


Abstract
Most unwanted RNA transcripts in the nucleus of eukaryotic cells, such as splicing-defective pre-mRNAs and spliced-out introns, are rapidly degraded by the nuclear exosome. In budding yeast, a number of these unwanted RNA transcripts, including spliced-out introns, are first recognized by the nuclear exosome cofactor Trf4/5p-Air1/2p-Mtr4p polyadenylation (TRAMP) complex before subsequent nuclear-exosome-mediated degradation. However, it remains unclear when spliced-out introns are recognized by TRAMP, and whether TRAMP may have any potential roles in pre-mRNA splicing. Here, we demonstrated that TRAMP is cotranscriptionally recruited to nascent RNA transcripts, with particular enrichment at intronic sequences. Deletion of TRAMP components led to further accumulation of unspliced pre-mRNAs even in a yeast strain defective in nuclear exosome activity, suggesting a novel stimulatory role of TRAMP in splicing. We also uncovered new genetic and physical interactions between TRAMP and several splicing factors, and further showed that TRAMP is required for optimal recruitment of the splicing factor Msl5p. Our study provided the first evidence that TRAMP facilitates pre-mRNA splicing, and we interpreted this as a fail-safe mechanism to ensure the cotranscriptional recruitment of TRAMP before or during splicing to prepare for the subsequent targeting of spliced-out introns to rapid degradation by the nuclear exosome.
INTRODUCTION
The coordinated action of the nuclear exosome and its cofactor Trf4/5p-Air1/2p-Mtr4p polyadenylation (TRAMP) complex contributes substantially to the rapid degradation of unwanted RNA transcripts in the nucleus (1–6). A great variety of different RNA substrates, such as cryptic unstable transcripts (CUTs), abnormally processed snRNAs, snoRNAs and rRNAs, hypomodified tRNAs, defective ribonucleoproteins (RNPs) and non-coding RNAs, are degraded via this nuclear RNA quality control pathway (5–17). In the current model (3–5), most RNA transcripts that should be degraded by the nuclear exosome are first recognized by TRAMP through its RNA-binding subunit Air1p or Air2p (18–20), which regulates the substrate recognition specificity of TRAMP (21). After that, a short poly(A) tail is added to the 3′ end of the TRAMP-bound RNA transcripts by its non-canonical poly(A) polymerase subunit Trf4p or Trf5p (3–5), with poly(A) tail length being controlled by its ATP-dependent 3′–5′ RNA helicase subunit Mtr4p (22). This poly(A) tail serves as the docking site for the nuclear exosome, which then binds to and finally degrades the RNA transcripts using its endoribonucleolytic (23,24) and 3′ to 5′ exoribonucleolytic activity (1,2,6) with the help from the RNA unwinding activity of Mtr4p (25). Cotranscriptional recruitment of TRAMP to a CUT (26) and several snoRNA genes (27), and that of the nuclear exosome to nascent transcripts synthesized by RNA polymerase II (28–30), has been previously described. These suggested the possibility that many unwanted RNA transcripts may already be bound by these nuclear RNA quality control factors during transcription.
Pre-mRNA splicing (31) is a relatively error-prone process which possibly leads to the production of splicing-defective pre-mRNAs, and must inevitably synthesize spliced-out introns as normal splicing by-products. Both splicing-defective pre-mRNAs and spliced-out introns are probably harmful to cells. While splicing-defective pre-mRNAs may be exported into cytoplasm and then translated into potentially deleterious proteins (32–34), the accumulation of spliced-out introns may lead to a severe growth defect, at least in fission yeast (35). To minimize their potentially harmful accumulation, splicing-defective pre-mRNAs are rapidly degraded in the nucleus by the nuclear exosome (36) and/or after export into the cytoplasm by both non-sense mediated decay (NMD) (37,38) as well as NMD-independent pathways (39). Likewise, effective RNA degradation systems should also exist for the removal of spliced-out introns, which are likely generated before transcription termination as splicing occurs cotranscriptionally (40–44). A previous RNA-IP-chip analysis demonstrated that Trf4p can bind to spliced-out introns (45). More recently, the physical interaction between U4/U6-U5 tri-small-nuclear-ribonucleoproteins (tri-snRNPs) splicing factors with TRAMP and nuclear exosome components has also been observed in mammalian cells (46). These findings implied that spliced-out introns are recognized by TRAMP before subsequent degradation by the nuclear exosome. However, it remains unclear whether intronic sequences are also bound by TRAMP in a cotranscriptional manner, and how the cotranscriptional recruitment of TRAMP (if any) is functionally linked to pre-mRNA splicing.
In this study, we found that TRAMP, together with the nuclear exosome component Rrp6p, is cotranscriptionally recruited to nascent transcripts synthesized from both an artificial intron-containing lacZ reporter construct (32) and a number of endogenous intron-containing genes, with significant enrichment at the intronic sequences. We infer that the cotranscriptional recruitment of TRAMP and Rrp6p to intronic sequences may serve an important role in committing the spliced-out introns, and very likely also the splicing-defective pre-mRNAs generated due to splicing errors, to nuclear-exosome-mediated degradation. Interestingly, we found that the cotranscriptional recruitment of TRAMP to intronic sequences (but not that of Rrp6p) has an additional function to facilitate pre-mRNA splicing. This novel finding is further supported by the genetic and physical interactions between the TRAMP component Trf4p and several splicing factors. We also showed that Trf4p can enhance the recruitment of Msl5p (one of the splicing factors that showed physical interaction with Trf4p) to intronic sequences, suggesting how TRAMP may directly promote pre-mRNA splicing. We propose that the requirement of TRAMP for an optimal pre-mRNA splicing efficiency may provide a fail-safe mechanism to ensure the timely recruitment of TRAMP to intronic sequences before or during splicing for the subsequent targeting of spliced-out introns to rapid degradation by the nuclear exosome.
MATERIALS AND METHODS
Yeast strains and plasmids
Yeast strains and plasmids used in this study are listed in Supplementary Tables S1 and S2, respectively. All gene deletions and epitope tagging were performed using standard homologous recombination techniques (47). Epitope switching was done using a previously described method (48). The gal10-Δ56 mutation was generated as described elsewhere (49).
Chromatin immunoprecipitation
Chromatin immunoprecipitations (ChIPs) were performed as previously described (50) with slight modifications. First, 30 ml of yeast cultures at a A600 of 0.6–0.8 were crosslinked with 1% formaldehyde at room temperature for 15 min, followed by centrifugation at 4000 rpm for 5 min, and then the cell pellets were washed twice with 30 ml of 1X TBS. The cells were subsequently lysed in FA lysis buffer (50 mM HEPES-KOH (pH 7.5), 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na deoxycholate and addition of protease inhibitors) with the help of glass beads by vigorous vortexing at 4°C for 45 min. Cell lysates were then sonicated using a Sonics Vibra-cell sonicator for a total of 5 min, with 9 s rest after every 9 s of sonication, followed by centrifugation at 13 200 rpm for 15 min. The supernatants were then used as the input for immunoprecipitations with either 40 µl of rabbit IgG agarose beads (Sigma, Cat #A2909) (for TAP-tagged proteins) or 1 µl of Anti-Rpb3p antibodies (Neoclone, Cat #W0012) (pre-incubated with 40 µl of Anti-mouse magnetic beads {Invitrogen, Cat #112-02D}) for 2 h at 4°C. After that, beads were washed twice with FA lysis buffer, followed by twice with wash buffer II (50 mM HEPES-KOH (pH 7.5), 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na deoxycholate), then twice with wash buffer III (10 mM Tris-HCl (pH 8.0), 250 mM LiCl, 1 mM EDTA, 0.5% NP-40, 0.5% Na deoxycholate), and finally once with TE (10 mM Tris-HCl (pH 8.0), 1 mM EDTA). Then 250 µl of elution buffer (10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 0.67% SDS) was added to the beads, while at the same time 50 µl of input cell lysates were also mixed with 200 µl of elution buffer, and then both input and IP samples were put into a 65°C water bath for 12 h to reverse crosslink. Following that, the chromatin fragments within the input and IP samples were purified by standard phenol-chloroform extraction procedures followed by ethanol precipitation, and the resulting DNA pellets were resuspended in 20 µl of H2O. Finally, the immunoprecipitated and input DNAs were analysed simultaneously by either radioactive PCR or quantitative PCR (qPCR). In the case of radioactive PCR, each reaction contained (33P)-labeled dCTP and two pairs of primers that amplified both a target region and the control region ChrV, in addition to standard PCR reaction components. PCR products were separated by 6% acrylamide gels, then exposed to phosphor screen after gel drying, and signals were detected by a phosphor imager with band intensities being later quantified using the associated software. Occupancies were then calculated by the following formula: (IPtarget/IPChrV) ÷ (Inputtarget/InputChrV). In the case of qPCR, diluted DNA samples were analysed using the SYBR Green method (SYBR® Premix Ex Taq™ from TaKaRa) with the StepOnePlus™ Real-Time PCR Systems from Applied Biosystems, according to manufacturer’s instructions. Occupancies were then calculated by the following formula: 2Ct(IP:ChrV) – Ct(IP:target) ÷ 2Ct(Input:ChrV) – Ct(Input:target). All primers used are listed in Supplementary Table S3.
