

Abstract
Leptin regulates energy homeostasis primarily by binding and activating its long form receptor (LRb). Deficiency of either leptin or LRb causes morbid obesity. Leptin stimulates LRb-associated JAK2, thus initiating multiple pathways including the Stat3 and phosphatidylinositol (PI) 3-kinase pathways that mediate leptin biological actions. Here we report that SH2-B, a JAK2-interacting protein, promotes activation of the PI 3-kinase pathway by recruiting insulin receptor substrate 1 (IRS1) and IRS2 in response to leptin. SH2-B directly bound, via its PH and SH2 domain, to both IRS1 and IRS2 both in vitro and in intact cells and mediated formation of a JAK2/SH2-B/IRS1 or IRS2 tertiary complex. Consequently, SH2-B dramatically enhanced leptin-stimulated tyrosine phosphorylation of IRS1 and IRS2 in HEK293 cells stably expressing LRb, thus promoting association of IRS1 and IRS2 with the p85 regulatory subunit of PI 3-kinase and phosphorylation and activation of Akt. SH2-B mutants with lower affinity for IRS1 and IRS2 exhibited reduced ability to promote association of JAK2 with IRS1, tyrosine phosphorylation of IRS1, and association of IRS1 with p85 in response to leptin. Moreover, deletion of the SH2-B gene impaired leptin-stimulated tyrosine phosphorylation of endogenous IRS1 in mouse embryonic fibroblasts (MEF), which was reversed by reintroduction of SH2-B. Similarly, SH2-B promoted growth hormone-stimulated tyrosine phosphorylation of IRS1 in both HEK293 and MEF cells. Our data suggest that SH2-B is a novel mediator of the PI 3-kinase pathway in response to leptin or other hormones and cytokines that activate JAK2.
Leptin is mainly produced and secreted from adipose tissues and regulates energy home ostasis primarily by activating its long isoform receptor (LRb)1 in the hypothalamic neurons (1). Deficiency of either leptin (ob/ob) or LRb (db/db) in mice causes morbid obesity, a primary risk factor for type 2 diabetes (2-6). LRb belongs to cytokine receptor family and binds to JAK2, a cytoplasmic tyrosine kinase that initiates and coordinates multiple pathways in response to leptin or other hormones and cytokines including growth hormone (GH), prolactin, erythropoietin, interferon-γ, and various interleukins. In response to leptin, JAK2 autophosphorylates as well as phosphorylates LRb at multiple tyrosines including Tyr985, Tyr1077, and Tyr1138 (7, 8). Phosphorylated Tyr985 binds to SHP2, promoting the mitogen-activated protein kinase pathway (7). Tyr985 also serves as an inhibitory site by binding to SOCS3 (8, 9). Phosphorylated Tyr1138 binds to Stat3, a cytoplasmic latent transcription factor, to allow JAK2 to phosphorylate and activate Stat3 (7, 10-12). Disruption of the Stat3 pathway causes severe leptin resistance and morbid obesity in mice, indicating that the JAK2/Stat3 pathway is required for leptin regulation of energy homeostasis (10-12).
Leptin stimulates tyrosine phosphorylation of both insulin receptor substrate 1 (IRS1) and IRS2 (13-18). Tyrosine phosphorylation of IRS proteins has been well characterized as an initial and rate-limiting step in the activation of the PI 3-kinase pathway in response to insulin and IGF-1 (19, 20). IRS1 and IRS2 bind directly to insulin receptor and are phosphorylated by insulin receptor at multiple YXXM motifs that bind specifically to the SH2 domain of the p85 regulatory subunit of PI 3-kinase (20). The interaction of p85 with IRS1 or IRS2 causes a conformational change, resulting in activation of the p110 catalytic subunit that is constitutively associated with p85 (21). PI 3-kinase phosphorylates the plasma membrane phospholipids that subsequently stimulate Akt by promoting phosphorylation at Thr308 and Ser473 (22-26). The IRS/PI 3-kinase/Akt pathway is required for insulin regulation of glucose homeostasis (27-29). Interestingly, leptin also stimulates the activation of the PI 3-kinase pathway in both hypothalamic neurons and peripheral target cells (15-18, 30-32). Inhibition of PI 3-kinase in the hypothalamus blocks leptin inhibition of food intake, whereas inhibition of PI 3-kinase in multiple peripheral tissues blocks various cellular responses to leptin (15, 16, 30-32). Moreover, deletion of IRS2 diminishes leptin-stimulated PI 3-kinase activity, resulting in leptin resistance and obesity (33, 34). These observations demonstrate that in addition to the JAK2/Stat3 pathway, the PI 3-kinase pathway is also required for leptin action in both the hypothalamus and peripheral tissues. IRS1 and IRS2 appear to mediate the PI 3-kinase pathway in response to leptin.
SH2-Bβ was originally identified as a JAK2-interacting protein (35). Alternative splicing of SH2-B mRNA generates at least four isoforms (α, β, γ, and δ) that differ in the C terminus following the SH2 domain (36). SH2-B binds directly via its SH2 domain to Tyr813 in JAK2 and enhances JAK2 autophosphorylation and activation in response to growth hormone (37-39). In addition to its SH2 domain, SH2-B contains multiple potential protein-protein interaction domain/motifs including a PH domain, multiple Pro-rich regions, and phosphorylation sites, suggesting that SH2-B may also act as an adaptor to recruit downstream signaling molecules as substrates for JAK2. In this work, we demonstrated that SH2-B binds simultaneously to both JAK2 and IRS proteins, thus promoting formation of a JAK2/SH2-B/IRS1 or IRS2 tertiary complex and subsequent tyrosine phosphorylation of IRS1 and IRS2 by JAK2. Our results suggest that SH2-B may mediate the IRS/PI 3-kinase/Akt pathway stimulated by leptin and multiple other hormones and cytokines that activate JAK2.
