


Abstract
A common feature of DNA repair proteins is their mobilization in response to DNA damage. The ability to visualizing and quantifying the kinetics of proteins localizing/dissociating from DNA double strand breaks (DSBs) via immunofluorescence or live cell fluorescence microscopy have been powerful tools in allowing insight into the DNA damage response, but these tools have some limitations. For example, a number of well-established DSB repair factors, in particular those required for non-homologous end joining (NHEJ), do not form discrete foci in response to DSBs induced by ionizing radiation (IR) or radiomimetic drugs, including bleomycin, in living cells. In this report, we show that time-dependent kinetics of the NHEJ factors Ku80 and DNA-dependent protein kinase catalytic subunits (DNA–PKcs) in response to IR and bleomycin can be quantified by Number and Brightness analysis and Raster-scan Image Correlation Spectroscopy. Fluorescent-tagged Ku80 and DNA–PKcs quickly mobilized in response to IR and bleomycin treatments consistent with prior reports using laser-generated DSBs. The response was linearly dependent on IR dose, and blocking NHEJ enhanced immobilization of both Ku80 and DNA–PKcs after DNA damage. These findings support the idea of using Number and Brightness and Raster-scan Image Correlation Spectroscopy as methods to monitor kinetics of DSB repair proteins in living cells under conditions mimicking radiation and chemotherapy treatments.
INTRODUCTION
The integrity of DNA is constantly under attack, with the most serious assault on DNA being the double strand break (DSB) (1). DSBs can be induced by a number of endogenous and exogenous agents including byproducts of cellular metabolism, ionizing radiation (IR) and radiomimetic agents such as bleomycin. If improperly repaired or left unrepaired, DSBs can lead to cell death, genomic instability or tumorigenesis (2). A number of efficient DSB repair pathways have evolved in mammalian cells with non-homologous end joining (NHEJ) likely playing the prominent role for the repair of DSBs in humans (3). NHEJ mediates the direct ligation of the broken DNA molecule, and unlike the other prominent DSB repair mechanism, homologous recombination, NHEJ does not require a homologous template to complete repair.
The general mechanism of NHEJ includes recognition and assembly of the NHEJ complex at the DSB, bridging of the DNA ends, DNA end processing if required and finally ligation of the broken ends (4). The DNA–PK complex, consisting of the Ku70/80 heterodimer and the DNA-dependent protein kinase catalytic subunit (DNA–PKcs), plays an essential role in NHEJ. The Ku heterodimer is the NHEJ factor, which initially recognizes and binds to the DSB (5,6). The dynamics of Ku70/80 at DSB sites in living cells showed that the Ku heterodimer does not bind rigidly to DNA ends, but that there is a continuous dynamic exchange between DNA-bound Ku70/80 and free unbound Ku70/80 (7). Once Ku70/80 is bound to the DSB it is then believed to act as a scaffold to recruit the other NHEJ factors to the DSB including DNA–PKcs. The recruitment of DNA–PKcs to DSBs in living cells is dependent on Ku70/80, and binding to the DNA–Ku complex stimulates the kinase activity of DNA–PKcs (8). Although the exact role the enzymatic activity of DNA–PKcs plays in NHEJ is unknown, it is known that the kinase activity of DNA–PKcs is essential for NHEJ, as inactivation of DNA–PKcs kinase activity results in radiosensitivity and a defect in DSB repair (9). The best-characterized substrate of DNA–PKcs is DNA–PKcs itself. A large number of the phosphorylation sites are clustered in different regions of DNA–PKcs (10–13). Two prominent phosphorylation clusters, which have been identified to be phosphorylated and autophosphorylated in response to IR, are the threonine 2609 (T2609) (10,14) and serine 2056 (S2056) cluster (15–17). Phosphorylation of S2056 is a bona fide autophosphorylation site (16), whereas phosphorylation of the T2609 cluster can be phosphorylated by DNA–PKcs itself, ATM and ATR (17,18). Phosphorylation of these two clusters is important for NHEJ, as mutation of phosphorylation sites causes increased radiosensitivity and less efficient DSB repair (11,14,16,19). Analysis of the dynamics of DNA–PKcs in which phosphorylation at the T2609 cluster and serine 2056 at DSB sites in vivo revealed that the exchange between DNA-bound and free DNA-PKCS progresses at a much lower rate than unphosphorylated DNA–PKCS, suggesting phosphorylation at these sites increases the rate of dissociation and re-association of DNA-PKCS at DSBs (6).
A central aspect of the response to DNA DSBs is the mobility of a large number of proteins to the site or near the DNA damage (20,21). A significant amount of work has been performed to characterize the spatiotemporal dynamics of the association and dissociation of repair proteins at the sites of DNA damage and the regulatory mechanisms involved in these processes (22). One key to studying the dynamics of repair proteins at DSBs is the fact that a number of these proteins self-assemble into nucleoplasmic repair foci (23). Repair foci appear shortly after creation of the DSB and resolve over the course of several hours. Visualization of foci is typically performed by indirect immunofluorescence or in real-time by tracking focus formation of fluorescent-tagged DSB repair proteins in living cells. Although these techniques have been powerful, there are limitations, including the fact that a large number of repair proteins, such as DNA–PKcs and Ku70/80, bind to DSBs in low numbers and thus their recruitment to the DSB is not easily discernible in fluorescence microscopy images over a background of freely moving protein. For this reason, the standard method of visualizing the recruitment of these repair proteins at DNA damage sites has been to induce damage by focused laser irradiation (8,20,22,23). The high density of damage sites created this way results in localized fluorescent hot spots that are visible over the fluorescent background of mobile protein moving through the same space in the nucleus (8). In addition, quantitative aspects of binding to damage sites have been obtained by Fluorescence Recovery After Photobleaching (FRAP) applied onto a laser damage hot spot (8,24). However, laser-induced DNA lesions are not well characterized and therefore, the response of DNA repair proteins on laser irradiation may not reflect their response on IR. It is, therefore, of interest to study DNA repair kinetics under conditions that resemble more closely cancer treatment, such as by γ-irradiation or chemotherapy.
In this work, we present the application of two quantitative microscopy methods known as Number and Brightness (N&B) analysis (25,26) and Raster-scan Image Correlation Spectroscopy (RICS) (27,28) as means to address this challenge and quantify the real-time DNA repair protein kinetics after sparse damage in living cells. N&B analysis quantifies the number of molecules per image pixel, effectively the concentration, and the fluorescence brightness per molecule for the mobile population fraction of a molecule (26). On the other hand, RICS quantifies the molecular concentration and apparent diffusion coefficients (Deff) of fluorescently tagged molecules within user-selected subcellular regions of interest (27). The N&B and RICS methods are relatively recent extensions of the Fluorescence Correlation Spectroscopy (FCS) technique that can quantify the Deff, binding kinetics, photo-physics and concentration of fluorescently tagged molecules (25–28). FCS has been previously applied to study nuclear protein interactions with DNA in vitro (29,30) and in living cells (31) but has not been applied to the quantification of DNA repair kinetics to date. Following DSBs induced by either γ-irradiation or bleomycin, N&B analysis was used to quantify relative changes in the mobile fraction of fluorescently tagged Ku70/80 and DNA–PKcs and RICS to quantify changes in the apparent Deffs of these two repair proteins. Changes in the protein mobile fraction and apparent Deffs were used as surrogate measurements of repair status. We also present control experiments verifying that the expected changes in kinetics are observed when DSB repair is inhibited. To our knowledge, we present the first application of N&B and RICS to the quantification of DNA repair kinetics after sparse damage, although RICS has been suggested as a means of identifying the presence of DNA damage after UV light exposure (32).
MATERIALS AND METHODS
Cell culture, γ-irradiation and bleomycin treatment
CHO V3 (DNA-PKcs null) cells stably expressing YFP-DNA-PKcs or its 7A phosphorylation mutant (8) and CHO Xrs6 (Ku80 null) cells stably expressing GFP-Ku80 (33) were maintained in Dulbecco's modified Eagle’s Medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% penicillin, 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) and 250 µg/ml of G418 (Invitrogen, Carlsbad, CA, USA). Cells were grown in 35-mm glass-bottom dishes (Mattek Cultureware, Ashland, MA, USA) and were maintained in a humidified 5% CO2 incubator at 37°C. Measurements were taken two days following cell splitting at a confluency of near 100%. During the measurements, cells were maintained in a CO2-independent medium (Invitrogen, Carlsbad, CA, USA) at 37°C. Cells were exposed to γ-rays using a 137Cs irradiator (Mark 1 irradiator, JL Shepherd & Associates, San Fernando, CA, USA) at doses of 1–10 Gy inside the 35-mm dishes, including the microscope stage dish holder, so as to identify the same cells post-irradiation more easily. DSB repair in GFP-Ku80 cells was inhibited by incubation with 20 µM Wortmannin (Sigma-Aldrich Corp., St. Louis, MO, USA) for 45 min after which the cells were washed and the media replaced. For the radiomimetic chemical treatments, 25 μg/ml or 100 μg/ml of bleomycin (Sigma-Aldrich Corp., St. Louis, MO, USA) was mixed into the CO2-independent media for 3 min and subsequently washed from the surfactant and replaced with fresh media.