RNA analysis
Total RNAs were extracted using standard hot phenol RNA preparation method (51). For RT-PCR analysis, 1.5 µg of total RNAs were subjected to reverse transcription (RT) using gene-specific primers, and then the resulting cDNAs were analysed by standard PCR method or qPCR. Primers used are listed in Supplementary Table S3. The half-life of pre-mRNAs was determined as described previously (52,53). For northern blotting, the procedures were as previously described (50) except that total RNAs (10 µg) were separated by 6% acrylamide gels. Sequences of oligonucleotide probes are listed in Supplementary Table S3. Band intensities were quantified using ImageJ densitometry software.
lacZ reporter assay
Overnight yeast cultures were diluted into 30 ml of SC medium containing 2% raffinose but lacking uracil or both uracil and leucine to a A600 of ∼0.15, and then induced with 2% galactose for 2 h when A600 reached 0.3–0.5. After that, yeast cells were lysed by standard freeze-thaw procedures using liquid nitrogen, followed by β-galactosidase activity measurement using chemiluminescent method (54).
GLAM assay
GLAM assay was conducted as described previously (50) using yeast cultures at an A600 of 0.3–0.5.
Coimmunoprecipitation
CoIP was performed as described previously (55), except that cell lysates were immunoprecipitated with 40 µl of rabbit IgG agarose beads (Sigma, Cat #A2909).
Western blotting
Western blotting was performed as described previously (56) except that yeast cells were lysed in FA lysis buffer.
RESULTS
TRAMP is required for optimal splicing of an intron-containing lacZ reporter transcript
To explore the possible role of TRAMP in pre-mRNA splicing, we used a previously described splicing-dependent lacZ reporter system (33,34,57) to measure splicing efficiency in yeast strains with or without a functional TRAMP complex. This system is composed of three reporter plasmids, with each of them bearing a GAL-promoter-driven lacZ reporter gene (Figure 1A) (32). The lacZ gene in pLGSD5 is intronless, and its encoded β-galactosidase activity serves as a control to normalize the differences in plasmid copy number and translation efficiency among different yeast strains. The lacZ gene in Acc° contains an intron close to the 5′ end, and due to differences in translational frame, only the spliced mRNAs, but not the unspliced pre-mRNAs, can be translated into functional β-galactosidase. The activity from Acc° after normalization to that from pLGSD5 provides a quantitative measure of the spliced mRNA level, which represents the efficiency of pre-mRNA splicing. The lacZ gene in Nde°Acc° contains one more nucleotide than Acc° at the intron–exon junction, with the ‘T’ nucleotide in Acc° being changed into ‘CG’ in Nde°Acc° (underlined), and as a result of translational frame-shift, only the unspliced pre-mRNAs from Nde°Acc° (if it is exported into the cytoplasm) can express β-galactosidase. After normalization to pLGSD5, the activity from Nde°Acc° represents the level of unspliced pre-mRNAs in the cytoplasm, which increases when there are splicing defects, pre-mRNA degradation defects, and/or pre-mRNA leakage from the nucleus. In theory, defects in pre-mRNA splicing should reduce the level of spliced mRNAs and elevate the level of unspliced pre-mRNAs, leading to a decreased activity from Acc° but an increased activity from Nde°Acc° after normalization to the activity from pLGSD5.
Figure 1.

Optimal splicing of an intron-containing lacZ reporter transcript is dependent on a functional TRAMP complex. (A) Structure of the three reporter constructs in the relevant lacZ reporter system, and the abilities of their encoded RNAs to be translated into functional β-galactosidase. (B–C) Results of the lacZ reporter assays. Yeast strains with the indicated genotypes transformed with the appropriate reporter plasmids were grown in SC medium containing 2% raffinose but lacking uracil until A600 reached 0.3–0.5, then induced with 2% galactose for 2 h, followed by measurement of β-galactosidase activity. The β-galactosidase activity from strains transformed with either Nde°Acc° (panel B) or Acc° (panel C) are shown relative to the activities in the same strain transformed with pLGSD5 after normalization to total protein concentration in the cell lysates. The differences in β-galactosidase activity between the indicated strains with wild-type were shown to be statistically significant by Student’s t-test (* represents P < 0.05 and ** represents P < 0.01). (D) Semi-quantitative RT-PCR analysis of the unspliced pre-mRNAs (Un) and spliced mRNAs (S) transcribed from the intron-containing lacZ gene on Acc°. A reverse primer located within exon 2 was used for RT, and the same primer was used together with a forward primer located within exon 1 for PCR amplification. The positions of unspliced pre-mRNAs and spliced mRNAs are indicated by the arrows on the right. Band intensities were quantified by ImageLab software, and the numbers below represent the ratio of PCR products produced from the unspliced pre-mRNAs to that of the spliced mRNAs after normalization to wild-type. (E–F) Similar to panel C but with cells being grown in SC medium lacking both uracil and leucine. All lacZ reporter results shown represent the average from at least four independent yeast cultures, with error bars showing the standard errors of the mean.
As expected, deletion of the gene encoding TRAMP components (Trf4p or both Air1p and Air2p) or the nuclear exosome component Rrp6p, which are involved in pre-mRNA degradation (36), and that encoding Cbp80p, which promotes the recruitment of splicing factors to introns (40,58), led to an increased relative β-galactosidase activity from Nde°Acc° to pLGSD5 (unspliced pre-mRNA level) (Figure 1B) due to the accumulation of unspliced pre-mRNAs. As a positive control, the reduced splicing efficiency in cbp80Δ cells (40,58) could be shown by a decreased relative β-galactosidase activity from Acc° to pLGSD5 (spliced mRNA level) (Figure 1C). Interestingly, deletion of RRP6, and that of TRF4 or both AIR1 and AIR2, resulted in distinct readouts of the Acc° reporter. The relative activity from Acc° to pLGSD5 (spliced mRNA level) was unchanged when RRP6 was deleted (Figure 1C). Thus, the increased relative activity from Nde°Acc° to pLGSD5 (Figure 1B), which represented an accumulation of unspliced pre-mRNAs in rrp6Δ cells, could be best attributed to a decreased pre-mRNA degradation rate. The absence of splicing defects in rrp6Δ cells also excluded the possibility that reduced turnover of abnormal splicing intermediates or unspliced pre-mRNAs (36) may indirectly compromise splicing by sequestrating the splicing machinery. In contrast, deletion of TRF4 or both AIR1 and AIR2 significantly reduced the relative activity from Acc° to pLGSD5 (spliced mRNA level) to an extent similar to that evoked by deletion of CBP80 (Figure 1C), indicating the presence of splicing defects in trf4Δ and air1Δair2Δ cells. To ensure that such observed changes of the activity of lacZ reporters were accurately reflecting the changes in RNA levels, we used RT-PCR analysis to directly measure the amount of the unspliced and spliced lacZ RNAs. Although both the deletion of TRAMP components and that of Rrp6p would give rise to an increased unspliced to spliced RNA ratio (Figure 1D), the underlying causes for this phenotype were different. The increased unspliced to spliced RNA ratio associated with the deletion of TRF4 or both AIR1 and AIR2 was due to the accumulation of unspliced pre-mRNAs and the decline of spliced mRNAs (Figure 1D), as also shown by the lacZ reporter results presented in Figures 1B and C, respectively, consistent with the presence of splicing defects. On the other hand, the increased unspliced to spliced RNA ratio in rrp6Δ cells was the consequence of pre-mRNA accumulation but not changes in spliced mRNA levels (Figure 1D), which agreed with the lacZ reporter results shown in Figures 1B and C and was consistent with the known pre-mRNA degradation defects in rrp6Δ cells. Furthermore, the reduced relative activity from Acc° to pLGSD5 (spliced mRNA level) in trf4Δ and air1Δair2Δ cells could be rescued by wild-type TRF4 (Figure 1E) or AIR1 (Figure 1F), respectively. Thus, although TRAMP is a well-defined cofactor of the nuclear exosome (3–5), our results suggested that TRAMP can facilitate the splicing of an intron-containing lacZ reporter transcript in a nuclear-exosome-independent manner.
TRAMP is cotranscriptionally recruited to the lacZ reporter transcript with enrichment over intronic sequences
To examine whether TRAMP directly facilitates splicing of the intron-containing lacZ reporter transcript, we used ChIP to analyse the cotranscriptional recruitment patterns of TAP-tagged TRAMP component Air2p at the galactose-inducible intron-containing lacZ reporter gene on Acc° and its intronless version on pLGSD5 (Figure 2A). Cotranscriptional recruitment of Air2p-TAP was detected along the entire coding sequences of both lacZ reporter genes (Figure 2B and C) when their transcription was induced in the presence of galactose. The high occupancy of the RNA polymerase II subunit Rpb3p (Figure 2B and D) confirmed that both genes were actively transcribing. Air2p-TAP occupancy observed at the intronless lacZ reporter gene on pLGSD5 (Figure 2C) closely followed that of Rpb3p (Figure 2D), leading to a uniform Air2p-TAP to Rpb3p occupancy ratio along the coding sequences (Figure 2E). Notably, the occupancy of Air2p-TAP at the intronic region (region A) on Acc° was significantly higher than the respective intronless region (region A) on pLGSD5 (Figure 2B and C), implying that the presence of an intron can enhance Air2p-TAP occupancy. This effect became even more obvious (Figure 2E) after normalization to the occupancy of Rpb3p (Figure 2D).