EXPERIMENTAL PROCEDURES
Reagents
Mouse leptin, porcine growth hormone, aprotinin, and leupeptin were purchased from Sigma. LipofectAMINE™ 2000 was purchased from Invitrogen. [125I]Leptin (human) was from PerkinElmer Life Sciences. Nonidet P-40 was purchased from Calbiochem (La Jolla, CA). Monoclonal anti-phosphotyrosine antibody (PY20) was purchased from Upstate Biotechnology, Inc. (Lake Placid, NY). Polyclonal anti-phospho-Akt (Thr308), anti-phospho-Stat3, and anti-phospho-Stat5b were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Monoclonal anti-Myc were purchased from Santa Cruz Inc. (Santa Cruz, CA). Polyclonal anti-SH2-B antibodies was raised against GST-SH2-B. Polyclonal anti-IRS1 and IRS2 antibodies were described previously (40). Protein A-agarose was purchased from Repligen (Waltham, MA).
Preparation of HEK293LRb
HEK293 cells were stably transfected with pcDNA3 expression plasmids encoding mouse LRb. G418-resistant clones were isolated and subjected to [125I]leptin binding assays as described previously (41). A stable clone (C6), designated HEK293LRb, was selected for the following experiments based on high [125I]leptin binding activity: 14069 ± 336 cpm/well in HEK293LRb versus 5791 ± 1399 cpm/well in untransfected parental HEK293 cells. Unlabeled cold leptin (2 μg/ml) reduced [125I]leptin binding to basal levels in HEK293LRb (4601 ± 44 cpm/well).
Cell Culture and Transfection
HEK293 cells were grown at 37 °C in 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 25 mm glucose, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% newborn calf serum. FAO cells were grown at 37 °C in 5% CO2 in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin.
HEK293 or HEK293LRb cells were split at 2 × 105 cells/well in a 6-well culture dish 24 h before transfection and transfected with indicated plasmids using LipofectAMINE™ 2000 reagents according to the manufacturer’s instructions. The cells were deprived of serum overnight 24 h after transfection and then treated with 100 ng/ml mouse leptin or 8 × 10−3 IU/ml GH for 10 min. The cell extracts were prepared and subjected to immunoprecipitation and immunoblotting.
Mouse embryo fibroblasts (MEF) were prepared and immortalized as described previously (42) and grown in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum, 25 mm glucose, 100 units/ml penicillin, and 100 μg/ml streptomycin. LRb was introduced into MEF cells using retrovirus-mediated gene transfer.
Immunoprecipitation and Immunoblotting
The cells were deprived of serum overnight in Dulbecco’s modified Eagle’s medium containing 0.5% bovine serum albumin and treated with leptin or GH at 37 °C. The cells were rinsed two times with ice-cold phosphate-buffered saline, solubilized in lysis buffer (50 mm Tris, pH 7.5, 1% Nonidet P-40, 150 mm NaCl, 2 mm EGTA, 1 mm Na3VO4, 100 mm NaF, 10 mm Na4P2O7, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin), and centrifuged at 14,000 × g for 10 min at 4 °C. The supernatant (cell extracts) was incubated with the indicated antibody on ice for 2 h. The immune complexes were collected on protein A-agarose during a 1-h incubation at 4 °C. The beads were washed three times with washing buffer (50 mm Tris, pH 7.5, 1% Nonidet P-40, 150 mm NaCl, 2 mm EGTA) and boiled for 5 min in SDS-PAGE sample buffer (50 mm Tris-HCl, pH 6.8, 2% SDS, 2% β-mercaptoethanol, 10% glycerol, 0.005% bromphenol blue). The solubilized proteins were separated by SDS-PAGE, transferred to nitrocellulose membrane (Amersham Biosciences), and detected by immunoblotting with the indicated antibody using ECL or Odyssey. Some membranes were subsequently incubated at 55 °C for 30 min in stripping buffer (100 mm β-mercaptoethanol, 2% SDS, 62.5 mm Tris-HCl, pH 6.7) to prepare them for a second round of immunoblotting.
RESULTS
SH2-B Binds Directly to IRS1 and IRS2 via Both Its PH and SH2 Domains
To determine whether SH2-B interacts with IRS proteins, SH2-Bβ was transiently coexpressed in HEK293 cells with either IRS1 or IRS2, and association of SH2-B with IRS1 or IRS2 was examined by coimmunoprecipitation assays. The cell extracts were immunoprecipitated with anti-SH2-B antibodies (αSH2-B) and immunoblotted with αIRS1 or αIRS2, respectively. SH2-Bβ was coimmunoprecipitated with both IRS1 and IRS2 (Fig. 1, A and B). Similarly, the cell extracts were immunoprecipitated with αIRS1 or αIRS2, respectively, and immunoblotted with αSH2-B. Both IRS1 and IRS2 were also coimmunoprecipitated with SH2-Bβ (Fig. 1, A and B).
