 2009;431:61–66. doi: 10.1016/j.gene.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 51.Merchan F, Boualem A, Crespi M, Frugier F. Plant polycistronic precursors containing non-homologous microRNAs target transcripts encoding functionally related proteins. Genome Biol. 2009;10:R136. doi: 10.1186/gb-2009-10-12-r136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu Q, Spriggs A, Matthew L, Fan L, Kennedy G, Gubler F, Helliwell C. A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res. 2008;18:1456–1465. doi: 10.1101/gr.075572.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAS and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006;57:19–53. doi: 10.1146/annurev.arplant.57.032905.105218. [DOI] [PubMed] [Google Scholar]
- 55.Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song J, Hammond SM, Joshua-Tor L, Hannon GJ. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- 56.Pillai RS, Artus CG, Filipowicz W. Tethering of human Ago proteins to mRNA mimics the miRNA-mediated repression of protein synthesis. RNA. 2004;10:1518–1525. doi: 10.1261/rna.7131604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wiesen JL, Tomasi TB. Dicer is regulated by cellular stresses and interferons. Mol. Immunol. 2009;46:1222–1228. doi: 10.1016/j.molimm.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khraiwesh B, Zhu J, Zhu J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta. 2012;1819:137–148. doi: 10.1016/j.bbagrm.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Q, Feng Y, Zhu Z. Dicer-like (DCL) proteins in plants. Funct. Integr. Genomics. 2009;9:277–286. doi: 10.1007/s10142-009-0111-5. [DOI] [PubMed] [Google Scholar]
- 60.Kapoor M, Arora R, Lama T, Nijhawan A, Khurana JP, Tyagi AK, Kapoor S. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genomics. 2008;9:451. doi: 10.1186/1471-2164-9-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li G, Luna C, Qiu J, Epstein DL, Gonzalez P. Targeting of integrin beta1 and kinesin 2alpha by microRNA 183. J. Biol. Chem. 2010;285:5461–5471. doi: 10.1074/jbc.M109.037127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 2008;283:1026–1033. doi: 10.1074/jbc.M707224200. [DOI] [PubMed] [Google Scholar]
- 63.Lindow M, Jacobsen A, Nygaard S, Mang Y, Krogh A. Intragenomic matching reveals a huge potential for miRNA-mediated regulation in plants. PLoS Comput. Biol. 2007;3:e238. doi: 10.1371/journal.pcbi.0030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kisliouk T, Yosefi S, Meiri N. MiR-138 inhibits EZH2 methyltransferase expression and methylation of histone H3 at lysine 27, and affects thermotolerance acquisition. Eur. J. Neurosci. 2010;33:224–235. doi: 10.1111/j.1460-9568.2010.07493.x. [DOI] [PubMed] [Google Scholar]
- 65.Vallejo DM, Caparros E, Dominguez M. Targeting Notch signalling by the conserved miR-8/200 microRNA family in development and cancer cells. EMBO J. 2011;30:756–769. doi: 10.1038/emboj.2010.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Le Bail A, Billoud B, Le Panse S, Chenivesse S, Charrier B. ETOILE regulates developmental patterning in the filamentous brown alga Ectocarpus siliculosus. Plant Cell. 2011;23:1666–1678. doi: 10.1105/tpc.110.081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dittami SM, Scornet D, Petit J, Ségurens B, Da Silva C, Corre E, Dondrup M, Glatting K, König R, Sterck L, et al. Global expression analysis of the brown alga Ectocarpus siliculosus (Phaeophyceae) reveals large-scale reprogramming of the transcriptome in response to abiotic stress. Genome Biol. 2009;10:R66. doi: 10.1186/gb-2009-10-6-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.