


Abstract
Overlap-directed DNA assembly methods allow multiple DNA parts to be assembled together in one reaction. These methods, which rely on sequence homology between the ends of DNA parts, have become widely adopted in synthetic biology, despite being incompatible with a key principle of engineering: modularity. To answer this, we present MODAL: a Modular Overlap-Directed Assembly with Linkers strategy that brings modularity to overlap-directed methods, allowing assembly of an initial set of DNA parts into a variety of arrangements in one-pot reactions. MODAL is accompanied by a custom software tool that designs overlap linkers to guide assembly, allowing parts to be assembled in any specified order and orientation. The in silico design of synthetic orthogonal overlapping junctions allows for much greater efficiency in DNA assembly for a variety of different methods compared with using non-designed sequence. In tests with three different assembly technologies, the MODAL strategy gives assembly of both yeast and bacterial plasmids, composed of up to five DNA parts in the kilobase range with efficiencies of between 75 and 100%. It also seamlessly allows mutagenesis to be performed on any specified DNA parts during the process, allowing the one-step creation of construct libraries valuable for synthetic biology applications.
INTRODUCTION
Synthetic biology is a rapidly growing interdisciplinary field that takes an engineering approach to biosciences research and to the development of new biotechnologies (1,2). The rise of synthetic biology has been triggered by the increasing use of quantitative and mathematical techniques in biological research and the explosion of biological data (e.g. genome sequences) provided by the ‘omics’ era (3,4), combined with impressive decreases in the cost of custom synthesized genes and DNA (5,6). The capacity of synthetic biology is now largely limited by our ability to design, assemble and screen new DNA-encoded constructs for their intended biological function (7–10).
One of the key engineering principles that synthetic biology researchers aim to apply to biology is standardization (11,12). Community efforts have made impressive steps toward standardization of measurement (13,14) and standardization of data and models (15,16); however, standardization of DNA assembly has fared less well. Early on in the emergence of synthetic biology, a standard for DNA assembly that defined DNA sequences as biological parts known as BioBricks (17), gained significant traction despite recognized shortcomings that include requiring all DNA parts to be absent of four common restriction enzyme recognition sites, and only permitting pairwise (two-at-a-time) assembly (9). Iterations of this standard have since appeared that address some of the limitations of BioBrick assembly and the BioBrick part format (18,19), but these have not been widely adopted. Instead, the DNA assembly methodologies that have seen the widest adoption within the synthetic biology community are those that do not require standardization of the parts involved. In particular, Gibson assembly, previously known as isothermal DNA assembly (20), has rapidly risen to being the most-used DNA assembly method in the field (21), despite being essentially a bespoke method, rather than a modular standard.
Gibson assembly, along with several other methods such as Circular Polymerase Extension Cloning (CPEC) (22), Sequence-Ligation Independent Cloning (SLIC) (23), Seamless Ligation Cloning Extract (SLiCE) (24), Overlap Extension Polymerase Chain Reaction (OE-PCR) (25), DNA Assembler (26) and direct assembly in yeast (27,28) is an overlap-directed DNA assembly method, where sequence homology at the 5′ and 3′ ends of DNA fragments directs their fusion, by end resection and DNA repair mechanisms (9). The advantages of overlap-directed methods are two-fold: (i) multiple parts can be assembled in a defined order at the same time in parallel (i.e. one-pot); and (ii) parts used within the assembly do not have to be free of specific ‘forbidden’ sequences. This second point is in contrast to many other assembly methods, such as the BioBrick standard (17), Golden Gate (29) and the recently described Standard European Vector Architecture (SEVA) standard (30). All of these methods use specific restriction enzymes for their DNA assembly and the recognition sequences of these are therefore forbidden within the sequences of the parts being assembled (9,31). From a synthetic biology standpoint, the core disadvantage of overlap-directed methods like Gibson assembly is that a DNA part from one assembly reaction cannot be used in an alternative assembly, without altering the homology sequences at its ends. This means that without a modification step, typically done by a PCR with newly synthesized primers, parts are not modular in the sense that they can easily be reused in different assembly reactions. Assemblies done with these overlap-directed methods are thus usually ‘bespoke’: done by scientists with a handful of parts, custom primers, and the goal of a single final construct.
Recent work has developed innovative software to automate the redesign of DNA parts to enable modularity in DNA assembly (32). However, we present here an alternative to this that does not require part modification when new constructs are required. The approach described here brings standardization to the powerful overlap-directed methods described above and enables rapid one-pot modular assembly through the use of designed overlap sequences that are compatible with a variety of long overlapping end assembly techniques. We call this strategy MODAL (Modular Overlap-Directed Assembly with Linkers) and we show here how it can be used to efficiently build and shuffle synthetic constructs made from gene-level parts (i.e. DNA parts encoding everything from promoter through to terminator). We demonstrate that the sequence of the overlap regions has a substantial impact on the accuracy of the assembly process, and thus to ensure efficient assembly and reduce unintended biological consequences, we accompany our strategy with a software tool for orthogonal overlap sequence design. Using the MODAL strategy and designed sequences, we go on to then demonstrate how our method can be used for efficient one-pot assembly of plasmid constructs and libraries in both yeast and Escherichia coli.
MATERIALS AND METHODS
Bacterial strains, media and reagents
Escherichia coli DH10B strain was used as the host to clone plasmid DNA assembled with Gibson and CPEC assembly methods. It was grown at 37°C in Luria-Bertani (LB) medium, with kanamycin (50 μg/ml) or ampicillin (50 μg/ml) added to select for cells transformed with the plasmid of interest. Saccharomyces cerevisiae YPH500 strain (33) was used as the host to clone plasmid DNA assembled through yeast in vivo recombination and was grown at 30°C in Yeast extract Peptone Dextrose (YPD) rich medium or synthetic complete drop-out medium lacking uracil (SC-Ura) to select for transformed cells. All enzymes used were purchased from New England Biolabs (NEB). Qiagen QiaQuick Spin and QiaPrep Spin were used, respectively, for PCR product and plasmid purifications.
PCR methods
All oligonucleotide primers used in this study are detailed in Supplementary Table S1 and all DNA regions used as parts are given in Supplementary Table S2. All parts used in the assembly reactions were first amplified using Phusion High-Fidelity DNA Polymerase (NEB). The reactions were prepared according to manufacturer's recommendations and the following cycles were performed: 30 s at 98°C followed by 25 cycles with 10 s at 98°C, 10 s at 65°C and 1 min at 72°C and a final extension step of 5 min at 72°C. The primers used for this PCR also add the adapter sequences to all fragments. These are then purified and ligated into pJET1.2 plasmids using the CloneJET PCR Cloning Kit (Fermentas), and transformed in E. coli cells. Clones were screened by either restriction digestion or PCR and sequence-verified using the primers provided with the kit.
Purified pJET1.2 plasmids containing the fragments flanked by adapters were used as template for the successive PCR, which adds the linker sequences to the fragments. The reactions were performed using Phusion High-Fidelity DNA Polymerase (NEB) according to manufacturer's recommendations, with the following steps: 30 s at 98°C, 25 cycles of 10 s at 98°C and 1 min at 72°C and a final extension step of 5 min at 72°C. Amplified fragments were then column-purified before use in DNA assembly reactions.