Confocal imaging
Fluorescence images were acquired using an LSM 510 META confocal microscope (Carl Zeiss, Jena, Germany). A 63× oil immersion Plan-Apochromat objective lens with a numerical aperture of 1.4 was used for all experiments. For the Xrs6 cells expressing GFP-Ku80, images were obtained using a 488-nm Argon laser. A dichroic mirror (HFT 488/543) was used to route laser excitation power onto the sample in an epifluorescence geometry. To exclude any excitation light leakage, a band pass filter (BA 505–530 IR) was used before a photomultiplier tube detector. For V3 cells expressing YFP-DNA-PKcs cells, a 514-nm argon laser was used. Detection of fluorescence was performed through a dichroic mirror (HFT 488/514) and a band pass filter (BA 530–600 IR) in front of the photomultiplier tube detector. For U2OS cells expressing YFP-53BP1, the same data acquisition settings were used as for YFP-DNA-PKcs. The relative intensity changes of foci with time were measured in 12-bit images by drawing a 2-µm diameter circular region of interest (ROI) around each focus.
N&B analysis
N&B analysis is based on the detection of time-series fluorescence signals as an image is raster-scanned to deduce information about the effective molecular concentration and the agglomeration status of fluorescently tagged proteins (25,34,35). In any given pixel obtained from the raster scan image, the fluorescence signal variance (σ2) and average intensity (<k>), the background-corrected number of fluorescent molecules (n) and molecular brightness (ε) are given by the following equations (25,34):
| (1) |
| (2) |
Confocal images for N&B analysis were acquired following previously published image acquisition parameters (26) with the aforementioned LSM 510 META microscope and ConfoCor3 attachment. The scan speed was set to 12.79 µs/pixel, corresponding to 1.38 s/frame, with each frame being 256 × 256 pixels (0.2 μm pixel size). A stack of 50 images was acquired for each measurement. Control experiments were performed to verify that N&B analysis could recover a known concentration of fluorescent bead solutions and verify that these were nearly monomeric after sonication. Green fluorescent beads of 25-nm diameter (Duke Scientific, Palo Alto, CA) at different dilutions from a stock solution of known concentration, placed in Lab-Tek chambered borosilicate coverglass wells (Nalge Nunc International, Rochester, NY), were used for these control experiments. An example of the images generated during the sequence of N&B analysis steps is shown in Figure 1A–C. Briefly, the mean intensity (Figure 1A) and variance images were used to obtain the values of n and ε (Figure 1B) as calculated by Equations 1 and 2, respectively. Mean values for these two parameters were then calculated by averaging results over a user-selected ROI. The histogram of brightness versus intensity values within the selected ROI was also plotted (Figure 1C) to check the uniformity of the molecular brightness distribution. The latter was indeed found to be rather uniform as evidenced from a single dominant cluster in that figure. Little molecular aggregation was present as indicated by a weak second cluster at higher intensity values taking ∼11% of the total histogram weight. Also, brightness values in Figure 1C were predominantly >1, which validated the presence of a mobile population fraction (26). For U2OS cells expressing YFP-53BP1, N&B analysis data acquisition used the same parameters as for YFP-DNA-PKcs. Multiple ROIs in-between YFP-53BP1 foci, each 16 × 16 pixels in size, were used to determine relative changes in the mobile fraction.
Figure 1.
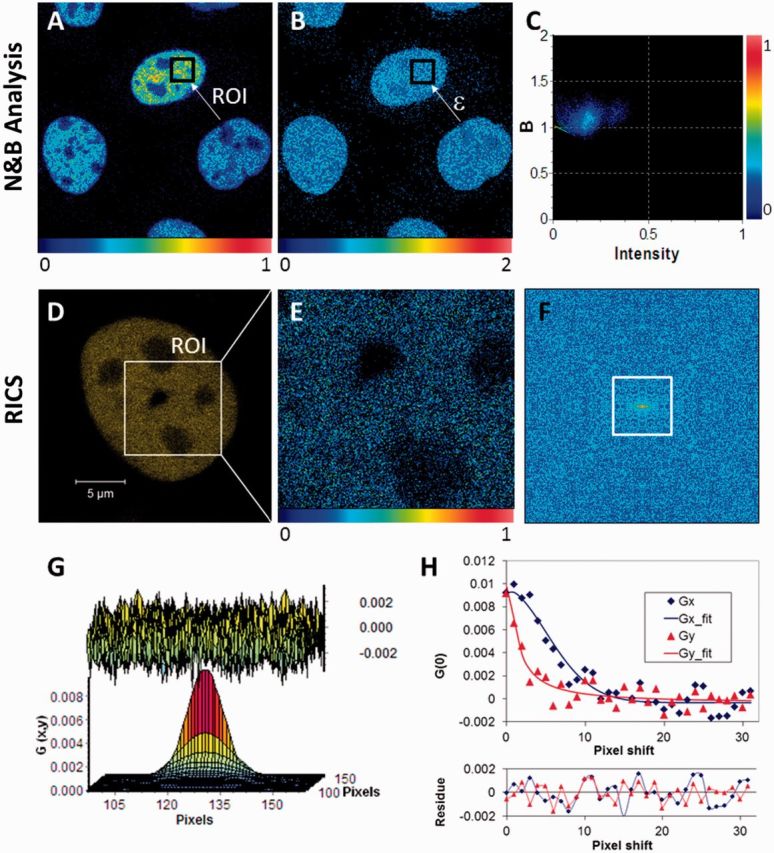
Pictorial description of the N&B analysis and RICS methods. (A) Relative fluorescence intensity (scale 0–1) image of Xrs6 cells expressing GFP-Ku80. The inset (arrow) indicates a typical user-selected ROI, the time series intensity data of which is processed by Equation 1 to determine the molecular number (n). (B) The molecular brightness (ε) map, obtained after processing the image in (A) with Equation 2, indicating an even distribution of molecular brightness throughout the nucleus despite differences in protein concentration between cells as seen in (A). These brightness values [B (scale 0–2)] can be converted to photon counts per molecule per second by calculating (B − 1)/(pixel dwell time). (C) The 2D histogram of (B) versus intensity for the ROI shown in (A) and (B). The spread of the points indicates pixel variance, which shows a predominantly unimodal distribution. (D) Confocal image of a V3 cell expressing YFP-DNA-PKcs with the inset showing a region of the nucleus selected for RICS measurement. (E) Snapshot of the time-averaged fluorescence intensity image of the selected ROI on which RICS analysis was then to be applied (relative intensity scale 0–1). (F) Image of the 2D RICS data (256 × 256 pixels) obtained after applying Equation 5. (G) Fitting of the 2D RICS data (64 × 64 pixels) obtained from inset in (F) using Equation 6 (top, the fit residuals). (H) Simultaneous plot of 1D cross-sections of the 2D RICS data along the x and y directions along with corresponding fit data (bottom, the fit residuals). The data extend for 32 pixels along each direction as indicated by the white square in (F).
Calculation of relative changes in the protein mobile fraction after DNA damage by combining N&B analysis with Strip-FRAP
The mobile protein fraction estimates obtained from N&B analysis could be contaminated and even overwhelmed by contributions from the immobile fraction if the latter was large. Therefore, a strategy was devised to minimize the immobile fraction contributions to the total signal by (i) pre-bleaching the cells and (ii) estimating the remaining immobile fraction with Strip-FRAP, so that its contribution could be applied as a correction to the N&B analysis estimates of the mobile fraction. These steps are outlined in the following paragraphs.
First, the immobile fraction of the fluorescently tagged proteins was pre-bleached by taking 50 images using the same parameters as the ones used for all measurements. For the case of γ-irradiation, a stack of 50 images was subsequently acquired just before exposure and at 5, 10, 15, 20, 30, 40 and 60 min post-irradiation. Ten cells were used for each time point with results being averaged from several experiments. For V3 cells, the kinetics of YFP-DNA-PKcs were quantified for 1, 5 and 7 Gy. For Xrs6 cells, the kinetics of GFP-Ku80 were quantified for 1, 5 and 10 Gy. The images acquired under these conditions were processed by N&B analysis using commercially available software (Globals, Laboratory for Fluorescence Dynamics, Irvine, CA). During data analysis, the photobleaching and cell movement effects were removed using a high-pass filter as described previously (34,36).