Figure 2.

TRAMP is cotranscriptionally recruited to the intron-containing lacZ reporter transcript with enrichment over intronic sequences. (A) Schematic diagram of the intron-containing lacZ gene on Acc° and the intronless lacZ gene on pLGSD5, showing regions subjected to ChIP analysis. The numbers shown represent the number of nucleotides away from the start codon. (B–E) Air2p-TAP strain harboring the indicated plasmids were grown in SC medium lacking uracil but containing 2% raffinose, with the addition of 2% galactose to the medium 1.5 h before crosslinking, and then subjected to ChIP analysis using radioactive PCR. Representative results for region A are shown in (B). Radiolabeled amplicons were quantified by phosphorimaging and the ratios of target to ChrV signals in the Air2p-TAP and Rpb3p immunoprecipitations (IPs) were normalized to that in the corresponding inputs to yield the occupancy values presented in (C) and (D), respectively. The Air2p-TAP occupancies shown in (C) were divided by the Rpb3p occupancies shown in (D) to yield the Air2p-TAP to Rpb3p occupancy ratios shown in (E). (F–G) ChIP analysis as in (B–E) but with cell lysates being treated with or without RNase A for 30 min at room temperature before immunoprecipitation with IgG agarose beads. Chromatin fragments of region A and ChrV were analysed by PCR without radiolabeling, and the amplicons were visualized by ethidium bromide (EtBr) staining followed by quantification using ImageLab software. Representative results are shown in (F), and Air2p-TAP occupancies relative to that at pLGSD5 without RNase A treatment were plotted in (G). The differences in Air2p-TAP occupancy and Air2p-TAP to Rpb3p occupancy ratio between pLGSD5 and Acc° were shown to be statistically significant using Student’s t-test (* represents P < 0.05 and ** represents P < 0.01). All results shown represent the average from three independent cultures, with error bars showing the standard errors of the mean.
We also investigated whether binding of TRAMP to the intronic region is RNA dependent. To address this question, ChIP analysis was performed using cell lysates being treated with or without RNase A before the immunoprecipitation step (59,60). Consistent with the result shown in Figure 2C, Air2p-TAP occupancy was significantly higher at region A on Acc° than that on pLGSD5 for cell lysates without RNase A treatment (Figure 2F and G). The enrichment of Air2p-TAP at region A on both Acc° and pLGSD5 was substantially decreased by the treatment of RNase A (Figure 2F and G), indicating that TRAMP binds directly to the nascent RNA transcript (but not the surrounding chromatin DNA) during transcription. The cotranscriptional recruitment of TRAMP to the nascent intron-containing lacZ reporter transcript with enrichment over intronic sequences probably implicated a direct stimulatory role of TRAMP in pre-mRNA splicing.
TRAMP recruitment to endogenous intron-containing transcripts is also enriched at intronic sequences
After demonstrating the cotranscriptional recruitment of TRAMP to the intron-containing lacZ reporter transcript, we asked whether TRAMP is also cotranscriptionally recruited to RNA transcripts transcribed from endogenous intron-containing genes. ChIP was performed to investigate the cotranscriptional recruitment of Air2p-TAP to the RNA transcripts synthesized from the intron-containing genes RPL18B and RPS9A (Figure 3A and E), in which their corresponding spliced-out introns have been previously shown to be recognized by TRAMP using RNA-IP-chip analysis (45). Air2p-TAP occupancy at all examined regions along both RPL18B and RPS9A was significantly higher than that of the no-tag control (Figure 3B and F), indicating that Air2p-TAP is also recruited to the RNA transcripts transcribed from these endogenous intron-containing genes. However, since both RPL18B and RPS9A genes are relatively short (∼1000 bp) and thus limited the resolution of the ChIP assay, there were no detectable effects of the presence of intron on Air2p-TAP recruitment (Figure 3A–H). To overcome this problem, the longer intron-containing genes ECM33 and DBP2 were also analysed. In addition, recruitment of the TRAMP components Air1p-TAP and Trf4p-TAP, as well as the nuclear exosome component Rrp6p-TAP, was also examined.
Figure 3.

TRAMP is cotranscriptionally recruited to nascent RNAs transcribed from endogenous intron-containing genes, with higher occupancy at intronic regions. (A, E, I, M) Schematic diagram of the intron-containing genes RPL18B (A), RPS9A (E), ECM33 (I) and DBP2 (M), showing regions subjected to ChIP analysis. The numbers shown represent the number of nucleotides away from the start codon. (B, F, J, N) Indicated yeast strains were grown in YPD medium to log-phase and then subjected to ChIP analysis using IgG agarose beads. The purified input and immunoprecipitated chromatin fragments were analysed by qPCR. The calculated occupancy values at the targeted regions (relative to a non-coding region in ChrV) for the indicated TAP-tagged proteins were plotted after normalization to the no-tag control. (C, G, K, O) The same lysates used in (B, F, J, N) were subjected to ChIP analysis using Anti-Rpb3p antibodies, yielding the plotted Rpb3p occupancy values. (D, H, L, P) The occupancies for the indicated TAP-tagged proteins shown in (B, F, J, N) were further normalized to the corresponding Rpb3p occupancies shown in (C, G, K, O) to calculate the TAP/Rpb3p occupancy ratios. The differences between Air2p-TAP and the untagged control in (B, D, F, H), and those between the indicated pairs of regions along ECM33 or DBP2 in (J, L, N, and P), were shown to be statistically significant using Student’s t-test (* represents P < 0.05 and ** represents P < 0.01). All results shown represent the average from three independent cultures, with error bars showing the standard errors of the mean.
At ECM33 (Figure 3I), in which the intron is located near the 5′ end just as that of the intron-containing lacZ reporter gene on Acc° (Figure 2A), the recruitment levels of TRAMP and Rrp6p were the highest (∼2.0–2.5-fold) at positions near the intron (regions A and B), but decreased to ∼1.5-fold at the middle (region C) or 3′ end (region D) of the long exon 2 (Figure 3J). The changes in TRAMP and Rrp6p occupancy along ECM33 were not due to differences in transcription activity (Figure 3K), as the occupancy of TRAMP and Rrp6p was still enriched at positions near the intron after normalization to Rpb3p occupancy (Figure 3L). At DBP2 (Figure 3 M), in which the intron is located at the middle of the gene closer to the 3′ end, both Air1p-TAP and Air2p-TAP occupancy remained at ∼1.5-fold along exon 1 (regions A and B), and then increased to ∼2.0–2.5-fold within (region C) or just downstream of the intron (region D) (Figure 3N). Both Trf4p-TAP and Rrp6p-TAP also showed similar recruitment pattern (Figure 3N), although the effect of the intron appeared to be smaller, probably because their binding to the intron should be mediated through Air1/2p. After normalization to Rpb3p occupancy (Figure 3O), the enrichment of TRAMP and Rrp6p occupancy within or just downstream of the DBP2 intron became even more prominent (Figure 3P). To summarize, these results clearly demonstrated that the cotranscriptional recruitment of both TRAMP and the nuclear exosome to nascent RNA transcripts are enriched at the intronic region of each gene, irrespective of its presence near the 5′ end [as for the intron-containing lacZ gene on Acc° (Figure 2) and ECM33 (Figure 3I–L)] or the 3′ end [as for DBP2 (Figure 3M–P)], suggesting that spliced-out introns, and probably also splicing-defective pre-mRNAs, may already be captured by TRAMP and committed to subsequent nuclear-exosome-mediated degradation during transcription (i.e. very likely before or during splicing). The cotranscriptional recruitment of TRAMP to RNA transcripts synthesized from endogenous intron-containing genes also led to the hypothesis that TRAMP may enhance the splicing of endogenous unspliced pre-mRNAs.
Pre-mRNA splicing is facilitated by TRAMP
To test whether TRAMP is needed for optimal splicing of endogenous unspliced pre-mRNAs, we performed RT-qPCR to monitor the level of the unspliced RPL18B and ECM33 pre-mRNAs in yeast strains with or without deletion of TRAMP components. Consistent with the lacZ reporter results (Figure 1B) and the requirement of nuclear exosome for unspliced pre-mRNA turnover (36), both RPL18B and ECM33 pre-mRNAs accumulated in yeast cells lacking Rrp6p (∼4.5-fold for RPL18B and ∼2.0-fold for ECM33) (Figure 4A and B). Deletion of the TRAMP components TRF4 or both AIR1 and AIR2 resulted in a moderately greater extent of pre-mRNA accumulation (∼6.0-fold for RPL18B and ∼3.0-fold for ECM33) (Figure 4A and B) than that of RRP6, suggesting that TRAMP may have an additional role in regulating pre-mRNA levels in addition to its plausible role in stimulating Rrp6p-dependent pre-mRNA turnover. Semi-quantitative RT-PCR analysis also revealed that the unspliced pre-mRNAs to spliced mRNAs ratio of RPL18B is significantly greater in trf4Δ and air1Δair2Δ as compared to wild-type or even the splicing-compromised cbp80Δ cells (40,58) (Figure 4C). There was no pre-mRNA accumulation in the trf5Δ strain (Figure 4C), probably because the presence of Trf4p may completely compensate for the loss of Trf5p, as suggested by previous studies (45,61). The observed increase in the RPL18B pre-mRNAs/mRNAs ratio in trf4Δ cells could be almost completely rescued by addition of the wild-type TRF4 gene (Figure 4D), while that in air1Δair2Δ cells could be partially complemented by either the AIR1 or the AIR2 gene (Figure 4D), further confirming that TRAMP is required for pre-mRNA metabolism.
