

Abstract
The active site of the Haloalkane Dehydrogenase (HaloTag) enzyme can be covalently attached to a chloroalkane ligand providing a mechanically strong tether, resistant to large pulling forces. Here we demonstrate the covalent tethering of protein L and I27 polyproteins between an AFM cantilever and a glass surface using HaloTag anchoring at one end, and thiol chemistry at the other end. Covalent tethering is unambiguously confirmed by the observation of full length polyprotein unfolding, combined with high detachment forces that range up to ~2000 pN. We use these covalently anchored polyproteins to study the remarkable mechanical properties of HaloTag proteins. We show that the force that triggers unfolding of the HaloTag protein exhibits a four-fold increase, from 131 pN to 491 pN, when the direction of the applied force is changed from the C-terminus to the N-terminus. Force-clamp experiments reveal that unfolding of the HaloTag protein is twice more sensitive to pulling force compared to protein L, and refolds at a slower rate. We show how these properties allow for the long-term observation of protein folding-unfolding cycles at high forces, without interference from the HaloTag tether.
Introduction
Specific covalent tethering of proteins to surfaces is of great importance in single molecule force spectroscopy techniques such as atomic force microscopy (AFM), where the attachment determines the direction and range of applicable forces. A typical AFM experiment consists of pressing a cantilever onto a thin layer of engineered polyproteins adsorbed on a surface 1–9. Under these conditions polyproteins adhere to the AFM tip and the surface by an unknown mechanism 1. Extension of a polyprotein generates a characteristic fingerprint, revealing the sequential unfolding of its domains 1. The polyproteins are randomly pulled from any two points along their length, resulting in a high variation of the number of unfolding domains from trace to trace 10. The uncertainty in the number of domains that are probed increases the difficulty in analyzing the data, requiring order statistics and controls that reduce the effectiveness of the technique 11,12. Moreover, this weak anchoring leads to a high probability of detachment, limiting the observation time and the range of forces that can be utilized 12. Hence, it is highly desirable to develop attachment chemistries that specifically tether single proteins between an AFM cantilever and a surface using covalent bonds. This attachment should allow a protein to be anchored at perfectly controlled points and to be mechanically pulled at high forces for extended periods of time.
To achieve such a goal we used thiol chemistry in combination with HaloTag, an engineered Haloalkane Dehalogenase capable of forming an ester bond with a chloroalkane-functionalized surface13. Attachment of proteins using HaloTag is becoming a popular approach to covalently anchor proteins to surfaces. For instance, fusion proteins with terminal HaloTag have been successfully used for purification of proteins with high yield and increased purity14,15 and for labeling live cells16–19. Furthermore, HaloTag has been successfully employed in single molecule force spectroscopy measurements to immobilize poly I27 polyprotein to mica surfaces20 and poly Filamin to beads through DNA spacers21.
In contrast to non-specific or antigen-antibody interactions, covalent anchoring has the advantage of high detachment forces and extended lifetime 22,23. The attachment strength can increase the range of forces to which a single molecule can be exposed and can also extend the possible tethering time under force. However, since little is known about the mechanical properties of HaloTag, its potential applications in single molecule experiments are limited. HaloTag is a protein that can denature under certain conditions. This feature poses both advantages and challenges. Contrary to other covalent attachment techniques24–27, HaloTag has the advantage of a mechanical fingerprint provided by its unfolding and extension up to its chloroalkane anchoring point. The response of HaloTag to denaturing forces must also be closely related to the direction of the pulling force and to the number of amino acids trapped within the protein fold. The main disadvantage of this attachment technique might come from possible interference of the HaloTag on the studied process. HaloTag might unfold at similar forces or might hinder the collapse and refolding of the protein under investigation.
Here we report several methods to attach proteins using HaloTag-chloroalkane anchoring and thiol chemistry and investigate the effects of HaloTag on the unfolding and refolding of two model proteins. To this end, we developed analytical tools to verify the specific HaloTag attachment to chloroalkane-functionalized surfaces. We study the unfolding mechanics of HaloTag pulled from different points using force-extension and force-clamp AFM. Based on these mechanical properties, we demonstrate the usage of HaloTag to investigate mechanical properties of other proteins by decoupling its unfolding and refolding from these proteins of interest. Below we also provide a roadmap for how to successfully employ covalent attachment of polyproteins though HaloTag anchoring and measure folding of HaloTag fused polyproteins.
Materials and Methods
Protein Expression and Purification
For our experiments we used several polyprotein constructs inserted in a pFN18A vector (Promega). Three polyproteins had the HaloTag at the N-end, followed by a specific polyprotein inserts (a construct with eight protein L domains – denoted Halo-L8-Cys, one with eight I27 C47/63A domains – denoted Halo-I278-Cys, and one with a GFP domain – denoted Halo-GFP-Cys), and terminated at the C-end with the (His)6 tag and one cysteine amino acid. One Halo-I278-Cys construct had a cysteine and the (His)6 tag at the N-end, followed by eight I27 domains and the HaloTag at the C-end. E.coli ERL cells transformed with the DNA vector were grown at 37 °C until OD600 ~ 0.6 in 400 mL LB broth in the presence of appropriate antibiotics. Polyprotein expression was induced with 1 mM IPTG at 25 °C overnight. Cells were harvested and resuspended in 50 mM sodium phosphate buffer pH 7.0, 300 mM NaCl, 10 % glycerol, 0.03 mM TCEP or 0.5 mM DTT, and lysed using a French press. The lysate was centrifuged at 30,000 g for 40 min and purified using Ni2+-NTA His GraviTrap affinity columns (GE Healthcare), by washing with the same resuspension buffer containing 7.5 mM imidazole and eluting with 250 mM imidazole (both buffers with 10% glycerol, 0.03 mM TCEP or 0.5 mM DTT). The proteins were further purified using a Superdex 200 fast protein liquid chromatography column (FPLC, Amersham Biosciences) with elution buffer (10 mM HEPES pH 7.2, 150 mM NaCl, 10% glycerol, 1 mM EDTA). Typical protein concentrations varied between 10 and 20 μM.