Even though pre-bleaching reduced significantly the contributions from the immobile fraction, it did not eliminate them completely. The residual immobile fraction could affect the number of molecules per pixel estimated from Equation 1, and a correction had to be applied to arrive at the true number of mobile molecules per pixel (nmobile) (35):
| (3) |
where R was the ratio between the intensity of immobile (Iimmobile) molecules to that of the mobile molecules per pixel. Iimmobile was calculated by multiplying image pixel intensity by the immobile fraction. The R value was calculated from strip-FRAP analysis as the ratio of immobile to mobile fraction after fluorescence recovery reached a plateau after photobleaching, as explained below.
Strip-FRAP (7,37) measurements were performed on fresh cells, i.e. not the same cells used for the N&B measurements but from the same cell population that was initially split, and R was deduced as the average value from 10 cells measured for each cell line. Specifically, a strip of 80 × 10 pixels (24) (0.1 μm pixel size) in the nucleus was selected from a 128 × 128 pixel (13 × 13 μm) image and photobleached for 0.58 s using the 488 nm line of the Argon/2-laser at a power of ∼0.55 mW at the focal spot for GFP-Ku80 cells and the 514 nm line from the same laser at ∼0.42 mW for YFP-DNA-PKcs cells. For GFP-Ku80, the pre-bleach and post-bleach fluorescence within the strip were measured by taking continuous confocal images, from 5 s pre-bleach to 20 s post-bleach in 20-ms time intervals. All other Strip-FRAP image acquisition settings on the LSM META 510 microscope for the two cell lines were identical to those used for confocal imaging, as described earlier.
Once the R value was known, the value for nmobile was computed using Equation 3. It is important to note that in this work cells were selected to have a fluorescently tagged protein concentration in the 50–500 nM range that corresponded to only a small variation in R values, namely, 2.15% for GFP-Ku80 and 2.69% for YFP-DNA-PKcs. The relative constancy in R values for these experimental conditions enabled applying a population-based correction to nmobile without having to do Strip-FRAP on each cell for which N&B measurements were performed. An additional correction to this parameter value was then performed due to a dose-dependent low-level fluorescence loss after irradiation. This fluorescence loss was measured on 10 cells per dose at 4 h post-irradiation where repair for most cells is known to be largely complete by that time point (11). No measurements were performed at prior time points for these cells to avoid any further loss of fluorescence owing to photobleaching from repeated N&B analysis measurements. This loss factor was then subtracted from nmobile in Equation 3 given earlier to yield a corrected value for this variable.
Finally, the normalized difference between the number of mobile molecules per pixel before and after irradiation was used to calculate the relative change in the mobile fraction of molecules per pixel at each time point post-irradiation, as shown in Equation 4:
| (4) |
where nr was the relative mobile fraction change, 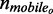 the number of mobile molecules per pixel before irradiation and
the number of mobile molecules per pixel before irradiation and 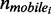 the number of mobile molecules per pixel at a particular time post-irradiation. The change in mobile fraction for GFP-Ku80 and YFP-DNA-PKcs as a function of time post-irradiation is the quantity that we have quantified in this work as a surrogate measure of DNA repair status in the cell lines expressing these two proteins.
the number of mobile molecules per pixel at a particular time post-irradiation. The change in mobile fraction for GFP-Ku80 and YFP-DNA-PKcs as a function of time post-irradiation is the quantity that we have quantified in this work as a surrogate measure of DNA repair status in the cell lines expressing these two proteins.
Similar to the γ-irradiation experiments, the same experimental procedures were also used to quantify the relative change in mobile fraction for GFP-Ku80 and YFP-DNA-PKcs at 5, 10, 15, 20, 30, 40 and 60 min post bleomycin treatment.
RICS
Spatiotemporal correlation images of RICS data were calculated with the Globals software package (Laboratory for Fluorescence Dynamics, Irvine, CA) that uses the following formula (27,28):
| (5) |
where the angled brackets indicate spatial averaging in both x and y directions, and where ξ and ψ are the spatial increments in the x and y directions, respectively (27,34). Each ξ and ψ increment value was 0.04 µm, and x and y values were 0–255. The data obtained from this correlation image were then fitted to the following equation (27,28):
| (6) |
Equation 6 is the product of two components, one due to correlation between neighbouring pixel intensity values from particle diffusion, GD (Equation 7), and one due to the reduction in correlation amplitude, relative to point measurements, as a result of the beam raster scanning, GS (Equation 8), with τp and τl being the pixel dwell times in x and y, respectively (27,28,38):
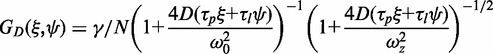 |
(7) |
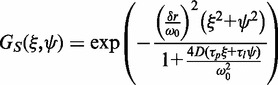 |
(8) |
In these equations, D is the molecular Deff, γ = 0.35 is a factor that accounts for the volume contrast in the assumed 3D Gaussian profile of the confocal volume (28), with ωo = 0.30 μm and ωz = 1.5 μm being the 1/e2 radii of this confocal volume in the radial and axial directions, respectively. N is the average number of molecules in the confocal volume given by N = CVeff, where C is the concentration and the effective confocal volume Veff = π3/2
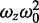 (39).
(39).
For the RICS experiments, stacks of images were acquired using the LSM 510 META microscope and ConfoCor3 attachment, similar to that described for the N&B analysis earlier in the text. In contrast to N&B analysis, the selection of pixel size and scan speed is critical to measuring correctly the Deff of a protein, which depends on its molecular weight (27,34). In this work, a 256 × 256 pixel area was used with pixel dwell times of 6.39 μs and 12.79 μs for GFP-Ku80 and YFP-DNA-PKcs, respectively. The pixel size, which was controlled by the electronic zoom, was set to 0.04 µm. This combination of spatiotemporal sampling settings resulted in frame rates of 0.98 s and 1.96 s for GFP-Ku80 and YFP-DNA-PKcs, respectively.
A visual example of how the RICS data were processed is shown in Figure 1D–H. For each cell to be measured, it was first checked that a square ROI of ∼10 μm per side (256 pixels in 0.04 µm steps) could fit into the cell nucleus (Figure 1D). This was the minimum ROI size found to produce good quality RICS curves and subsequent model fits for our system. A stack of images was then acquired over this ROI with the microscope settings described earlier (30–50 images acquired, corresponding to measurement times of 59–98 s per time point). The fluorescence intensity fluctuations acquired over this ROI (time-averaged intensity image shown in Figure 1E) were background-corrected and used to compute the 2D spatial correlation function using Equation 5 (Figure 1F). The background correction was performed to remove stationary or slow-moving objects and this entailed subtracting the average value of all pixels in the field of view from the fluorescence intensity value of each pixel, as described previously (40). Then the computed 2D autocorrelation surface was fitted to the diffusion model of Equation 6 (Figure 1G). An example model fit along the x-axis and y-axis cross-sections of the 2D autocorrelation curve is shown in Figure 1H.
Statistical analysis
The statistical significance of any changes in the relative mobile fraction for GFP-Ku80 and YFP-DNA-PKcs after different γ-irradiation and bleomycin doses, across the ensemble of time-points where measurements were performed, was tested by linear mixed model analysis (41) to take into account the possible correlation structure between repeated measures. Specifically, the F-test [F(n,N); n = degrees of freedom; N = data points] compared all pair-wise combinations of data in time for different doses and adjusted the P-value yielding significance depending on the number of degrees of freedom and data points in each set of experiments. The data for each separate experimental condition as a function of time post-treatment were fitted to smooth curves (log normal distribution with four fitting parameters) in Sigma Plot (Systat Software, Inc., Chicago, IL, USA) to guide visual interpretation of results.
It is also important to note that for both γ-irradiation and bleomycin treatments, ∼30% of cells did not show any detectable kinetics for GFP-Ku80 or YFP-DNA-PKcs. Measurements for these cells were not included in any subsequent analyses.
RESULTS AND DISCUSSION
N&B analysis and RICS enabled repeat measurements of DNA repair protein kinetics on the same cell while also giving consistent results with standard kinetics quantification methods
The standard method for studying the real-time DNA repair protein kinetics at DSBs in living cells has been the use of laser micro-irradiation combined with time-lapse fluorescence microscopy. The laser micro-irradiation allows one to follow the time-dependent intensity changes of a readily visible hot spot of fluorescently tagged proteins as they accumulate and subsequently dissociate from the focused DNA damage area (8). In this work, we aimed to find a quantitative microscopy method that would enable monitoring the kinetics of these repair proteins following sparse DNA damage, including damage that is produced via ionizing radiation and radiomimetic agents. We first attempted to measure DNA repair protein kinetics by the FCS method, which has been previously used to quantify protein–DNA interactions in the cell nucleus (33). The practical challenge of this work with respect to prior studies was that quantification of kinetics over time post DNA damage requires repeated measurements on the same cell. Unfortunately, we found that these repeated measurements resulted in significant photobleaching when the FCS technique was used due to the presence of a low mobility protein fraction (Supplementary Figure S1A–E). On the other hand, we found that use of the N&B analysis and RICS techniques, where the focal volume was repeatedly scanned over a wider user-selected area of the cell nucleus, resulted in significantly less photobleaching after repeat measurements (Supplementary Figure S1F–J) (26,27). Our findings were supported by the constancy of molecular brightness values (Equation 2) with time for both types of repair proteins studied in this work (Supplementary Figure S2A and B).
