


Abstract
Transfer RNA (tRNA) methylation is necessary for the proper biological function of tRNA. The N1 methylation of guanine at Position 9 (m1G9) of tRNA, which is widely identified in eukaryotes and archaea, was found to be catalyzed by the Trm10 family of methyltransferases (MTases). Here, we report the first crystal structures of the tRNA MTase spTrm10 from Schizosaccharomyces pombe in the presence and absence of its methyl donor product S-adenosyl-homocysteine (SAH) and its ortholog scTrm10 from Saccharomyces cerevisiae in complex with SAH. Our crystal structures indicated that the MTase domain (the catalytic domain) of the Trm10 family displays a typical SpoU-TrmD (SPOUT) fold. Furthermore, small angle X-ray scattering analysis reveals that Trm10 behaves as a monomer in solution, whereas other members of the SPOUT superfamily all function as homodimers. We also performed tRNA MTase assays and isothermal titration calorimetry experiments to investigate the catalytic mechanism of Trm10 in vitro. In combination with mutational analysis and electrophoretic mobility shift assays, our results provide insights into the substrate tRNA recognition mechanism of Trm10 family MTases.
INTRODUCTION
Transfer RNA (tRNA), which is typically 73–94 nt in length, serves as the carrier of activated amino acids to the ribosome for the biological synthesis of proteins (translation). The specific nucleotide sequence of an mRNA specifies the amino acids that are incorporated into the protein product of the gene from which the mRNA is transcribed, and the role of tRNA is to specify the corresponding amino acid from the genetic code. Therefore, tRNA plays an indispensable role in the process of protein translation. During tRNA maturation, extensive specific posttranscriptional modifications are introduced by many different enzymes to ensure proper tRNA structure and function (1,2). To date, over 100 different chemical modifications in tRNA from the three domains of life have been identified (3). Based on their affects on tRNA functions, posttranscriptional modifications can be generally divided into two groups: some modifications are thought to stabilize the 3D structure of tRNAs and are extremely important for in vivo stability, while others that are present within or in proximity to anticodon are well known to affect the efficiency or fidelity of translation (4–6).
A common tRNA modification is the methylation of ribonucleosides, including the N1 methylation of guanine (m1G). However, thus far, only a few m1G modifications have been identified in tRNA. The m1G37 modification, which is present at Position 37 (adjacent to 3′ of the anticodon) of the tRNA, is found to be essential in maintaining the reading frame fidelity (7). This modification is catalyzed by TrmD in bacteria and Trm5 in archaea and eukaryotes (8,9). Another modification is the m1G9 modification, which occurs at the junction of the D and acceptor stems. In Saccharomyces cerevisiae, this modification was identified in 10 tRNA species out of 34 sequenced tRNAs and was reported to be catalyzed by Trm10 (10). The Trm10 gene knockout strain of S. cerevisiae exhibited elevated sensitivity to 5-fluorouracil (5-FU) compared with the wild-type strain (11), indicating that loss of the m1G9 modification might result in the destabilization of tRNAs and growth defects of the strain. Additionally, another study provided genetic evidence supporting an important role for the m1G9 modification in translation termination efficiency (12). Moreover, Position 9 (G or A) of human tRNA was methylated by Trm10 homolog (13). Helm et al. reported that the in vitro transcript of human (mt)tRNALys that was deprived of the m1A9 modification did not fold into a cloverleaf, suggesting that the presence of a methyl group at A9 would prevent the Watson–Crick interaction A9–U64 and consequently hinder the formation of the extended amino acid-accepting stem (14).
RNA methyltransferases (MTases) often use the S-adenosyl-l-methionine (SAM) as a methyl donor to catalyze the methyl group transfer to various positions of the nucleotide bases, resulting in the formation of methylated RNA and S-adenosyl-homocysteine (SAH) as products (15). Thus far, all known RNA MTases can be classified into at least four unrelated superfamilies, including Rossmann-fold (RFM), SpoU-TrmD (SPOUT), radical-SAM and FAD/NAD(P)-dependent MTases (16,17). Most of the known MTases belong to the RFM family, which frequently exist as monomers (16). The SPOUT family includes SpoU and TrmD MTases and represents the second largest RNA MTase family. One structural characteristic of SPOUT MTases is that they form tightly bound homodimers that are mediated by the interaction between their MTase domains. An additional feature is the presence of a trefoil knot at the C-terminus of the MTase domain that is responsible for SAM binding (18,19).
Trm10 homologs are widely found in eukaryotes and archaea but not in eubacteria. Orthologs of Trm10 in archaea act at Position 9 of tRNA and catalyze both m1A and m1G formation (20). In vertebrates such as Homo sapiens, three Trm10 isoforms have been identified: TrmT10A, TrmT10B and TrmT10C. The human homologs TrmT10A and TrmT10B demonstrate tRNA m1G MTase activity at Position 9. TrmT10C was first identified as one subunit (MRPP1) of the human mitochondrial (mt)RNase P complex that is necessary to process tRNA precursors and even the maturation of human mitochondrial (21). However, TrmT10C requires another protein, SDR5C1 (MRPP2), to execute its m1R9 (G9 and A9) MTase and additional RNase P activity (13). There is no identifiable sequence homology between the Trm10 family and previously characterized MTases, including m1G MTases, such as Trm5 and TrmD. Thus far, there is no available structural information for the Trm10 family, although a bioinformatics study predicted that Trm10 may be a member of the SPOUT superfamily (19). Knowledge of the 3D structure of Trm10 family MTases would aid in the understanding of the tRNA substrate recognition and catalytic mechanism of Trm10 for tRNA modification.
Here, we report the crystal structures of the Schizosaccharomyces pombe tRNA m1G9 MTase Trm10 in the presence and absence of SAH in addition to the crystal structure of S. cerevisiae Trm10 in complex with SAH, which represent the first crystal structures of the Trm10 family. Using small angle X-ray scattering (SAXS), we further demonstrate that Trm10 functions as a monomer in solution. In vitro tRNA MTases assays and electrophoretic mobility shift assays (EMSA) provide a better understanding of the structure–function relationship of the Trm10 family.
MATERIALS AND METHODS
Protein expression and purification
The Trm10 gene was PCR amplified from yeast genomic DNA using PrimeSTAR HS DNA Polymerase (TaKaRa). The PCR fragments were inserted into the expression vector pET28b (Novagen, modified) to generate the recombinant plasmid that encodes an N-terminally His-tagged Trm10 for expression in Escherichia coli. The plasmid was then transformed into E. coli BL21/Gold (DE3) (Novagen) competent cells. Selenomethionine-labeled spTrm10_74 (residues 74-281) was expressed in the B834 strain. Cells were grown at 37°C to an OD600 of 0.8–1.0, and protein expression was induced through the addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were collected by centrifugation after 24 h of incubation at 16°C and were then lysed by sonication in lysis buffer A (20 mM Tris–HCl, pH 7.0 and 1 M NaCl) containing 10 µg/ml DNase and 10 µg/ml RNase. The crude lysate was then centrifuged at 14 000 rpm for 30 min at 4°C. The supernatant was applied to a Ni-NTA column (QIAGEN) followed by size exclusion chromatography using a Superdex 200 (GE Healthcare) column and cation exchange chromatography using an SP Sepharose Fast Flow column. The purified protein was then dialyzed against buffer B (containing 20 mM Tris–HCl, pH 7.0 and 150 mM NaCl). Mutations of Trm10 were generated using PCR and MutanBEST kit (TaKaRa). Mutant proteins were expressed under identical conditions as those used for the expression of wild-type Trm10.
TrmT10C (residues 40–403) was cloned into expression vector pET21b, and the plasmid for the expression of SDR5C1 was a gift from Dr Walter Rossmanith (Medical University of Vienna). The purification of the recombinant TrmT10C-SDR5C1 complex was performed as previously described (13). Purified His-tagged SDR5C1 was saturated with an excess of native untagged TRMT10C (in the form of a crude bacterial lysate), and the mixture of the two proteins was applied on a Ni–NTA column. A TrmT10C Q226A mutant in complex with SDR5C1 was prepared using the identical procedures as previously described.
Crystallization and data collection
The native Trm10 protein and the Trm10–SAH complex (which was prepared by mixing Trm10 with SAH at a 1:3 molar ratio) were concentrated to 25 mg/ml prior to crystallization trials. In initial crystallization attempts, Crystal Screen, Crystal Screen 2, Index, Grid Screen 1, Grid Screen 2 (Hampton Research) and protein–protein complex crystallization screening kits (22) were used to screen for crystallization conditions. Subsequently, 1 μl of protein and 1 μl of reservoir solution were mixed together, and single crystals of all proteins were grown using the hanging drop vapor diffusion method at 20°C. SpTrm10_74-apo (residues 74–281) and selenomethionine (Se-Met)-derivatized spTrm10_74-SAH were crystallized using an identical reservoir solution containing 0.1 M NaAc, pH 5.0 and 15% PEG 6000 (w/v). SpTrm10_FL (full-length)-SAH was crystallized in a buffer consisting of 0.1 M NaAc, pH 5.0 and 15% PEG 8000 (w/v). The crystal of scTrm10_84 (residues 84–276)-SAH was obtained in 0.1 M Bis–Tris–HCl, pH 7.0 and 1.5 M Li2SO4 monohydrate. The N-terminal deletions of Trm10 (spTrm10_74 and scTrm10_84) were chosen based on multiple sequence alignment, fold prediction and limited proteolysis (trypsin). The result of limited proteolysis analysis is shown in Supplementary Figure S7G.