Preparation of Chloroalkane Surfaces
Clean glass and cantilevers were activated using oxygen-plasma (Harrick Palsma) and silanized with (3-aminopropyl)triethoxysilane (Sigma-Aldrich), as further described in the Supplementary Information. These amine-terminated surfaces were then reacted for 1 hour with 10 mM NHS-PEG-Maleimide Crosslinker (SM(PEG)24, Thermo Scientific) dissolved in 50 mM Borax buffer pH 8.5. After washing with double-distilled water, the surfaces were further reacted overnight with 7.5 mM Thiol-PEG4-Chloroalkane ligand (HaloTag Thiol O4 ligand, Promega) dissolved in 50 mM Borax buffer pH 8.5. The reaction was quenched with 50 mM 2-mercaptoethanol (Sigma-Aldrich).
Single Molecule AFM Experiments
The AFM instrument was custom-made and described elsewhere 28. For each condition 4–10 different experiments were performed. The solution containing the polyproteins was left in contact with the chloroalkane or gold surface for ~30 min. The surface was then washed with HEPES buffer (10 mM HEPES pH 7.2, 150 mM NaCl, 1 mM EDTA) to remove non-specifically bound protein, and the measurements were performed in the same buffer. During a typical experiment the surface was moved laterally over 500 nm2 to insure that several surface sites were probed. The cantilevers were calibrated using the thermal noise method based on the equipartition theorem. Standard silicon nitride and chloroalkane functionalized cantilevers had spring constants of 16 ± 1 pN/nm, and gold coated cantilevers had a slightly higher constant of 17 ± 2 pN/nm. Force-extension traces were acquired at a rate of 400 nm/s. Traces showing one HaloTag unfolding fingerprint were used to estimate the mechanical properties of the proteins and the strength of the attachment.
Results and discussion
While covalent anchoring is often assumed after a given protocol has been followed, distinguishing a specific from a non-specific attachment can be a challenging task. Furthermore, a clear fingerprint is often missing to indicate with certainty if the experimental data report on the protein or process under investigation. Here we implemented two approaches to probe the formation of covalent attachment between HaloTag and chloroalkane-functionalized surfaces. These approaches are needed to verify that the surface functionalization protocol has been successful and that the HaloTag terminated proteins are active. To this end, we constructed polyproteins that included a recognition site for Tobacco Etch Virus (TEV) protease (EDLYFQS). This site was inserted between HaloTag and the other protein domains. In a first approach, two drops containing HaloTag-GFP were adsorbed to a chloroalkane surface. TEV protease was added to one of the drops. As shown in Figure 1A, both drops showed strong fluorescence due to the presence of the GFP linked protein. Washing the surface removed unbound proteins. The region where the HaloTag-GFP was adsorbed alone displayed a strong fluorescence signal, while most of the GFP was cleaved from the surface and washed away in the region where TEV was added. Very little non-specific adsorption can be seen after a first wash, with the surface where TEV was absent being ~16 times brighter than the one where TEV was present. In a second experiment, we adsorbed a HaloTag-(I27)8 polyprotein construct to the chloroalkane surface. After extensive washing for 1–2 h, to remove all non-specifically adsorbed proteins, we incubated the surface with TEV protease and collected the released proteins. As seen in SDS-PAGE gels (Figure 1B), the collected protein from the surface wash showed a band at ~ 82 kDa, which corresponds to the (I27)8 alone. The HaloTag-(I27)8 control showed a molecular mass of ~117 kDa, while TEV migrated to ~ 27 kDa. The lane corresponding to the reaction in solution between HaloTag-(I27)8 and TEV showed the (I27)8 and TEV band, together with a third band at ~ 35 kDa, characteristic for the HaloTag. These two experiments prove that the attachment is highly specific and takes place at the HaloTag end of the polyprotein construct. These fluorescence and gel-based methods provide an easy assay to evaluate the success of the chloroalkane surface chemistry.
Figure 1. Verification of HaloTag anchoring to glass surfaces functionalized with chloroalkane.
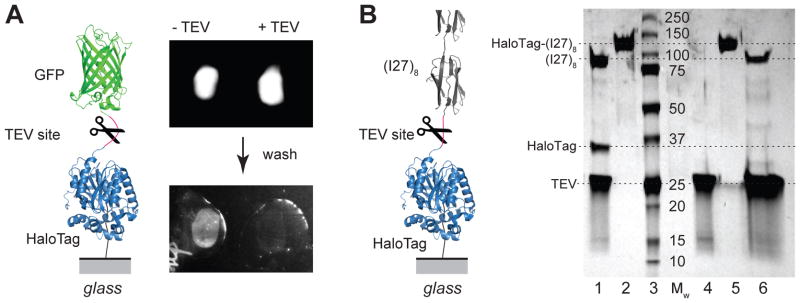
A) Fluorescence assay: two drops of HaloTag-GFP fusion protein solution deposited on a glass surface functionalized with the chloroalkane ligand in the presence and absence of TEV protease. Washing the glass surface removes unbound proteins, leaving a layer of surface anchored GFP (left) that was absent when TEV was added to the solution (right). The inset on the left shows a diagram of the HaloTag-GFP construct, the location of the TEV cleavage site, and the attachment point to the glass surface. B) SDS-PAGE gel assay: lane 1 – HaloTag-(I27)8 protein digested with TEV in solution shows (I27)8 at ~82 kDa, HaloTag at ~35 kDa and TEV at ~27 kDa; lanes 2 and 5: HaloTag-(I27)8 control ~117 kDa; lane 3 – molecular weight marker; lane 4: TEV control ~27 kDa; lane 6 – protein collected by washing four glass coverslides functionalized with chloroalkane, and reacted with HaloTag-(I27)8 polyprotein. The wash solution contained 2 mg/ml TEV protease. The wash contains (I27)8 at ~82 kDa and TEV at ~27 kDa and does not contain HaloTag protein which remains covalently anchored to the surface. The inset on the left shows a diagram of the HaloTag-(I27)8 experiment.