Figure 4.

Deletion of TRAMP components resulted in accumulation of endogenous unspliced pre-mRNAs largely independent of impaired Rrp6p or Dis3p activity in the absence of TRAMP components. (A–B) RT-qPCR analysis of the unspliced pre-mRNAs transcribed from RPL18B (panel A) and ECM33 (panel B) in yeast strains with the indicated genotypes. Total RNAs extracted from log-phase yeast cultures were subjected to DNase I treatment and then RT using gene-specific primers located within exon 2, followed by qPCR analysis of cDNAs using a forward primer located within the intron and a reverse primer identical to that used in RT. The PMA1 transcript was also analysed in parallel to serve as an internal control. The relative unspliced pre-mRNA levels after normalization to that of PMA1 were shown. (C–D) The cDNAs originating from yeast strains with the indicated genotypes were analysed by semi-quantitative PCR to detect the levels of the unspliced (Un) and spliced (S) RPL18B transcripts (with their positions shown on the right). Band intensities were quantified using ImageLab software, and the relative ratios of unspliced to spliced transcripts were shown at the bottom. (E–H) Same as (A–B) but using primers that can amplify the indicated unspliced pre-mRNAs. (I–J) Same as (C–D) but using primers that can amplify the indicated unspliced pre-mRNAs (Un) and spliced mRNAs (S). (K–L) Same as (E) and (G) but using yeast strains with the indicated genotypes that were grown either in the absence (-DOX) or presence (+DOX) of doxycycline (20 µg/ml) for 8 h from the early-log phase. All yeast strains with the Tet-Off DIS3 allele also contained a plasmid expressing a mutant form of Dis3p with simultaneous mutations in both catalytic domains (D171N-D551N). Student’s t-test was applied to confirm that the differences in pre-mRNA levels between the indicated yeast strains and wild-type (A–B, E–H), or between the indicated pairs of yeast strains (K–L), were statistically significant (* represents P < 0.05 and ** represents P < 0.01). All qPCR results shown represent the average from three independent cultures, with error bars showing the standard errors of the mean.
In order to determine whether pre-mRNA accumulation observed in trf4Δ and air1Δair2Δ cells was due at least partially to the involvement of TRAMP in Rrp6p-independent pathways, we also examined in these cells the level of several other unspliced pre-mRNAs which do not require Rrp6p for their normal turnover (37). The deletion of RRP6 had no effects on the level of the RPL16B, RPL39, RPL21A and RPS18A pre-mRNAs (Figure 4E–H), consistent with previous report that Rrp6p is not involved in their degradation (37). Importantly, the deletion of TRF4 or both AIR1 and AIR2 led to significant increments (∼1.5–2.0-fold) in the level of these unspliced pre-mRNAs (Figure 4E–H), with the accumulation of RPL16B and RPL21A pre-mRNAs being further confirmed using semi-quantitative RT-PCR analysis of their pre-mRNAs/mRNAs ratio (Figure 4I and J). These results suggested that pre-mRNA accumulation observed in trf4Δ and air1Δair2Δ cells was largely independent of the role of TRAMP in enhancing Rrp6p-mediated pre-mRNA turnover.
In addition to Rrp6p, the nuclear exosome contains another catalytic subunit known as Dis3p (or Rrp44p) (1,2,6), which is also involved in the degradation of pre-mRNAs and CUTs such as NEL025c (13). As DIS3 is an essential gene, we used a doxycycline-repressible promoter system to deplete the expression of endogenous DIS3, which effectiveness could be shown by the huge accumulation of NEL025c transcripts upon the addition of doxycycline (Supplementary Figure S1). Using the same experimental system, we observed a slight increase (∼1.5-fold) in RPL16B and RPL21A pre-mRNA levels (Figure 4K and L) upon DIS3 depletion. Note that in order to rule out the possibility that depletion of Dis3p might affect its non-catalytic functions, a catalytically inactive Dis3p mutant (D171N-D551N) was constitutively expressed in the endogenous Dis3p-depleted strains. We next asked whether the accumulation of RPL16B and RPL21A pre-mRNAs in TRAMP-defective cells (Figure 4E and G) was simply due to the loss of the theoretically possible stimulation of Dis3p activity by TRAMP, although this was very unlikely as it has been previously reported that TRAMP can facilitate the action of Rrp6p but not that of Dis3p or the core exosome (62). We found that further deletion of TRF4 from yeast strain already carrying both deletion of RRP6 and doxycycline-repressible DIS3 gene still resulted in significant RPL16B and RPL21A pre-mRNA accumulation even when DIS3 expression was depleted by doxycycline treatment (Figure 4K and L). This finding indicated that pre-mRNA accumulation associated with TRAMP component deletion was largely unattributed to the possible role of TRAMP in enhancing Rrp6p- or Dis3p-mediated pre-mRNA degradation. Therefore, we concluded that most of the accumulated RPL16B, RPL39, RPL21A and RPS18A pre-mRNAs, and at least a small portion of the accumulated RPL18B and ECM33 pre-mRNAs, observed in trf4Δ and air1Δair2Δ cells were due to splicing defects. These results provided strong evidences implicating the moonlighting function of TRAMP in promoting efficient splicing of endogenous pre-mRNAs.
Pursuing these, we further analysed the effect of deletion of TRAMP components on the half-life of pre-mRNAs. Since a newly synthesized pre-mRNA should either be spliced into mature mRNA (31) or degraded into ribonucleotides [may occur in the nucleus (36) or after export into cytoplasm (37–39)], half-life of the pre-mRNA is dependent on both its splicing efficiency and the rate of its degradation. After transcriptional shut-off with thiolutin (52,53), the RPL18B pre-mRNAs are spliced and/or degraded with a half-life of ∼7.4 min (Figure 5A). Consistent with its role in the degradation of RPL18B pre-mRNAs, deletion of RRP6 significantly prolonged its half-life to ∼11.0 min (Figure 5A). In agreement with a stimulatory role of TRAMP in both the splicing of RPL18B pre-mRNAs and their Rrp6p-mediated degradation, trf4Δ and air1Δair2Δ cells showed an even longer RPL18B pre-mRNA half-life (∼12.1 min for trf4Δ and ∼16.3 min for air1Δair2Δ) as compared to rrp6Δ cells (∼11.0 min) (Figure 5A). The half-life of RPL16B and RPL21A pre-mRNAs in these yeast strains was also consistent with their steady-state levels. Both RPL16B and RPL21A pre-mRNAs had a half-life of ∼8.2 min in wild-type cells, and their half-life remained unaffected in rrp6Δ (Figure 5B and C), in accordance with that Rrp6p is not required for their normal turnover (Figure 4E–J) (37). In contrast, their half-life was significantly prolonged to >10.0 min in trf4Δ and air1Δair2Δ cells (Figure 5B and C), suggesting that their splicing efficiency (but not degradation rate since their half-life was not affected in rrp6Δ) was compromised. Thus, the analysis of endogenous unspliced pre-mRNA half-life after transcription inhibition with thiolutin also supported that pre-mRNA splicing is facilitated by TRAMP.
Figure 5.

Analysis of the half-life of endogenous unspliced pre-mRNAs and the accumulation rate of spliced lacZ mRNAs in yeast strains lacking TRAMP components or Rrp6p. (A–C) Yeast cells with the indicated genotypes were harvested at different time points after the addition of thiolutin (4 µg/ml) to inhibit transcription. The cDNAs prepared from these cells were subjected to qPCR analysis to determine the levels of the indicated pre-mRNAs and the internal control SCR1. The amount of pre-mRNAs remaining in the cells after normalization to SCR1 were expressed in terms of the percentage of the pre-mRNA level at t = 0. The estimated pre-mRNA half-life and its standard errors of the mean are shown at the bottom. (D) The indicated yeast strains transformed with the Acc° reporter plasmid were first grown in SC medium containing 2% raffinose but lacking uracil, and then harvested at different time points after the addition of 2% galactose to induce expression of the intron-containing lacZ gene on Acc°. After that, RT-qPCR analysis was performed to measure the levels of the spliced lacZ mRNAs (with the forward primer being complementary to the exon 1–exon 2 junction) and also SCR1. The spliced lacZ mRNA levels at the indicated time points were shown after normalization to SCR1, with the relative amount of spliced mRNAs at t = 0 being set at 1.0 for each yeast strain. Student’s t-test was used to demonstrate that the indicated differences were statistically significant (* represents P < 0.05 and ** represents P < 0.01). All results shown represent the average from three independent cultures, with error bars showing the standard errors of the mean.