Furthermore, we verified that N&B analysis results were consistent with the ones found from γ-irradiation of U2OS cells expressing YFP-53BP1, a protein that forms visible DNA damage foci (42). The time-dependent decrease in fluorescence intensity of these foci as a function of time after irradiation mirrored the increase in mobile protein fraction at the corresponding time points (Supplementary Figure S3). N&B analysis was possible in these cells because the field of view size was chosen to be small enough to fit between foci. Otherwise, the very high intensity of the foci would have overshadowed the fluorescence fluctuation signal of the mobile fraction on which N&B analysis is based. Unfortunately, it was not possible to also perform RICS measurements in YFP-53BP1 expressing cells after irradiation because the field of view size requirement for this method meant that foci could not be avoided. Nevertheless, before performing measurements in living cells, it was verified by using measurements in fluorescent bead solutions (described in Methods) that FCS, N&B analysis and RICS could all measure the same bead concentration, and FCS and RICS could measure the same Deff to within a few percentage. Furthermore, the baseline Deff value of 1.42 × 10−11 m2/s that was previously determined by FCS for GFP-Ku80 (33) was also found to be within a few percentage of the value obtained here by RICS.
Quantification of GFP-Ku80 kinetics after γ-irradiation
Previous studies showed that fluorescent-tagged Ku80 quickly localizes to laser-generated DSBs where it serves as a sensor of the DNA damage and subsequently recruits the other NHEJ factors to the DSB (7,8,43). However, observing the ability of the Ku heterodimer to localize to γ-irradiation-induced DSBs in living cells has been unsuccessful (data not shown) (44). Therefore, it was tested if N&B analysis could be used to calculate the change in the mobile protein fraction of GFP-tagged Ku80 expressed in the Ku80 deficient cell line Xrs6 following irradiation and correlate this mobility as a surrogate measurement for monitoring the kinetics of Ku80 at DSBs and the progress of DNA repair. For each cell measurement, changes in the mobile fraction concentration, nmobile, as a function of time post-irradiation were quantified by N&B analysis as described in the Methods and in Figure 1A–C. The computation of nmobile included the immobile fraction correction for these cells, as measured by Strip-FRAP, which was found to be ∼12% irrespective of time before and after γ-irradiation (Supplementary Figure S4A), or of the dose used (Supplementary Figure S4B). The nmobile value was further corrected for the radiation-induced fluorescent loss, as described in the Methods, which however was only a few percentage of the pre-irradiation GFP-Ku80 fluorescence intensity. Once all corrections to nmobile were completed, the change in mobile fraction was normalized to the mobile fraction of the pre-irradiation time point, i.e. nr was computed using Equation 4, so as to compensate for differences in protein expression levels between cells and thus be able to pool results from multiple cells. The change in relative mobile fraction, nr, of GFP-Ku80 in Xrs6 cell nuclei is shown as a function of time post-irradiation using doses of 1, 5 and 10 Gy (Figure 2A). The resulting curves showed a continuous decrease in the protein mobile fraction that had changed mobility relative to pre-irradiation levels, reaching ∼60% of the early peak value at 1 h post-irradiation. Results in Figure 2A were normalized to unity at the time point with the maximum detected value, which was ∼5 min post-damage, i.e. at the shortest time possible for locating and measuring the same cells post-irradiation. The normalization to the first measurement time point was used to demonstrate the γ-ray dose independence of GFP-Ku80 kinetics, which was proven by linear mixed model analysis (41). Specifically, the F-test comparing all pair-wise combinations of curves for different doses was not significant [F(2.24) = 0.089, P = 0.956]. Furthermore, the relative change in the mobile fraction produced similar shaped curves at the different doses of IR (Figure 2A), but the higher doses produced larger fractional changes in mobile protein concentration, which correlated with increasing γ-irradiation doses (Figure 2B). These data illustrate that as the dose of IR goes up, so do the number of DSBs that are produced and a larger number of GFP-tagged Ku80 molecules mobilize. These results are qualitatively similar to the association/dissociation kinetics of Ku70/80 hot spots created by laser micro-irradiation (7,45). However, it should be noted that DNA laser-generated damage is very concentrated and may produce lesions that are more complicated than those created by γ-ray damage (46). Therefore, although the laser-generated kinetics for DNA-PKcs (8) and Ku70/80 (7) are similar to those found in this work for IR, these kinetics may not be strictly comparable.
Figure 2.
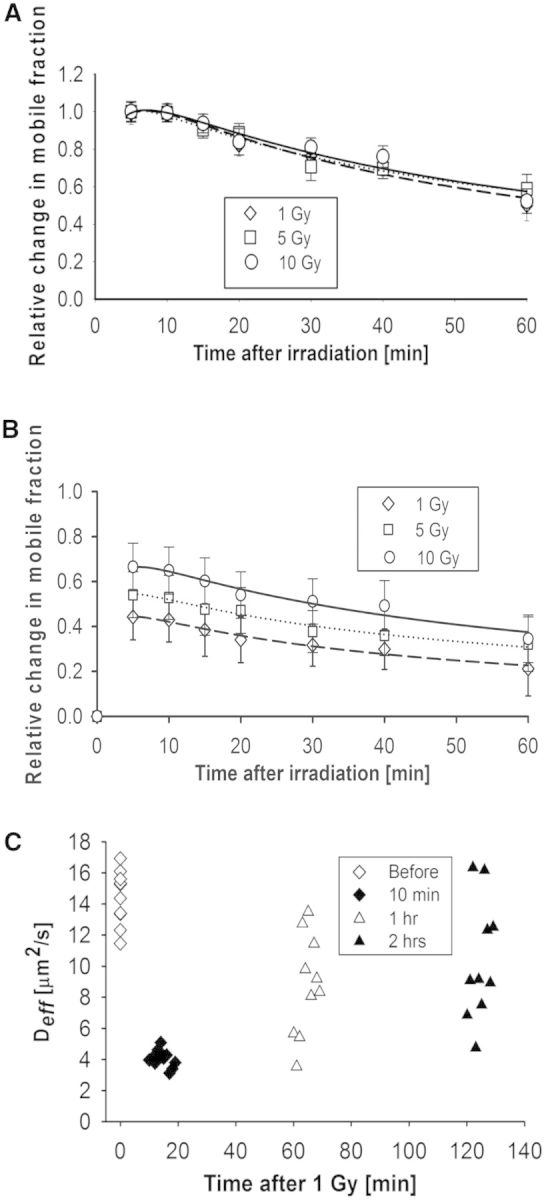
Quantification of GFP-Ku80 kinetics by N&B analysis and RICS. (A) Change in GFP-Ku80 relative mobile fraction as a function of time inside the nucleus of Xrs6 cells that were irradiated at different doses (1 Gy: diamonds, 5 Gy: squares, 10 Gy: circles). The error bars represent the standard error to the mean calculated from 10 different cells. Smooth curve fits to the data points for each dose (1 Gy: dashes, 5 Gy: dots, 10 Gy: solid) are also shown. (B) As in (A), but without normalizing data to the ∼5 min peak so as to illustrate the dose dependence. (C) Variation of Deff for GFP-Ku80 in Xrs6 cells; the average value was 14.42 ± 1.75 μm2/s before 1 Gy of γ-irradiation, 4.02 ± 0.57 μm2/s at 10 min, 8.77 ± 3.68 μm2/s at 1 h and 10.36 ± 3.88 μm2/s at 2 h post-irradiation.
In addition to N&B analysis, the RICS method (27,28) was also applied as an alternate approach to monitoring DNA repair kinetics after γ-irradiation. In RICS, the change in Deff of the fluorescently tagged repair protein was used as a surrogate measure of repair status, with complete repair being represented by a return of this Deff to pre-irradiation values. For the case of GFP-Ku80, the change in Deff was monitored at 10 min, 1 h and 2 h after 1 Gy of γ-irradiation (Figure 2C). At 10 min post-irradiation, all cells showed low Deff values with little variation (4.02 ± 0.57 μm2/s) compared with pre-irradiation values (14.42 ± 1.75 μm2/s). However, with increasing time post-damage, it was seen that some cells repair faster than others, as evidenced by the increasing spread in Deff values at 1 h (8.77 ± 3.68 μm2/s) versus at 2 h (10.36 ± 3.88 μm2/s). As γ-irradiation produces mixed DNA damage, the slower repairing cells may be those with more complex damage (47–50). Furthermore, part of this variation could be due to differences in where the damage was produced, as it has been shown that damage produced in the euchromatin is repaired faster than damage produced in the heterochromatin (51). A linear mixed model with repeated measurements performing all the possible pair-wise combinations of Deff values at different time points indicated significant differences between some periods [F(3.27) = 188.92, P < 0.0001]. Specifically, the Deff value changes for the 10-min [F(3.27) = 23.70, P < 0.0001], 1 h [F(3.27) = 8.98, P < 0.0001] and 2 h [F(3.27) = 8.91, P < 0.0001] time points, relative to pre-irradiation, were all significant.