Having the certainty of the specific attachment of our HaloTag terminated proteins to the chloroalkane functionalized surfaces, we used AFM to investigate the mechanics of polyproteins containing eight repeats of either protein L or I27, terminated with a HaloTag protein at one end and a cysteine at the opposite end (Halo–L8–Cys and Halo–I278–Cys). In a first configuration, the Halo–L8–Cys polyprotein construct was attached covalently to the surface through the HaloTag–chloroalkane reaction (Figure 2A and Figure S1). A gold-coated cantilever tip was then pressed onto the protein layer for ~ 1 s. Retracting the tip with an attached molecule led to the sequential unfolding and extension of the component domains of the polyprotein. The unfolding events result in a saw-tooth pattern of the measured force, while the final detachment peak represents the failing of the weakest anchoring bond. Unlike other covalent attachments, HaloTag produces an unfolding fingerprint by partially unraveling under force. In most cases, the unfolding of HaloTag protein preceded that of the other proteins in the construct (Figure S3). Alignment of the force-extension curves with the final unfolding event shows a narrow contact point distribution with a spread of ~ 40 nm (Figure 2B). This spread in length is likely the result of different attachment points of the polyprotein to the tip of the cantilever, which has a radius of 20–60 nm (ref. 29). Hence, tethering of polyproteins to the ends of the construct showed a narrow variation, which can be attributed to the physical dimensions of the cantilever.
Figure 2. Mechanical fingerprint of covalently anchored polyproteins.
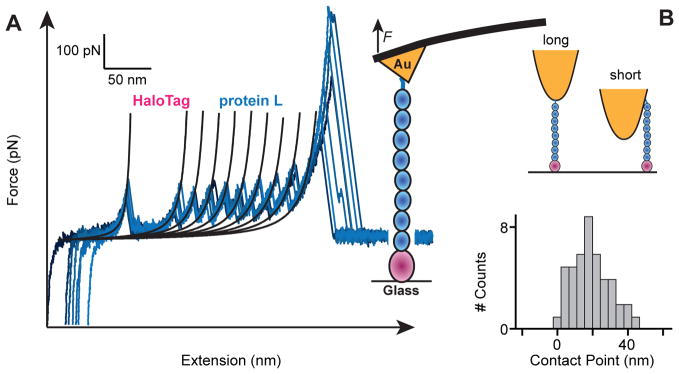
A) Several superimposed force-extension traces of single HaloTag–L8–Cys polyproteins. Unfolding of each chloroalkane-anchored HaloTag shows a characteristic increase in contour length of ~66 nm. In all cases we observed eight protein L unfolding events identified by contour length increases of ~19 nm and unfolding forces of ~140 pN. A high detachment force of 535 ± 245 pN is measured, indicating successful thiol-gold anchoring. The unfolding of the HaloTag can occur at any position along the sawtooth pattern (Figure S3). However, for the sake of simplicity, only curves where HaloTag unfolds first were included in this figure. B) Perfectly anchored polyproteins should all have exactly the same length when pulled vertically. However, given that thiol-gold anchoring can occur at random locations throughout the AFM tip, we observe a variation in length of up to ~40 nm, from the longest trace (marked as zero), in excellent agreement with the radius of curvature of AFM cantilevers.
Force leads to the unfolding of HaloTag protein, followed by the immediate extension of the previously trapped amino acids between its chloroalkane attachment point and the neighboring proteins. By changing the position of the HaloTag from the N-end of the polyprotein construct to the C-end, we are exposing different regions inside this protein fold to force (Figure 3 and Figure S4). When placed at the N-end of the construct, the part of the HaloTag between the chloroalkane site and the C-terminus of the protein was exposed to force. In this case, HaloTag displayed an unfolding fingerprint given by a contour length increase of 66 ± 2 nm (s.d., n = 436) and an unfolding force of 131 ± 31 pN (Figure 3). This change in contour length closely reflects the number of amino acids under force 20. Force-extension curves of Halo–I278–Cys with C-terminus of the HaloTag protein under force showed the unfolding of HaloTag, followed by eight I27 domains, which are mechanically more stable (Figure 3). Placing the HaloTag at the C-end of the construct exposed a smaller part of the protein to force, from its N-terminus to the chloroalkane site. In this case N-terminus HaloTag had a contour length increment of 26.5 ± 0.6 nm (s.d., n = 359) and an unfolding force of 491 ± 129 pN (Figure 3 and Figure S2). This increase in mechanical stability was obtained solely by exposing different parts of the HaloTag protein to force and can be exploited to investigate proteins with a wide range of mechanical stabilities. In the case of C-terminus HaloTag, force is applied between residues 106 and 296, while N-terminus HaloTag has the residues 1 to 106 exposed to force. We30 and others31,32 have shown that the mechanical stability of a protein strongly depends on the points where the force is applied. HaloTag protein showed an increase in mechanical stability from ~131 to ~491 pN when the direction of the applied force was changed from the C to the N-terminus.
Figure 3. The mechanical strength of the HaloTag-chloroalkane molecule depends on the direction of the applied force.

A) Cartoon diagrams of the force distribution throughout the structure of the HaloTag-chloroalkane molecule when force is applied through the C-terminus (top, red) or the N-terminus (bottom, red). B) Force extension traces of a single covalently anchored Halo-I278-Cys polyprotein, with the HaloTag placed at the N terminus of the polyprotein (top) or at the C terminus (bottom). C) An immobilized HaloTag protein unfolds at 131 ± 31 pN when force is applied to its C terminus, increasing to 491 ± 129 pN when force is applied through its N terminus. D) We observe unfolding contour length increases of 66 ± 2 nm when force is applied to its C terminus, decreasing to 26.5 ± 0.6 nm when force is applied through its N terminus.