Fig. 1. SH2-B directly binds to IRS1 and IRS2.
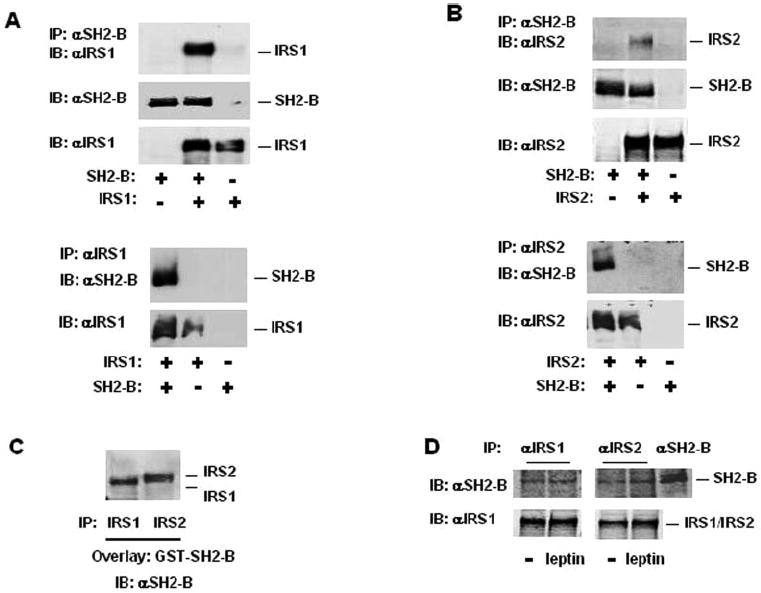
A, HEK293 cells were transiently cotransfected with expression plasmids encoding IRS1 (0.8 μg) and SH2-Bβ (1 μg). The cell extracts were prepared 48 h after transfection, immunoprecipitated (IP) with anti-SH2-B (αSH2-B), and immunoblotted (IB) with αIRS1. The same blot was reprobed with αSH2-B. The cell extracts were also immunoblotted with αIRS1. In a parallel experiment, the cell extracts were immunoprecipitated with αIRS1 and immunoblotted with αSH2-B. The same blot was reprobed with αIRS1. B, HEK293 cells were transiently cotransfected with expression plasmids encoding IRS2 (0.8 μg) and SH2-Bβ (1 μg). The cell extracts were prepared 48 h after transfection, immunoprecipitated αSH2-B, and immunoblotted with αIRS2. The same blot was reprobed with αSH2-B. The cell extracts were also immunoblotted with αIRS2. In a parallel experiment, the cell extracts were immunoprecipitated with αIRS2 and immunoblotted with αSH2-B. The same blot was reprobed with αIRS2. C, HEK293 cells were transiently transfected with expression plasmids encoding IRS1 (1 μg) or IRS2 (1 μg). The cell extracts were prepared 48 h after transfection and immunoprecipitated with αIRS1 or αIRS2, respectively. Immunopurified IRS1 and IRS2 were resolved by SDS-PAGE and transferred to nitrocellulose membrane. The blot was incubated with GST-SH2-B, and subsequently immunoblotted with αSH2-B. D, FAO cells were treated with 100 ng/ml leptin for 10 min. The cell extracts were immunoprecipitated with αIRS1, αIRS2, or αSH2-B, respectively, and immunoblotted with αSH2-B. The same blots were reprobed with αIRS1 or αIRS2 as indicated.
To determine whether SH2-B binds directly to IRS1 and IRS2, immunopurified IRS1 and IRS2 were resolved by SDS-PAGE and transferred to a nitrocellulose membrane. IRS1 and IRS2 immobilized on the nitrocellulose membrane were incubated with GST-SH2-B to allow GST-SH2-B binding to targets and visualized by immunoblotting with αSH2-B (Far Western analysis). SH2-B bound directly to both IRS1 and IRS2 (Fig. 1C).
To determine the interaction of endogenous SH2-B with endogenous IRS1 and IRS2, FAO cells (derived from rat liver) were treated with leptin, and the proteins were immunoprecipitated with αIRS1 or αIRS2 and immunoblotted with αSH2-B. Both endogenous IRS1 and IRS2 bound constitutively to endogenous SH2-B (Fig. 1D). FAO cells express endogenous leptin receptor at a very low level.
SH2-B contains multiple protein-protein interaction domains including a PH domain and a SH2 domain. To determine whether the SH2 domain of SH2-B is involved in its interaction with IRS proteins, the essential Arg555 within the SH2 domain of SH2-Bβ was replaced with Glu (SH2-B(R555E)). IRS1 was transiently coexpressed with SH2-Bβ or SH2-B(R555E) and immunoprecipitated with αIRS1. Coimmunoprecipitated proteins were immunoblotted with αSH2-B. IRS1 bound to SH2-Bβ as expected, whereas its interaction with SH2-B(R555E) was significantly reduced (Fig. 2A). Both SH2-B and SH2-B(R555E) were expressed at a similar level (data not shown). Similarly, disruption of the SH2 domain of SH2-Bβ also reduced its ability to bind to IRS2 (data not shown). These observations suggest that the SH2 domain is required for a full interaction of SH2-B with IRS1 or IRS2; however, other sites also contribute to the interaction.