In summary, we have found that both N&B analysis and RICS can be used to quantify changes in GFP–Ku80 kinetics as a function of time after sparse DNA damage is induced by different doses of γ-irradiation. The quantification of DNA repair protein kinetics by these scanning correlation spectroscopy methods is a novel concept that enables deriving surrogate measures of sparse DNA damage repair. In contrast, the current standard methods cannot quantify sparse damage kinetics and depend on visualizing intensity changes in areas of focused DNA damage created by laser micro-irradiation (45). The fact that the N&B analysis yielded curves that were qualitatively similar to those reported for laser-induced damage hot spots (7) supported the notion that changes in the GFP-Ku80 mobile fraction with time post-irradiation can serve as surrogate markers of the localization and subsequent dissociation of this protein from DSBs even though that was not directly observable.
Quantification of YFP-DNA-PKcs repair kinetics after γ-irradiation
Similar to Ku70/80, determining the kinetics of DNA–PKcs following γ-irradiation in living cells has been unsuccessful (data not shown) (44,52). Therefore, we were also motivated to apply our methods to this protein to compare and contrast its kinetics following sparse DNA damage. First, N&B analysis was performed to quantify kinetics YFP-DNA-PKcs in V3 cells after γ-irradiation at experimental conditions similar to the ones described earlier for GFP-Ku80. The only difference was that the highest dose used was 7 Gy and not 10 Gy. This was necessitated by the increased sensitivity of the YFP-tagged protein to the loss of fluorescence after γ-irradiation, compared with that of the GFP-tagged Ku80, which amounted to ∼33% for 1 Gy, 42% for 5 Gy and 50% for 7 Gy. Despite these dose-dependent fluorescence losses, there was enough fluorescence signal post-irradiation to enable quantification of repair kinetics once this loss factor was compensated for in Equation 4, as was done for GFP-Ku80 earlier. Comparison of Figure 3A with that of Figure 2A indicates a steeper initial descending slope for the change in the YFP-DNA-PKcs mobile fraction in the first 30 min post-irradiation, which suggests faster disengagement of this protein compared with GFP-Ku80. Also, the fact that all peak-normalized kinetics curves are similar in Figure 3A for all doses used suggests a dose independence for the YFP-DNA-PKcs kinetics similar to what was observed with GFP-Ku80 (Figure 2A). A linear mixed model with repeated measurements verified that the differences in relative mobile fraction change with dose were not significant [F(2,24) = 0.051, P = 0.950]. Dose independence indicated the similarity in shape for the different dose curves in Figure 3A, although there were larger fractional changes in mobile protein concentration with increasing γ-irradiation doses as shown in Figure 3B, which were similar to those observed with Ku80 (Figure 2B).
Figure 3.
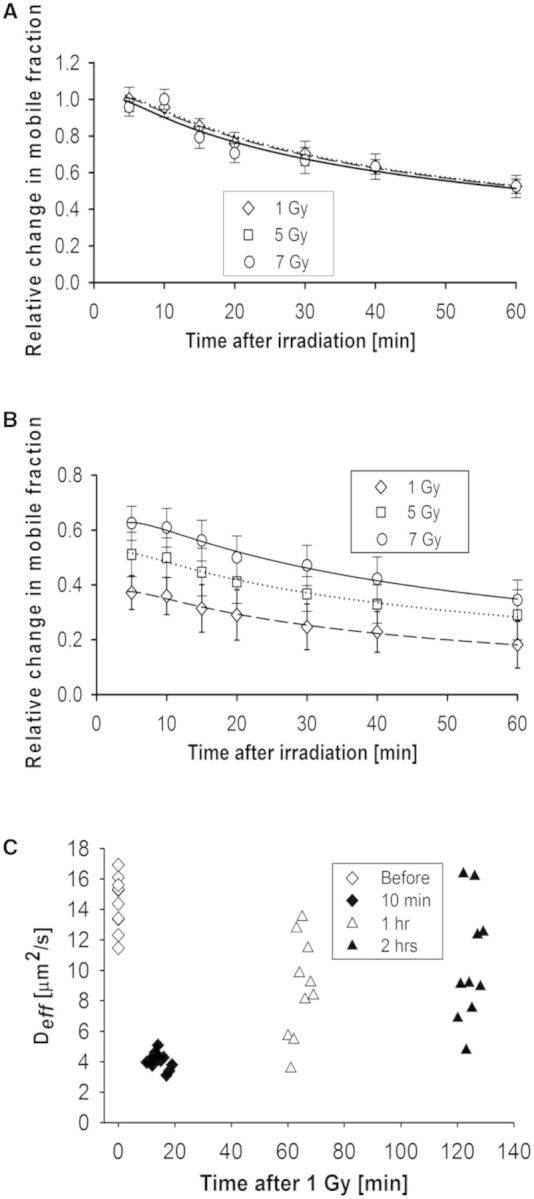
Quantification of YFP-DNA-PKcs kinetics by N&B analysis and RICS. (A) Change in YFP-DNA-PKcs relative mobile fraction as a function of time post-irradiation for V3 cells irradiated at different doses (1 Gy: diamonds, 5 Gy: triangles, 7 Gy: circles). The error bars represent the standard error to the mean calculated from 10 different cells. Smooth curve fits to the data points for each dose (1 Gy: dashes, 5 Gy: dots, 10 Gy: solid) are also shown. (B) As in (A), but without normalizing data to ∼5 min peak so as to illustrate the dose dependence. (C) Variation of Deff for YFP-DNA-PKcs in V3 cells; the average value was 6.17 ± 0.49 μm2/s before 1 Gy of γ-irradiation, 3.35 ± 1.08 μm2/s at 10 min, 4.80 ± 1.09 μm2/s at 1 h and 5.39 ± 1.23 μm2/s at 2 h post-irradiation.
In addition, RICS measurements were performed for YFP-DNA-PKcs in V3 cells for a dose of 1 Gy. Figure 3C shows that Deff values progressively returned towards pre-irradiation levels, with mean values decreasing to 3.35 ± 0.99 μm2/s at 15 min post-irradiation compared with the higher pre-irradiation values (6.17 ± 0.49 μm2/s). The mean Deff values at the 1 h and 2 h time points were 4.80 ±1.09 μm2/s and 5.39 ±1.23 μm2/s, respectively, and had a wider spread with respect to pre-irradiation values, reflecting ongoing repair. Interestingly, the spread of Deff values seen at 15 min post-irradiation in Figure 3C for YFP-DNA-PKcs was higher compared with the corresponding spread of Deff values at 15 min for GFP-Ku80 seen in Figure 2C. This observation is qualitatively consistent with the steeper descending slope for the change in mobile fraction within the first 30 min post-irradiation seen in Figure 3A, compared with Figure 2A, which indicates faster kinetics for YFP-DNA-PKcs at this initial period. These observations are consistent with the known notion that Ku70/80 is the first molecule to arrive at a DSB, binds tightly onto it and is necessary for the subsequent recruitment of DNA-PKcs at the damage site (8). A linear mixed model with repeated measures was fitted to the YFP-DNA-PKcs diffusion data after γ-irradiation to ascertain whether there was any difference between mean diffusion at different times. The F-test indicated that there are significant differences between Deff value means between some periods [F(3.27) = 33.34, P < 0.0001]. Specifically, the Deff value changes for the 10-min [F(3.27) = 10.65, P < 0.0001], 1-h [F(3.27) = 14.68, P < 0.0001] and 2-h [F(3.27) = 14.54, P < 0.0001] time points, relative to pre-irradiation, were all significant.
Overall, the application of N&B analysis and RICS to the quantification of YFP-DNA-PKcs in addition to GFP-Ku80 supported the notion that these methods could be applied to quantify the kinetics of other DNA repair proteins after sparse DNA damage and could also be used for exploring differences in kinetics between proteins that are known to be part of the same repair pathway.