Before unfolding, a mechanically stable protein extends under force up to its mechanical transition state 33,34. The measured contour length increment is given by the length of the polypeptide released by unfolding from the mechanical transition state structure to the fully extended chain. The transition state of most stable proteins involves parallel beta stands with hydrogen bonds perpendicular to the pulling direction 35,36. HaloTag is an alpha-beta protein that has at its core six parallel beta strands and the ligand-binding site at the end of the third parallel beta strand (amino acid number 106; Figure S4). An unfolding contour length increment of 26.5 ± 0.6 nm for N-terminus HaloTag suggests that a mechanically stable transition state forms roughly between beta strands B and C of the protein (Figure S4). Rupture of such a transition state would release amino acids 35 through 106 upon unfolding with a resulting contour length increase of ~27.1 nm, which is close to the measured value of ~27 nm (see Supplementary Information). Most proteins studied with force spectroscopy have a number of amino acids that “peel off” at low force, before the extending protein encounters its transition state structure at its beta sheet core. Similarly, in this case amino acids 1–35 do not contribute to the contour length increase of unfolding (Figure S4). In the case of C-terminus unfolding, the experimental change in contour length is ~66 nm. This value is consistent with a mechanically stable transition state clamp somewhere between beta strands C – F, which would release amino acids 106 through 270 upon unfolding, resulting in a contour length increase of ~64 nm, in close agreement with our experimental observations (see also Supplementary Information). The large difference in the mechanical stability of these two pulling geometries must arise from the particular transition state structures involved. Detailed molecular dynamics studies of the transition state structures of the HaloTag protein will be necessary before we fully understand these differences.
Attachment at the opposite end (from the HaloTag) of our polyprotein constructs was done using thiol chemistry, as detailed below. Covalent attachment is essential to obtain end-to-end tethering which can withstand high-pulling forces. The extension of covalently anchored polyproteins should manifest in several ways: the extended polyproteins should fully unfold (9 peaks in the present case), extend up to the same length, and detach with a high force. Our data closely fulfills these criteria. The majority of the traces containing the HaloTag unfolding fingerprint had a total of nine unfolding peaks with the expected contour length increments, marking specific covalent attachment to the termini (Figure 2–5 and Figure S5). These peaks include the unfolding of the HaloTag protein plus that of eight protein L or I27 domains. The contact point distribution was narrow and in the range of our cantilever radius (Figure 2B). Finally, we measured high detachment forces, different for different chemistries, as further discussed below.
Figure 5. Unfolding kinetics of protein L and HaloTag probed with force-clamp.

A) Unfolding at constant force of the Halo–L8–Cys construct results in stair-case like increase in the measured length of ~16 nm for protein L (blue part of the traces) and ~58 nm for HaloTag (magenta part of the traces). The lower black traces show the measured force under feed-back control. B) Normalized histograms of the unfolding order for the component proteins. HaloTag is the last protein to unfold at low forces (L8H) and the first to unfold (HL8) at high forces. C) Averaged (Protein L)8 (blue) and HaloTag (magenta) unfolding time courses obtained at different constant stretching forces. The black lines correspond to single exponential fits assuming a two-state model. D) Semi-logarithmic plot of the rate of unfolding of (Protein L)8 (blue circles) and HaloTag (magenta squares) as a function of force and measured from the same traces. The gray points represent the measured unfolding rate for protein L from a stand-alone construct, as reported by Liu et al44.
Figure 4 shows a summary of the results from similar experiments using alternative attachment strategies. We only included for further analysis traces that show unfolding of the C-terminus HaloTag protein, which is marked by a ~66 nm contour length increase, which unambiguously identifies covalently attached polyproteins to the chloroalkane functionalized surface 20. What we call specificity refers to the percentage of recordings showing the full end-to-end polyprotein unfolding fingerprint (nine unfolding events) out of the traces that showed HaloTag unfolding (Figure S1).
Figure 4. Histograms of detachment forces of covalently tethered Halo–L8–Cys polyproteins.

Several attachment configurations were investigated using Halo–L8–Cys polyprotein. A) Polyproteins attached to the surface through HaloTag – chloroalkane chemistry and pulled with silicon-nitride cantilevers show a detachment force distribution toward low forces (top) and a variable number of unfolding peaks (bottom). B) Same surface attachment as in in the previous configuration, but pulled using gold coated cantilevers, which are reactive toward the terminal cysteine. We observe a distribution of detachment forces of 535 ± 245 pN. In this configuration, 86% of the selected traces show nine unfolding peaks, characteristic of the specific attachment at the termini. C) Polyproteins attached to the surface through gold–thiol reaction and pulled through the HaloTag – chloroalkane chemistry. This arrangement has a detachment force of 1013 ± 347 pN and a specificity of 66%. D) Polyproteins attached to the surface through maleimide–thiol reaction and pulled using the HaloTag – chloroalkane chemistry. This arrangement shows the highest detachment force of 2001 ± 585 pN and the measured specificity is 86%. The inserts represent schematics of the considered attachment configurations. The black lines are Gaussian fits to the data.