Fig. 2. SH2-B binds to IRS1 and IRS2 via multiple sites.
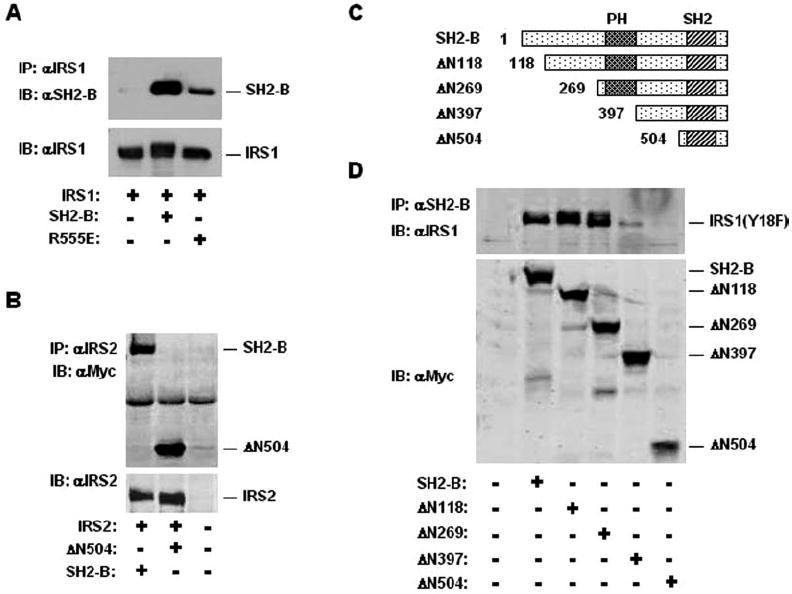
A, HEK293 cells were transiently cotransfected with expression plasmids encoding IRS1 (1 μg) and SH2-Bβ (0.8 μg) or SH2-B(R555E) (0.8 μg). The cell extracts were prepared 48 h after transfection, immunoprecipitated (IP) with αIRS1, and immunoblotted (IB) with αSH2-B. The same blot was reprobed with αIRS1. B, HEK293 cells were transiently cotransfected with expression plasmids encoding IRS2 (0.7 μg) and Myc-tagged ΔN504 (1.2 μg). The cell extracts were prepared 48 h after transfection, immunoprecipitated with αIRS2, and immunoblotted with αMyc. The same blot was reprobed with αIRS2. C, schematic representation of full-length and N-terminal truncated SH2-Bβ. D, HEK293 cells were transiently cotransfected with expression plasmids encoding IRS1(Y18F) (1 μg) and Myc-tagged full-length or truncated SH2-Bβ (1 μg). The cell extracts were prepared 48 h after transfection, immunoprecipitated with αSH2-B, and immunoblotted with αIRS1. The cell extracts were immunoblotted with αMyc to estimate the expression of full-length or various N-terminally truncated SH2-Bβ.
To determine whether the SH2 domain is sufficient to bind to IRS proteins, an SH2-Bβ mutant (ΔN504) was generated by deleting the N-terminal 1–504 amino acids. ΔN504, which contains the entire SH2 domain plus additional C-terminal 44 amino acids, was transiently coexpressed with either IRS1 or IRS2. ΔN504 coimmunoprecipitated with both IRS1 (data not shown) and IRS2 (Fig. 2B), whereas deletion of the C-terminal 44 amino acids alone did not affect interaction of SH2-Bβ with IRS1 or IRS2 (data not shown). Moreover, both IRS1 and IRS2 were tyrosine-phosphorylated in these experimental conditions (data not shown). Replacements of potential phosphorylation sites of 18 tyrosines with Phe in IRS1(Y18F) abolished interaction of ΔN504 with IRS1(Y18F) (Fig. 2D). These results suggest that the SH2 domain of SH2-B may bind directly to phosphorylated tyrosines in IRS proteins.
To identify other regions involved in the interaction, SH2-Bβ was truncated progressively at its N terminus (Fig. 2C). IRS1 or IRS1(Y18F) were transiently coexpressed individually with each mutant, and its interaction with Myc-tagged SH2-B mutants was examined by coimmunoprecipitation assays. Wild type IRS1 interacted with all mutants as expected (data not shown), because each mutant contains an intact SH2 domain (Fig. 2C). IRS1(Y18F) bound similarly to SH2-B, ΔN118, and ΔN269, indicating that the N-terminal 1–269 amino acids are dispensable for SH2-B interaction with IRS1 (Fig. 2D). In contrast, the deletion of additional 128 amino acids (ΔN397) dramatically impaired the interaction (Fig. 2D). The region of amino acids 269–397 contains the entire PH domain (Fig. 2C); therefore, the PH domain may mediate SH2-B binding to non-tyrosine-phosphorylated IRS proteins. ΔN504 bound to wild type IRS1 and IRS2 as described above but not to IRS1(Y18F) (Fig. 2D). These data suggest that the PH and SH2 domain of SH2-B may bind IRS1 or IRS2 independently, and a full interaction may require both domains.