For data collection, all crystals were soaked in a cryoprotectant solution consisting of the respective reservoir solution supplemented with 20% (v/v) glycerol and then flash frozen in liquid nitrogen. Datasets for all aforementioned crystals were collected on beamline 17U at the Shanghai Synchrotron Radiation Facility (SSRF) at 100 K.
Structure determination and refinement
A SAD dataset was integrated using iMosflm (23) and scaled with SCALA (24) from the CCP4 program suite (25), and the calculation of initial phase was performed using AutoSol and AutoBuild in PHENIX (26). The native datasets were processed using HKL2000 (27) and the structures were determined by molecular replacement using Phaser (28) in the CCP4 package. All models were further built and refined using Coot (29) and Refmac5 (30), respectively. The quality of the structures was validated using PROCHECK (31). The details regarding the data collection and processing of these crystal structures are presented in Table 2.
Table 2.
Crystal parameters, data collection and structure refinement
| Se-spTrm10-74-SAH | spTrm10-74-apo | spTrm10-FL (full length)-SAH | scTrm10-84-SAH | |
|---|---|---|---|---|
| SAD | ||||
| Crystal parameters | ||||
| Space group | P 21 21 21 | C 2 2 21 | P 21 | C 2 |
| a, b, c (Å) | 51.57, 73.16, 119.56 | 51.67, 69.56, 121.07 | 57.82, 45.65, 66.41 | 131.58, 58.86, 99.76 |
| α, β, γ (°) | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 | 90.00, 100.18, 90.00 | 90.00, 131.13, 90.00 |
| Data collection statistics | ||||
| Wavelength | 0.97917 | 0.97915 | 0.97922 | 0.97922 |
| Resolution | 51.57–2.40 (2.40–2.53)a | 50–2.50 (2.50–2.59) | 50–2.04 (2.04–2.11) | 50–1.76 (1.76–1.82) |
| Used reflections | 17384 | 7469 | 20378 | 53987 |
| Rmergeb | 0.113 (0.404) | 0.090 (0.492) | 0.066 (0.435) | 0.066 (0.507) |
| I/σI | 16.6 (7.8) | 24.35 (6.44) | 20.24 (3.65) | 19.43 (3.26) |
| Completeness | 99.9 (100.0) | 99.8 (100.0) | 97.3 (95.1) | 99.7 (100.0) |
| Redundancy | 13.6 | 7.0 | 3.6 | 3.7 |
| Refinement statistics | ||||
| Protein molecules in ASU | 2 | 1 | 2 | 2 |
| Residues | 370 | 175 | 365 | 386 |
| Water molecules | 115 | 36 | 147 | 328 |
| Total no. of atoms | 3230 | 1591 | 3192 | 3685 |
| Rfactor/Rfreec | 0.2164/0.2619 | 0.2111/0.2839 | 0.1979/0.2567 | 0.2187/0.2511 |
| RMSD bond lengths (Å) | 0.0084 | 0.0167 | 0.0128 | 0.0132 |
| RMSD bond angles (°) | 1.2153 | 1.7272 | 1.3674 | 1.3955 |
| Mean B factor (Å2) | 23.37 | 44.82 | 31.29 | 30.31 |
| Ramachandran plot (% residues) | ||||
| Most favored | 87.6 | 92.6 | 89.6 | 91.5 |
| Additional allowed | 12.4 | 7.4 | 10.4 | 8.5 |
| Disallowed regions | 0 | 0 | 0 | 0 |
aValues in parentheses are for highest-resolution shell. bRmerge = ∑|Ii − <I>|/∑|I|, where Ii is the intensity of an individual reflection and <I> is the average intensity of that reflection. cRfactor = ∑||Fobs| − |Fcalc||/∑|Fobs| for all reflections, Rfree was calculated on the 5% of data excluded from refinement. ASU means asymmetric units.
In vitro transcription of tRNA
The DNA templates of tRNA containing a T7 RNA polymerase promoter were generated by a two-step PCR. The primer sequences are provided in Supplementary Table S1, and the cloverleaf structures of the tRNAs are presented in Supplementary Figure S1. All tRNAs were prepared by in vitro transcription using T7 RNA polymerase. Transcription was performed at 37°C for 6 h using 200 mM HEPES–Na pH 8.0, 6 mM of each nucleotide triphosphate, 5 mM DTT, 1.25 mM spermidine, 30–50 mM MgCl2, 0.01% (v/v)Triton X-100, 20 µg of template DNA and T7 RNA polymerase in a 5-ml volume, followed by DNase I (RNase free) treatment. Subsequently, the transcription products were purified using denaturing polyacrylamide gel electrophoresis. The gel containing the tRNA transcripts was first treated using a crush and soak method in NaAc buffer (0.3 M NaAc, pH 5.3 and 2 mM EDTA). The tRNA product in the collected buffer was then dialyzed into Tris buffer (50 mM Tris–HCl, pH 8.0 and 50 mM NaCl) and quantified using the absorbance at 260 nm. Prior to use, the tRNA substrates were heat-denatured at 85°C for 2 min and annealed at room temperature for an additional 20 min in the presence of 2 mM MgCl2.
In vitro methyltransferase activity assay
The reaction mixture for the tRNA MTase assay (40 μl) consisted of 50 mM Tris–HCl, pH 8.0, 3 mM MgCl2, 1 mM DTT, 50 mM NH4Ac, 1 mM spermidine, 2 µCi 3H-SAM, 0.5 µM enzyme and 3.2 µM tRNA. The reaction mixture was then incubated at 30°C for 2 h (Supplementary Figure S2A). The reaction was stopped through the addition of 100 µl of water-saturated phenol. The aqueous phase was carefully separated from phenol phase and further extracted with 40 µl of chloroform/isoamyl alcohol (24:1 v/v). The tRNAs present in the supernatant were precipitated through the addition of cold ethanol. Two methods were used to examine the in vitro methyltransferase activity: (i) the precipitates were dissolved in DEPC-treated water and analyzed using Urea–PAGE and autoradiography; (ii) the precipitates were collected by filtration through a nitrocellulose membrane (Millipore), washed with cold ethanol, and air-dried before scintillation counting (32,33). For each counting assay, control experiments were performed and background radioactivity was subtracted from the data.
The apparent kinetic parameters of spTrm10 and scTrm10 for tRNA were determined from a Lineweaver–Burk plot under saturating 3H-SAM condition (25 μM). Increasing concentrations of sptRNAGly (0, 1, 1.5, 2, 4 and 10 μM for spTrm10; and 0, 1, 1.5, 2, 3, 6 and 15 μM for scTrm10) were premixed with saturating 3H-SAM at 30°C. The reaction was initiated through the addition of the enzyme at a final active concentration of 100 nM for spTrm10 and scTrm10.
SAXS measurement and data processing
The spTrm10 protein was characterized at 1, 3 and 5 mg/ml in Tris buffer (30 mM Tris–HCl, pH 7.0, 200 mM NaCl and 5 mM DTT). SAXS experiments were performed at the beamline 12ID-B of the Advanced Photon Sources (APS) at Argonne National Laboratory (Chicago site) with a wavelength of 1.033 Å. The data were analyzed using the ATSAS package (34) following the standard procedures. After subtracting buffer scattering, the data curves from different concentrations were scaled and merged using PRIMUS (35). GNOM (36) was employed to estimate the particle maximum dimension (Dmax) and calculate of the pair distance distribution function (PDDF). The radius of gyration (Rg) of the protein was derived in real space using the PDDF. The solute molecular mass was evaluated using an online tool called SAXS MOW (37). Chain-compatible dummy residues (DR) models of spTrm10_FL were constructed ab initio using the program GASBOR (38). The determined X-ray structure of spTrm10_74 was superposed onto the DR model by SUPCOMB (39) and visualized using VMD (40).
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) measurements were performed using an ITC-200 titration calorimeter (MicroCal, Northampton, MA, USA). The measurements were performed at 20°C in buffer B (20 mM Tris–HCl, pH 7.0 and 150 mM NaCl). SAM at a concentration of 1.5 mM was titrated into wild-type or mutant Trm10 (100 μM). Control experiments were performed under identical conditions by titrating SAM into buffer alone. The ITC data were subsequently analyzed using MicroCal Origin 7.0 software. The ITC-binding isotherms are presented in Supplementary Figure S9.
IR-EMSA assays
The sptRNAGly transcript was labeled with Alexa Fluor® 680 Maleimide (Invitrogen) for IR-EMSA (infrared EMSAs). A thiol group was modified to the 5′-end of the in vitro-transcribed sptRNAGly through cystamine modification of 5′-phosphate groups using EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride; Thermo), imidazole and additional DTT (dithiothreitol). The SH-labeled tRNA was purified by size exclusion chromatography in PBS buffer (10 mM sodium phosphate, pH 7.2, 150 mM NaCl and 10 mM EDTA) (41). The SH-activated tRNA was then labeled using Alexa Fluor® 680 Maleimide according to manufacturer’s protocol. Labeled tRNA was isopropanol precipitated and further purified using size-exclusion chromatography. The concentration of tRNA was determined by measuring the absorbance at 260 nm.