Quantification of dose-dependent changes in the mobile fraction of repair proteins
The relative changes in mobile protein fraction at ∼5 min post-irradiation, where the maximum change was seen relative to pre-irradiation for the time points measured in this work for GFP-Ku80 (Figure 2B) and YFP-DNA-PKcs (Figure 3B), were examined as a function of dose (Figure 4). It is clear from these data that there was a strong linear dependence with dose for both proteins. Furthermore, the slopes of these two lines were comparable. By multiplying the slopes of these lines with the estimated number of proteins per nucleus, which was obtained by multiplying number densities with an approximate nuclear volume of ∼600 µm3, and dividing by an estimated ∼30 DSBs per Gy (53,54), resulted in an estimated ∼300 molecules changing mobility per DSB for both proteins. The fact that many proteins change mobility is the key factor that enables the N&B analysis and RICS to detect sparse damage repair kinetics. Interestingly, although it is known that only two Ku heterodimer molecules bind per DSB (55), and indirect evidence exists for the same for DNA-PKcs (8,56), our data suggest that many more molecules are mobilized in response to DNA damage.
Figure 4.

The change in repair protein mobile fraction is linearly dependent with γ-irradiation dose. The change in mobile fraction at the ∼5 min peak time post-irradiation is shown for YFP-DNA-PKcs (squares) and GFP-Ku80 (diamonds) along with the corresponding linear fits and R2 values. The error bars represent the standard error to the mean calculated from 10 different cells.
Quantification of DNA repair protein kinetics under repair-inhibiting conditions
As the application of N&B analysis to the quantification of repair protein kinetics after sparse DNA damage is novel, it was important to verify that the kinetics of DNA repair proteins changed and did not return as rapidly to pre-irradiation levels when their ability to affect DSB repair was inhibited. Control experiments were performed to validate the expected reduction in mobile fraction change as a function of time post-irradiation under inhibited repair conditions for GFP-Ku80 and YFP-DNA-PKcs. Specifically, the kinetics of GFP-Ku80 were quantified after exposure to Wortmannin, a covalent inhibitor of phosphoinositide 3-kinases known to inhibit the activity of DNA–PKcs and thus NHEJ-mediated DSB repair (7). As shown in Figure 5A, treatment of Xrs6 cells expressing GFP-tagged Ku80 with Wortmannin resulted in a significant increase in the mobile fraction. This result is similar to a previous study, which showed that dissociation of GFP-tagged Ku80 from DSBs was attenuated when cells were treated with Wortmannin (7). The kinetics of YFP-tagged DNA-PKcs in which phosphorylation at the threonine 2609 cluster and serine 2056 (7A) was ablated also resulted in significant increase in the mobile fraction (Figure 5B). Similarly, 7A was previously shown to be retained at DSBs following micro-irradiation, suggesting phosphorylation at these seven sites is required for the ability of DNA–PKcs to dissociate from DSBs (8). Taken together, these results clearly show that N&B analysis is a powerful method to observe the mobility of DNA repair proteins in response to DNA damage and that this mobility strongly correlates with the kinetics of repair proteins binding and dissociating from DSBs.
Figure 5.
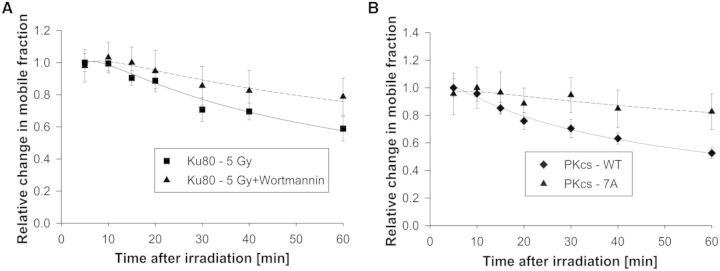
Quantification of GFP-Ku80 and YFP-DNA-PKcs kinetics by N&B analysis under repair-inhibited conditions. (A) Change in GFP-Ku80 relative mobile fraction as a function of time post-irradiation for Xrs6 cells irradiated with 5 Gy (squares) and for the same cells irradiated with the same dose after exposure to Wortmannin (triangles). The error bars represent the standard error to the mean calculated from 10 different cells. (B) Change in relative mobile fraction of wild-type (WT) YFP-DNA-PKcs as a function of time post-irradiation for V3 cells irradiated with 1 Gy and corresponding kinetics for the 7A-DNA-PKcs phosphorylation mutant for the same dose.
Quantification of GFP-Ku and YFP-DNA-PKcs repair kinetics after bleomycin treatment
In addition to IR, radiomimetic chemicals can also produce sparse DNA damage. We were thus motivated to explore if N&B analysis and RICS could also quantify repair protein kinetics after exposure to a radiomimetic chemical. To that end, the GFP-Ku80 and YFP-DNA-PKcs expressing cell lines were exposed to bleomycin, which is known to produce DSBs (57–59). Similar to following γ-irradiation (44) YFP-53BP1 formed foci after bleomycin treatment, but the cells expressing GFP-Ku80 and YFP-DNA-PKcs did not (Supplementary Figure S5). Figure 6A shows the kinetics of GFP-Ku80 for two different bleomycin dose treatments (25 and 100 μg/ml). Although in absolute terms there was a larger change in the mobile fraction of GFP-Ku80 for the higher bleomycin dose, the data for both doses were normalized to the first time point so that the shape of the curves could be compared. Interestingly, the lower of the two doses showed faster repair kinetics, whereas the higher dose showed slower kinetics, as DNA damage creation and chemical turnover persisted for longer. The difference in repair kinetics with respect to γ-irradiation was that DNA damage continued to occur even after the chemical was washed out of the cell media. This was because some of the chemical was already inside the cell and could not be removed by washing. As a result, the kinetics observed for bleomycin represented a continuous repair response to DNA damage that was ongoing, although ever less so with increasing time post-washing as the drug was turned over and excreted from the cell. Also in Figure 6A, it is seen that error bars were larger compared with the γ-irradiation experiments (Figures 2A and 3A). We surmise that this is because of the greater variability in repair kinetics between cells as bleomycin uptake and turnover was an additional variable that was not present in the irradiation experiments where DNA damage happened all at once. Similar observations were made for cells expressing YFP-DNA-PKcs, although the overall kinetics were slower after exposure to the same doses of bleomycin (Figure 6B).
Figure 6.

Quantification of GFP-ku80 and YFP-DNA-PKcs kinetics by N&B analysis and RICS after bleomycin treatment. (A) Change in GFP-Ku80 relative mobile fraction as a function of time inside the nucleus of Xrs6 cells that were treated with different bleomycin doses (25 μg/ml: circles, 100 μg/ml: triangles). (B) Change in YFP-DNA-PKcs relative mobile fraction as a function of time inside the nucleus of V3 cells that were treated with different bleomycin doses (25 μg/ml: circles, 100 μg/ml: triangles). The error bars in both top panels represent the standard error to the mean calculated from 10 different cells. (C) Variation of Deff for GFP-Ku80 as a function of time after bleomycin treatment; the average value was 14.57 ± 1.41 μm2/s before treatment, 2.17 ± 1.58 μm2/s at 10 min, 7.75 ± 4.35 μm2/s at 1 h and 9.02 ± 5.78 μm2/s at 2 h post-treatment. (D) Variation of Deff for YFP-DNA-PKcs as a function of time after bleomycin treatment; the average value was 6.13 ± 1.19 μm2/s before bleomycin treatment, 3.13 ± 0.78 μm2/s at 10 min, 4.64 ± 1.38 μm2/s at 1 h and 5.41 ± 1.71 μm2/s at 2 h post-treatment.
RICS analysis presented patterns of time-dependent changes in Deff values after bleomycin treatment (100 μg/ml) with a heterogeneous increase towards pre-treatment values with time for both GFP-Ku80 (Figure 6C) and YFP-DNA-PKcs (Figure 6D), which is similar to the results observed in the γ-irradiation experiments. Interestingly, a larger relative change in Deff was seen compared with pre-treatment values at 10-min post-treatment for GFP-Ku80 compared with YFP-DNA-PKcs, similar to corresponding observations for the 10-min time point in Figures 2B and 3B for γ-irradiation. The spread in Deff values at subsequent times after bleomycin treatment was very wide, although YFP-DNA-PKcs seemed to return to pre-treatment values faster than GFP-Ku80, similar to what was observed for γ-irradiation. The Deff value changes for the 10-min time point, relative to pre-treatment, were significant for both YFP-DNA-PKcs [F(3.27) = 6.24, P < 0.0001] and GFP-Ku80 [F(3.27) = 17.09, P < 0.0001], as were the pre-treatment value comparisons with respect to 1 h [F(3.27) = 2.79, P = 0.0096; (F(3.27) = 4.94, P < 0.0001)] and 2 h [(F(3.27) = 1.16, P = 0.2546); (F(3.27) = 3.02, P = 0.0055)] for YFP-DNA-PKcs and GFP-Ku80, respectively.
In conclusion, the above experiments showed that N&B analysis and RICS could quantify DNA repair protein kinetics after sparse damage was created not only by γ-irradiation but also by treatment with a radiomimetic chemical. It was seen that there was a more heterogeneous dose-dependent response with the latter, which was possibly due to variations in the chemical’s uptake and turnover between cells. Nevertheless, these types of measurements provided indirect information about how long a given dose of a radiomimetic chemical continued to produce DNA damage in a given cell, which was not possible to assess with prior methods.