SH2-B Promotes Formation of a JAK2/SH2-B/IRS1 or IRS2 Tertiary Complex
We have shown previously that SH2-B binds directly to JAK2 via multiple sites (35, 39). Because it binds to both JAK2 and IRS1 or IRS2, SH2-B may mediate association of JAK2 with IRS1 or IRS2, thus promoting tyrosine phosphorylation of IRS1 and IRS2 by JAK2. To determine formation of a JAK2/SH2-B/IRS1 tertiary complex, JAK2 was transiently coexpressed with IRS1 in the presence or absence of coexpression of SH2-Bβ, and its association with IRS1 was examined by coimmunoprecipitation assays. JAK2 association with IRS1 was barely detectable in the absence of SH2-Bβ, whereas SH2-B dramatically increased the association (Fig. 3A). SH2-Bβ was coimmunoprecipitated with both JAK2 and IRS1 simultaneously as predicted (Fig. 3A).
Fig. 3. SH2-B mediates a JAK2/SH2-B/IRS1 or IRS2 tertiary complex.
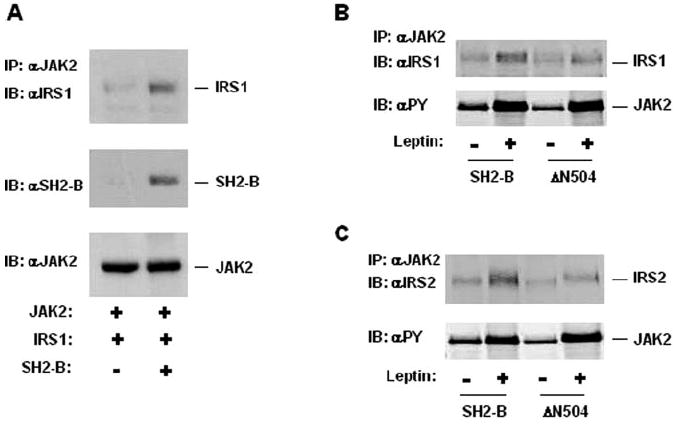
A, HEK293 cells were transiently cotransfected with expression plasmids encoding IRS1 (1 μg), JAK2 (0.8 μg), and SH2-Bβ (0.6 μg) as indicated. The cell extracts were prepared 48 h after transfection, immunoprecipitated (IP) with αJAK2, and immunoblotted (IB) sequentially with αIRS1, αSH2-B, and αJAK2. B, HEK293LRb cells were transiently cotransfected with expression plasmids encoding IRS1 (1.2 μg), JAK2 (0.6 μg), and SH2-Bβ or ΔN504 (0.6 μg) as indicated. The cells were treated with 100 ng/ml leptin for 10 min. The cell extracts were immunoprecipitated with αJAK2 and immunoblotted with αIRS1. The same blot was reprobed with anti-phosphotyrosine (αPY). C, HEK293LRb cells were transiently cotransfected with expression plasmids encoding IRS2 (1.2 μg), JAK2 (0.6 μg), and SH2-Bβ or ΔN504 (0.6 μg) as indicated. The cells were treated with 100 ng/ml leptin for 10 min. The cell extracts were immunoprecipitated with αJAK2 and immunoblotted with αIRS2. The same blot was reprobed with anti-phosphotyrosine.
Leptin binds and activates LRb, stimulating autophosphorylation and activation of LRb-bound JAK2. Because the SH2 domain is the primary binding site of SH2-B for tyrosine-phosphorylated JAK2, leptin is predicted to stimulate interaction of SH2-B with JAK2, thus promoting association of JAK2 with SH2-B-bound IRS proteins. To test this hypothesis, SH2-B and JAK2 were transiently coexpressed with IRS1 or IRS2 in HEK293LRb cells stably expressing LRb. The cells were treated with 100 ng/ml mouse leptin, and the cell extracts were immunoprecipitated with αJAK2 and immunoblotted with αIRS1 or IRS2, respectively. Leptin promoted association of JAK2 with both IRS1 and IRS2 (Fig. 3, B and C). Leptin stimulated tyrosine phosphorylation of JAK2 as expected (Fig. 3, B and C). In the absence of SH2-B, association of JAK2 with IRS1 or IRS2 was barely detectable (Fig. 3A and data not shown).
Because the PH domain of SH2-B binds IRS1 or IRS2, deletion of the PH domain is predicted to impair ability of the SH2-B mutant ΔN504 to mediate association of JAK2 with IRS1 or IRS2. To test this possibility, IRS1 and JAK2 were transiently coexpressed with ΔN504 in HEK293LRb cells. The ability of ΔN504 to promote coimmunoprecipitation of JAK2 with IRS1 or IRS2 was significantly reduced, although both SH2-Bβ and ΔN504 enhanced leptin-stimulated tyrosine phosphorylation of JAK2 to a similar extent (Fig. 3, B and C). The residual association of JAK2 with IRS1 or IRS2 in ΔN504-expressing cells might be mediated by endogenous SH2-B. Alternatively, the SH2 domain of ΔN504 may bind to both JAK2 and IRS proteins simultaneously, although with reduced affinity.