IR-EMSA were performed in a 20-µl reaction mixture containing 50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 3 mM MgCl2, 30 nM tRNA and the enzyme at different concentrations. Reaction mixtures were incubated at 37°C for 30 min, immediately applied to a 5% nondenaturing polyacrylamide gel composed of 1× TBE buffer, and electrophoresed at 4°C for 30 min at a constant voltage of 100 V. Subsequently, the gel was directly scanned by an Odyssey® Infrared Imaging System to quantitate bound (shifted) and unbound RNA. The bound fraction was plotted against the protein concentration, and the binding curves were fit according to a one-site binding mode using Origin software.
CD spectroscopy
The purified Trm10 and its mutants (0.1 mg/ml) in 30 mM sodium phosphate buffer (pH 7.0) were loaded into a quartz cuvette (d = 0.1 cm path length) and CD spectra from 190 to 260 nm were recorded on a JASCO-J810 spectropolarimeter at 25°C. A buffer-only sample was used as a reference. All CD spectra represent the average of three successive spectra. The molar ellipticities (θ) were plotted versus wavelength, and the reference curve was subtracted from each curve. The resulting spectra are presented in Supplementary Figure S10. The percentages of secondary structure were estimated using the protein secondary structure estimation program provided with the JASCO J-810 spectropolarimeter.
Molecular modeling of the spTrm10–SAH–GMP complex
Molecular docking was performed using AutoDock 4.2 (42). AutoDock uses a scoring function based on the AMBER force field and estimates the binding free energy of a ligand for its target protein. First, all bound waters were removed from the spTrm10–SAH complex structure. The protein was checked for polar hydrogens, and partial atomic charges were assigned. Torsion bonds of the small molecule ligand GMP were selected and defined. Second, a 3D grid box was created using AutoGrid (part of the AutoDock package). The grid maps representing the intact ligand in the actual docking target site were calculated using AutoGrid. A 30-Å grid size (x, y, z) for a 3D grid box and a default spacing of 0.375 Å were used. Eventually, cubic grids encompassed the binding site where the intact ligand was embedded. Finally, AutoDock was used to calculate the binding free energy of given ligand conformations in the macromolecular structure. At the end of the docking run, among the 10 ligand conformations (named 1–10), we found that 4 conformations are similar (clusters, conformations 6, 7, 8 and 9) with relatively low binding free energies (Supplementary Figure S6A and S6B). Therefore, these clusters can be considered the best conformations.
Limited proteolysis
The limited proteolysis of spTrm10 in the absence and presence of sptRNAGly was performed using trypsin. In 30-µl reactions, 1 mg/ml of spTrm10 in the presence or absence of equivalent sptRNAGly was incubated at room temperature with trypsin at an enzyme to substrate molar ratio of 1:100. At time points 0 min (before trypsin addition), 1, 2 and 3 min, 5-µl aliquots were removed from the sample and immediately mixed with 1× SDS loading buffer. Each aliquot was then boiled at 95°C to quench proteolysis before gel analysis.
RESULTS
Trm10 from S. pombe demonstrates identical tRNA MTase activity as Trm10 from S. cerevisiae
Previous studies have reported that Trm10 family is an m1R9 (m1G9 and m1A9) tRNA MTase. ScTrm10 from S. cerevisiae was found to be required for m1G9 modification of tRNAGly (10). Orthologs of Trm10 in archaea can catalyze both m1A and m1G formation at Position 9 of tRNAs (43). Additionally, the human mitochondrial maturation subcomplex TrmT10C–SDR5C1 was found to be responsible for the formation of m1G9 and m1A9 in (mt)tRNAs (13).
Our multiple sequence alignment analysis revealed that spTrm10 from the fission yeast S. pombe exhibits a domain architecture that is similar to the orthologous scTrm10 (Figures 1A and 2C). To investigate whether spTrm10 can catalyze the modification of tRNA, we prepared sptRNAGly (from S. pombe) transcript and subsequently determined the in vitro tRNA MTase activity. Similar to scTrm10, spTrm10 was able to methylate the in vitro-transcribed sptRNAGly. Additionally, the MTase domains of the two proteins display MTase activity as well, although the activity is relatively low when compared with full-length Trm10 (Figure 1B).
Figure 1.

Trm10 from yeast specifically catalyzes G9 methylation of tRNAGly. (A) A schematic representation of Trm10 from S. pombe and S. cerevisiae. (B) MTase activity of the following Trm10 constructs using sptRNAGly as the substrate: spTrm10_FL (full length), spTrm10_74 (residues 74–281), scTrm10_FL (full length) and scTrm10_84 (residues 84–276). (C) MTase activity of spTrm10 using wild-type sptRNAGly and tRNAGly with mutations at Position 9 and MTase activity of scTrm10 with sptRNAGly(G9) and sctRNAGly(A9). tRNA_G9, tRNA_G9A, tRNA_G9C and tRNA_G9U originate from S. pombe, and tRNA_A9 originates from S. cerevisiae. In (B) and (C), the upper panels represent the results of autoradiography analysis, and the lower panels represent the results of scintillation counting. Error bars indicate the standard deviation of three separate measurements.
Figure 2.

Overall structure of Trm10. (A) The crystal structure of the spTrm10_74–SAH complex. The product SAH is shown as a ball-and-stick model and is colored yellow. (B) Topology diagram of the Trm10 MTase domain with the C-terminal trefoil knot indicated by a gray box. (C) Multiple sequence alignment of the following members of the Trm10 family: SpTRM10 from S. pombe (UniProtKB entry O14214); scTRM10 from S. cerevisiae (Q12400); caTRM10 from Candida albicans (Q59Q39); and hsTRMT10A, hsTRMT10B, and hsTRMT10C from H. sapiens (Q8TBZ6, Q6PF06 and Q7L0Y3, respectively). The secondary structures of the spTrm10 MTase domain are shown above the sequences. Strictly conserved residues and highly conserved residues are colored orange and yellow, respectively. Residues that were mutated in this study are indicated by stars; blue stars indicate the residues that are involved in tRNA binding, and red stars indicate the residues that are necessary for MTase activity. Motif I (β4–β5), motif II (β5–α4) and motif III (β6–α5) are boxed in cyan.
Furthermore, to confirm that the MTase activity of yeast Trm10 is specific for G9 of tRNAGly, we designed three sptRNAGly mutants at Position 9 (G9A, G9C and G9U) and determined the MTase activity. Our results, as shown in Figure 1C, indicated that the MTase activities were completely abolished for all three tRNAGly mutants. Additionally, we cannot detect the m1A9 MTase activity of scTrm10 for sctRNAGly (from S. cerevisiae) in vitro, indicating that yeast Trm10 is specifically involved in the m1G9 modification of tRNA, which is consistent with a previous study (13).
Moreover, to better explore the kinetic mechanism of Trm10, we further determined the apparent kinetic parameters (Km and Kcat) of the full-length Trm10 for sptRNAGly. The results are summarized in Table 1 and Supplementary Figure S2B and S2C, respectively. The apparent Km values of spTrm10 and scTrm10 were determined to be 3.2 and 5.3 μM, respectively, which are similar to those of TrmD and Trm5 (2.8 and 1.2 μM, respectively) (44), indicating that m1G MTases possess a similar affinity for substrate tRNA from different species. In addition, similar values of Kcat were obtained for spTrm10 and scTrm10 (0.16 and 0.17 min−1, respectively), which are consistent with a very recent study of Trm10 family (0.14–0.72 min−1) (45). And the Kcat/Km values of spTrm10 and scTrm10 are 0.05 and 0.032 μM−1 min−1, respectively. Compared with the Kcat values of TrmD and Trm5 (1.2–1.5 min−1) (44,46–47), our results indicated that Trm10 does not exhibit such high catalytic efficiency towards substrate tRNAs as TrmD and Trm5.
Table 1.
Kinetic parameters of Trm10 for substrate sptRNAGly
| Kcat (min−1) | Km (μM) | Kcat/Km (μM−1 min−1) | |
|---|---|---|---|
| spTrm10 | 0.16 ± 0.022 | 3.2 ± 0.12 | 0.050 ± 0.0018 |
| scTrm10 | 0.17 ± 0.035 | 5.3 ± 0.52 | 0.032 ± 0.0071 |
Overall structures of Trm10 from S. pombe and S. cerevisiae
To gain insights into the catalytic mechanism of Trm10, we solved four crystal structures in this study. The structure of spTrm10_74 in complex with SAH was determined by single wavelength anomalous dispersion (SAD) using selenomethionine-labeled protein and was refined to 2.4 Å resolution. The structures of the apo spTrm10_74 and spTrm10_FL–SAH complex were determined at 2.5 and 2.0 Å, respectively, by molecular replacement. However, some regions of spTrm10_FL were not visible in the electron density and could not be modeled. These regions include the N-terminus (residues 1–83), C-terminus (residues 271–304) and one loop spanning residues 236–240. The N- and C-terminal regions are likely disordered or highly flexible and are apparently dispensable for the core structure. Subsequently, the structure of scTrm10_84 in complex with SAH was determined at a resolution of 1.8 Å by molecular replacement. The details of the data collection and refinement statistics are summarized in Table 2.