Methodological limitations encountered during quantification of DNA repair protein kinetics
As this work presents the use of new methods for the quantification of DNA repair protein kinetics, it is important that some details of the method limitations are discussed. For both N&B analysis and RICS measurements, cells were selected to have a fluorescently tagged protein concentration in the 50–500 nM range, as verified by sample FCS measurements. Higher concentrations produced very low amplitude RICS curves and very low N&B analysis signals. The lower bound in this concentration range was set by the expression levels of the cells and was well measurable by both techniques. It should be noted that an additional correlation component due to triplet state blinking (28) was not considered in this analysis because it occurred at shorter time scales than the protein kinetics measured and was dealt with by removing the early correlation data, which was advantageous to fitting stability over having the triplet lifetime as an additional fitting component. It should also be mentioned that other existing RICS models including a second diffusion component or binding (27), which involved a higher number of fitting parameters, could not be supported by the data acquired in this work. RICS analysis was therefore limited to single-component diffusion that yielded only an effective Deff representing a population-averaged value of the mobility of proteins in the nucleus. A fraction of these proteins moved more slowly, with this fraction reducing with time post-damage as DNA repair progressed, while the remainder of repair proteins diffused freely. As a result, the value of Deff was time-dependent.
Importantly, the RICS data acquisition conditions required a small pixel size to capture the Deff-dependent spatiotemporal correlations of fluorescence. As a result, the RICS pixel size (0.04 µm) was a lot smaller than the radial beam waist (ωo = 0.30 μm) and the N&B analysis pixel size (0.2 μm), which translated to longer RICS scan times relative to N&B analysis for the same field of view size. As longer scan times would make RICS measurements more susceptible to cell movement, these spatiotemporal sampling constraints restricted the field of view size to ∼7 × 7 μm and 20 × 20 μm, for GFP-Ku80 and YFP-DNA-PKcs, respectively, which resulted in measuring the Deff time course for only one cell nucleus at a time (Figure 1E and F). This requirement made RICS experiments much more time-consuming than N&B ones, which are not as sensitive to pixel size and therefore enabled having 3–4 cells in the field of view at the same time (Figure 1A–D). However, the choice of larger pixel size in N&B analysis for the sake of measuring more cells at once came with a compromise: larger pixel sizes contained larger numbers of molecules that in turn reduced the variance amplitude of fluorescence fluctuations, which is the basis of the N&B analysis signal (Equations 1 and 2). Owing to this choice of pixel size for N&B analysis in this work, the mobile fraction at time points beyond 1 h post-irradiation started becoming comparable with photobleaching-corrected pre-irradiation values and, as a result, the change in mobile fraction was no longer detectable. Had the choice of pixel size been smaller, as was the case for RICS, the N&B analysis could have also been carried out to 2 h post-irradiation, but at the cost of measuring only one cell at a time.
CONCLUSIONS
This work demonstrates that it is feasible to study the real-time kinetics of different DNA repair proteins in living cells after DSBs are induced by exposure to γ-irradiation or after exposure to the radiomimetic chemical bleomycin. Specifically, the N&B analysis and RICS methods were used in combination with Strip-FRAP, to quantify DSB-induced changes in kinetics for two key NHEJ DNA repair pathway proteins that were fluorescently tagged, namely, GFP-Ku80 and YFP-DNA-PKcs, after DSBs were induced by exposure to γ-irradiation or after exposure to the radiomimetic chemical bleomycin. To our knowledge, this is the first time that the feasibility of quantifying repair kinetics after γ-irradiation or exposure to a radiomimetic chemical was shown for proteins that do not bind to DSBs in large numbers. The advantage of the proposed approaches is that they can quantify sparse DNA damage repair protein kinetics in the living cell in response to IR and bleomycin treatment, thus avoiding potential issues associated with repair of the more complex laser-induced damage. They could also be used in cases where laser-induced repair protein recruitment does not work (46). Furthermore, there was a qualitative resemblance between the IR-induced kinetics found here with previous reports on laser-induced damage for Ku70/80 and DNA–PKcs (7,8), On the other hand, conventional methods cannot quantify the repair protein kinetics for exposure to a radiomimetic chemical in vivo. The results of this work suggest the possible applicability of N&B and RICS analysis to the study of kinetics for numerous other DNA repair and signalling proteins after sparse DNA damage under conditions that resemble more closely the radiation therapy or chemotherapy of cancer.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [1–7].
FUNDING
The National Cancer Institute [5R21CA154958-02]; Cancer Prevention and Research Institute of Texas [RP110465-P1, RP110465-C2]. Funding for open access charge: The National Cancer Institute [5R21CA154958-02]; Cancer Prevention and Research Institute of Texas [RP110465-P1, RP110465-C2].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Hirohiko Yajima, Shih-Ya Wang, Kazi Fattah, Schichuan Zhang, Nozomi Tomimatsu, Yu-Fen Lin, Kyung-Jong Lee and Burong Hu for their help with cell irradiation.
REFERENCES
- 1.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 2.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 3.Burma S, Chen BP, Chen DJ. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair. 2006;5:1042–1048. doi: 10.1016/j.dnarep.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 4.Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013;2:130–143. doi: 10.3978/j.issn.2218-676X.2013.04.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mari PO, Florea BI, Persengiev SP, Verkaik NS, Bruggenwirth HT, Modesti M, Giglia-Mari G, Bezstarosti K, Demmers JA, Luider TM, et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc. Natl Acad. Sci. USA. 2006;103:18597–18602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari PO, van Gent DC, Chen BP, Chen DJ. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell. Biol. 2007;177:219–229. doi: 10.1083/jcb.200608077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mari PO, Florea BI, Persengiev SP, Verkaik NS, Brueggenwirth HT, Modesti M, Giglia-Mari G, Bezstarosti K, Demmers JAA, Luider TM, et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc. Natl Acad. Sci. USA. 2006;103:18597–18602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari PO, van Gent DC, Chen BPC, Chen DJ. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell. Biol. 2007;177:219–229. doi: 10.1083/jcb.200608077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung CS, Peterson SR, Chen DJ. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol. Cell. Biol. 1999;19:3877–3884. doi: 10.1128/mcb.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merkle D, Douglas P, Moorhead GB, Leonenko Z, Yu Y, Cramb D, Bazett-Jones DP, Lees-Miller SP. The DNA-dependent protein kinase interacts with DNA to form a protein-DNA complex that is disrupted by phosphorylation. Biochemistry. 2002;41:12706–12714. doi: 10.1021/bi0263558. [DOI] [PubMed] [Google Scholar]
- 11.Douglas P, Cui X, Block WD, Yu Y, Gupta S, Ding Q, Ye R, Morrice N, Lees-Miller SP, Meek K. The DNA-dependent protein kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Mol. Cell. Biol. 2007;27:1581–1591. doi: 10.1128/MCB.01962-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 13.Ma Y, Pannicke U, Lu H, Niewolik D, Schwarz K, Lieber MR. The DNA-dependent protein kinase catalytic subunit phosphorylation sites in human artemis. J. Biol. Chem. 2005;280:33839–33846. doi: 10.1074/jbc.M507113200. [DOI] [PubMed] [Google Scholar]
- 14.Chan DW, Chen BP, Prithivirajsingh S, Kurimasa A, Story MD, Qin J, Chen DJ. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002;16:2333–2338. doi: 10.1101/gad.1015202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, Meek K. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol. Cell. Biol. 2005;25:10842–10852. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K, Botvinick E, Qin J, Chen DJ. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J. Biol. Chem. 2005;280:14709–14715. doi: 10.1074/jbc.M408827200. [DOI] [PubMed] [Google Scholar]
- 17.Chen BP, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, Yajima H, Lobrich M, Shiloh Y, Chen DJ. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 2007;282:6582–6587. doi: 10.1074/jbc.M611605200. [DOI] [PubMed] [Google Scholar]