DNA assembly and cell transformation protocols
All DNA assembly reactions were performed using 0.1 pmol of DNA for each part and were set up on ice. Gibson assembly was performed as described (20), with a 1-h incubation time. CPEC was performed as described by Quan et al. (22) using 30 cycles and a final annealing/extension step of 3 min at 72°C and 5 μl of the assembly reaction solution were transformed in chemically competent E. coli cells. Transformation reactions were run following NEB's High Efficiency Transformation Protocol (C3019), except using LB medium and plating half of the final culture on a single plate. Where necessary, plasmids were verified by gel electrophoresis following analytical restriction digestion or by colony PCR across the assembly junctions.
Saccharomyces cerevisiae transformations were performed using a modified version of the ‘quick and easy’ protocol by Gietz et al. (34). A YPH500 colony grown on a YPD agar plate was used to inoculate 5 ml of YPD liquid medium: this starter culture was incubated at 30°C overnight and used the next day to inoculate a 3-ml YPD culture for each transformation. These cultures were grown for 4 h at 30°C and then used in the previously described protocol. After the heat-shock step, the cells were pelleted and resuspended in 1 ml of water. Five hundred microliters were then taken aside, pelleted again, resuspended in 75 μl water and plated on SC-Ura agar plates for selective growth.
Saccharomyces cerevisiae pADH1 promoter library
Yeast plasmids with mutated ADH1 promoters were generated simply by modifying the PCR step that adds linkers to assembly parts. Primers used were the same, but a mutagenic PCR was used where a two-step PCR was performed as described previously by Zaccolo et al. (35) with addition of 200 μM of nucleotide analogues 2′-deoxy-P-nucleoside-5′-triphosphate (dPTP) and 8-oxo-dGTP (TriLink BioTechnologies). To select and characterize a library of promoters from the mutagenic assembly, colonies were randomly picked from the yeast transformation plate, and their green and red fluorescence were simultaneously measured with flow cytometry as described below. From the initial screen, 20 colonies covering a range of GFP expression were selected to form the characterized library. For these, flow cytometry analysis was repeated in triplicate, taking readings of cells in exponential growth in SC-Ura liquid media. The ADH1-derived promoter DNA of these 20 colonies was then determined by Sanger sequencing of both strands.
Flow cytometry
Single-cell measurements were taken with a modified Becton Dickinson FACScan flow cytometer capable of parallel measurement of green and red fluorescent proteins (GFP and RFP, respectively). Escherichia coli grown in liquid culture to exponential phase were diluted in water and passed through the cytometer for 30 s. A 488-nm laser was used for excitation of green fluorescence detecting through a 530 nm band pass filter (FL1) with gain 890. Red fluorescence was excited with a 561 nm laser and 610 nm filter (FL5) with gain 850. Analysis of E. coli plasmids was performed using Cyflogic software (CyFlo Ltd.), applying a gate on forward and side scatter to capture readings from appropriately sized single bacteria. The means of FL1 and FL5 were used as the measure of per cell GFP and RFP, respectively. For gene expression from yeast, data analysis was performed in FlowJo (Treestar Inc.), gating forward and side scatter for appropriately sized yeast cells. GFP per cell was measured as the geometric mean of FL1 for cells that had been gated for a mid-range of RFP expression per cell (FL5). RFP-based gating was used to normalize for plasmid copy number variation within the population as described previously (36). An example of this gating is shown in Supplementary Figure S1.
Colony counting and visualization
All assembly reactions and cell transformations for a single DNA assembly method were done on the same day, using the same batch of competent cells and plates to ensure comparability. Following colony growth, all transformation plates were scanned using a Fuji FLA-5000 scanner: GFP expressing colonies were visualized by scanning with a blue (473 nm) laser and Fluorescein isothiocyanate (FITC) filter; RFP expressing colonies were visualized by scanning with a green (532 nm) laser and a long pass green (LPG) filter. Images were overlaid and aligned to correct for chromatic aberration using ImageJ software (NIH). The total numbers of colonies on each plate were counted manually. The assembly accuracy of each reaction was calculated as the percentage of total colonies for a reaction that showed the correct expression profile.
Software: R2oDNA designer
Initial sequences were generated using a Monte Carlo algorithm, and were optimized using a scoring function with a Monte Carlo Simulated Annealing algorithm (MCSA) (37,38). Given a degenerate IUPAC sequence specification, a starting sequence was generated at random (e.g. the degenerate sequence NNATNN may result in the random starting sequence AGATCT). Variable regions in the starting sequence were then subject to allowable random point mutations; if the random mutation produced a lower scoring sequence (where lower scores are better), then this mutated sequence was accepted. If the sequence produced a higher scoring sequence (i.e. a worse sequence) then this sequence had a probability of acceptance based on the Metropolis criterion (39).
Where ΔS is the change in score of the sequence and T is the ‘temperature’. Each simulated annealing run consisted of 4650 accepted mutations (steps). During each MCSA run, T was linearly ramped from 10 000 to 1000 over 100 steps, ramped from T = 1000 to T = 10 over 1000 steps, ramped from T = 10 to T = 1 over 1000 steps, ramped from T = 1 to T = 0.001 over 1000 steps, maintained at T = 0.001 for 1500 steps and finally maintained at T = 0.0001 for 50 steps. The lowest scoring sequence during the entire run was retained. If the lowest scoring sequence was within the allowed tolerances for each score term, then it was output and stored in a pool of initial sequences. This MCSA procedure was repeated until the pool of initial sequences reached a specified number.
R2oDNA designer: scoring function
The scoring function consisted of a weighted linear sum of five terms:
Where Shp is the hairpin score. This is the sum of the lengths of inverted repeats over a minimum length (4). Sr is the repeat score and is the sum of the lengths of repeats over a minimum length (6). Sf is the forbidden sequences score and is a sum of the number of matches to a list of undesirable sequences. The undesirable sequences are defined as Java regular expressions allowing the specification of complicated sequence motifs and both strands of the DNA were searched. SGC penalizes GC content straying from the target value and is equal to the length of the specified region multiplied by the square of the difference between the target GC content and actual GC content. STm is the same as SGC but targets Tm rather than GC content. Tm was calculated using the base stacking algorithm of SantaLucia (40) with 50 mM monovalent salt concentration, 0 mM divalent salt concentration and 200 nM primer concentration. The weights, whp, wr and wf were set to 10.0 and the weights wGC and wTm were set to 1.0. The Tm and GC terms could contribute nothing to the scoring function if no target regions were defined.
R2oDNA designer: elimination of sequences
Sequences in the initial pool were first aligned by BLAST (41) against the genome sequence of possible host organisms (saccharomyces_cerevisiae_genome, eschericichia_coli_k12_dh10b, eschericichia_coli_k12_mg1655, eschericichia_coli_k12_w3110 and bacillus_subtilis_168) using the software program blastn with a word size of 8. Sequences with hits below a certain e-value cutoff (in this case 1) were eliminated from the pool of sequences. Sequences with the potential to mis-anneal to other sequences in the pool were then eliminated using a network elimination algorithm (42). An undirected graph was created by carrying out all-against-all pairwise alignments of the sequences in the pool. Edges were drawn between two sequences if (i) there is an exact 8-character subsequence match between the sequences or if (ii) the sequence identity in the global alignment reached above a certain threshold (50%). A random sequence was picked and all connected sequences were eliminated. This process was iterated until a completely disconnected graph was obtained. The remaining sequences then formed the final set of orthogonal sequences.