The determined structures all adopt a globular α/β structure consisting of six-stranded β-sheets sandwiched by α-helices at both sides (Figure 2A). The topology of the globular domain is shown in Figure 2B, and the N-terminal half resembles an RFM MTase fold, whereas the C-terminal half of Trm10 exhibits a deep trefoil knot, which forms a deep crevice to accommodate the product SAH and represents a possible active site of the enzyme. Moreover, multiple sequence alignment of the Trm10 family revealed that the MTase domain is highly conserved (Figure 2C). Overall, the four Trm10 structures are highly similar, displaying a root mean square deviation (RMSD) of 0.6 Å for the aligned 140 Cα atoms (Supplementary Figure S3).
In addition, considering that the Trm10 family does not exhibit obvious sequence homology to other MTases, the Dali server (48) was used to search for the protein structures that are similar to Trm10. The results revealed that the MTase domain of Trm10 was structurally homologous to the SPOUT family MTases, such as YibK (PDB code 1MXI) (49), with a Dali Z-score of 11.8 and an RMSD of 3.2 Å for 156 Cα atoms, and TrmH (PDB code 1V2X) (50), with a Z-score of 9.6 and an RMSD of 2.8 Å for 192 Cα atoms. Collectively, based on the Dali results and the characteristic trefoil knot that is present in Trm10, we propose that Trm10 may be a new member of the SPOUT MTase family.
Trm10 functions as a monomer in solution
SPOUT family MTases studied to date are known to functionally exist as homodimers, in which the catalytic site is formed at the interaction interface between two monomers (19,50–52). The interface of these interactions buries a solvent accessible surface area of 1511.3 Å2 in the TrmH dimer (PDB code 1V2X) and 1599.5 Å2 in the YibK dimer (PDB code 1MXI). However, a homodimer was not observed in the Trm10 crystal structures, and the interface between the two molecules of spTrm10_74 in the asymmetric unit only buries a solvent accessible surface area of 338.4 Å2, indicating that these molecules do not correspond to a biological dimer.
SAXS was utilized to investigate the structure of spTrm10_FL in solution. Figure 3A shows the experimental SAXS curve of spTrm10_FL, and the corresponding PDDF calculated by GNOM is shown in the top-right inset. The Rg and Dmax of the protein are 30.0 ± 0.6 Å and 98.5 Å, respectively. In addition, the molecular weight that was determined using SAXS MoW is ∼37.0 kDa, which is consistent with its theoretical molecular weight (36.6 kDa) and suggests that spTrm10 exists as a monomer in solution. The shape of the PDDF indicates that the solution structure of spTrm10_FL is quite extended. Ten DR models were generated using GASBOR (four of them are shown in Figure 3B), with final χ-values against the raw SAXS data ranging from 0.23 to 0.25. From these DR models, we can clearly observe an extended region and a globular region (Figure 3B), which may represent the N-terminus and MTase domain of Trm10, respectively. The X-ray structure of spTrm_74 can be superimposed onto the globular region of the DR models quite well, with normalized spatial discrepancy values from 0.94 to 1.07. The orientation of the extended region in different DR models varies (Figure 3B), which may suggest that the N-terminal extension of Trm10 is quite flexible in solution.
Figure 3.

Monomeric state of spTrm10 in solution. (A) The experimental SAXS curve of spTrm10_FL including data up to q = 0.513 Å−1. The PDDF that was calculated using GNOM is shown in the top-right inset. (B) DR models of spTrm10_FL that were generated using GASBOR. The X-ray structure of spTrm10_74 was superimposed onto the globular region of each DR model, respectively. The figure was created using VMD. (C) The dimer interface of TrmD (1P9P, left) and TrmH (1V2X, right) that is formed between two different subunits. One monomer is colored pink and the other is colored sky blue. (D) Structural superposition of spTrm10 (green and red) and TrmH monomer (gray, 1V2X). The two α-helices (α1 and α5) and motif III in spTrm10 are colored lime green. The C-terminal α6 helix in spTrm10 is colored red.
In our crystal structures, Trm10 contains an α6 helix at the C-terminus, which can interact with the α5 helix, β4–β5 loop and β6–α5 loop via hydrogen bonds and hydrophobic interactions, forming a hydrophobic core in the Trm10 MTase domain (Supplementary Figure S4A). Thus, we suggest that the core Trm10 MTase domain includes the α6 helix, which was confirmed by limited proteolysis and mass spectrometry analysis (Supplementary Figure S7G). Structural comparison revealed that there is a C-terminal α-helix that is adjacent to the MTase domain present in TrmH but not in YibK or TrmD. However, it has been reported that the core MTase domain of TrmH does not include the C-terminal α-helix (53) (Supplementary Figure S4A). Multiple sequence alignment with other core SPOUT MTase domains (YibK, TrmH and TrmD) indicated that the unique α6 helix is only present in Trm10 (Supplementary Figure S4B).
Further structural comparisons between Trm10 and other SPOUT MTases (TrmH and TrmD) indicated that two α-helices that are involved in dimerization are also present in spTrm10 (α1 and α5) (Figure 3C and D). The α6 helix of Trm10 would sterically clash with the dimer interface of TrmH (Figure 3D). Therefore, the presence of the α6 helix may explain the monomeric behavior of Trm10, in contrast to the homodimers observed for other SPOUT MTases. Taken together, Trm10 is the first MTase that functions as a monomer but adopts a SPOUT MTase fold, suggesting that Trm10 may act through a different catalytic mechanism than other members of the SPOUT family.
SAM-binding pocket and conformational rearrangement of the active site
In the structures of the complexes, the methyl-donor product SAH is clearly visible in the electron density map and adopts a bent conformation in a deep knotted region of Trm10 (Figure 4A and Supplementary Figure S5A). The SAH-binding regions consist of three motifs, motif I (β4–β5), motif II (β5–α4) and motif III (β6–α5), which are highly conserved among members of the Trm10 family (Figure 2C). The adenine moiety of SAH is buried in a hydrophobic cavity that is formed by motif I and III. The 2′-OH and 3′-OH groups of the SAH ribose moiety form hydrogen bonds with the main chain of G203 of spTrm10. The 2′-OH group also forms tight hydrogen bond with the carbonyl group of L183. In addition, the homocysteine moiety of SAH interacts via its α-COO− group with the main chain amide group of C215 and via its α- group with the side chains of D207 and N209 (D210 and N212, respectively, in scTrm10) (Figure 4A and Supplementary Figure S5B). A schematic diagram of the interaction between spTrm10 and SAH is depicted in Figure 4B.
group with the side chains of D207 and N209 (D210 and N212, respectively, in scTrm10) (Figure 4A and Supplementary Figure S5B). A schematic diagram of the interaction between spTrm10 and SAH is depicted in Figure 4B.
Figure 4.

Conformational rearrangement of the active site. (A) Structural superposition of spTrm10_74–apo and spTrm10_74–SAH. The SAH-binding regions consist of three motifs, motif I, motif II and motif III, which are colored green in spTrm10_74–SAH. The right figure shows the conformations of the side chains of D207, K208 and N209 in motif II of spTrm10 before (gray) and after (green) binding the SAH. The difference electron density map (2Fo–Fc contoured at 1.5σ) of SAH shows that it adopts a clear bent conformation. (B) A schematic diagram of the SAH-binding site of spTrm10. Green dashed lines represent hydrogen bonds. A red dashed line represents the interaction between SAH and the side chain of D207 through a hydrogen bond or ionic interaction. Black dashed curves represent the hydrophobic portions of the side chains of residues that interact with the SAH. (C) MTase activity of wild-type Trm10 (WT) and its mutants. (D) Dissociation constants of the interactions between Trm10 and SAM determined using ITC. The N value represents the binding stoichiometry of the reaction at a 1:1 ratio. ΔG was calculated using the equation ΔG = −RT ln Ka. The ITC-binding isotherms are presented in Supplementary Figure S9. (E) Structural superposition of four spTrm10 molecules indicates that motif III is highly flexible. (F) MTase activity of spTrm10 M1 (residues 237–240 deleted), M2 (residues 237–240 mutated to 4 Ala) and M3 (residues 237–240 mutated to 4 Pro) mutants.