Uncoated silicon-nitride cantilevers, which are standard in AFM force spectroscopy experiments, showed low detachment forces as well as low specificity of attachment (Figure 4A). Furthermore, the number of unfolding events had a large variation and sometimes exceeded nine peaks, probably due to the presence of dimers linked through a disulfide bond. Repeating the same experiment with cantilevers coated with freshly evaporated gold yielded a large increase in the specificity of attachment to the termini, varying from 78% for Halo–I278–Cys (Figure S5) to 86% for the Halo–L8–Cys (Figure 4B, lower panel). A histogram of the detachment forces reveals a broad distribution with a value of 535 ± 245 pN (s.d., n = 222) for the Halo–L8–Cys polyproteins and 533 ± 375 pN (n = 260) for the Halo–I278–Cys. Given the high specificity observed in this configuration, it is tempting to conclude that we are observing the actual detachment forces of a single thiol, bonded to the gold surface. While this detachment force seems low for a covalent bond, the detachment of a bonded thiol from a gold covered surface is a complex process. This process is now known to be dependent on the disposition of adsorbed atoms (adatoms) and vacancies, resulting in a high level of complexity with unknown effects on its mechanical properties 23,37. Furthermore, similarly low values were measured for gold-methylsulfide 38 and ferric-thiolate bonds 39. Interestingly, the inverse configuration of attachment – with the proteins adsorbing to a gold coated surface and picked-up by chloroalkane functionalized cantilever – yielded higher detachment forces of 1013 ± 347 pN (n = 195, Figure 4C), albeit at a lower specificity of 66%. It is possible that these differences of the measured detachment forces are the result of the different reaction times allowed for the gold-thiol reaction 40 and/or the different structures of the gold adatoms on the evaporated surfaces 37. Zhang and collaborators have calculated the breaking force of the gold-thiol junction before and after the hydrogen atom leaves the complex and predicted rupture forces of 600 ± 200 pN and 1500 ± 200 pN respectively 41. Furthermore, the strength of the S-Au bond was predicted to be higher than the Au-Au bond itself, leading to nanowire extrusion of Au atoms upon pulling 42. The force measured in this second configuration agrees with the value calculated for breaking the Au-Au bond, of ~1200 pN 42. Finally, polyproteins added to a maleimide-covered surface showed similar specificity and the highest detachment forces (Figure 4D). Thiols react with maleimide groups, forming stable thioether bonds. Of all the approaches examined, the maleimide-thiol chemistry led to the highest observed average detachment force of 2001 ± 585 pN (n = 108) and a similarly high specificity of 86%. However, while initially high, the success rate of the maleimide-thiol experiments rapidly decayed over time, yielding fewer recordings after ~ 1h of measurement. All three covalent-based attachments showed high specificity toward tethering the HaloTag-fused polyproteins to exactly their ends. The measured detachment force from ~530 to ~2000 pN suggests, at least for the low force detachment, that the bond at the thiol-end of the construct was being broken.
Our preferred method is to adsorb HaloTag containing polyproteins onto a chloroalkane coated glass surface and pull them with a gold-coated cantilever. This method results in a very high specificity of attachment (~86 %) obtained with a remarkably high pick-up frequency (see also Figure S1). In this configuration, detachment forces average at ~530 pN, sufficient to probe the majority of proteins studied to date 43. Furthermore, functionalization of glass slides is much simpler than that of cantilevers and all experiments are free of laser interference. Therefore, we chose this method to investigate the unfolding kinetics of Halo–L8–Cys polyproteins. In force-extension mode, the Halo–L8–Cys construct showed similar unfolding forces for protein L (~140 pN) and HaloTag (~130 pN) and a preferential order of unfolding of the HaloTag as the first protein to denature at the used pulling rate of 400 nm/s (Figure S3). In force-clamp mode, this polyprotein was exposed to a constant pulling force, and each unfolding domain displayed a step increase in the measured extension. The measured size of each step directly reflects the number of amino acids released when that single domain unfolds. As shown in Figure 5A, the measured extension showed steps of ~16 nm, characteristic to unfolding of protein L, and of ~ 58 nm, characteristic to denaturation of HaloTag. The pulling force was varied between 60 and 160 pN. The variations in the mechanical stabilities of protein L and HaloTag with force were different and crossed in the measured force range at 120 pN. At forces below 120 pN HaloTag was mechanically more stable than protein L. At forces above 120 pN this trend reversed (Figure 5A). This variation in force-dependency reflects on both the unfolding order and the unfolding rate of the measured proteins. Histograms of the unfolding order measured at different constant forces mark the 120 pN value as a crossing point where both HaloTag and protein L have equal probabilities to unfold (Figure 5B). Below 120 pN protein L unfolded first, while above this point HaloTag denatured first. Figure 5C shows the rates of unfolding of HaloTag and protein L, extracted from the same traces. The final extension of the averaged unfolding traces of eight protein L domains or a HaloTag increased with force, as predicted by the worm-like-chain model for polymer elasticity 44. The unfolding rate and the error of the measurement were evaluated assuming a single exponential behavior and applying bootstrapping 45. The measured unfolding rate of protein L as a function of force superimposes on the measured rate for the same protein alone (Figure 5D, blue and grey circles). The unfolding rates in the absence of force were estimated by assuming a simple two–state process: k(F) = k0 · exp(− FΔx/kBT), where k(F) is the measured rate at the applied force F, k0 is the rate in the absence of force, Δx is the distance to transition state, kB the Boltzmann’s constant, T the temperature. The extrapolated rates in the absence of force are 5.9·10−2 s−1 for protein L and 1.0·10−4 s−1 for HaloTag, while the measured distances to transition state were Δx = 1.6 Å for protein L and Δx = 3.6 Å for HaloTag. These two values of Δx were measured from two different proteins within the same polyprotein construct, where they were exposed to the same pulling force simultaneously, eliminating the possibility of errors arising from the calibration of the cantilevers 46,47. These results show that protein L and HaloTag respond differently to force and that choosing a certain force and exposure time can lead to the unfolding of only one of the two proteins.
We took advantage of the different force dependency of HaloTag and protein L to separate their folding. As seen in Figure 5D, at 80 pN protein L unfolded on a higher rate than HaloTag. When Halo–L8–Cys construct was exposed to 80 pN of force for 5 s, only protein L unfolded (Figure 6A). We then quenched the force to 0 pN for varying periods of time. When the force was ramped back up, we observed unfolding of protein L domains that refolded during the quench time, followed by the HaloTag protein – a signature for covalent attachment. For low quench times we also observed formation of stable collapsed states, a precursor of the folded state 48. Applying a procedure described elsewhere 49, we estimated the folding rate by comparing the number of folded domains as a function of quench time. For protein L, we found a folding rate of 1.4 ± 0.8 s−1 (Figure 6B). This experiment demonstrates how the different force-sensitivities of protein L and HaloTag can be used to effectively study the folding properties of mechanically weak proteins, such as protein L.
Figure 6. Measuring protein folding in HaloTag tethered proteins.