To further confirm the stimulatory role of TRAMP in splicing, we also measured the accumulation rate of the spliced lacZ mRNAs synthesized from the intron-containing lacZ gene on the Acc° reporter plasmid when its expression was induced by galactose. As expected, the spliced lacZ mRNA level increased sharply in wild-type cells after galactose addition (Figure 5D). The deletion of RRP6 did not significantly affect its rate of accumulation (Figure 5D), consistent with our finding that Rrp6p is not involved in pre-mRNA splicing (Figure 1C). Importantly, there was a severe delay in the accumulation of the spliced lacZ mRNAs in trf4Δ and air1Δair2Δ cells (Figure 5D). This effect was not due to defective induction of the GAL promoter, as there were no significant changes in the accumulation rate of GAL10 mRNAs in the absence of TRAMP components (Supplementary Figure S2A). Thus, the induction time profile of the spliced lacZ mRNAs remained significantly delayed in the TRAMP-defective cells even after normalization to that of GAL10 mRNAs (Supplementary Figure S2B), which again, indicated that TRAMP is required for optimal splicing of pre-mRNAs.
Splicing defects associated with deletion of TRAMP components are not due to defects in transcription elongation
Pre-mRNA splicing and transcription elongation are tightly linked processes (63–65). Since TRAMP components physically associate with subunits of the THO complex (27) and the Ccr4-Not complex (66), which are both involved in transcription elongation (67,68), we first examined whether TRAMP may indirectly regulate splicing by affecting transcription elongation rate.
The Δ56 mutation within the GAL10 3′-UTR of normal cells results in transcriptional read-through into the downstream GAL7 gene (Figure 6A), rendering a Gal– phenotype (unable to grow on galactose-containing medium) that can be rescued by the deletion of transcription elongation factors (49) or anti-termination factors (69). The deletion of both Air1p and Air2p did not detectably rescue the Gal– phenotype (Figure 6B), suggesting that TRAMP is not involved in transcription elongation. In addition, transcription elongation defects could only be detected in the positive control npl3Δ (70) but not in trf4Δ and air1Δair2Δ cells (Figure 6C) using a well-established assay for gene-length dependent accumulation of mRNA (GLAM) to measure transcription elongation rate (71), thus further excluding the possibility that TRAMP may facilitate pre-mRNA splicing by participating in transcription elongation.
Figure 6.

Splicing defects in trf4Δ and air1Δair2Δ are not due to defects in transcription elongation or snRNA synthesis. (A) Schematic diagram explaining the rationale of the gal10Δ56 assay. In yeast strains without transcription elongation and/or anti-termination defects, the transcriptional read-through from the GAL10 gene containing the Δ56 deletion in its 3′-UTR interferes with the proper transcription of the downstream GAL7 gene, thus preventing the cells from growing on a medium that contains galactose as the only carbon source (Gal– phenotype). In yeast strains with defects in transcription elongation and/or anti-termination, the impaired transcriptional read-through into GAL7 partially restores GAL7 transcription, thus allowing the cells to grow on the galactose-containing medium (Gal + phenotype). (B) Results of the gal10Δ56 assay. Ten-fold serial dilutions of yeast cultures with the indicated genotypes were spotted onto SC-Leu agar plates containing either glucose or galactose as the only carbon source, and then incubated at 30°C or 37°C for 4 days. (C) Results of the GLAM assay. The acid phosphatase activity from the indicated yeast strains being transformed with pSCh209-LAC4 were divided by that from the same strain being transformed with pSCh202 to yield the GLAM ratio shown. The reduced GLAM ratio in npl3Δ cells was statistically significant by Student’s t-test (* represents P < 0.05). The results represent the average from three independent cultures, with error bars showing the standard errors of the mean. (D) Northern blot analysis of both mature and precursor forms of U1, U4 and U5 snRNAs, and the loading control SCR1, on total RNAs extracted from log-phase cultures of the indicated yeast strains grown in YPD using (32P)-labeled oligonucleotide probes. For each snRNA, two images with different exposure time are shown to enable clear visualization of both mature and precursor forms, with their positions indicated on the right. Band intensities were quantified using ImageJ software, and the numbers at the bottom of each image represent the amount of mature or precursor forms of U1, U4 or U5 snRNAs, as indicated, relative to wild-type after normalization to the loading control SCR1. Note that mature U5 snRNA has two forms: U5L represents the long form, while U5S represents the short form.
Splicing defects associated with deletion of TRAMP components are not due to defective snRNA processing
It has been previously demonstrated that the nuclear exosome is involved in the synthesis of U1, U4 and U5 small nuclear ribonucleic acids (snRNAs) from their precursors by 3′ end processing (72), and that the TRAMP complex component Trf4p also contributes to U4 snRNA synthesis (73). This raised the possibility that defects of pre-mRNA splicing caused by deletion of TRAMP components may be indirectly due to snRNA processing defects. However, northern blot analysis of U1, U4 and U5 snRNAs as well as their precursors revealed that the snRNA processing defects observed in cells lacking TRF4 or both AIR1 and AIR2 were in all cases less severe than those observed in rrp6Δ cells. There was no detectable accumulation of U1 precursor in trf4Δ or air1Δair2Δ (Figure 6D, compare lanes 2 and 3 versus lane 1); and although both U4 and U5 precursors did accumulate in these cells, the phenotypes were significantly less severe than those in rrp6Δ cells, with an accumulation of only ∼2.0–3.0-fold for both U4 and U5 precursors in trf4Δ or air1Δair2Δ strains (Figure 6D, compare lanes 2 and 3 versus lane 1) compared to >4.0-fold in rrp6Δ cells (Figure 6D, compare lane 4 with lane 1). As deletion of RRP6 did not detectably affect splicing efficiency (Figure 1C), it is very unlikely that the splicing defects in trf4Δ and air1Δair2Δ strains arise from their less pronounced snRNA processing defects. Furthermore, the levels of the mature forms of U1, U4 and U5 snRNAs in these TRAMP knockout strains were in no cases less than the wild-type levels (Figure 6D, compare lanes 2 and 3 versus lane 1), indicating that there are sufficient or even excess snRNAs available for the splicing machinery. These results excluded the possibility that defective snRNA processing is the primary cause of the splicing defects observed in trf4Δ and air1Δair2Δ cells, and suggested a more direct stimulatory role for TRAMP in pre-mRNA splicing.
TRF4 shows negative genetic interactions with the splicing factor genes MUD2 and MSL1
To explore whether there are any direct interactions between the TRAMP complex and the splicing machinery, we characterized the genetic interactions between TRF4 and splicing factor genes. A previous genome-wide genetic interactions study suggested that TRF4 may have a negative genetic interaction with MUD2 (74), which encodes the U2AF65 protein involved in polypyrimidine tract binding during splicing (31). To verify this, we constructed the TRF4 and MUD2 deletion strains and characterized the genetic interaction between these mutations. The growth rate of yeast strain containing single deletion of TRF4 (doubling time ∼3.9 h) was moderately slower than that of wild-type cells (doubling time ∼2.9 h), while the single deletion of MUD2 (doubling time ∼2.9 h) had no detectable effect on growth rate (Figure 7A). However, the double deletion of both TRF4 and MUD2 resulted in a synthetic growth defect, with a growth rate (doubling time ∼4.6 h) much slower than that of trf4Δ (Figure 7A), indicating that negative genetic interaction exists between TRF4 and MUD2. RT-PCR analysis revealed that while the single deletion of MUD2 (∼1.1-fold) from wild-type cells did not result in any significant accumulation of the RPL21A pre-mRNAs, MUD2 deletion from the trf4Δ background (∼1.4-fold) could led to a more severe RPL21A pre-mRNA accumulation phenotype (∼1.9-fold) (Figure 7B), which further demonstrated the negative genetic interaction between the two genes. In addition, we also uncovered the negative genetic interaction between TRF4 and the U2 snRNP gene MSL1. Double knockout yeast strains lacking both TRF4 and MSL1 showed a much more severe growth defect than either of the corresponding single knockout strains (Figure 7C). Although MSL1 deletion from wild-type cells already resulted in a significant increase (∼1.5-fold compared to wild-type) in the level of RPL21A pre-mRNAs, the effect of its deletion was moderately greater (∼2.7-fold compared to wild-type; ∼1.9-fold compared to trf4Δ) in the trf4Δ background (Figure 7D), suggesting that negative genetic interaction also exists between TRF4 and MSL1. The negative genetic interactions between TRF4 and splicing factor genes implied a linkage between the TRAMP complex and the pre-mRNA splicing machinery.
Figure 7.

Trf4p shows genetic and physical interactions with splicing factors. (A and C) Ten-fold serial dilutions of yeast cultures with the indicated genotypes were spotted onto YPD agar plates and then incubated at 30°C for 2 days. (B and D) RT-PCR analysis of the unspliced (Un) and spliced (S) RPL21A transcripts in the indicated yeast strains. The numbers shown at the bottom represent the ratio of unspliced to spliced transcripts relative to that of the wild-type cells. (E) Yeast strains expressing Trf4p-myc together with or without a TAP-tagged protein, as indicated, were grown in YPD medium to log-phase and then subjected to immunoprecipitation using IgG agarose beads. Both the input (1% of the amount used for immunoprecipitation) and immunoprecipitated proteins (10% of the eluted proteins for Air2p-TAP and 50% for others) were subsequently analysed by western blotting.