A comparison of apo spTrm10 and spTrm10 in complex with SAH revealed that conformational rearrangements primarily occur in two regions. One of the regions is motif II, including residues D207, K208 and N209. Of these three residues, D207 and N209 rotate their side chains toward the SAH, whereas K208 exhibits the largest structural rearrangements (∼13 Å) with its side chain pointing outward from the SAH. Previous studies of Trm10 from H. sapiens and archaea have suggested that the corresponding Asp residue, which is absolutely conserved in the Trm10 family, plays an important role in the Trm10 active site (13,43). Considering Asp may function as a general base for proton abstraction from the tRNA, the substitution of this Asp with Asn would, therefore, abolish tRNA MTase activity (15). In our complex structure, the side chain of D207 (D210 in scTrm10) is ∼4.5 Å (4.7 Å) from the sulfonium atoms of SAH (Figure 4A and Supplementary Figure S5B) and could potentially serve as a catalytic base during methyl group transfer. To investigate the actual contributions of D207 and N209 on spTrm10 activity, we generated D207N and N209A mutants and determined the respective tRNA MTase activities. The catalytic activity of the N209A mutant was reduced to ∼10% of the wild-type spTrm10 activity, whereas the activity of the D207N mutant was completely abolished (Figure 4C), indicating that N209 may play an essential role in stabilizing the SAM through direct interaction, while D207 is very likely a candidate for the catalytic base.
To further study the catalytic role of D207 and N209, the binding affinity of Trm10 for SAM was assessed using ITC. ITC-binding isotherms were best fitted using a single-site model with a binding stoichiometry of the reaction (N-value) of 1 (Supplementary Figure S9). The Kd for SAM for wild-type spTrm10 was determined to be 4.46 μM, whereas the binding free energy change ΔG was calculated to be −7.11 Kcal mol−1. Similar binding affinities for SAM have been previously reported for other SPOUT MTases. For example, Kd values of 26 and 52 μM were determined for YibK (54) and OrfX (55), respectively. Compared with wild-type spTrm10, the spTrm10 N209A mutant exhibited much weaker affinity for SAM (Kd = 83.3 μM), which resulted in an increase of ΔG of 1.69 Kcal/mol. These changes suggested that spTrm10 N209A mutant broke the interaction between N209 with SAM. In contrast, no affinity of spTrm10 D207N for SAM was detected (Figure 4D), suggesting that the D207N mutation may abolish the enzymatic activity of spTrm10 by not only preventing the ability of D207 to act as the general base but also disrupting the direct interaction between D207 and SAM. Identical mutations were also introduced at the corresponding residues D210 and N212 in scTrm10, and similar results were obtained (Figure 4C and D).
In addition to the conformational changes observed for motif II, motif III also exhibits different rearrangements in different molecules of the spTrm10 structures (Figure 4E). SPOUT family MTases deeply bury the product SAH using a topological knot, and the enzymes may undergo isomerization during catalysis to cycle SAM in and out of the catalytic center (56). Collectively, Trm10 contains flexible regions (motif II and motif III) near the active site, suggesting that the binding of the SAM to Trm10 may also occur through an induced-fit mechanism.
Furthermore, portions of motif III (residues 236–240 with the sequence KITDR) in spTrm10_FL are not observed in the electron density likely due to its high structural flexibility, raising the question of the influence of this region on Trm10 MTase activity. We, therefore, designed three spTrm10 mutants, spTrm10 M1 (residues 237–240 deleted), spTrm10 M2 (residues 237–240 mutated to 4 Ala) and spTrm10 M3 (residues 237–240 mutated to 4 Pro), and determined their activities. Our results indicated that spTrm10 M1 exhibited significantly diminished enzymatic activity compared with wild-type, whereas the mutants spTrm10 M2 and spTrm10 M3 exhibited no major difference from spTrm10 wild-type in terms of enzyme activity (Figure 4F), indicating that the presence of residues 237–240, not their flexibility, is necessary for tRNA MTase activity. These mutations did not affect protein stability or folding as assessed by CD analysis (Supplementary Figure S10A and S10B).
A possible guanine-binding pocket
Formation of m1G in tRNAs can occur at two different positions, namely, Position 9 and 37. The m1G9 modification is catalyzed by Trm10, whereas the m1G37 modification is synthesized by Trm5 in archaea and eukaryotes and by TrmD in bacteria (8,9). Although the three m1G MTases (Trm5, TrmD and Trm10) possess similar functions, the absence of overall sequence homology and structural similarity suggests that they could present different catalytic strategies.
Both Trm5 and TrmD recognize SAM as the methyl donor and G37 of the tRNA as the methyl acceptor, but they share no structural homology. Trm5 functions as a monomer and belongs to the RFM MTase superfamily (57). The crystal structure of Trm5 in complex with its substrate tRNA and SAM reveals that the SAM in Trm5 exists in an extended open conformation, and the G37 of the tRNA is located next to the methyl moiety of SAM (Figure 5A). The NPPY motif (N265–L266–P267–K268) positions the target nitrogen atom (N1) of G37, and the first residue, N265, in the NPPY motif stabilizes the guanine base, permitting E185 to abstract the proton from the N1 of G37. R145 lowers the transition-state free energy by interacting with the incipient negative charge on the O6 of the tRNA (46). In contrast, TrmD functions as a homodimer and binds SAM using a rare trefoil knot fold (SPOUT MTase family), and the SAM exists in an L-shaped bent conformation (52,56). Unlike RFM MTases, the crystal structure of TrmD shows that it does not contain a conserved NPPY motif. The active site of TrmD is located at the subunit interface (Figure 5A), and one phosphate ion in the active site may mimic the phosphate that is 5′ of G37 in the tRNA. Two strictly conserved residues, Glu116 and Arg154′ (from another subunit), which are located around the reactive methyl group of SAM, may recognize the guanine ring of G37 in the tRNA. Another highly conserved residue, D169′, which is located in the SAM-binding region, may act as a general base that accepts a proton from the N1 of G37. TrmH possesses a similar active site and catalyzes the 2′-O-methylation of G18 in tRNA. In the crystal structure of TrmH (Figure 5A), a sulfate ion mimics the 5′-phosphate group of guanine in the tRNA, and a strictly conserved Arg41′ enters into a pocket formed by residues of subunit 1 (K32, H34, E124, K125 and N152) to constitute the active site (50). Taken together, location of the substrate nucleotide in the tRNA should stay close to the methyl group of the SAM to facilitate the reaction; therefore, we presume that the guanine-binding site of Trm10 lies within the vicinity of the product SAH.
Figure 5.
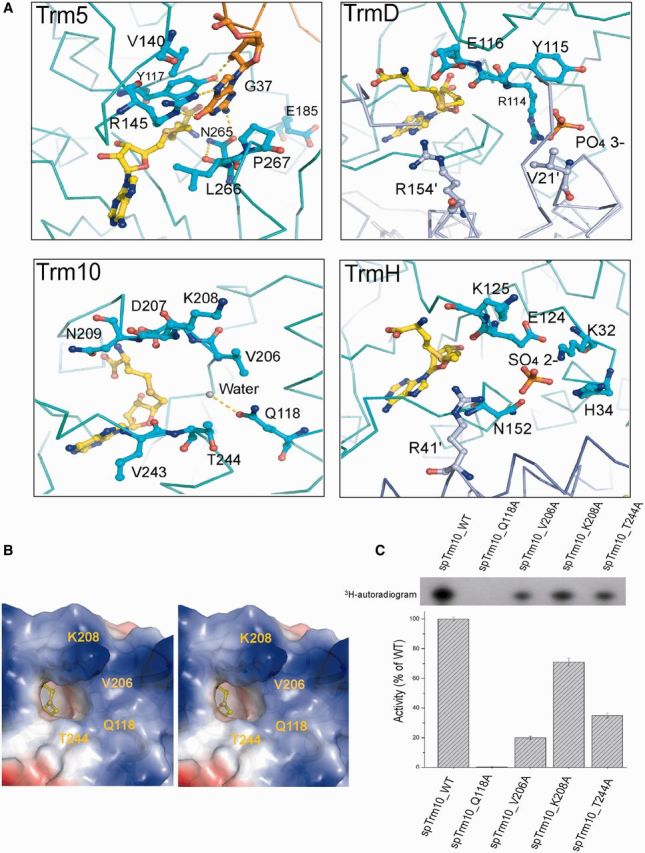
A possible guanine-binding pocket in spTrm10. (A) Comparison of the active sites in Trm5 (2ZZM), TrmD (1UAM), TrmH (1V2X) and Trm10, respectively. (B) Stereoview of the surface electrostatic potential of the spTrm10 active site, which reveals a deep pocket adjacent to the buried SAH. The surrounding residues (Q118, V206, K208 and T244) are potentially involved in G9 binding. K208 points towards the reader. (C) MTase activities of spTrm10 mutants containing mutations of residues that are involved in guanine binding.
An examination of the active site reveals a deep pocket that is adjacent to the buried SAH, which appears to be optimal to accommodate the G9 of a substrate tRNA (Figure 5B). To confirm this possibility, we docked GMP into the spTrm10–SAH structure. Our results indicated that the docked guanine does not sterically clash with the surrounding residues (Q118, V206, K208 and T244) (Supplementary Figure S6A), which are potentially involved in the enzymatic activity and G9 binding.