A) Force-quench traces showing first the unfolding of covalently tethered Halo–L8–Cys polyproteins at a constant force of 80 pN. All eight protein L domains unfold during the five second pulse to 80 pN. The force was then quenched to 0 pN for a variable time, Δt. We measured the refolded fraction by counting the number of protein L unfolding steps (N, 16 nm steps) measured during a force-ramp. B) Plot of the refolded fraction N/8 as a function of Δt. The solid line is an exponential fit that measures a folding rate of 1.4 ± 0.8 s−1. In all traces, the HaloTag protein does not unfold during the five-second pulse to 80 pN. The HaloTag protein in these traces unfolds last, at a higher force during the final force-ramp (red). This experimental protocol allows for the study of refolding, without interference of an unfolded HaloTag protein.
Exposing the Halo-I278-Cys with N-terminus HaloTag (placed at the C-end of the construct) to force-clamp conditions led to unfolding steps of 25 nm, specific for I27. These steps were followed by a final extension of ~22nm, specific for the denaturation of the HaloTag protein (Figure 7). In order to denature the N-terminus HaloTag, the force was ramped up either after a given dwell time or all the way from the beginning of the trace. This attachment geometry could allow measuring of more stable proteins such as I27, without interference from the HaloTag protein. Nevertheless, the high unfolding force of N-terminus HaloTag prevented us from seeing its unfolding fingerprint in all traces, which is a direct proxy for covalent attachment to the C-end of the construct (see also Figure S2).
Figure 7. N-terminus anchoring allows for high force unfolding of a polyprotein under force-clamp conditions, without HaloTag interference.

A) Force-clamp trace of unfolding at 200 pN, of eight mechanically stable I27 domains without interference from N terminus anchored HaloTag, which unfolds only when the forces rises to ~400 pN during the final ramp of this protocol. B) Similar experiment using a force ramp.
The proven high mechanical strength of HaloTag anchoring and gold-thiol covalent attachments increases the life-time of the tethers under force. We have developed a protocol for AFM force-clamp to continuously unfold and refold a single polyprotein chain. We managed to record several long traces and, most often, instrumental drift rather than detachment of the tethered molecule was the limiting temporal factor. For example, we exposed a single Halo–L8–Cys polyprotein to alternating pulling cycles of 100 pN for 2.5 s and pushing cycles for 4.5 s, which led to the unfolding and refolding of the proteins from the construct (Figure 8). In the chosen pull time, the same polyprotein displayed full or partial unfolding of the component protein domains. When the force was quenched, some protein L domains refolded. Interestingly, the measured fraction of folded domains of 0.6 ± 0.2 is close to the value measured from individual force-quench experiments (Figure 6B). This resemblance suggests that the folding of the HaloTag protein does not significantly interfere with the refolding of protein L domains. To the best of our knowledge, these traces are also the longest recorded for such high pulling forces. These long traces of the same polyprotein repeatedly undergoing a biophysical process or a chemical reaction under force offer unique prospects by expanding the possibilities of force spectroscopy to gather data not only from a large number of molecules, but also from a single molecule probed over a long period. This approach eliminates a source of heterogeneity given by sampling several molecules, which might reflect on different local environment, different attachment sites and pulling geometries 1, or different posttranslational modifications of proteins 50.
Figure 8. Thirteen minutes long folding-unfolding recording from a single covalently tethered polyprotein.

A) Force clamp trace of length as a function of time for 82 unfolding and refolding cycles of a single Halo–L8–Cys protein. The force pulse protocol consisted of repeated cycles of a 100 pN pulse to unfold the polyprotein (B, 2.5 s) followed by a negative force of −50 pN to contact the surface to verify and correct for drift, while allowing the protein to fold. B) Three unfolding traces, shown at an expanded time scale, corresponding to the regions marked by the dashed lines in (A). In some of the unfolding staircases we observed the unfolding of the HaloTag protein (magenta) and of protein L (blue), indicating that the proteins refold during the pulse protocol.
Conclusions
We have demonstrated covalent tethering of HaloTag-fused polyproteins, sensitive to the placement of the HaloTag protein in the construct and to used thiol chemistry. The chloroalkane surfaces to which HaloTag terminated proteins were attached were obtained after following a four-step protocol, prone to errors. We have implemented fluorescence and gel-based assays to verify the chloroalkane surfaces, which will become standard in experiments using HaloTag attachment. We have developed HaloTag-fused polyproteins, which provided an exclusive mechanical fingerprint embedded in their molecular design. This mechanical fingerprint was marked by the unfolding of the proteins in the engineered construct, with specific contour length change and unfolding force. Our experimental design revealed novel characteristics specific to single molecule measurements. The end-to-end attachment showed a variation of ~40 nm in the initial extension measured before the unfolding of the first protein in the construct. The physical dimensions of the cantilever and the geometry of attachment are likely responsible for this variation in initial extension, which is often used as a proxy for mechanically unstable proteins. Unlike other covalent tethering methods, attachment of HaloTag to chloroalkane ligand showed a specific mechanical fingerprint. This fingerprint consisted of the partial unfolding of the HaloTag up to its attachment point to the surface. This fingerprint was given by a contour length increment of ~ 66 nm at a force of ~131 pN when the C-terminus part of the HaloTag denatured under force and of ~27 nm and ~491 pN when N-terminus HaloTag unfolded. N-HaloTag unfolding is distinguished by its contour length increase of ~26 nm and high unfolding force. In the majority of the traces, we observed the N-HaloTag fingerprint, followed by detachment (e.g. Figure 3B). Detachment takes place after a given lifetime, and since it is probabilistic, it does not occur at a single, predetermined force. Hence detachment can take place at a force lower than that of the N-HaloTag (e.g. traces in Figure 3B). In about a third of the traces containing the full unfolding fingerprint of eight I27 domains, the molecule detached without showing N-HaloTag unfolding, suggesting the failure of the attachment before N-HaloTag had a chance to unfold. In these cases, N-HaloTag may have been much more stable than we were able to measure. Such events result in an underestimate of the average N-HaloTag unfolding force. Thus, the unfolding force of 500 pN or higher of N-terminus HaloTag is higher than most of the stable proteins studied to date 35. This extraordinary mechanical stability can be exploited to unfold and study other proteins under force without interference from the HaloTag.