Trf4p physically interacts with splicing factors
Cotranscriptional recruitment of TRAMP complex is enriched at intronic regions (Figures 2 and 3) with recruitment patterns similar to factors involved in the early stage of spliceosome assembly (40–44). However, several previous large-scale pull-down of TRAMP components failed to identify any physically interacting splicing factors (3–5). We hypothesized that the absence of any detectable physical interactions between TRAMP components and splicing factors in these experiments was due to their relatively weak and transient interactions inside cells. To overcome this difficulty, we adapted a previously used coimmunoprecipitation protocol to capture transient protein–protein interactions by formaldehyde crosslinking (55) for examining the physical interactions between Trf4p and the early splicing factors Prp42p (U1-snRNP component), Msl5p (branch-point binding protein), and Lea1p (U2-snRNP component). The possible physical interactions between Trf4p and the late splicing factors Prp43p (required for spliceosome disassembly after splicing) and Prp16p (required for the second catalytic step of splicing) were also analysed.
We constructed yeast strains capable of expressing myc-tagged Trf4p together with or without a TAP-tagged splicing factor. Trf4p-myc could be detected in the immunoprecipitate when any one of the five tested splicing factors (Figure 7E, lanes 5–9) was pulled-down, while no Trf4p-myc could be observed in the negative controls when either the eIF3 subunit Tif32p-TAP (Figure 7E, lane 3) or no TAP-tagged proteins (Figure 7E, lane 1) was immunoprecipitated. This result demonstrated that at least transient physical interactions exist between Trf4p and these splicing factors, suggesting the possibility that TRAMP may enhance pre-mRNA splicing by facilitating splicing factor recruitment.
Trf4p contributes to the cotranscriptional recruitment of Msl5p to intronic regions
The branch-point binding protein Msl5p was the strongest Trf4p-interacting early splicing factor in our coimmunoprecipitation experiment (Figure 7E). Therefore, we examined whether Msl5p-TAP recruitment is affected in trf4Δ cells by ChIP. The cotranscriptional recruitment of Msl5p-TAP to both RPL18B (regions A and B) and ECM33 (region B) (Figure 8A) was significantly reduced in trf4Δ cells as compared to TRF4-complemented cells (Figure 8B), even despite a generally higher Rpb3p recruitment level in trf4Δ (Figure 8C). As a result, the Msl5p-TAP to Rpb3p occupancy ratios at RPL18B (regions A and B) and ECM33 (regions A and B) increased substantially after the TRF4 gene was introduced into the corresponding trf4Δ cells (Figure 8D). Similar results could also be obtained by the analysis of RPL18B (region A) intronic chromatin fragments (Figure 8A) using PCR with radiolabeling (Figure 8E and F). All observed differences in Msl5p-TAP recruitment were not due to changes in protein expression levels, as Msl5p-TAP showed equal expression in both the presence and absence of Trf4p (Figure 8G). Our results indicated that Trf4p can facilitate the cotranscriptional recruitment of Msl5p, and that impaired Msl5p recruitment at least partially contributes to the splicing defects observed in yeast strains with deletion of TRAMP complex components.
Figure 8.

Trf4p is required for optimal recruitment of the splicing factor Msl5p. (A) Schematic diagram of the intron-containing genes RPL18B and ECM33, showing regions subjected to ChIP analysis. (B–D) trf4Δ strain expressing TAP-tagged Msl5p was transformed with either empty vector or a TRF4-containing plasmid, and then subjected to ChIP analysis with cells grown in SC medium lacking leucine. The purified input and immunoprecipitated chromatin fragments were analysed by qPCR, yielding the Msl5p-TAP occupancies, Rpb3p occupancies and Msl5p-TAP to Rpb3p occupancy ratios plotted in (B), (C) and (D), respectively. (E–F) The RPL18B intron (region A) fragments, together with the non-coding region control ChrV, were analysed by radioactive PCR in the presence of (33P)-dCTP. Representative results and the calculated Msl5p-TAP to Rpb3p occupancy ratios are shown in (E) and (F), respectively. Student’s t-test was applied to confirm that the indicated differences in Msl5p-TAP occupancies or Msl5p-TAP to Rpb3p occupancy ratios between trf4Δ cells transformed with empty vector and those carrying a TRF4-containing plasmid were statistically significant (* represents P < 0.05 and ** represents P < 0.01). All results shown represent the average from at least three independent cultures, with error bars showing the standard errors of the mean. (G) Whole-cell extracts from cells with the same genotypes as those used for the ChIP analysis in (B–F) were subjected to western blot analysis using antibodies against the TAP tag of Msl5p-TAP and the loading control Pgk1p.
DISCUSSION
In this work, we demonstrated that both the TRAMP complex and the nuclear exosome component Rrp6p are cotranscriptionally recruited to intron-containing transcripts, particularly to intronic regions (Figures 2 and 3), and uncovered a novel role of TRAMP in promoting optimal pre-mRNA splicing efficiency (Figures 1, 4 and 5). Since TRAMP is also cotranscriptionally recruited to the intronless lacZ reporter transcript (Figure 2), and it can facilitate recruitment of the splicing factor Msl5p to intronic sequences (Figure 8), it is very likely that TRAMP is recruited to intron-containing transcripts before or during splicing. Based on our findings, we propose a revised model for the coupling between nuclear RNA quality control and pre-mRNA splicing (Figure 9). First, TRAMP, together with the nuclear exosome, are recruited to the nascent pre-mRNAs, particularly to the introns, in a cotranscriptional manner. After that, TRAMP stimulates splicing of the pre-mRNAs by enhancing Msl5p (and possibly other splicing factors) recruitment. After splicing, the spliced-out introns will remain associated with TRAMP and then degraded by the nuclear exosome to prevent their accumulation in the cells. The cotranscriptional recruitment of TRAMP and the nuclear exosome to pre-mRNAs may also be important to commit splicing-defective pre-mRNAs to rapid degradation if a functional spliceosome fails to be assembled.
Figure 9.
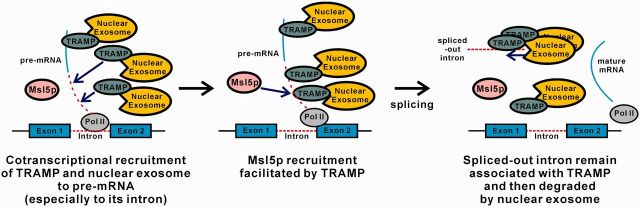
A working model for the coupling between nuclear RNA quality control and pre-mRNA splicing. (Left) During transcription of an intron-containing gene by RNA polymerase II, TRAMP, together with the nuclear exosome, is recruited to the nascent pre-mRNA, especially to the intronic region. (Middle) TRAMP then stimulates splicing of the pre-mRNA by enhancing Msl5p recruitment. (Right) After splicing, TRAMP still remains associated with the spliced-out intron, which is then degraded by the nuclear exosome to prevent its accumulation. The cotranscriptional recruitment of TRAMP and the nuclear exosome may also function to recognize and degrade the splicing-defective pre-mRNA produced as a result of splicing errors.
We envision that the stimulatory role of TRAMP in pre-mRNA splicing may have important functional significance to its primary role in spliced-out intron recognition. Efficient degradation of spliced-out introns probably requires the binding of TRAMP to intronic sequences before or during splicing, that is, before the introns are spliced-out. Thus, the facilitation of splicing by TRAMP may serve as a safety mechanism to ensure the timely (before or during splicing) recruitment of TRAMP to intronic sequences. As a result, pre-mRNA splicing only proceeds at the optimal efficiency after the binding of TRAMP. This mechanism may serve to prevent introns that are not yet bound by TRAMP from being spliced-out.
Recently, it has been suggested that negative splicing regulation triggers the degradation of pre-mRNAs by the nuclear exosome, providing another layer of expression control of intron-containing genes (75). Since TRAMP is probably involved in both pre-mRNA degradation (by being a cofactor of the nuclear exosome) (36) and splicing (this study), it is likely that this kind of gene regulatory mechanism may also require TRAMP. It is potentially interesting to examine how TRAMP may regulate the expression of intron-containing genes through its roles in pre-mRNA degradation and/or splicing.
It remains to be determined how the nuclear RNA quality control system distinguishes introns from the surrounding exonic sequences. It has been proposed that the kinetic competition between proper formation of a functional RNP and commitment to degradation by the RNA quality control system determines the fate of nascent RNA transcripts (1,2,6). The enrichment of TRAMP and nuclear exosome at introns may also act by a similar kinetics-based mechanism. As splicing may delay the assembly of an export-competent RNP, an extended time interval is probably available for further recruitment of TRAMP and the nuclear exosome to introns. It would be of great interest to experimentally delineate the exact mechanism underlying the specific enrichment of their recruitment at intronic sequences.
The human homologs of both the nuclear exosome and TRAMP in budding yeast have already been identified (76,77). Recently, the human homologs of yeast Trf4p, Mtr4p and Rrp6p, have been shown to interact with U4/U6-U5 tri-snRNP components, suggesting that association between the nuclear RNA quality control machinery and splicing complexes may also occur in human cells (46). Given that a higher frequency of introns can be found in the human genome, an efficient nuclear RNA quality control system is particularly important for the rapid degradation of an increased amount of spliced-out introns. It is tempting to determine how the recognition and degradation of spliced-out introns is coupled to the nuclear RNA quality control pathway in human cells, and this remains a major goal for future research.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [1R01TW00829801A1 to C.M.W.]; National Natural Science Foundation of China [31271361 to C.M.W.]; This research was supported in part by the Intramural Program of the National Institutes of Health. Funding for open access charge: NIH [1R01TW00829801]; National Natural Science Foundation of China [31271361].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank P. Legrain, M. Rosbash and S. Chávez for plasmids, B. Séraphin and D. Libri for yeast strains, and members of Jin’s and Wong’s laboratories for critical reading of manuscript.