To better validate the binding pocket for the G9 nucleoside in spTrm10, we individually mutated the residues Q118, V206, K208 and T244 in spTrm10 to Ala and then determined the respective tRNA MTase activities. All of these mutants exhibited reduced activities compared to the wild-type. The mutation of the highly conserved Q118, which is located in the α1 helix, to Ala abolished tRNA MTase activity, whereas V206A, which is located in motif II, demonstrated a loss of activity that was 19% of the wild-type spTrm10 activity. Moreover, K208A and T244A demonstrated a loss of activity to 72% and 35%, respectively, of the wild-type spTrm10 activity (Figure 5C). These residues are conserved in scTrm10 (Supplementary Figure S6C), and similar activities were obtained for the corresponding scTrm10 mutants (Supplementary Figure S6D). Together, these results confirmed that four conserved residues that lie adjacent to the SAH-binding pocket all contribute to the accommodation of G9 during catalysis with in an order of influence of Q118 > V206 > T244 > K208.
tRNA interaction site
Many RNA MTases contain additional domains fused to the N- or C-terminus, most of which represent various nucleic acid-binding domains, such as PUA, THUMP, OB-fold or L30e (19,58–60). These additional domains can enhance the binding affinity for the RNA substrate, thereby increasing the activity of the enzyme. SpTrm10 contains an N-terminal extension as well, but it is a highly flexible region, as suggested by SAXS analysis, which is consistent with our observations in the spTrm10_FL crystal structure. Moreover, multiple sequence alignment analysis indicated that this region is not well conserved in the Trm10 family and is rich in basic residues. Therefore, it appears that this N-terminal extension may functionally assist Trm10 by recruiting substrate tRNAs.
To confirm this hypothesis, the substrate sptRNAGly-binding ability of Trm10 was analyzed using EMSA. As expected, Trm10_FL demonstrates strong affinity for tRNA (Kd = 0.24 μM for scTrm10 and Kd = 0.2 μM for spTrm10) (Figure 6A and B), and the spTrm10 N-terminal extension (residues 1–73) also exhibits strong affinity for tRNA (Kd = 0.86 μM) (Supplementary Figure S7A and B), which is consistent with our results of the MTase assays that indicated that the N-terminal region of Trm10 largely contributes to its enzymatic activity.
Figure 6.

Analysis of the interaction between Trm10 and the sptRNAGly substrate. (A) EMSA of Trm10_FL (full length) and the Trm10 MTase domain with dye-labeled sptRNAGly. The protein concentrations are indicated below the gel in the figure. (B) The Kd of Trm10 for tRNA as assessed using EMSA; the detailed procedures are described in the ‘Materials and Methods’ section. (C) Electrostatic potential surface of spTrm10. A basic region is present in the Trm10 MTase domain. (D) Five positively charged residues (K110, R121, R127, R147 and K153) in the basic region (left figure). EMSA of wild-type spTrm10_74 and its mutants using sptRNAGly as the substrate (right figure).
To further study the recognition of the tRNA substrate by the spTrm10 MTase domain, we generated two combinatorial spTrm10 MTase domain mutants, spTrm10_74 M4 (K110E/R121E/R127E) and spTrm10_74 M5 (K153E/R147E), which were based on the electrostatic potential surface of the structure and conserved positively charged residues (Figure 6C), and then determined their binding affinities for tRNA using EMSA. Among the mutated residues, K110, R121 and R127 in α1 helix, which compose a positive charged surface, are highly conserved in the Trm10 family according to multiple sequence alignment analysis. Therefore, it is not surprising that the interaction between the spTrm10_74 M4 and spTrm10_74 M5 mutants and tRNA is completely abolished (Figure 6D). Two additional mutants, spTrm10_74 M6 (K110A/R121A/R127A) and spTrm10_74 M7 (K153A/R147A), also lost their ability to bind tRNA, further confirming the aforementioned results (Supplementary Figure S7C). These observations are also consistent with the determined tRNA MTase activities (Supplementary Figure S7D). CD analysis of these four mutants indicated that the mutations did not affect the overall protein structure (Supplementary Figure S10C). Although the secondary structure content that is determined using CD analysis exhibits some differences from those determined from the atomic-resolution coordinates, the results obtained using CD analysis exhibit a correlation with those determined from the crystal structures (61). The secondary structure content of wild-type spTrm10_74 was further estimated from the CD spectra using the JASCO software and Raussens’s method (62). Similar to previous analysis, our results closely resemble the secondary structure of spTrm10_74 as assigned using DSSP (63) (Supplementary Figure S10D).
Generally, the basic surface on the Trm10 MTase domain is essential to bind the substrate tRNA, which suggests that the MTase domain of Trm10 not only plays an important role in its enzymatic activity but also recognizes the substrate tRNA.
DISCUSSION
Trm10 is a unique tRNA MTase that adopts a SPOUT fold
Herein, we present for the first time a detailed structural investigation of the Trm10 family. Our findings revealed that Trm10 is a new member of the SPOUT MTase superfamily. Both X-ray crystallographic and SAXS studies indicate that Trm10 functions as a monomer. The monomeric state of the Trm10 family differentiates from other SPOUT MTases in certain aspects, such as the catalytic mechanism and substrate tRNA recognition. It has been reported that SPOUT RNA MTase Nucleolar Essential Protein 1 (Nep1) binds a 14-base RNA fragment using the cleft between two monomers (64). However, Trm10 recognizes the substrate tRNA using an N-terminal extension and a basic surface on the MTase domain. A detailed understanding of RNA MTase catalytic and substrate recognition mechanisms generally requires the high-resolution structure of an RNA MTase in complex with its substrate RNA.
One might wonder concerning the biological significance of the monomeric state of Trm10, whereas other known tRNA MTases that possess SPOUT topology all exist as homodimers. It is well known that tRNA folds into a compact L-shaped 3D structure, and its G9 site is localized at the junction between the D stem and acceptor stem, which is not easily accessible for large functional proteins. It has been reported that RNase H cannot completely cleave the G9 site due to poor accessibility (33). We, therefore, propose that the monomeric feature is evolutionarily selected to enable Trm10 to access the G9 site more easily to fulfill its role. Thus, we speculate that Trm10 in other species would most likely function as a monomer.
The evolutionary relationship between Trm10 and other tRNA m1G MTases
Until now, three tRNA m1G MTases (Trm5, TrmD and Trm10) have been identified and characterized. Although these three enzymes exhibit similar catalytic activities, they display different topologies and possess various catalytic active sites (52,57). Trm10 and TrmD belong to the SPOUT family, whereas Trm5 belongs to the RFM family of MTases. The m1G37 MTases TrmD and Trm5 independently evolved from a nonoverlapping pathway. In contrast, TrmD and Trm10 may have evolved from a common SPOUT ancestor. These differences imply that m1G methylation independently appeared at least two times in the evolution of SPOUT MTases; thus, an ancestral MTase might give rise to the SPOUT and RFM family followed by an m1G SPOUT MTase ancestor to give rise to TrmD and Trm10 (19).
Implications for the catalytic mechanism of Trm10
The residue D207 in the spTrm10 catalytic site is strictly conserved within the Trm10 family. Recent studies indicated that the substitution of the corresponding Asp with Asn abolished the tRNA MTase activities in both eukaryotes and archaea (13,43). Based on our MTase assays and ITC experiments, we conclude that D207 may play a bifunctional role in the catalytic site of Trm10: as a catalytic base during methyl group transfer and as a stabilizer of interactions between Trm10 and SAM.
Another conserved residue is Q118 in spTrm10, which contributes most significantly, among the residues (Q118, V206, K208 and T244) that constitute the G9-binding pocket, to the recognition by Trm10 of the substrate tRNA and may directly hydrogen bond with GMP. In our spTrm10–SAH complex structure, a water molecule in active site (Figure 5A) might mimic an H or O atom of GMP and form a hydrogen bond with Q118. Therefore, Q118 in spTrm10 likely plays a similar role as the conserved Arg in the active site of other SPOUT MTases.
The N-terminal extension of Trm10 is necessary for substrate tRNA recognition
Many RNA MTases or other RNA modification enzymes contain additional extension domains that are involved in RNA binding and are even essential for enzymatic activity, such as in Pus10, TruB and AspRS (19,59,65–67). Although these additional extension domains display different topologies, the interaction between them and RNA is largely dependent on electrostatic interactions. Sequence analysis of the Trm10 family indicates that there is a short positively charged polypeptide in the N-terminus. A similar polypeptide is found in AspRS (eukaryotic class IIb aminoacyl-tRNA synthetase), and it was shown that its N-terminal region adopts a structure composed of amphiphilic helices that interact with the tRNA substrate and is necessary for the catalytic efficiency (65,68). The N-terminal extension of Trm10 displays the similar role as well, however, unlike AspRS, the extension of Trm10 adopts a possible local secondary structure consisting of β-strands as determined using CD analysis (Supplementary Figure S7E).
To investigate whether conformational changes of the N-terminal extension occur upon tRNA binding, we performed limited proteolysis of spTrm10 in the presence and absence of sptRNAGly (Supplementary Figure S7F). The analysis indicated that free Trm10 likely exists in a more open conformation in solution and Trm10 in complex with tRNA becomes compact and is stabilized.