We used gold-thiol and maleimide-thiol chemistry to attach the opposite end of the polyprotein construct. Breaking these tethers took place after unfolding the proteins in the constructs and at large separations between cantilever and surface, without interference coming from other short-ranged interfacial forces 23,27. Covalent tethering was characterized by a 66 to 86 % specificity of attachment to exactly the two ends of the polyprotein construct. Furthermore, depending on the thiol chemistry, the detachment forces averaged between 530 and 2000 pN, forces much higher than that measured for non-specific detachment. This variation in anchoring strength also implies that, at least in the low force case, the bond formed at the thiol-end of the construct was the one braking.
Kinetic measurements reporting on the unfolding of HaloTag and protein L were obtained using force-clamp AFM. Due to the different force sensitivities of Δx = 1.6 Å for protein L and Δx = 3.6 Å for HaloTag, the unfolding rates of these two proteins crossed at 120 pN. This difference in Δx allowed us to unfold only protein L from the Halo–L8–Cys construct and study its refolding kinetics. Mechanically weak proteins such as protein L could be studied at forces where C-terminus HaloTag unfolds slowly, while stronger proteins such as I27 required the use of the more stable N-terminus HaloTag. In this case, I27 was the first protein to unfold, but some traces lacked the N-terminus HaloTag fingerprint, which occurred at forces comparable to the detachment from the cantilever.
The reported covalent anchoring allowed tethering of proteins at high forces for long periods. We have exposed single HaloTag-fused polyproteins to repeated unfolding-refolding cycles and have found little interference from HaloTag in the folding behavior of protein L.
The methods described here will have a significant impact on single protein force-spectroscopy by opening new vistas in single molecule experiments, where a protein can be tethered covalently to exactly its ends at high forces and for extended times, and unfolded and refolded repeatedly under force. These recordings will allow to construct folding energy landscapes of proteins, to study dynamic disorder of folding, to probe intermediate folding states and to expose the same polyprotein to different interacting molecules and study their influence on the mechanical behavior of the substrate.
Supplementary Material
Acknowledgments
This work was funded by National Institutes of Health to J.M.F. (grants HL66030 and HL61228). I.P. was supported through a Swiss National Science Foundation fellowship. J.A.C. was supported by grant 1K99AI06072 from the National Institutes of Health. We acknowledge Pallav Kosuri for his help with analyzing the experiment from Figure S1 and David Giganti for his help with HaloTag topography (Figure S4). We thank all Fernandez group members for helpful discussions.
Footnotes
Supporting Information. Methods and additional results. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Carrion-Vazquez M, Oberhauser AF, Fisher TE, Marszalek PE, Li HB, Fernandez JM. Prog Biophys Mol Bio. 2000;74:63. doi: 10.1016/s0079-6107(00)00017-1. [DOI] [PubMed] [Google Scholar]
- 2.Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE. Science. 1997;276:1109. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 3.Clausen-Schaumann H, Seitz M, Krautbauer R, Gaub HE. Current Opinion in Chemical Biology. 2000;4:524. doi: 10.1016/s1367-5931(00)00126-5. [DOI] [PubMed] [Google Scholar]
- 4.Oesterhelt F, Oesterhelt D, Pfeiffer M, Engel A, Gaub HE, Muller DJ. Science. 2000;288:143. doi: 10.1126/science.288.5463.143. [DOI] [PubMed] [Google Scholar]
- 5.Sandal M, Valle F, Tessari I, Mammi S, Bergantino E, Musiani F, Brucale M, Bubacco L, Samori B. Plos Biology. 2008;6:99. doi: 10.1371/journal.pbio.0060006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benedetti F, Micheletti C, Bussi G, Sekatskii SK, Dietler G. Biophys J. 2011;101:1504. doi: 10.1016/j.bpj.2011.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller DJ, Dufrene YF. Nat Nanotechnol. 2008;3:261. doi: 10.1038/nnano.2008.100. [DOI] [PubMed] [Google Scholar]
- 8.Fisher TE, Oberhauser AF, Carrion-Vazquez M, Marszalek PE, Fernandez JM. Trends Biochem Sci. 1999;24:379. doi: 10.1016/s0968-0004(99)01453-x. [DOI] [PubMed] [Google Scholar]
- 9.Ikai A. Philosophical Transactions of the Royal Society B-Biological Sciences. 2008;363:2163. doi: 10.1098/rstb.2008.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li HB, Oberhauser AF, Fowler SB, Clarke J, Fernandez JM. P Natl Acad Sci USA. 2000;97:6527. doi: 10.1073/pnas.120048697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Manyes S, Brujic J, Badilla CL, Fernandez JM. Biophys J. 2007;93:2436. doi: 10.1529/biophysj.107.104422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brujic J, Hermans RIZ, Garcia-Manyes S, Walther KA, Fernandez JM. Biophys J. 2007;92:2896. doi: 10.1529/biophysj.106.099481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohane RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. Acs Chemical Biology. 2008;3:373. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 14.Motejadded H, Kranz B, Berensmeier S, Franzreb M, Altenbuchner J. Applied Biochemistry and Biotechnology. 2010;162:2098. doi: 10.1007/s12010-010-8985-1. [DOI] [PubMed] [Google Scholar]
- 15.Locatelli-Hoops S, Sheen FC, Zoubak L, Gawrisch K, Yeliseev AA. Protein Expression and Purification. 2013;89:62. doi: 10.1016/j.pep.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, So MK, Loening AM, Yao HQ, Gambhir SS, Rao JH. Angew Chem Int Edit. 2006;45:4936. doi: 10.1002/anie.200601197. [DOI] [PubMed] [Google Scholar]
- 17.Neklesa TK, Tae HS, Schneekloth AR, Stulberg MJ, Corson TW, Sundberg TB, Raina K, Holley SA, Crews CM. Nature Chemical Biology. 2011;7:538. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takemoto K, Matsuda T, McDougall M, Klaubert DH, Hasegawa A, Los GV, Wood KV, Miyawaki A, Nagai T. Acs Chemical Biology. 2011;6:401. doi: 10.1021/cb100431e. [DOI] [PubMed] [Google Scholar]
- 19.So MK, Yao HQ, Rao JH. Biochem Bioph Res Co. 2008;374:419. doi: 10.1016/j.bbrc.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taniguchi Y, Kawakami M. Langmuir. 2010;26:10433. doi: 10.1021/la101658a. [DOI] [PubMed] [Google Scholar]
- 21.Aubin-Tam ME, Olivares AO, Sauer RT, Baker TA, Lang MJ. Cell. 2011;145:257. doi: 10.1016/j.cell.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beyer MK. J Chem Phys. 2000;112:7307. [Google Scholar]
- 23.Beyer MK, Clausen-Schaumann H. Chem Rev. 2005;105:2921. doi: 10.1021/cr030697h. [DOI] [PubMed] [Google Scholar]
- 24.Wang T, Arakawa H, Ikai A. Biochem Bioph Res Co. 2001;285:9. doi: 10.1006/bbrc.2001.5118. [DOI] [PubMed] [Google Scholar]
- 25.Kufer SK, Dietz H, Albrecht C, Blank K, Kardinal A, Rief M, Gaub HE. Eur Biophys J Biophy. 2005;35:72. doi: 10.1007/s00249-005-0010-1. [DOI] [PubMed] [Google Scholar]
- 26.Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M. P Natl Acad Sci USA. 2012;109:E690. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grandbois M, Beyer M, Rief M, Clausen-Schaumann H, Gaub HE. Science. 1999;283:1727. doi: 10.1126/science.283.5408.1727. [DOI] [PubMed] [Google Scholar]
- 28.Popa I, Fernandez JM, Garcia-Manyes S. J Biol Chem. 2011;286:31072. doi: 10.1074/jbc.M111.264093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MLCTBruker. Bruker AFM Probes. http://www.brukerafmprobes.com.