SH2-B Mediates Tyrosine Phosphorylation of IRS1 and IRS2 in Response to Leptin
To determine whether SH2-B mediates tyrosine phosphorylation of IRS1 or IRS2 by JAK2 in response to leptin, IRS1 or IRS2 was transiently coexpressed with Myc-tagged SH2-Bβ, SH2-B(R555E), or ΔN504 in HEK293LRb cells. The cells were treated with 100 ng/ml leptin for 10 min, and the cell extracts were immunoblotted with anti-phosphotyrosine antibodies. Leptin slightly stimulated tyrosine phosphorylation of IRS1 and IRS2 in the absence of SH2-Bβ, whereas SH2-Bβ dramatically enhanced tyrosine phosphorylation of both proteins (Fig. 4, A and B). In contrast, SH2-B(R555E) failed to promote IRS1 tyrosine phosphorylation. ΔN504 promoted leptin-stimulated tyrosine phosphorylation of IRS1 to a lesser extent (Fig. 4A), although both SH2-Bβ and ΔN504 stimulated tyrosine phosphorylation and activation of JAK2 to a similar extent (Fig. 3, B and C) (38, 39). SH2-B also enhanced basal levels of IRS1 tyrosine phosphorylation (Fig. 4A). These results suggest that both the PH and SH2 domains of SH2-B are involved in mediating tyrosine phosphorylation of IRS1 and IRS2 by JAK2 in response to leptin, consistent with involvement of both domains in interaction of SH2-B with IRS1 or IRS2.
Fig. 4. SH2-B mediates leptin-stimulated tyrosine phosphorylation of IRS1 and IRS2.
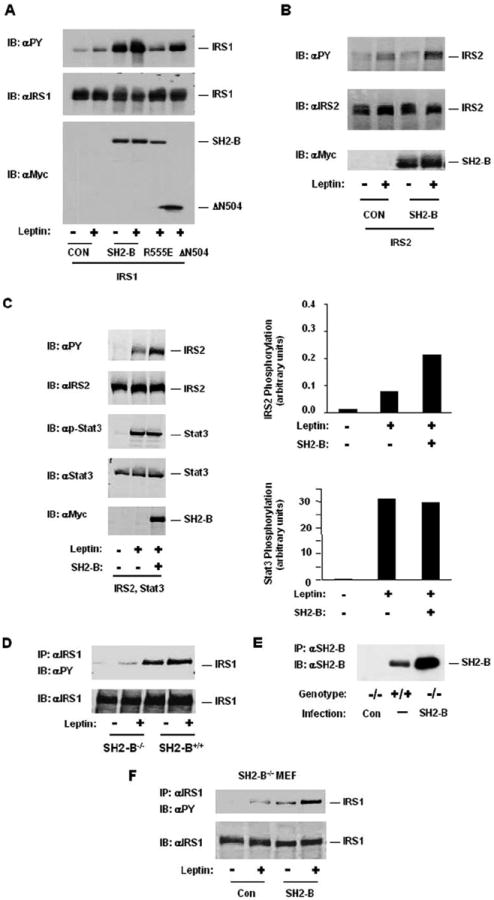
A, HEK293LRb cells were transiently cotransfected with expression plasmids encoding IRS1 (1 μg) and SH2-Bβ, SH2-B(R555E), or ΔN504 (0.8 μg) as indicated. The cells were treated with 100 ng/ml leptin for 10 min. The cell extracts were immunoblotted (IB) with anti-phosphotyrosine. The positions of IRS1, SH2-Bβ, SH2-B(R555E), and ΔN504 were marked. CON, control. B, HEK293LRb cells were transiently cotransfected with expression plasmids encoding IRS2 (1 μg) and SH2-Bβ (0.8 μg). The cells were treated with 100 ng/ml leptin for 10 min. The cell extracts were immunoblotted with αPY. The positions of IRS2 and SH2-Bβ were marked. C, HEK293LRb cells were transiently cotransfected with expression plasmids encoding IRS2 (0.8 μg), Stat3 (0.4 μg), and Myc-tagged SH2-Bβ (0.3 μg) as indicated. The cells were treated with 200 ng/ml leptin for 10 min. The cell extracts were immunoblotted with anti-phosphotyrosine (αPY), anti-IRS2, anti-phospho-Stat3, anti-Stat3, and anti-Myc as indicated. The positions of IRS2, Stat3, and SH2-Bβ were marked. The phosphorylation of IRS2 and Stat3 was quantitated and normalized to total IRS2 or Stat3, respectively. D, SH2-B−/−/LRb and SH2-B+/+/LRb MEFs stably expressing LRb were treated with 100 ng/ml leptin for 10 min. The cell extracts were immunoprecipitated (IP) with αIRS1 and immunoblotted with anti-phosphotyrosine. The same blot was reprobed with αIRS1. E, SH2-B−/−/LRb MEFs were infected with control (Con) or SH2-Bβ retroviruses, and stable clones were selected. The cell extracts were prepared from SH2-B+/+/LRb and SH2-B−/−/LRb MEFs infected with control or SH2-Bβ retroviruses, immunoprecipitated with αSH2-B, and immunoblotted with αSH2-B. F, control or SH2-Bβ retroviruses-infected SH2-B−/−/LRb MEFs were treated with 100 ng/ml leptin for 10 min. The cell extracts were immunoprecipitated with αIRS1 and immunoblotted with αPY. The same blot was reprobed with αIRS1.