REFERENCES
- 1.Doma MK, Parker R. RNA quality control in eukaryotes. Cell. 2007;131:660–668. doi: 10.1016/j.cell.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 2.Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–776. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 3.LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, Tollervey D. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell. 2005;121:713–724. doi: 10.1016/j.cell.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 4.Vanacova S, Wolf J, Martin G, Blank D, Dettwiler S, Friedlein A, Langen H, Keith G, Keller W. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 2005;3:e189. doi: 10.1371/journal.pbio.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME, Boulay J, Regnault B, Devaux F, Namane A, Seraphin B, et al. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell. 2005;121:725–737. doi: 10.1016/j.cell.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 6.Schmid M, Jensen TH. The exosome: a multipurpose RNA-decay machine. Trends Biochem. Sci. 2008;33:501–510. doi: 10.1016/j.tibs.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 2004;18:1227–1240. doi: 10.1101/gad.1183804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadaba S, Wang X, Anderson JT. Nuclear RNA surveillance in Saccharomyces cerevisiae: Trf4p-dependent polyadenylation of nascent hypomethylated tRNA and an aberrant form of 5S rRNA. RNA. 2006;12:508–521. doi: 10.1261/rna.2305406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider C, Kudla G, Wlotzka W, Tuck A, Tollervey D. Transcriptome-wide analysis of exosome targets. Mol. Cell. 2012;48:422–433. doi: 10.1016/j.molcel.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wlotzka W, Kudla G, Granneman S, Tollervey D. The nuclear RNA polymerase II surveillance system targets polymerase III transcripts. EMBO J. 2011;30:1790–1803. doi: 10.1038/emboj.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Libri D, Dower K, Boulay J, Thomsen R, Rosbash M, Jensen TH. Interactions between mRNA export commitment, 3′-end quality control, and nuclear degradation. Mol. Cell Biol. 2002;22:8254–8266. doi: 10.1128/MCB.22.23.8254-8266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis CA, Ares M., Jr Accumulation of unstable promoter-associated transcripts upon loss of the nuclear exosome subunit Rrp6p in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. 2006;103:3262–3267. doi: 10.1073/pnas.0507783103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gudipati RK, Xu Z, Lebreton A, Seraphin B, Steinmetz LM, Jacquier A, Libri D. Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol. Cell. 2012;48:409–421. doi: 10.1016/j.molcel.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hieronymus H, Yu MC, Silver PA. Genome-wide mRNA surveillance is coupled to mRNA export. Genes Dev. 2004;18:2652–2662. doi: 10.1101/gad.1241204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neil H, Malabat C, d'Aubenton-Carafa Y, Xu Z, Steinmetz LM, Jacquier A. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature. 2009;457:1038–1042. doi: 10.1038/nature07747. [DOI] [PubMed] [Google Scholar]
- 16.van Hoof A, Lennertz P, Parker R. Yeast exosome mutants accumulate 3′-extended polyadenylated forms of U4 small nuclear RNA and small nucleolar RNAs. Mol. Cell Biol. 2000;20:441–452. doi: 10.1128/mcb.20.2.441-452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue K, Mizuno T, Wada K, Hagiwara M. Novel RING finger proteins, Air1p and Air2p, interact with Hmt1p and inhibit the arginine methylation of Npl3p. J. Biol. Chem. 2000;275:32793–32799. doi: 10.1074/jbc.M004560200. [DOI] [PubMed] [Google Scholar]
- 19.Fasken MB, Leung SW, Banerjee A, Kodani MO, Chavez R, Bowman EA, Purohit MK, Rubinson ME, Rubinson EH, Corbett AH. Air1 zinc knuckles 4 and 5 and a conserved IWRXY motif are critical for the function and integrity of the Trf4/5-Air1/2-Mtr4 polyadenylation (TRAMP) RNA quality control complex. J. Biol. Chem. 2011;286:37429–37445. doi: 10.1074/jbc.M111.271494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holub P, Lalakova J, Cerna H, Pasulka J, Sarazova M, Hrazdilova K, Arce MS, Hobor F, Stefl R, Vanacova S. Air2p is critical for the assembly and RNA-binding of the TRAMP complex and the KOW domain of Mtr4p is crucial for exosome activation. Nucleic Acids Res. 2012;40:5679–5693. doi: 10.1093/nar/gks223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidt K, Xu Z, Mathews DH, Butler JS. Air proteins control differential TRAMP substrate specificity for nuclear RNA surveillance. RNA. 2012;18:1934–1945. doi: 10.1261/rna.033431.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia H, Wang X, Liu F, Guenther UP, Srinivasan S, Anderson JT, Jankowsky E. The RNA helicase Mtr4p modulates polyadenylation in the TRAMP complex. Cell. 2011;145:890–901. doi: 10.1016/j.cell.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lebreton A, Tomecki R, Dziembowski A, Seraphin B. Endonucleolytic RNA cleavage by a eukaryotic exosome. Nature. 2008;456:993–996. doi: 10.1038/nature07480. [DOI] [PubMed] [Google Scholar]
- 24.Schneider C, Leung E, Brown J, Tollervey D. The N-terminal PIN domain of the exosome subunit Rrp44 harbors endonuclease activity and tethers Rrp44 to the yeast core exosome. Nucleic Acids Res. 2009;37:1127–1140. doi: 10.1093/nar/gkn1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia H, Wang X, Anderson JT, Jankowsky E. RNA unwinding by the Trf4/Air2/Mtr4 polyadenylation (TRAMP) complex. Proc. Natl Acad. Sci. 2012;109:7292–7297. doi: 10.1073/pnas.1201085109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Houseley J, Kotovic K, El Hage A, Tollervey D. Trf4 targets ncRNAs from telomeric and rDNA spacer regions and functions in rDNA copy number control. EMBO J. 2007;26:4996–5006. doi: 10.1038/sj.emboj.7601921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larochelle M, Lemay JF, Bachand F. The THO complex cooperates with the nuclear RNA surveillance machinery to control small nucleolar RNA expression. Nucleic Acids Res. 2012;40:10240–10253. doi: 10.1093/nar/gks838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrulis ED, Werner J, Nazarian A, Erdjument-Bromage H, Tempst P, Lis JT. The RNA processing exosome is linked to elongating RNA polymerase II in Drosophila. Nature. 2002;420:837–841. doi: 10.1038/nature01181. [DOI] [PubMed] [Google Scholar]
- 29.Hessle V, Bjork P, Sokolowski M, Gonzalez de Valdivia E, Silverstein R, Artemenko K, Tyagi A, Maddalo G, Ilag L, Helbig R, et al. The exosome associates cotranscriptionally with the nascent pre-mRNP through interactions with heterogeneous nuclear ribonucleoproteins. Mol. Biol. Cell. 2009;20:3459–3470. doi: 10.1091/mbc.E09-01-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hessle V, von Euler A, Gonzalez de Valdivia E, Visa N. Rrp6 is recruited to transcribed genes and accompanies the spliced mRNA to the nuclear pore. RNA. 2012;18:1466–1474. doi: 10.1261/rna.032045.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 32.Legrain P, Rosbash M. Some cis- and trans-acting mutants for splicing target pre-mRNA to the cytoplasm. Cell. 1989;57:573–583. doi: 10.1016/0092-8674(89)90127-x. [DOI] [PubMed] [Google Scholar]
- 33.Rutz B, Seraphin B. A dual role for BBP/ScSF1 in nuclear pre-mRNA retention and splicing. EMBO J. 2000;19:1873–1886. doi: 10.1093/emboj/19.8.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rain JC, Legrain P. In vivo commitment to splicing in yeast involves the nucleotide upstream from the branch site conserved sequence and the Mud2 protein. EMBO J. 1997;16:1759–1771. doi: 10.1093/emboj/16.7.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nam K, Lee G, Trambley J, Devine SE, Boeke JD. Severe growth defect in a Schizosaccharomyces pombe mutant defective in intron lariat degradation. Mol. Cell Biol. 1997;17:809–818. doi: 10.1128/mcb.17.2.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bousquet-Antonelli C, Presutti C, Tollervey D. Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell. 2000;102:765–775. doi: 10.1016/s0092-8674(00)00065-9. [DOI] [PubMed] [Google Scholar]
- 37.Sayani S, Janis M, Lee CY, Toesca I, Chanfreau GF. Widespread impact of nonsense-mediated mRNA decay on the yeast intronome. Mol. Cell. 2008;31:360–370. doi: 10.1016/j.molcel.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawashima T, Pellegrini M, Chanfreau GF. Nonsense-mediated mRNA decay mutes the splicing defects of spliceosome component mutations. RNA. 2009;15:2236–2247. doi: 10.1261/rna.1736809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hilleren PJ, Parker R. Cytoplasmic degradation of splice-defective pre-mRNAs and intermediates. Mol. Cell. 2003;12:1453–1465. doi: 10.1016/s1097-2765(03)00488-x. [DOI] [PubMed] [Google Scholar]