- 30.Carrion-Vazquez M, Li HB, Lu H, Marszalek PE, Oberhauser AF, Fernandez JM. Nat Struct Biol. 2003;10:738. doi: 10.1038/nsb965. [DOI] [PubMed] [Google Scholar]
- 31.Mickler M, Dima RI, Dietz H, Hyeon C, Thirumalai D, Rief M. P Natl Acad Sci USA. 2007;104:20268. doi: 10.1073/pnas.0705458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brockwell DJ, Paci E, Zinober RC, Beddard GS, Olmsted PD, Smith DA, Perham RN, Radford SE. Nat Struct Biol. 2003;10:731. doi: 10.1038/nsb968. [DOI] [PubMed] [Google Scholar]
- 33.Marszalek PE, Lu H, Li H, Carrion-Vazquez M, Oberhauser AF, Schulten K, Fernandez JM. Nature. 1999;402:100. doi: 10.1038/47083. [DOI] [PubMed] [Google Scholar]
- 34.Ainavarapu RK, Brujic J, Huang HH, Wiita AP, Lu H, Li LW, Walther KA, Carrion-Vazquez M, Li HB, Fernandez JM. Biophys J. 2007;92:225. doi: 10.1529/biophysj.106.091561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sikora M, Sulkowska JI, Cieplak M. Plos Comput Biol. 2009:5. doi: 10.1371/journal.pcbi.1000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu H, Schulten K. Biophys J. 2000;79:51. doi: 10.1016/S0006-3495(00)76273-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cossaro A, Mazzarello R, Rousseau R, Casalis L, Verdini A, Kohlmeyer A, Floreano L, Scandolo S, Morgante A, Klein ML, Scoles G. Science. 2008;321:943. doi: 10.1126/science.1158532. [DOI] [PubMed] [Google Scholar]
- 38.Frei M, Aradhya SV, Hybertsen MS, Venkataraman L. J Am Chem Soc. 2012;134:4003. doi: 10.1021/ja211590d. [DOI] [PubMed] [Google Scholar]
- 39.Zheng P, Li HB. J Am Chem Soc. 2011;133:6791. doi: 10.1021/ja200715h. [DOI] [PubMed] [Google Scholar]
- 40.Matthiesen JE, Jose D, Sorensen CM, Klabunde KJ. J Am Chem Soc. 2012;134:9376. doi: 10.1021/ja302339d. [DOI] [PubMed] [Google Scholar]
- 41.Qi YH, Qin JY, Zhang GL, Zhang T. J Am Chem Soc. 2009;131:16418. doi: 10.1021/ja902573e. [DOI] [PubMed] [Google Scholar]
- 42.Kruger D, Fuchs H, Rousseau R, Marx D, Parrinello M. Phys Rev Lett. 2002:89. doi: 10.1103/PhysRevLett.89.186402. [DOI] [PubMed] [Google Scholar]
- 43.Sulkowska JI, Cieplak M. J Phys-Condens Mat. 2007:19. [Google Scholar]
- 44.Liu RC, Garcia-Manyes S, Sarkar A, Badilla CL, Fernandez JM. Biophys J. 2009;96:3810. doi: 10.1016/j.bpj.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiita AP, Perez-Jimenez R, Walther KA, Grater F, Berne BJ, Holmgren A, Sanchez-Ruiz JM, Fernandez JM. Nature. 2007;450:124. doi: 10.1038/nature06231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Manyes S, Kuo TL, Fernández JM. J Am Chem Soc. 2011;133:3104. doi: 10.1021/ja109865z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Popa I, Kosuri P, Alegre-Cebollada J, Garcia-Manyes S, Fernandez JM. Nat Protocols. doi: 10.1038/nprot.2013.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia-Manyes S, Dougan L, Badilla CL, Brujic J, Fernandez JM. P Natl Acad Sci USA. 2009;106:10534. doi: 10.1073/pnas.0901213106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kosuri P, Alegre-Cebollada J, Feng J, Kaplan A, InglÈs-Prieto A, Badilla CL, Stockwell BR, Sanchez-Ruiz JM, Holmgren A, Fernández JM. Cell. 2012;151:794. doi: 10.1016/j.cell.2012.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Min W, English BP, Luo GB, Cherayil BJ, Kou SC, Xie XS. Accounts of Chemical Research. 2005;38:923. doi: 10.1021/ar040133f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.