RESULTS
Overlap-directed assembly methods rely on 20–50 bp of shared sequence between DNA parts that conjoin, depending on the precise method used (9). Research suggested that a minimum of 40 bp is required for efficient assembly by yeast homologous recombination, and that a similar length of DNA is optimal for the Gibson assembly method (26,43,44). To enable our MODAL standard to be compatible with Gibson, CPEC and yeast assembly, we selected 45 bp to be the length of our overlap regions, which we call linkers and act as ‘guiding ends’ in the assembly reactions. To be able to easily attach these to any DNA part using a standard protocol, the parts need to go through a one-time standardization process. This consists of a PCR where the DNA part is amplified from any source (i.e. plasmid or genomic DNA) using part-specific primers that contain 15-bp ‘adapter’ sequences (Figure 1A). These two sequences (prefix adapter and suffix adapter) are universal and flank the DNA parts after the amplification. Note that this step can be replaced by direct DNA synthesis. The guiding end linkers can be then attached to any such DNA part using a standardized PCR: this uses primers from a set of reusable universal primers (the linker set), which anneal to the adapter sequences and encode the guiding end linkers (Figure 1A). The position of the DNA parts in the final constructs is determined by the linkers, since they guide the assembly reaction: for example, if the desired assembly is part A followed by part B, then A is amplified using a reverse primer that is the reverse complement of the linker sequence plus the suffix adapter sequence, and B is amplified using a forward primer, that is, the linker sequence plus the prefix adapter sequence. However, if the desired order is reversed (i.e. part B followed by part A) then the primers used to amplify the parts are swapped around, so that B is amplified with the aforementioned reverse primer and A is amplified with the forward primer.
Figure 1.

Overlap-directed assembly of DNA parts by addition of modular linker sequences can enable standardized assembly of genes in different orders and orientations without context effects. (A) Schematic of the MODAL strategy. Using an initial PCR, selected DNA parts are amplified from their source to be flanked by defined 15-bp adapter sequences (P = prefix adapter, S = suffix adapter), which can then be cloned, stored and sequence verified in the pJET vector. Linker sequences (numbered 1–4) are then added 5′ and 3′ of the adapters by PCR and these guide homology-mediated overlap assembly into plasmids or other constructs. The bifurcation in the diagram shows an example of how changing which linkers are added to part A and part B changes their order in the final construct. (B) Using a single set of four random 45-bp linkers and four standardized E. coli parts (constitutive GFP expression, constitutive RFP expression, kanamycin resistance and a pUC origin of replication), Gibson assembly was used to construct plasmids with parts in a variety of orders. GFP and RFP expression per cell as measured by flow cytometry did not show significant variation when parts were shuffled to different positions in the plasmid and contained different linker sequences upstream and downstream of genes. Mean fluorescence per cell was calculated from mean FL1 (GFP) and mean FL5 (RFP) measurements (n = 5). Error bars indicate standard error.
To mitigate against PCR-borne errors, we took three steps: (i) all PCRs were performed using a high-fidelity DNA polymerase, (ii) standardized parts generated in PCR step 1 were cloned into the pJET vector and sequence verified before further use, and (iii) the number of cycles in PCR step 2 did not exceed 25 cycles.
Modular overlap sequences enable one-pot assembly of multigene constructs with variable contexts
To validate our approach, we took a set of four gene-level DNA parts and generated a small preliminary library of MODAL primers with which to assemble them. Four parts require four linkers to be assembled, so we generated eight 60-mer primers: each forward primer consists of a 45-base guiding end linker sequence followed by the 15-base prefix adapter sequence, and each reverse primer consists of the reverse complement of the linker sequence followed by the 15-base suffix adapter. The linker sequences were randomly generated to have ∼50% GC content over the 45-bp overlap region. Using the MODAL strategy (Figure 1A) with the one-pot Gibson assembly method, we generated E. coli plasmids encoding constitutive GFP and RFP expression, DNA replication and kanamycin resistance. Our method allowed the same starting materials to combine our four gene-level parts into functional plasmids that have their genes in a variety of different orders and orientations. To determine if our synthetic linker sequences impart any context-specific effects (i.e. modulating local gene expression), we also determined the GFP and RFP output from plasmids constructed from parts in different orders. Quantification of GFP and RFP revealed remarkably reliable gene expression regardless of the local context and the presence of the linker regions (Figure 1B). Thus, not only does the MODAL strategy enable the order and orientation of genes within constructs to be rapidly rearranged, but the synthetic sequences encoding the adapters and linkers were well-tolerated between gene-level DNA parts (i.e. when intergenic).
With prefix adapter, suffix adapter and 45-bp linkers, our modular approach leaves 75 bp of synthetic DNA between parts. While this may be tolerable at the gene-level of DNA assembly, such long sequences are unlikely to be completely neutral when placed between the sub-gene-level parts that constitute gene expressing units, such as promoters, 5′ untranslated regions and open reading frames (ORFs). We confirmed this by constructing a series of bicistronic expression cassettes consisting of a promoter, GFP and RFP and assessing the relative GFP and RFP expression levels when different linkers were used. Unsurprisingly, gene expression varied significantly depending on the linkers selected and the order of the bicistron (Supplementary Figure S2). Such context-dependency is likely due to differences in local RNA folding within transcripts, modulating the efficiencies of elements such as ribosome binding site (RBS) sequences (45) and adjusting the stability of the mRNA.
R2oDNA designer: online software for the design of orthogonal linker sequences for overlap-directed DNA assembly
These initial experiments demonstrated the potential for standardizing overlap-directed DNA assembly. However, when assembling the plasmids in Figure 1B, almost half of the colonies either did not express any GFP or RFP, or only expressed a single fluorescent reporter. This reflects our broader experience of Gibson assembly at the plasmid level, where we see significant variations in efficiency depending on the parts being assembled. We therefore chose to develop a software tool, R2oDNA Designer, as a means to both test the constraints required for efficient DNA assembly and to facilitate the generation of orthogonal DNA sequences suitable for overlaps. In so doing we recognized that any DNA could contain sequences that make them inefficient for overlap DNA assembly methods (e.g. polyA runs or sequences that form strong hairpins when single-stranded), or could contain restriction enzyme sequences that impede planned downstream handling. In addition, certain DNA sequences could be deleterious to the biological function of the system (e.g. transcription factor binding sites) or to the host via unpredicted homologous recombination with the genome.
To address these issues, R2oDNA Designer has been designed to generate synthetic DNA sequences of a defined length and composition that are suitable for overlap assembly and can exclude predefined forbidden sequences. The software uses a MCSA to randomly sample DNA sequence space and score the sequences generated, converging on a set that meet the defined criteria of predetermined length and GC-content or melting temperature (Figure 2A). These sequences are compared with the genome sequences of a provided list of organisms (here we have used E. coli, S. cerevisiae and Bacillus subtilis) using the BLAST algorithm (41), to eliminate any that could cross-anneal with chromosomal sequences. Finally, all remaining sequences are then compared with one another using a network elimination algorithm to further eliminate any that may cross-anneal and therefore would not be orthogonal and suitable for defined one-pot assembly.