- 40.Gornemann J, Kotovic KM, Hujer K, Neugebauer KM. Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol. Cell. 2005;19:53–63. doi: 10.1016/j.molcel.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 41.Lacadie SA, Rosbash M. Cotranscriptional spliceosome assembly dynamics and the role of U1 snRNA:5′ss base pairing in yeast. Mol. Cell. 2005;19:65–75. doi: 10.1016/j.molcel.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 42.Gunderson FQ, Johnson TL. Acetylation by the transcriptional coactivator Gcn5 plays a novel role in co-transcriptional spliceosome assembly. PLoS Genet. 2009;5:e1000682. doi: 10.1371/journal.pgen.1000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gunderson FQ, Merkhofer EC, Johnson TL. Dynamic histone acetylation is critical for cotranscriptional spliceosome assembly and spliceosomal rearrangements. Proc. Natl Acad. Sci. 2011;108:2004–2009. doi: 10.1073/pnas.1011982108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kotovic KM, Lockshon D, Boric L, Neugebauer KM. Cotranscriptional recruitment of the U1 snRNP to intron-containing genes in yeast. Mol. Cell Biol. 2003;23:5768–5779. doi: 10.1128/MCB.23.16.5768-5779.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.San Paolo S, Vanacova S, Schenk L, Scherrer T, Blank D, Keller W, Gerber AP. Distinct roles of non-canonical poly(A) polymerases in RNA metabolism. PLoS Genet. 2009;5:e1000555. doi: 10.1371/journal.pgen.1000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nag A, Steitz JA. Tri-snRNP-associated proteins interact with subunits of the TRAMP and nuclear exosome complexes, linking RNA decay and pre-mRNA splicing. RNA Biol. 2012;9:334–342. doi: 10.4161/rna.19431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 48.Sung MK, Ha CW, Huh WK. A vector system for efficient and economical switching of C-terminal epitope tags in Saccharomyces cerevisiae. Yeast. 2008;25:301–311. doi: 10.1002/yea.1588. [DOI] [PubMed] [Google Scholar]
- 49.Kaplan CD, Holland MJ, Winston F. Interaction between transcription elongation factors and mRNA 3′-end formation at the Saccharomyces cerevisiae GAL10-GAL7 locus. J. Biol. Chem. 2005;280:913–922. doi: 10.1074/jbc.M411108200. [DOI] [PubMed] [Google Scholar]
- 50.Wong CM, Tang HM, Kong KY, Wong GW, Qiu H, Jin DY, Hinnebusch AG. Yeast arginine methyltransferase Hmt1p regulates transcription elongation and termination by methylating Npl3p. Nucleic Acids Res. 2010;38:2217–2228. doi: 10.1093/nar/gkp1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmitt ME, Brown TA, Trumpower BL. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 1990;18:3091–3092. doi: 10.1093/nar/18.10.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Das B, Butler JS, Sherman F. Degradation of normal mRNA in the nucleus of Saccharomyces cerevisiae. Mol. Cell Biol. 2003;23:5502–5515. doi: 10.1128/MCB.23.16.5502-5515.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuai L, Das B, Sherman F. A nuclear degradation pathway controls the abundance of normal mRNAs in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. 2005;102:13962–13967. doi: 10.1073/pnas.0506518102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Campbell KS, Buder A, Deuschle U. Interactions between the amino-terminal domain of p56lck and cytoplasmic domains of CD4 and CD8 alpha in yeast. Eur. J. Immunol. 1995;25:2408–2412. doi: 10.1002/eji.1830250842. [DOI] [PubMed] [Google Scholar]
- 55.Gong F, Fahy D, Smerdon MJ. Rad4-Rad23 interaction with SWI/SNF links ATP-dependent chromatin remodeling with nucleotide excision repair. Nat. Struct. Mol. Biol. 2006;13:902–907. doi: 10.1038/nsmb1152. [DOI] [PubMed] [Google Scholar]
- 56.Tang HM, Siu KL, Wong CM, Jin DY. Loss of yeast peroxiredoxin Tsa1p induces genome instability through activation of the DNA damage checkpoint and elevation of dNTP levels. PLoS Genet. 2009;5:e1000697. doi: 10.1371/journal.pgen.1000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Galy V, Gadal O, Fromont-Racine M, Romano A, Jacquier A, Nehrbass U. Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell. 2004;116:63–73. doi: 10.1016/s0092-8674(03)01026-2. [DOI] [PubMed] [Google Scholar]
- 58.Izaurralde E, Lewis J, McGuigan C, Jankowska M, Darzynkiewicz E, Mattaj IW. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell. 1994;78:657–668. doi: 10.1016/0092-8674(94)90530-4. [DOI] [PubMed] [Google Scholar]
- 59.Abruzzi KC, Lacadie S, Rosbash M. Biochemical analysis of TREX complex recruitment to intronless and intron-containing yeast genes. EMBO J. 2004;23:2620–2631. doi: 10.1038/sj.emboj.7600261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dermody JL, Buratowski S. Leo1 subunit of the yeast paf1 complex binds RNA and contributes to complex recruitment. J. Biol. Chem. 2010;285:33671–33679. doi: 10.1074/jbc.M110.140764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Houseley J, Tollervey D. Yeast Trf5p is a nuclear poly(A) polymerase. EMBO Rep. 2006;7:205–211. doi: 10.1038/sj.embor.7400612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Callahan KP, Butler JS. TRAMP complex enhances RNA degradation by the nuclear exosome component Rrp6. J. Biol. Chem. 2010;285:3540–3547. doi: 10.1074/jbc.M109.058396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kornblihtt AR, de la Mata M, Fededa JP, Munoz MJ, Nogues G. Multiple links between transcription and splicing. RNA. 2004;10:1489–1498. doi: 10.1261/rna.7100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lacadie SA, Tardiff DF, Kadener S, Rosbash M. In vivo commitment to yeast cotranscriptional splicing is sensitive to transcription elongation mutants. Genes Dev. 2006;20:2055–2066. doi: 10.1101/gad.1434706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perales R, Bentley D. “Cotranscriptionality”: the transcription elongation complex as a nexus for nuclear transactions. Mol. Cell. 2009;36:178–191. doi: 10.1016/j.molcel.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Azzouz N, Panasenko OO, Colau G, Collart MA. The CCR4-NOT complex physically and functionally interacts with TRAMP and the nuclear exosome. PLoS ONE. 2009;4:e6760. doi: 10.1371/journal.pone.0006760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dominguez-Sanchez MS, Barroso S, Gomez-Gonzalez B, Luna R, Aguilera A. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 2011;7:e1002386. doi: 10.1371/journal.pgen.1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kruk JA, Dutta A, Fu J, Gilmour DS, Reese JC. The multifunctional Ccr4-Not complex directly promotes transcription elongation. Genes Dev. 2011;25:581–593. doi: 10.1101/gad.2020911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong CM, Qiu H, Hu C, Dong J, Hinnebusch AG. Yeast cap binding complex impedes recruitment of cleavage factor IA to weak termination sites. Mol. Cell Biol. 2007;27:6520–6531. doi: 10.1128/MCB.00733-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dermody JL, Dreyfuss JM, Villen J, Ogundipe B, Gygi SP, Park PJ, Ponticelli AS, Moore CL, Buratowski S, Bucheli ME. Unphosphorylated SR-like protein Npl3 stimulates RNA polymerase II elongation. PLoS one. 2008;3:e3273. doi: 10.1371/journal.pone.0003273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morillo-Huesca M, Vanti M, Chavez S. A simple in vivo assay for measuring the efficiency of gene length-dependent processes in yeast mRNA biogenesis. FEBS J. 2006;273:756–769. doi: 10.1111/j.1742-4658.2005.05108.x. [DOI] [PubMed] [Google Scholar]
- 72.Allmang C, Kufel J, Chanfreau G, Mitchell P, Petfalski E, Tollervey D. Functions of the exosome in rRNA, snoRNA and snRNA synthesis. EMBO J. 1999;18:5399–5410. doi: 10.1093/emboj/18.19.5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Egecioglu DE, Henras AK, Chanfreau GF. Contributions of Trf4p- and Trf5p-dependent polyadenylation to the processing and degradative functions of the yeast nuclear exosome. RNA. 2006;12:26–32. doi: 10.1261/rna.2207206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lemieux C, Marguerat S, Lafontaine J, Barbezier N, Bahler J, Bachand F. A Pre-mRNA degradation pathway that selectively targets intron-containing genes requires the nuclear poly(A)-binding protein. Mol. Cell. 2011;44:108–119. doi: 10.1016/j.molcel.2011.06.035. [DOI] [PubMed] [Google Scholar]
- 76.Allmang C, Petfalski E, Podtelejnikov A, Mann M, Tollervey D, Mitchell P. The yeast exosome and human PM-Scl are related complexes of 3′ –> 5′ exonucleases. Genes Dev. 1999;13:2148–2158. doi: 10.1101/gad.13.16.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Andersen JS, Dziembowski A, Jensen TH. Interaction profiling identifies the human nuclear exosome targeting complex. Mol. Cell. 2011;43:624–637. doi: 10.1016/j.molcel.2011.06.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.