SH2-B could mediate leptin-stimulated tyrosine phosphorylation of IRS1 and IRS2 by two distinct mechanisms. First, SH2-B potentiates JAK2 activation (38), thus globally enhancing tyrosine phosphorylation of all substrates of JAK2 including IRS1, IRS2, and transcription factor Stat3. Second, SH2-B specifically promotes tyrosine phosphorylation of IRS1 and IRS2 by mediating interaction of JAK2 with IRS1 or IRS2. In supporting the second mechanism, ΔN504, which lacks one site for IRS1, exhibited a reduced ability to promote leptin-stimulated tyrosine phosphorylation of IRS1 (Fig. 4A), although both ΔN504 and SH2-Bβ stimulate JAK2 activation and autophosphorylation similarly (38, 39). To provide additional evidence for the second mechanism, both IRS2 and Stat3 were transiently coexpressed with SH2-Bβ in HEK293LRb cells. The cells were treated with 200 ng/ml leptin for 10 min, and phosphorylation of IRS1 and Stat3 were measured by immunoblotting with anti-phosphotyrosine or anti-phospho-Stat3 antibodies, respectively. Leptin stimulated phosphorylation of both IRS2 and Stat3 (Fig. 4C). SH2-B enhanced tyrosine phosphorylation of IRS2 by approximate ~3-fold but did not enhance Stat3 phosphorylation in response to leptin (Fig. 4C). These results suggest that recruiting IRS1 and IRS2 to JAK2 might be the primary mechanism by which SH2-B mediates leptin-stimulated tyrosine phosphorylation of IRS1 and IRS2.
To determine whether SH2-B is required for leptin-stimulated tyrosine phosphorylation of IRS proteins, LRb were stably expressed in wild type or SH2-B−/− knockout MEFs. The levels of the plasma membrane LRb were similar between SH2-B+/+ and SH2-B−/− MEF cells based on the [125I]leptin binding analysis (data not shown). The cells were treated with 100 ng/ml leptin for 10 min, and immunopurified IRS1 were immunoblotted with anti-phosphotyrosine antibodies. Tyrosine phosphorylation of IRS1 was easily detected and slightly stimulated by leptin in SH2-B+/+ MEF cells (Fig. 4D). In contrast, IRS1 tyrosine phosphorylation was dramatically reduced in SH2-B−/− cells (Fig. 4D). To confirm the role of SH2-B, SH2-Bβ was reintroduced into SH2-B−/− MEF cells (Fig. 4E). Leptin slightly stimulated tyrosine phosphorylation of IRS1 in control SH2-B−/− cells, presumably mediated by other members of SH2-B family such as APS. Restoration of SH2-B dramatically increased basal as well as leptin-stimulated tyrosine phosphorylation of IRS1 (Fig. 4F). These data suggest that endogenous SH2-B mediates leptin-stimulated tyrosine phosphorylation of endogenous IRS proteins.
SH2-B Promotes Leptin-stimulated Activation of the IRS/PI 3-Kinase/Akt Pathway
Tyrosine phosphorylation of IRS1 and IRS2 initiates activation of the PI 3-kinase/Akt pathway (20, 21). To determine whether SH2-B enhances IRS1 binding to p85, IRS1 was transiently coexpressed with SH2-Bβ, SH2-B(R555E), or ΔN504 in HEK293LRb cells, and its interaction with p85 was examined by coimmunoprecipitation assays. Leptin slightly stimulated coimmunoprecipitation of p85 with IRS1 in control cells, and SH2-B dramatically enhanced the association (Fig. 5A). ΔN504 enhanced the coimmunoprecipitation to a much lesser extent, whereas the stimulation by SH2-B(R555E) was barely detectable (Fig. 5A). Similarly, SH2-B also promoted association of p85 with IRS2 in response to leptin (data not shown).
Fig. 5. SH2-B enhances leptin-induced activation of the PI 3-kinase pathway.
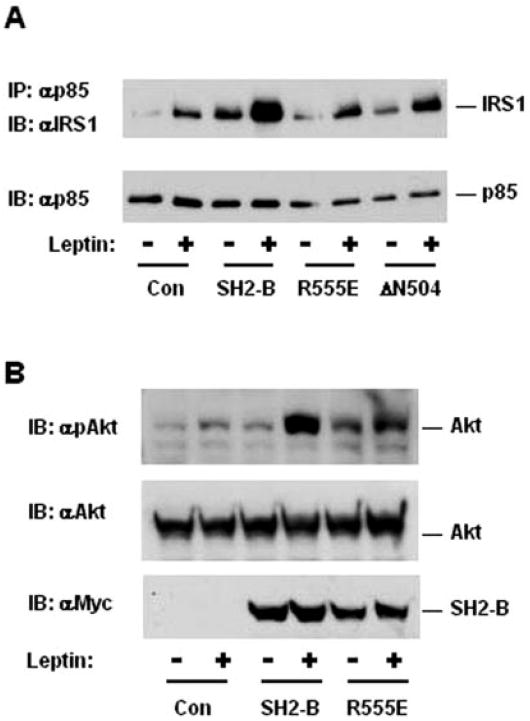
A, HEK293LRb cells were transiently cotransfected with expression plasmids encoding IRS1 (1 μg) and SH2-Bβ, SH2-B(R555E), or ΔN504 (0.8 μg) as indicated. The cells were treated with 100 ng/ml leptin for 10 min. The cell extracts were immunoprecipitated (IP) with αp85 and immunoblotted (IB) with αIRS1. The same blot was reprobed with αp85. B, HEK293LRb cells were transiently cotransfected with expression plasmids encoding IRS1 (1 μg), Akt1 (0.8 μg), and SH2-Bβ or SH2-B(R555E) (0.8 μg) as indicated. The cells were treated with 100 ng/ml leptin for 10 min. The cell extracts were immunoblotted with αphospho-Akt (Thr308), αAkt, and αSH2-B as indicated. Con, control.