Figure 2.
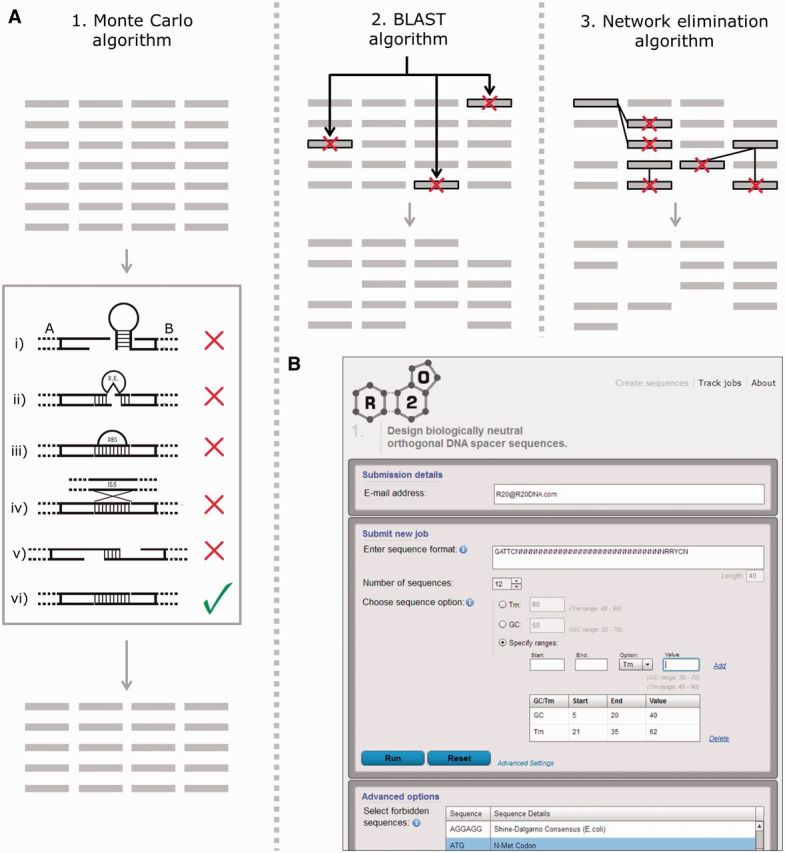
R2oDNA Designer is an online software tool for the design of synthetic linker sequences for the MODAL strategy. (A) Process overview. An initial pool of random sequences are generated that match primary user requirements (length- and position-specific nucleotide constraints). These are then optimized using MCSA using a scoring function that penalizes (i) single-stranded DNA secondary structures, (ii) forbidden sequence motifs including restriction enzyme sites, (iii) functional motifs like ribosome binding sequences and (iv) insertion element sequences, (v) unwanted self-annealing and (vi) off-target GC content or Tm. All sequences containing forbidden motifs or medium to strong secondary structures or self-annealing are optimized out. Any sequences with significant BLAST hits to selected genomes are eliminated. Finally, any remaining sequences that cross-anneal to one another are removed using a network elimination algorithm. (B) R2oDNA Designer user interface is shown. Users can input linker length, sequence requirements and preferences for GC content or melting temperature in the first window. A list of the forbidden sequences and genomes to compare with can be adjusted in the advanced settings window.
R2oDNA Designer software, available free online at http://www.r2odna.com, allows a user to select from a set of default forbidden sequences (Supplementary Table S3) or upload their own (Figure 2B). Similarly, the list of genome sequences that the synthetic sequences are compared with can be customized. A further feature of the software is the ability to score existing sequences for their ‘acceptability’ to be used as an overlap linker. More details of the software and its features are provided in the Supplementary Materials.
Designed overlap sequences enable efficient one-pot modular DNA assembly by a variety of techniques
To demonstrate how linker sequences generated by R2oDNA Designer can improve the cloning efficiency of modular plasmids for a variety of one-pot DNA assembly methods, we tested the DNA assembly efficiency of two 4-part plasmids: an E. coli plasmid constitutively expressing GFP and RFP and an S. cerevisiae 2 -μ plasmid also constitutively expressing GFP and RFP variants (yEGFP and mCherry, respectively, see Figure 1B and Supplementary Figure S1). DNA assembly of the E. coli plasmid was tested using the Gibson assembly and CPEC methods, transforming assembly products into DH10B E. coli and quantifying assembly efficiency as the percentage of colonies that were both red and green when scanned for fluorescence (Figure 3A). DNA assembly of the yeast plasmid was tested using direct assembly in yeast (in vivo recombination), transforming the modular parts into YPH500 S. cerevisiae and quantifying assembly efficiency as the percentage of colonies that were both red and green when scanned for fluorescence.
Figure 3.

Assessment of designed linkers used within the MODAL strategy with Gibson, CPEC and S. cerevisiae recombination DNA assembly methods. (A) To assess modular linker sequences, four parts were assembled by Gibson assembly into a plasmid encoding constitutive GFP and RFP expression, following the scheme illustrated in Figure 1A. DH10B E. coli were transformed with assembly reactions and grown on LB + kanamycin agar plates overnight and then scanned for green and red fluorescence the following day. This was repeated three times on separate days, and a single set is shown here. Assembly was done using six different linker sets: random, designed with 40% GC content, designed with 50% GC content, designed with 40% GC content, functional and scarless. Correct assembly gives colonies appearing yellow due to simultaneous green and red fluorescence. (B) The total number of colonies and the percentage of those containing correctly assembled plasmids (the ‘accuracy’) were calculated from image analysis of each plate for DNA assemblies using different linker sequences and using the Gibson, CPEC and S. cerevisiae recombination DNA assembly methods. Equivalent E. coli parts and competent cells were used for Gibson and CPEC (n = 3), but for S. cerevisiae assembly, YPH500 cells were used and DNA parts encoded constitutive yEGFP and mCherry RFP expression, uracil selection and a 2 -μ plasmid origin (n = 2). Error bars indicate standard error. (C) Schematic illustrating the difference between the assembly process and intergenic regions formed by the MODAL strategy and scarless assembly.
Four different sets of R2oDNA Designer-generated linkers were assessed for Gibson assembly of the E. coli plasmid, comparing these with a standard bespoke Gibson assembly that produces a seamless product (‘Scarless’) and to randomly generated linkers, as used in Figure 1 (‘Random’). Two sets of the four linkers were also assessed for CPEC and direct assembly in yeast. The four sets consisted of three designed by typical runs of R2oDNA Designer, with 40, 50 and 60% GC content specified during the design process (see Supplementary Table S3 for design parameters). The fourth set, ‘Functional’, was designed to be illustrative of the problems of an approach suggested previously where short functional parts are encoded within the sequences of the overlap regions (26). This set of four linker sequences, encoding a promoter, a terminator, a peptide tag sequence and an RNAse III site, was selected using R2oDNA Designer’s ability to score existing sequences for suitability as use as linkers, which is described further in the Supplementary Materials. For all assembly reactions, the overlap region was designed to be 45 bp.