Implications for the function of H. sapiens Trm10
Trm10 homologs are conserved throughout yeast and other eukaryotes, but the reason for the presence of multiple isoforms of the protein in higher order eukaryotes such as H. sapiens, which are named TrmT10A, TrmT10B and TrmT10C, remains unclear. TrmT10A and TrmT10B demonstrate high sequence homology with yeast Trm10, and TrmT10C contains a mitochondrial targeting sequence and is located in mitochondria (13). On the one hand, from an evolutionary perspective, Jackman and colleagues have shown that the archaea and eukaryotic homologs clearly branch separately from one another using phylogenetic analysis. Furthermore, mitochondrial TrmT10C branched separately from other members (TrmT10A, TrmT10B and yeast Trm10) within eukaryotic group during the evolution of the Trm10 family (10,21). Archaea orthologs of Trm10 have recently been identified: Saci_1677p from the crenarchaeon S. acidocaldarius catalyzes m1A9 formation in tRNA and TK0422p from the euryarchaeon T. kodakaraensis catalyzes m1R9 (G9 and A9) formation in tRNA (43). The evolution of the Trm10 family and the dual m1R9 (G9 and A9) MTase activity of the ancestor of Trm10 might explain the reasonable presence of different isoforms of Trm10 in higher order eukaryotes. On the other hand, Elisa and colleagues have demonstrated that the presence of multiple isoforms of Trm10 accounted for substrate tRNA selectivity and tissue specificity (13), which may also explain the presence of multiple isoforms of Trm10 in H. sapiens.
Among the examined eukaryotic Trm10 homologs, TrmT10A and TrmT10B contain the highly conserved active site and may share a common mechanism for substrate recognition and catalysis with yeast Trm10. In the TrmT10C–SDR5C1 MTase complex, SDR5C1 is identified as a member of the ubiquitous short-chain dehydrogenase or reductase family and utilizes NAD(H) as a cofactor (69). However, SDR5C1 does not play a direct role in tRNA recognition, whereas TrmT10C plays an essential role in tRNA binding and specificity and appears to be the catalytic subunit of the MTase. According to the structure of yeast Trm10 and multiple sequence alignment analysis, the N-terminal region of human TrmT10C is rich in basic residues, and its MTase domain also contains a conserved basic surface (K218, R235, R236, R256 and K260). Therefore, we speculate that the tRNA-binding region of TrmT10C may consist of the N-terminus and the basic surface on the MTase domain. A similar MTase complex is also found in the tRNA m7G46 MTase Trm8–Trm82 complex, in which the noncatalytic subunit Trm82 is not involved in tRNA binding (70), suggesting that SDR5C1 could be involved in stabilizing the catalytic TrmT10C subunit or activating and fine tuning its activity.
Multiple sequence alignment of the Trm10 family from archaea to H. sapiens illustrates that the active site and purine-binding pocket are highly conserved. The corresponding residues include Q226, V313, D314, K314 and T352 in TrmT10C and Q122, V205 and D206 in TK0422p (Supplementary Figure S8A); these two Trm10 homologs have been found to methylate the two purine bases at an identical position. Similar to the yeast Trm10, the loss of TrmT10C m1A9 MTase activity in the D314N mutant suggests that D314 acts as the general base during methyl group transfer (13). Additionally, the Q226A mutation in TrmT10C abolished the m1A9 MTase activity as well (Supplementary Figure S8B), suggesting that Q226 may also directly interact with the purine. Collectively, these findings indicate that the corresponding residues Gln and Asp (Q118 and D207 in spTrm10; Q226 and D314 in TrmT10C) are strictly conserved and essential for the m1R9 (m1G9 and m1A9) MTase activity.
In the human (mt)RNase P complex, TrmT10C-SDR5C1 constitutes a stable subcomplex, and the catalytic subunit PRORP (MRPP3) contains two tandem pentatricopeptide repeat (PPR) motifs at the N-terminus and a metallo-nuclease domain at the C-terminus. PPR motifs may be involved in protein-protein or protein–RNA interactions, whereas the metallo-nuclease domain enables RNA hydrolysis (71). Previous studies have indicated that the interaction of PRORP with the stable subcomplex is very weak, and none of the three proteins alone demonstrated any pre-tRNA processing activity (21). Although methylation and tRNA processing are not coupled (13), it appears reasonable to postulate a common mechanism for the TrmT10C–SDR5C1 complex upon substrate tRNA binding. The TrmT10C–SDR5C1 complex may induce conformational changes in the pre-tRNA upon association and activate PRORP activity. Such a mechanism could be explored through future biochemical characterization and the determination of a high-resolution structure for the complex.
PROTEIN DATA BANK ACCESSION CODES
Coordinates have been submitted to the RCSB Protein Data Bank with the following accession codes: 4JWF for Se-Met-spTrm10-74-SAH, 4JWG for spTrm10_74-apo, 4JWH for spTrm10-FL-SAH and 4JWJ for scTrm10_84-SAH.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The National Basic Research Program of China (973 Program) [grants 2011CB911104 and 2009CB825500]; Chinese National Natural Science Foundation [grants 31270760, 31270014 and 31130018]; the Fundamental Research Funds for the Central Universities [2070000020]; Chinese Ministry of Science and Technology [grant 2012CB917200]; the “Outstanding Technical Talent” project of the Chinese Academy of Sciences; Anhui Natural Science Foundation [grant 1208085MC38]; and the Science and Technological Fund of Anhui Province for Outstanding Youth [Grant 1308085JGD08]. Funding for open access charge: National Basic Research Program of China (973 Program) [grants 2011CB911104].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Jianye Zang, Minhao Wu and Hejun Liu for their assistance with the X-ray data collection and data processing. The authors also thank the staff at BL17U of the Shanghai Synchrotron Radiation Facility (SSRF) for their assistance with the X-ray data collection. The authors thank Chongyuan Wang, Yiyang Jiang, Pengzhi Wu and Lin Cheng for their valuable suggestions throughout this project. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.
REFERENCES
- 1.Bjork GR. Genetic dissection of synthesis and function of modified nucleosides in bacterial transfer-Rna. Prog. Nucleic Acid Res. 1995;50:263–338. doi: 10.1016/s0079-6603(08)60817-x. [DOI] [PubMed] [Google Scholar]
- 2.Agris PF. Decoding the genome: a modified view. Nucleic Acids Res. 2004;32:223–238. doi: 10.1093/nar/gkh185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang XN, Vendeix FAP, Fabris D, Agris PF. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–D201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Helm M. Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res. 2006;34:721–733. doi: 10.1093/nar/gkj471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gustilo EM, Vendeix FA, Agris PF. tRNA's modifications bring order to gene expression. Curr. Opin. Microbiol. 2008;11:134–140. doi: 10.1016/j.mib.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackman JE, Alfonzo JD. Transfer RNA modifications: nature's combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA. 2013;4:35–48. doi: 10.1002/wrna.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li JN, Bjork GR. Structural alterations of the tRNA(m1G37)methyltransferase from Salmonella typhimurium affect tRNA substrate specificity. RNA. 1999;5:395–408. doi: 10.1017/s1355838299980834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bjork GR, Jacobsson K, Nilsson K, Johansson MJ, Bystrom AS, Persson OP. A primordial tRNA modification required for the evolution of life? EMBO J. 2001;20:231–239. doi: 10.1093/emboj/20.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christian T, Evilia C, Williams S, Hou YM. Distinct origins of tRNA(m1G37) methyltransferase. J. Mol. Biol. 2004;339:707–719. doi: 10.1016/j.jmb.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 10.Jackman JE, Montange RK, Malik HS, Phizicky EM. Identification of the yeast gene encoding the tRNA m1G methyltransferase responsible for modification at position 9. RNA. 2003;9:574–585. doi: 10.1261/rna.5070303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gustavsson M, Ronne H. Evidence that tRNA modifying enzymes are important in vivo targets for 5-fluorouracil in yeast. RNA. 2008;14:666–674. doi: 10.1261/rna.966208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torabi N, Kruglyak L. Variants in SUP45 and TRM10 underlie natural variation in translation termination efficiency in Saccharomyces cerevisiae. Plos Genet. 2011;7:e1002211. doi: 10.1371/journal.pgen.1002211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vilardo E, Nachbagauer C, Buzet A, Taschner A, Holzmann J, Rossmanith W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012;40:11583–11593. doi: 10.1093/nar/gks910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helm M, Brule H, Degoul F, Cepanec C, Leroux JP, Giege R, Florentz C. The presence of modified nucleotides is required for cloverleaf folding of a human mitochondrial tRNA. Nucleic Acids Res. 1998;26:1636–1643. doi: 10.1093/nar/26.7.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou YM, Perona JJ. Stereochemical mechanisms of tRNA methyltransferases. FEBS Lett. 2010;584:278–286. doi: 10.1016/j.febslet.2009.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motorin Y, Helm M. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA. 2011;2:611–631. doi: 10.1002/wrna.79. [DOI] [PubMed] [Google Scholar]
- 17.Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 2003;28:329–335. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anantharaman V, Koonin EV, Aravind L. SPOUT: a class of methyltransferases that includes spoU and trmD RNA methylase superfamilies, and novel superfamilies of predicted prokaryotic RNA methylases. J. Mol. Microbiol. Biotechnol. 2002;4:71–75. [PubMed] [Google Scholar]
- 19.Tkaczuk KL, Dunin-Horkawicz S, Purta E, Bujnicki JM. Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases. BMC Bioinformatics. 2007;8:73. doi: 10.1186/1471-2105-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kempenaers M, Roovers M, Oudjama Y, Tkaczuk KL, Bujnicki JM, Droogmans L. New archaeal methyltransferases forming 1-methyladenosine or 1-methyladenosine and 1-methylguanosine at position 9 of tRNA. Nucleic Acids Res. 2010;38:6533–6543. doi: 10.1093/nar/gkq451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holzmann J, Frank P, Loffler E, Bennett KL, Gerner C, Rossmanith W. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008;135:462–474. doi: 10.1016/j.cell.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 22.Radaev S, Li S, Sun PD. A survey of protein-protein complex crystallizations. Acta Crystallogr. D Biol. Crystallogr. 2006;62:605–612. doi: 10.1107/S0907444906011735. [DOI] [PubMed] [Google Scholar]
- 23.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 25.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 32.Grosjean H, Droogmans L, Roovers M, Keith G. Detection of enzymatic activity of transfer RNA modification enzymes using radiolabeled tRNA substrates. Methods Enzymol. 2007;425:55–101. doi: 10.1016/S0076-6879(07)25003-7. [DOI] [PubMed] [Google Scholar]
- 33.Hou YM. High-purity enzymatic synthesis of site-specifically modified tRNA. Methods Mol. Biol. 2012;941:195–212. doi: 10.1007/978-1-62703-113-4_15. [DOI] [PubMed] [Google Scholar]
- 34.Konarev PV, Petoukhov MV, Volkov VV, Svergun DI. ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2006;39:277–286. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003;36:1277–1282. [Google Scholar]
- 36.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992;25:495–503. [Google Scholar]
- 37.Fischer H, Neto MD, Napolitano HB, Polikarpov I, Craievich AF. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 2010;43:101–109. [Google Scholar]
- 38.Svergun DI, Petoukhov MV, Koch MHJ. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 2001;34:33–41. [Google Scholar]
- 40.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38, 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 41. Hermanson G. T. (2008) Bioconjugate Techniques (Second Edition), 27, 1012–1013.