- 18.Yajima H, Lee KJ, Chen BP. ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol. Cell. Biol. 2006;26:7520–7528. doi: 10.1128/MCB.00048-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding Q, Reddy YV, Wang W, Woods T, Douglas P, Ramsden DA, Lees-Miller SP, Meek K. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol. Cell. Biol. 2003;23:5836–5848. doi: 10.1128/MCB.23.16.5836-5848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukas C, Bartek J, Lukas J. Imaging of protein movement induced by chromosomal breakage: tiny ‘local' lesions pose great ‘global' challenges. Chromosoma. 2005;114:146–154. doi: 10.1007/s00412-005-0011-y. [DOI] [PubMed] [Google Scholar]
- 22.Bekker-Jensen S, Mailand N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair. 2010;9:1219–1228. doi: 10.1016/j.dnarep.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 23.Nagy Z, Soutoglou E. DNA repair: easy to visualize, difficult to elucidate. Trends. Cell Biol. 2009;19:617–629. doi: 10.1016/j.tcb.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Essers J, Houtsmuller AB, Kanaar R. Analysis of DNA recombination and repair proteins in living cells by photobleaching microscopy. Methods Enzymol. 2006;408:463–485. doi: 10.1016/S0076-6879(06)08029-3. [DOI] [PubMed] [Google Scholar]
- 25.Dalal RB, Digman MA, Horwitz AF, Vetri V, Gratton E. Determination of particle number and brightness using a laser scanning confocal microscope operating in the analog mode. Microsc. Res. Tech. 2008;71:69–81. doi: 10.1002/jemt.20526. [DOI] [PubMed] [Google Scholar]
- 26.Digman MA, Dalal R, Horwitz AF, Gratton E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008;94:2320–2332. doi: 10.1529/biophysj.107.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown CM, Dalal RB, Hebert B, Digman MA, Horwitz AR, Gratton E. Raster image correlation spectroscopy (RICS) for measuring fast protein dynamics and concentrations with a commercial laser scanning confocal microscope. J. Microsc. 2008;229:78–91. doi: 10.1111/j.1365-2818.2007.01871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Digman MA, Gratton E. Analysis of diffusion and binding in cells using the RICS approach. Microsc. Res. Tech. 2009;72:323–332. doi: 10.1002/jemt.20655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merkle D, Block WD, Yu Y, Lees-Miller SP, Cramb DT. Analysis of DNA-dependent protein kinase-mediated DNA end joining by two-photon fluorescence cross-correlation spectroscopy. Biochemistry. 2006;45:4164–4172. doi: 10.1021/bi0524060. [DOI] [PubMed] [Google Scholar]
- 30.Roche Y, Zhang D, Segers-Nolten GM, Vermeulen W, Wyman C, Sugasawa K, Hoeijmakers J, Otto C. Fluorescence correlation spectroscopy of the binding of nucleotide excision repair protein XPC-hHr23B with DNA substrates. J. Fluoresc. 2008;18:987–995. doi: 10.1007/s10895-008-0390-6. [DOI] [PubMed] [Google Scholar]
- 31.Michelman-Ribeiro A, Mazza D, Rosales T, Stasevich TJ, Boukari H, Rishi V, Vinson C, Knutson JR, McNally JG. Direct measurement of association and dissociation rates of DNA binding in live cells by fluorescence correlation spectroscopy. Biophys. J. 2009;97:337–346. doi: 10.1016/j.bpj.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watson NB, Nelson E, Digman M, Thornburg JA, Alphenaar BW, McGregor WG. RAD18 and associated proteins are immobilized in nuclear foci in human cells entering S-phase with ultraviolet light-induced damage. Mutat. Res. 2008;648:23–31. doi: 10.1016/j.mrfmmm.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merkle D, Zheng D, Ohrt T, Crell K, Schwille P. Cellular dynamics of Ku: characterization and purification of Ku-eGFP. Chembiochem. 2008;9:1251–1259. doi: 10.1002/cbic.200700750. [DOI] [PubMed] [Google Scholar]
- 34.Digman MA, Brown CM, Sengupta P, Wiseman PW, Horwitz AR, Gratton E. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys. J. 2005;89:1317–1327. doi: 10.1529/biophysj.105.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ossato G, Digman MA, Aiken C, Lukacsovich T, Marsh JL, Gratton E. A two-step path to inclusion formation of huntingtin peptides revealed by number and brightness analysis. Biophys. J. 2010;98:3078–3085. doi: 10.1016/j.bpj.2010.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Digman MA, Wiseman PW, Choi C, Horwitz AR, Gratton E. Stoichiometry of molecular complexes at adhesions in living cells. Proc. Natl Acad. Sci. USA. 2009;106:2170–2175. doi: 10.1073/pnas.0806036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warmerdam DO, Kanaar R, Smits VA. Differential dynamics of ATR-mediated checkpoint regulators. J. Nucleic Acids. 2010;2010:pii: 319142. doi: 10.4061/2010/319142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Digman MA, Brown CM, Horwitz AR, Mantulin WW, Gratton E. Paxillin dynamics measured during adhesion assembly and disassembly by correlation spectroscopy. Biophys. J. 2008;94:2819–2831. doi: 10.1529/biophysj.107.104984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwille P, Haupts U, Maiti S, Webb WW. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys. J. 1999;77:2251–2265. doi: 10.1016/S0006-3495(99)77065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rossow MJ, Sasaki JM, Digman MA, Gratton E. Raster image correlation spectroscopy in live cells. Nat. Protoc. 2010;5:1761–1774. doi: 10.1038/nprot.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muller KE, Stewart PW. Linear Model Theory: Univariate, Multivariate, and Mixed Models. Hoboken, NJ: Wiley-Interscience; 2006. [Google Scholar]
- 42.Lukas J, Lukas C, Bartek J. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell. Biol. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 43.Koike M, Koike A. Accumulation of Ku80 proteins at DNA double-strand breaks in living cells. Exp. Cell. Res. 2008;314:1061–1070. doi: 10.1016/j.yexcr.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 44.Asaithamby A, Chen DJ. Cellular responses to DNA double-strand breaks after low-dose gamma-irradiation. Nucleic Acids Res. 2009;37:3912–3923. doi: 10.1093/nar/gkp237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong X, Mohanty SK, Stephens J, Heale JT, Gomez-Godinez V, Shi LZ, Kim JS, Yokomori K, Berns MW. Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res. 2009;37:e68. doi: 10.1093/nar/gkp221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams ES, Stap J, Essers J, Ponnaiya B, Luijsterburg MS, Krawczyk PM, Ullrich RL, Aten JA, Bailey SM. DNA double-strand breaks are not sufficient to initiate recruitment of TRF2. Nat. Genet. 2007;39:696–698. doi: 10.1038/ng0607-696. author reply 698–699. [DOI] [PubMed] [Google Scholar]
- 47.Reynolds P, Anderson JA, Harper JV, Hill MA, Botchway SW, Parker AW, O'Neill P. The dynamics of Ku70/80 and DNA-PKcs at DSBs induced by ionizing radiation is dependent on the complexity of damage. Nucleic Acids Res. 2012;40:10821–10831. doi: 10.1093/nar/gks879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Lobrich M, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011;30:1079–1092. doi: 10.1038/emboj.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Lobrich M. ATM and artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28:3413–3427. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Asaithamby A, Uematsu N, Chatterjee A, Story MD, Burma S, Chen DJ. Repair of HZE-particle-induced DNA double-strand breaks in normal human fibroblasts. Radiat. Res. 2008;169:437–446. doi: 10.1667/RR1165.1. [DOI] [PubMed] [Google Scholar]
- 51.Jakob B, Splinter J, Conrad S, Voss KO, Zink D, Durante M, Lobrich M, Taucher-Scholz G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011;39:6489–6499. doi: 10.1093/nar/gkr230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weterings E, van Gent DC. The mechanism of non-homologous end-joining: a synopsis of synapsis. DNA Repair. 2004;3:1425–1435. doi: 10.1016/j.dnarep.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 53.Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl Acad. Sci. USA. 2003;100:5057–5062. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Britton S, Coates J, Jackson SP. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell. Biol. 2013;202:579–595. doi: 10.1083/jcb.201303073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rivera-Calzada A, Spagnolo L, Pearl LH, Llorca O. Structural model of full-length human Ku70-Ku80 heterodimer and its recognition of DNA and DNA-PKcs. EMBO Rep. 2007;8:56–62. doi: 10.1038/sj.embor.7400847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei Z, Jin LF, He JL, Lou JL, Wang BH, Deng HP. Detecting DNA repair capacity of peripheral lymphocytes from cancer patients with UVC challenge test and bleomycin challenge test. Mutagenesis. 2005;20:271–277. doi: 10.1093/mutage/gei037. [DOI] [PubMed] [Google Scholar]
- 58.Benitez-Bribiesca L, Sanchez-Suarez P. Oxidative damage, bleomycin, and gamma radiation induce different types of DNA strand breaks in normal lymphocytes and thymocytes–a comet assay study. Ann. N. Y. Acad. Sci. 1999;887:133–149. doi: 10.1111/j.1749-6632.1999.tb07928.x. [DOI] [PubMed] [Google Scholar]
- 59.Grigaravicius P, Rapp A, Greulich KO. A direct view by immunofluorescent comet assay (IFCA) of DNA damage induced by nicking and cutting enzymes, ionizing (137)Cs radiation, UV-A laser microbeam irradiation and the radiomimetic drug bleomycin. Mutagenesis. 2009;24:191–197. doi: 10.1093/mutage/gen071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.