All Gibson assembly reactions of the four-part E. coli plasmid gave a high number of colonies per transformation except for two: when the designed linkers with 60% GC content were used and with the functional linkers (Figure 3B). Given the poor suitability for overlap assembly of the functional linkers (as assessed by our software) it is unsurprising that they were inefficient; however, we were surprised that 60% GC designed linkers also performed badly. Repetition of this DNA assembly but using a further set of four more linker sequences designed to have 60% GC content did not change the number of colonies per transformation, demonstrating that the low efficiency was not a specific feature of one or more of the sequences used as a linker.
The highest accuracy for Gibson assembly, with the greatest percentage of correct clones, was seen with the MODAL strategy with the 40% GC linkers. Scarless assembly generated many colonies, but a large percentage of these expressed only one fluorescent protein. This is likely due to incorrect assembly directed by partial sequence homologies between the promoter and terminator sequences of the GFP- and RFP-encoding parts (Figure 3C).
Having determined that 40% GC linkers designed by R2oDNA Designer gave the highest accuracy for Gibson assembly and that 60% GC linkers were problematic, we next examined whether these observations also held for two other commonly used overlap-directed assembly methods, CPEC and direct assembly in yeast. For CPEC, both 40 and 60% GC linkers gave a high percentage (85%) of correct colonies but much lower numbers of transformants than with Gibson assembly. In this instance, significantly more colonies were obtained using 60% GC linkers compared with 40% GC linkers (Figure 3B). For direct assembly in yeast, both 40 and 60% GC linkers gave 100% accuracy, meaning that every colony was visible as a correct assembly each time the experiment was repeated. In contrast to CPEC, but as with Gibson, 40% GC linkers were preferable to 60% GC linkers, yielding substantially more colonies (Figure 3B). It is interesting to note that the 60% GC linkers are preferred by the assembly method that occurs cycling at high (≥60°C) temperatures (CPEC), whereas the 40% GC linkers are preferred when the assembly reaction is isothermal and performed at lower temperatures.
Mutagenic PCR within MODAL allows rapid construction of libraries with diversity targeted to specific parts
The MODAL strategy of using PCR to connect modular parts to designed linker sets allows almost any amplifiable sequence to be used within our assembly approach, which is in contrast to many restriction enzyme-dependent techniques [such as GoldenBraid (46) and BioBricks (17)], that specify that certain sequences must be absent from parts. While we normally keep PCR to a low number of cycles and use a high-fidelity polymerase to avoid errors being introduced into parts, it is also straightforward at this stage to replace high-fidelity PCR with mutagenic PCR for one or more selected parts of a construct to be diversified (35,47). With only a slight modification, our approach thus becomes a rapid method for generating construct libraries within the process of DNA assembly.
To demonstrate this variation of our MODAL strategy, we modified our four-part yeast GFP and RFP expression plasmid design, so that the constitutive promoter for GFP (the ADH1 promoter) and the transcribed GFP region were split and treated as two separate modular parts, meaning that our design was now a five-part plasmid assembly using five 40% GC linker sequences. DNA assembly was done directly into yeast as before, but during the PCR to add the linker regions to parts, we amplified the ADH1 promoter part in two different reactions in parallel; a standard reaction using a high-fidelity polymerase, and mutagenic reaction, optimized to incorporate ∼10 mutations per 100 bp (Figure 4A). To examine the efficiency of this approach to rapidly generate libraries, we picked four colonies from the non-mutated assembly and 52 colonies from the mutated assembly. These colonies were grown overnight in liquid media and then characterized for GFP and RFP expression using two-color flow cytometry. GFP expression per plasmid was seen to vary significantly between colonies selected from the mutagenic PCR assembly, but remained constant between the colonies from the standard assembly (Figure 4B, inset). From the mutagenic assembly, we selected a library of 20 mutated ADH1 promoters that gave a variety of different GFP outputs. We repeated their characterization in triplicate and determined their nucleotide sequences (Figure 4B and Supplementary Table S4). Thus, within the simple steps of our MODAL strategy, we were able to direct mutagenesis to a specific part in the final construct and create a constitutive yeast promoter library useful for synthetic biology and metabolic engineering projects (47,48).
Figure 4.
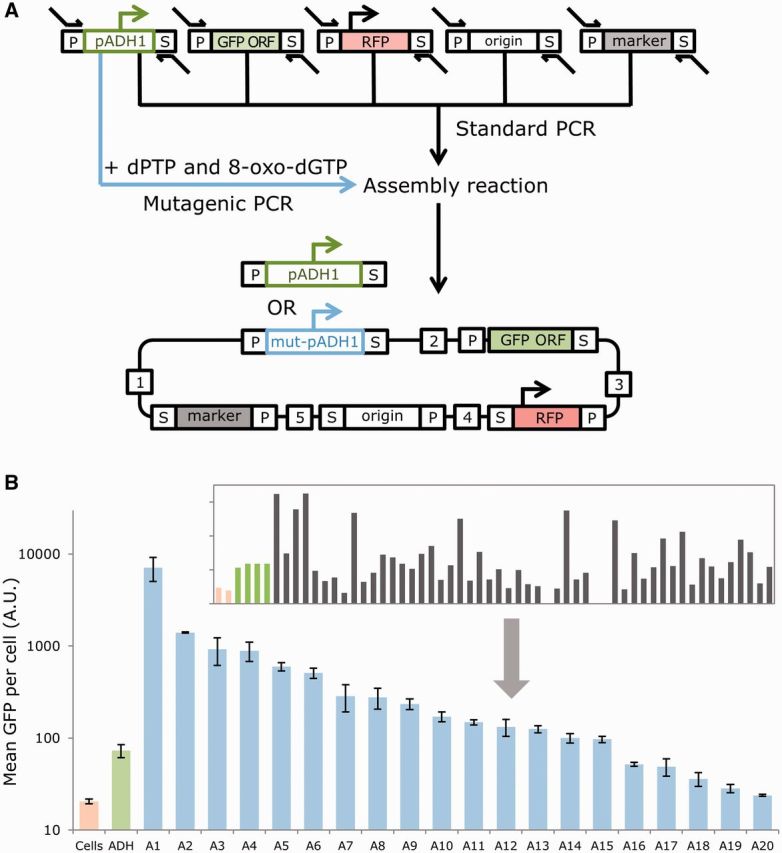
Incorporation of mutation within the MODAL strategy process enables rapid creation of construct libraries. (A) As well as performing standard assembly, selected parts can also be readily mutated as part of the assembly workflow by the addition of nucleotide analogues in the PCR amplification that adds the linker regions, as in the scheme displayed. Mutagenic PCR (35) uses dPTP and 8-oxo-dGTP to mis-incorporate a high percentage of sequence errors into PCR. (B) Direct assembly of a five-part modular plasmid in yeast with mutation applied to the ADH1 promoter part results in hundreds of yeast colonies displaying different mean GFP expression levels. Initial screening of 52 colonies by flow cytometry (inset) allows a graded library of 20 mutant ADH1 promoters (A1–A20) to be selected that covers a 3 orders of magnitude expression range, above and below the output provided by the unmutated ADH1 promoter (ADH). Error bars indicate standard error (n = 3).