- 42.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kempenaers M, Roovers M, Oudjama Y, Tkaczuk KL, Bujnicki JM, Droogmans L. New archaeal methyltransferases forming 1-methyladenosine or 1-methyladenosine and 1-methylguanosine at position 9 of tRNA. Nucleic Acids Res. 2010;38:6533–6543. doi: 10.1093/nar/gkq451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christian T, Hou YM. Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J. Mol. Biol. 2007;373:623–632. doi: 10.1016/j.jmb.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swinehart WE, Henderson JC, Jackman JE. Unexpected expansion of tRNA substrate recognition by the yeast m1G9 methyltransferase Trm10. RNA. 2013;19:1137–1146. doi: 10.1261/rna.039651.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Christian T, Lahoud G, Liu C, Hoffmann K, Perona JJ, Hou YM. Mechanism of N-methylation by the tRNA m1G37 methyltransferase Trm5. RNA. 2010;16:2484–2492. doi: 10.1261/rna.2376210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christian T, Lahoud G, Liu C, Hou YM. Control of catalytic cycle by a pair of analogous tRNA modification enzymes. J. Mol. Biol. 2010;400:204–217. doi: 10.1016/j.jmb.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lim K, Zhang H, Tempczyk A, Krajewski W, Bonander N, Toedt J, Howard A, Eisenstein E, Herzberg O. Structure of the YibK methyltransferase from Haemophilus influenzae (HI0766): a cofactor bound at a site formed by a knot. Proteins. 2003;51:56–67. doi: 10.1002/prot.10323. [DOI] [PubMed] [Google Scholar]
- 50.Nureki O, Watanabe K, Fukai S, Ishii R, Endo Y, Hori H, Yokoyama S. Deep knot structure for construction of active site and cofactor binding site of tRNA modification enzyme. Structure. 2004;12:593–602. doi: 10.1016/j.str.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 51.Michel G, Sauve V, Larocque R, Li Y, Matte A, Cygler M. The structure of the RlmB 23S rRNA methyltransferase reveals a new methyltransferase fold with a unique knot. Structure. 2002;10:1303–1315. doi: 10.1016/s0969-2126(02)00852-3. [DOI] [PubMed] [Google Scholar]
- 52.Ahn HJ, Kim HW, Yoon HJ, Lee BI, Suh SW, Yang JK. Crystal structure of tRNA(m1G37)methyltransferase: insights into tRNA recognition. EMBO J. 2003;22:2593–2603. doi: 10.1093/emboj/cdg269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hori H, Suzuki T, Sugawara K, Inoue Y, Shibata T, Kuramitsu S, Yokoyama S, Oshima T, Watanabe K. Identification and characterization of tRNA (Gm18) methyltransferase from Thermus thermophilus HB8: domain structure and conserved amino acid sequence motifs. Genes Cells. 2002;7:259–272. doi: 10.1046/j.1365-2443.2002.00520.x. [DOI] [PubMed] [Google Scholar]
- 54.Mallam AL, Morris ER, Jackson SE. Exploring knotting mechanisms in protein folding. Proc. Natl Acad. Sci. USA. 2008;105:18740–18745. doi: 10.1073/pnas.0806697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boundy S, Safo MK, Wang L, Musayev FN, O'Farrell HC, Rife JP, Archer GL. Characterization of the Staphylococcus aureus rRNA methyltransferase encoded by orfX, the gene containing the staphylococcal chromosome Cassette mec (SCCmec) insertion site. J. Biol. Chem. 2013;288:132–140. doi: 10.1074/jbc.M112.385138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elkins PA, Watts JM, Zalacain M, van Thiel A, Vitazka PR, Redlak M, Andraos-Selim C, Rastinejad F, Holmes WM. Insights into catalysis by a knotted TrmD tRNA methyltransferase. J. Mol. Biol. 2003;333:931–949. doi: 10.1016/j.jmb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 57.Goto-Ito S, Ito T, Kuratani M, Bessho Y, Yokoyama S. Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat. Struct. Mol. Biol. 2009;16:1109–1115. doi: 10.1038/nsmb.1653. [DOI] [PubMed] [Google Scholar]
- 58.Murzin AG. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 1993;12:861–867. doi: 10.1002/j.1460-2075.1993.tb05726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aravind L, Koonin EV. Novel predicted RNA-binding domains associated with the translation machinery. J. Mol. Evol. 1999;48:291–302. doi: 10.1007/pl00006472. [DOI] [PubMed] [Google Scholar]
- 60.Aravind L, Koonin EV. THUMP–a predicted RNA-binding domain shared by 4-thiouridine, pseudouridine synthases and RNA methylases. Trends Biochem. Sci. 2001;26:215–217. doi: 10.1016/s0968-0004(01)01826-6. [DOI] [PubMed] [Google Scholar]
- 61.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raussens V, Ruysschaert JM, Goormaghtigh E. Protein concentration is not an absolute prerequisite for the determination of secondary structure from circular dichroism spectra: a new scaling method. Anal. Biochem. 2003;319:114–121. doi: 10.1016/s0003-2697(03)00285-9. [DOI] [PubMed] [Google Scholar]
- 63.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 64.Thomas SR, Keller CA, Szyk A, Cannon JR, Laronde-Leblanc NA. Structural insight into the functional mechanism of Nep1/Emg1 N1-specific pseudouridine methyltransferase in ribosome biogenesis. Nucleic Acids Res. 2011;39:2445–2457. doi: 10.1093/nar/gkq1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frugier M, Moulinier L, Giege R. A domain in the N-terminal extension of class IIb eukaryotic aminoacyl-tRNA synthetases is important for tRNA binding. EMBO J. 2000;19:2371–2380. doi: 10.1093/emboj/19.10.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kamalampeta R, Keffer-Wilkes LC, Kothe U. tRNA binding, positioning, and modification by the pseudouridine synthase Pus10. J. Mol. Biol. 2013;425:3863–3874. doi: 10.1016/j.jmb.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 67.Pan H, Agarwalla S, Moustakas DT, Finer-Moore J, Stroud RM. Structure of tRNA pseudouridine synthase TruB and its RNA complex: RNA recognition through a combination of rigid docking and induced fit. Proc. Natl Acad. Sci. USA. 2003;100:12648–12653. doi: 10.1073/pnas.2135585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Agou F, Waller JP, Mirande M. Expression of rat aspartyl-tRNA synthetase in Saccharomyces cerevisiae. Role of the NH2-terminal polypeptide extension on enzyme activity and stability. J. Biol. Chem. 1996;271:29295–29303. doi: 10.1074/jbc.271.46.29295. [DOI] [PubMed] [Google Scholar]
- 69.Bray JE, Marsden BD, Oppermann U. The human short-chain dehydrogenase/reductase (SDR) superfamily: a bioinformatics summary. Chem. Biol. Interact. 2009;178:99–109. doi: 10.1016/j.cbi.2008.10.058. [DOI] [PubMed] [Google Scholar]
- 70.Leulliot N, Chaillet M, Durand D, Ulryck N, Blondeau K, van Tilbeurgh H. Structure of the yeast tRNA m7G methylation complex. Structure. 2008;16:52–61. doi: 10.1016/j.str.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 71.Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl Acad. Sci. USA. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.