DISCUSSION
In this study, a DNA assembly strategy that utilizes standardized linker sequences was developed to enable modular construction via a variety of overlap-directed assembly methods. A variation of the MODAL strategy was also introduced that facilitates the rapid production of construct libraries with mutation applied only to defined parts. To assist our strategy, an online software tool, R2oDNA Designer, was developed that produces sets of efficient linker sequences suitable for different projects.
Modular linker regions are tolerated in intergenic regions of plasmids but affect expression levels when within genes
When combined with the standard prefix and suffix adapter sequences used for amplification, the linkers we describe here add a region of 75 bp of synthetic DNA between assembled parts. Between gene-level parts, this synthetic DNA appears to be well-tolerated and does not affect expression levels of assembled parts. However, at the sub-gene-level DNA assembly, where ORFs and promoters are linked to make genes and operons, significant differences in expression levels were observed. This observation ties in with those made recently by several groups investigating the role local flanking DNA sequences have on defined biological parts. DNA sequences immediately at the junctions between the promoter, RBS and ORF parts of E. coli operons appear to dramatically affect part function (45,49–52). Similar effects have also been observed to some extent in yeast (53). These local context effects all occur at DNA sequences that are transcribed as part of the mRNA, as we have seen here. While this feature of varied gene expression from the same modular parts can be embraced as a rapid means to provide diversity in gene expression, it also prevents predictability. Fortunately, three recent studies have developed new classes of RNA processing biological parts that act to alleviate these effects to regain predictable gene expression (50–52).
Outside of the transcribed regions of genes within the intergenic space, the addition of synthetic DNA sequences such as those given here may actually provide some level of insulation against local context effects of neighboring parts simply by providing spacing. A recent cautionary tale has shown how an unwanted promoter can be unintentionally created within a construct by BioBrick assembly of four non-promoter parts (54). Previous work has also shown that reliability in the reuse of promoters in different constructs can be improved by adding spacer sequence upstream of the promoter (55). Synthetic linker sequences designed specifically to be free of unwanted sequence motifs may offer a valuable resource as intergenic insulators within engineered constructs.
Increased GC content in linkers decreases overlap-directed assembly efficiency for isothermal methods
We assessed designed linkers with 40, 50 and 60% GC content with Gibson assembly of four-part plasmids in E. coli and saw both the highest total number of colonies and the highest accuracy achieved with 40% GC content. Compared with 60% GC, 40% GC linkers gave 28 times more correct colonies using Gibson assembly into E. coli and also gave 4.5 times more correct colonies when plasmid assembly was done directly in yeast. In contrast to this, the CPEC assembly method gave equivalent accuracies with 40 and 60% GC linkers and returned nearly nine times more colonies with the latter. We attribute these findings to the nature of the methods used for DNA assembly. Both Gibson and yeast assembly are isothermal methods occurring at 50 and 30°C, respectively, whereas CPEC is a cycled reaction similar to PCR that includes a melting step at 98°C. At this high temperature, all base pairing regardless of GC content can be melted and during the cycling any mis-folding or mis-annealing of single-stranded DNA is removed. High GC content then acts to improve the accuracy of the linkers coming together during CPEC. During Gibson assembly, the lower temperature and isothermal conditions prevent the DNA overlaps from being melted; a high GC content is thus more likely to lead to the overlap sequences being caught in thermodynamic traps that inhibit the search process for the correct partner and this prevents mismatched linker pairings from being resolved. With lower GC content overlaps, the thermodynamic barrier to sampling different pairings is reduced, thus facilitating the search process. The calculated melting temperatures of the linker regions also provide a further way to assess their suitability for different assembly methods, and for our linkers these values are provided in the Supplementary Table S1. In anticipation of this, we have also included into R2oDNA Designer the ability to design linkers to a specific calculated melting temperature rather than GC content.
We expect that linkers with higher GC-content and melting temperature will generally be preferable for assembly by methods similar to CPEC, such as OE-PCR (25) and successive hybridization assembly (56). Linkers with lower GC content and melting temperatures should be preferable for all other isothermal DNA assembly methods, such as SLICE, SLIC, uracil specific excision reaction (USER) and direct assembly in B. subtilis, all of which occur at 37°C (23,24,57,58). Importantly, R2oDNA Designer can easily design linker sets optimized for certain DNA assembly techniques or can be used to generate linkers compatible with many overlap assembly methods (e.g. 50% GC content linkers).
PCR-based DNA assembly enables rapid adoption of new parts and targeted mutation
The MODAL strategy described here standardizes parts through the addition of 15-bp prefix and suffix adapter sequences. While this is not strictly necessary for overlap-directed assembly, as with previous standards, it significantly increases the interoperability and reusability of DNA parts. Furthermore, it radically improves the workflow and cost per assembly since a single batch of oligonucleotide primers can be used in hundreds of assembly reactions, and any lab only needs to maintain a limited set of such primers for the purposes of DNA assembly. It therefore saves reagent costs, but more importantly it saves time in the design and ordering of primers. Our MODAL strategy can also be adopted for other adapter sequences that are compatible with being used as PCR annealing sites (18).
The MODAL strategy requires at least one step of PCR to attach standardized linkers to parts before DNA assembly. PCR is often seen as problematic to include in DNA assembly protocols, as even with high-fidelity DNA polymerases, errors in replication occur. However, the advantage of PCR is that it is a rapid and cheap method to take any DNA of known sequence from almost any source and convert it into a linear DNA part to go straight into an assembly. It is also a technique available to almost every bioscience lab in the world. To minimize error propagation, our strategy recommends cloning and sequence-verifying each part after initial amplification from its source using adapter attaching primers. Following this, only a standardized 25-cycle high-fidelity PCR is needed to generate assembly parts. Even for parts as long as 18 kb, amplification errors with this PCR can be estimated to occur only in <1 in 5 molecules (as calculated at www.thermoscientificbio.com/webtools/fidelity). The strategy thus fits well with the expected size of all gene-level parts that may need to be assembled, with the longest yeast gene, MDN1, being ∼15 kb (59) and the longest E. coli operon, dcw, being 18 kb (60).
We have also demonstrated here that inaccuracies in PCR can be amplified intentionally using mutagenic PCR to generate diversity at a specified part while performing assembly. The mutagenic PCR protocol used here yielded ∼10 mutations per 100 bp (Supplementary Figure S3), which proved ideal for creating a yeast promoter library covering a wide range of expression levels. The inherent G to A and C to T bias of the analogues used proved to be particularly favorable for creating high output promoters (61). For protein-coding regions, such a high error rate is unlikely to yield functional proteins, but the inaccuracy of mutagenic PCR can be tuned by adding less nucleotide analogues, thus allowing error incorporation to be less frequent where necessary, for example, in enzyme-directed evolution experiments.
Future developments
The MODAL strategy offers reliable modular DNA assembly of gene-level parts via a variety of overlap-directed methods. This could in the future be integrated into a full parts-to-chromosome workflow, where methods that quickly combine small parts with minimal scars such as Golden Gate (29) are used to generate gene-level parts, which can feed into the modular assembly strategy we describe here for large construct assembly (5–20 kb; pathway/system). Subsequently, in vivo DNA assembly methods more suited to ‘Big DNA’, such as recombinase- or TALEN-mediated recombination (62–64) may then be used to assemble chromosomes from the large constructs assembled with our designed linkers. In this scenario, a megabase chromosome built from 2-kb gene-level parts would need ∼500 linker sequences designed by our software. While each would be orthogonal within the chromosome, so unlikely to unintentionally recombine, the chromosome would still contain hundreds of copies the 15-bp prefix and suffix adapter sequences. While these sequences are shorter than the >30-bp length thought to trigger homologous recombination in yeast (26,44), further work is needed to assess the long-term stability of constructs containing multiple copies of these. Previously, scar sequences found within E. coli plasmids assembled using the BioBrick method were identified as hot spots for deleterious recombination (65). To avoid similar situations arising with our methods, a future iteration that leaves behind shorter adapter sequences would be ideal.
Assembly at the sub-gene-level could also be enabled in the future with our method as teams develop new parts and approaches that address the local context issues that occur at part junctions. The assessment algorithm of R2oDNA Designer could be used to determine parts encoded by short sequences that can themselves be used as reliable linkers for overlap assembly. As well as the software rejecting any unwanted sequences from these linkers, it could be used to design linkers to include functional sequences such as RBSs. The software could also be modified to compare sequences against part registries alongside its current ability to compare and reject sequences homologous with genomes. By making the Designer available for the community, we hope to learn what sequences are desired and which are unwanted for linkers and thus improve it over time.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
EPSRC [EP/G036004/1, EP/H019154/1]; EU Framework Program 7 [ST-FLOW; KBBE.2011.5-289326]. Funding for open access charge: EPSRC [EP/G036004/1].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to acknowledge the help and expertise of Chris Hirst, Dr James Chappell, Tom Adie, Marianeve Quartuccio, Valentina Gnassi and Professor Dick Kitney.
REFERENCES
- 1.Heinemann M, Panke S. Synthetic biology–putting engineering into biology. Bioinformatics. 2006;22:2790–2799. doi: 10.1093/bioinformatics/btl469. [DOI] [PubMed] [Google Scholar]
- 2.Andrianantoandro E, Basu S, Karig DK, Weiss R. Synthetic biology: new engineering rules for an emerging discipline. Mol. Syst. Biol. 2006;2:2006 0028. doi: 10.1038/msb4100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett CL, Kim TY, Kim HU, Palsson BO, Lee SY. Systems biology as a foundation for genome-scale synthetic biology. Curr. Opin. Biotechnol. 2006;17:488–492. doi: 10.1016/j.copbio.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Church GM. From systems biology to synthetic biology. Mol. Syst. Biol. 2005;1 doi: 10.1038/msb4100007. 2005 0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma S, Tang N, Tian J. DNA synthesis, assembly and applications in synthetic biology. Curr. Opin. Chem. Biol. 2012;16:260–267. doi: 10.1016/j.cbpa.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munnelly K. Engineering for the 21st century: synthetic biology. ACS Synth. Biol. 2013;2:213–215. doi: 10.1021/sb400039g. [DOI] [PubMed] [Google Scholar]
- 7.MacDonald JT, Barnes C, Kitney RI, Freemont PS, Stan GB. Computational design approaches and tools for synthetic biology. Integr. Biol. (Camb) 2011;3:97–108. doi: 10.1039/c0ib00077a. [DOI] [PubMed] [Google Scholar]
- 8.Purnick PE, Weiss R. The second wave of synthetic biology: from modules to systems. Nat. Rev. Mol. Cell Biol. 2009;10:410–422. doi: 10.1038/nrm2698. [DOI] [PubMed] [Google Scholar]
- 9.Ellis T, Adie T, Baldwin GS. DNA assembly for synthetic biology: from parts to pathways and beyond. Integr. Biol. (Camb) 2011;3:109–118. doi: 10.1039/c0ib00070a. [DOI] [PubMed] [Google Scholar]
- 10.Lu TK, Khalil AS, Collins JJ. Next-generation synthetic gene networks. Nat. Biotechnol. 2009;27:1139–1150. doi: 10.1038/nbt.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Endy D. Foundations for engineering biology. Nature. 2005;438:449–453. doi: 10.1038/nature04342. [DOI] [PubMed] [Google Scholar]
- 12.Arkin A. Setting the standard in synthetic biology. Nat. Biotechnol. 2008;26:771–774. doi: 10.1038/nbt0708-771. [DOI] [PubMed] [Google Scholar]
- 13.Kelly JR, Rubin AJ, Davis JH, Ajo-Franklin CM, Cumbers J, Czar MJ, de Mora K, Glieberman AL, Monie DD, Endy D. Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 2009;3:4. doi: 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canton B, Labno A, Endy D. Refinement and standardization of synthetic biological parts and devices. Nat. Biotechnol. 2008;26:787–793. doi: 10.1038/nbt1413. [DOI] [PubMed] [Google Scholar]
- 15.Peccoud J, Anderson JC, Chandran D, Densmore D, Galdzicki M, Lux MW, Rodriguez CA, Stan GB, Sauro HM. Essential information for synthetic DNA sequences. Nat. Biotechnol. 2011;29:22. doi: 10.1038/nbt.1753. [DOI] [PubMed] [Google Scholar]
- 16.Galdzicki M, Rodriguez C, Chandran D, Sauro HM, Gennari JH. Standard biological parts knowledgebase. PLoS One. 2011;6:e17005. doi: 10.1371/journal.pone.0017005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shetty RP, Endy D, Knight TF., Jr Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2008;2:5. doi: 10.1186/1754-1611-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson JC, Dueber JE, Leguia M, Wu GC, Goler JA, Arkin AP, Keasling JD. BglBricks: a flexible standard for biological part assembly. J. Biol. Eng. 2010;4:1. doi: 10.1186/1754-1611-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sleight SC, Bartley BA, Lieviant JA, Sauro HM. In-fusion BioBrick assembly and re-engineering. Nucleic Acids Res. 2010;38:2624–2636. doi: 10.1093/nar/gkq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 21.Kahl LJ, Endy D. A survey of enabling technologies in synthetic biology. J. Biol. Eng. 2013;7:13. doi: 10.1186/1754-1611-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quan J, Tian J. Circular polymerase extension cloning of complex gene libraries and pathways. PLoS One. 2009;4:e6441. doi: 10.1371/journal.pone.0006441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li MZ, Elledge SJ. SLIC: a method for sequence- and ligation-independent cloning. Methods Mol. Biol. 2012;852:51–59. doi: 10.1007/978-1-61779-564-0_5. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Werling U, Edelmann W. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 2012;40:e55. doi: 10.1093/nar/gkr1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 26.Shao Z, Zhao H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 2009;37:e16. doi: 10.1093/nar/gkn991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibson DG, Benders GA, Axelrod KC, Zaveri J, Algire MA, Moodie M, Montague MG, Venter JC, Smith HO, Hutchison CA., 3rd One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome. Proc. Natl Acad. Sci. USA. 2008;105:20404–20409. doi: 10.1073/pnas.0811011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larionov V, Kouprina N, Graves J, Chen XN, Korenberg JR, Resnick MA. Specific cloning of human DNA as yeast artificial chromosomes by transformation-associated recombination. Proc. Natl Acad. Sci. USA. 1996;93:491–496. doi: 10.1073/pnas.93.1.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One. 2008;3:e3647. doi: 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silva-Rocha R, Martinez-Garcia E, Calles B, Chavarria M, Arce-Rodriguez A, de Las Heras A, Paez-Espino AD, Durante-Rodriguez G, Kim J, Nikel PI, et al. The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 2013;41:D666–D675. doi: 10.1093/nar/gks1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen WH, Qin ZJ, Wang J, Zhao GP. The MASTER (methylation-assisted tailorable ends rational) ligation method for seamless DNA assembly. Nucleic Acids Res. 2013;41:e93. doi: 10.1093/nar/gkt122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hillson NJ, Rosengarten RD, Keasling JD. j5 DNA assembly design automation software. ACS Synth. Biol. 2012;1:14–21. doi: 10.1021/sb2000116. [DOI] [PubMed] [Google Scholar]
- 33.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gietz RD, Schiestl RH. Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007;2:35–37. doi: 10.1038/nprot.2007.14. [DOI] [PubMed] [Google Scholar]
- 35.Zaccolo M, Williams DM, Brown DM, Gherardi E. An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol. 1996;255:589–603. doi: 10.1006/jmbi.1996.0049. [DOI] [PubMed] [Google Scholar]
- 36.Liang JC, Chang AL, Kennedy AB, Smolke CD. A high-throughput, quantitative cell-based screen for efficient tailoring of RNA device activity. Nucleic Acids Res. 2012;40:e154. doi: 10.1093/nar/gks636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, Gustafsson C. Design parameters to control synthetic gene expression in Escherichia coli. PLoS One. 2009;4:e7002. doi: 10.1371/journal.pone.0007002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoover DM, Lubkowski J. DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 2002;30:e43. doi: 10.1093/nar/30.10.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953;21:1087–1092. [Google Scholar]
- 40.SantaLucia J., Jr A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl Acad. Sci. USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Q, Schlabach MR, Hannon GJ, Elledge SJ. Design of 240,000 orthogonal 25mer DNA barcode probes. Proc. Natl Acad. Sci. USA. 2009;106:2289–2294. doi: 10.1073/pnas.0812506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gibson DG. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 2011;498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hua SB, Qiu M, Chan E, Zhu L, Luo Y. Minimum length of sequence homology required for in vivo cloning by homologous recombination in yeast. Plasmid. 1997;38:91–96. doi: 10.1006/plas.1997.1305. [DOI] [PubMed] [Google Scholar]
- 45.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarrion-Perdigones A, Falconi EE, Zandalinas SI, Juarez P, Fernandez-del-Carmen A, Granell A, Orzaez D. GoldenBraid: an iterative cloning system for standardized assembly of reusable genetic modules. PLoS One. 2011;6:e21622. doi: 10.1371/journal.pone.0021622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc. Natl Acad. Sci. USA. 2005;102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ellis T, Wang X, Collins JJ. Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nat. Biotechnol. 2009;27:465–471. doi: 10.1038/nbt.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mutalik VK, Guimaraes JC, Cambray G, Mai QA, Christoffersen MJ, Martin L, Yu A, Lam C, Rodriguez C, Bennett G, et al. Quantitative estimation of activity and quality for collections of functional genetic elements. Nat. Methods. 2013;10:347–353. doi: 10.1038/nmeth.2403. [DOI] [PubMed] [Google Scholar]
- 50.Mutalik VK, Guimaraes JC, Cambray G, Lam C, Christoffersen MJ, Mai QA, Tran AB, Paull M, Keasling JD, Arkin AP, et al. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods. 2013;10:354–360. doi: 10.1038/nmeth.2404. [DOI] [PubMed] [Google Scholar]
- 51.Lou C, Stanton B, Chen YJ, Munsky B, Voigt CA. Ribozyme-based insulator parts buffer synthetic circuits from genetic context. Nat. Biotechnol. 2012;30:1137–1142. doi: 10.1038/nbt.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qi L, Haurwitz RE, Shao W, Doudna JA, Arkin AP. RNA processing enables predictable programming of gene expression. Nat. Biotechnol. 2012;30:1002–1006. doi: 10.1038/nbt.2355. [DOI] [PubMed] [Google Scholar]
- 53.Crook NC, Freeman ES, Alper HS. Re-engineering multicloning sites for function and convenience. Nucleic Acids Res. 2011;39:e92. doi: 10.1093/nar/gkr346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao AI, Fenton TA, Owsley K, Seitzer P, Larsen DJ, Sit H, Lau J, Nair A, Tantiongloc J, Tagkopoulos I, et al. Promoter element arising from the fusion of standard BioBrick parts. ACS Synth. Biol. 2013;2:111–120. doi: 10.1021/sb300114d. [DOI] [PubMed] [Google Scholar]
- 55.Davis JH, Rubin AJ, Sauer RT. Design, construction and characterization of a set of insulated bacterial promoters. Nucleic Acids Res. 2011;39:1131–1141. doi: 10.1093/nar/gkq810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang X, Yang J, Zhang H, Zou H, Wang C, Xian M. In vitro assembly of multiple DNA fragments using successive hybridization. PLoS One. 2012;7:e30267. doi: 10.1371/journal.pone.0030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nour-Eldin HH, Geu-Flores F, Halkier BA. USER cloning and USER fusion: the ideal cloning techniques for small and big laboratories. Methods Mol. Biol. 2010;643:185–200. doi: 10.1007/978-1-60761-723-5_13. [DOI] [PubMed] [Google Scholar]
- 58.Tsuge K, Matsui K, Itaya M. One step assembly of multiple DNA fragments with a designed order and orientation in Bacillus subtilis plasmid. Nucleic Acids Res. 2003;31:e133. doi: 10.1093/nar/gng133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garbarino JE, Gibbons IR. Expression and genomic analysis of midasin, a novel and highly conserved AAA protein distantly related to dynein. BMC Genomics. 2002;3:18. doi: 10.1186/1471-2164-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vicente M, Gomez MJ, Ayala JA. Regulation of transcription of cell division genes in the Escherichia coli dcw cluster. Cell. Mol. Life Sci. 1998;54:317–324. doi: 10.1007/s000180050158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lubliner S, Keren L, Segal E. Sequence features of yeast and human core promoters that are predictive of maximal promoter activity. Nucleic Acids Res. 2013;41:5569–5581. doi: 10.1093/nar/gkt256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi Z, Wedd AG, Gras SL. Parallel in vivo DNA assembly by recombination: experimental demonstration and theoretical approaches. PLoS One. 2013;8:e56854. doi: 10.1371/journal.pone.0056854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muyrers JP, Zhang Y, Stewart AF. Techniques: recombinogenic engineering–new options for cloning and manipulating DNA. Trends Biochem. Sci. 2001;26:325–331. doi: 10.1016/s0968-0004(00)01757-6. [DOI] [PubMed] [Google Scholar]
- 65.Sleight SC, Bartley BA, Lieviant JA, Sauro HM. Designing and engineering evolutionary robust genetic circuits. J. Biol. Eng. 2010;4:12. doi: 10.1186/1754-1611-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.