To determine whether SH2-B enhances activation of Akt, SH2-Bβ or SH2-B(R555E) were coexpressed with Akt1 in HEK293LRb cells. Akt1 activation was estimated by immunoblotting with anti-phospho-Akt(Thr308) antibodies that specifically recognize phosphorylated and active Akt1. Leptin slightly stimulated phosphorylation of Akt1 at Thr308, which was dramatically enhanced by SH2-B but to a much lesser extent by SH2-B(R555E) (Fig. 5B).
SH2-B Mediates GH-stimulated Tyrosine Phosphorylation of IRS1
JAK2 mediates cell signaling in response to a variety of hormones and cytokines including GH, prolactin, erythropoietin, interferon-γ, and various interleukins in addition to leptin. To determine whether SH2-B also promotes the PI 3-kinase pathway by enhancing tyrosine phosphorylation of IRS1 and IRS2 in a similar fashion in response to other hormones and cytokines, GH receptor, IRS1, and Stat5b were transiently coexpressed in HEK293 cells in the presence or absence of coexpression of SH2-Bβ. Stat5b is a physiological substrate of JAK2 required for GH action (43). Phosphorylation of IRS1 and Stat5b were determined by immunoblotting with anti-phosphotyrosine or anti-phospho-Stat5b antibodies, respectively. GH stimulated Stat5b phosphorylation similarly regardless of co-expression of SH2-Bβ (Fig. 6A). In contrast, tyrosine phosphorylation of IRS1 was undetectable in the absence of SH2-B, and coexpression of SH2-Bβ increased both basal and GH-stimulated tyrosine phosphorylation of IRS1 (Fig. 6A). Moreover, deletion of SH2-B dramatically reduced GH-stimulated tyrosine phosphorylation of endogenous IRS1 mediated by endogenous GH receptor in MEF cells (data not shown). Restoration of SH2-B rescued GH-stimulated IRS1 phosphorylation (Fig. 6B). These results suggest that SH2-B mediates GH-stimulated tyrosine phosphorylation of IRS1 primarily by recruiting IRS1 to JAK2.
Fig. 6. SH2-B mediates GH-stimulated tyrosine phosphorylation of IRS1.
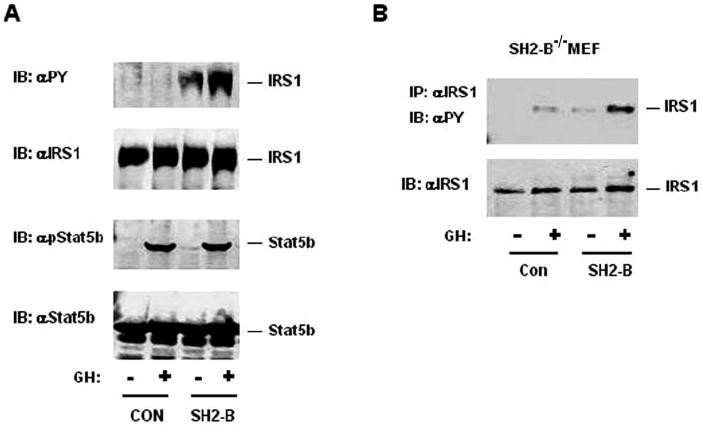
A, HEK293 cells were transiently cotransfected with expression plasmids encoding GH receptor (0.7 μg) IRS1 (1.2 μg), Stat5b (0.5 μg), and SH2-Bβ (0.6 μg) as indicated. The cells were treated with 8 × 10−3 IU/ml GH for 10 min. The cell extracts were immunoblotted (IB) with anti-phosphotyrosine (αPY), anti-IRS1, anti-phospho-Stat5b, and anti-Stat5b as indicated. The positions of IRS1 and Stat5b are marked. CON, control. B, control (Con) or SH2-Bβ retrovirus-infected SH2-B−/−/LRb MEFs were treated with 8 × 10−3 IU/ml GH for 10 min. The cell extracts were immunoprecipitated (IP) with αIRS1 and immunoblotted with αPY. The same blot was reprobed with αIRS1.
DISCUSSION
In this study, we found that SH2-B mediates leptin-stimulated phosphorylation of IRS proteins, resulting in activation of the PI 3-kinase pathway. Five lines of evidence support this conclusion. First, SH2-B bound directly to both IRS1 and IRS2 in vitro in Far Western analysis. Second, SH2-B was coimmunoprecipitated with both IRS1 and IRS2. Both the PH and SH2 domains were involved in the interaction. Third, SH2-B mediated a JAK2/SH2-B/IRS1 or IRS2 tertiary complex, which was promoted by leptin. Fourth, expression of SH2-B dramatically promoted leptin-stimulated tyrosine phosphorylation of IRS1 and IRS2, whereas deletion of the SH2-B gene impaired IRS1 tyrosine phosphorylation. Fifth, SH2-B specifically enhanced tyrosine phosphorylation of IRS1 and IRS2 but not Stat3 and Stat5 by JAK2, suggesting that physical interaction of SH2-B with IRS proteins contributes mainly to increased phosphorylation of IRS1 and IRS2. Finally, SH2-B enhanced leptin-stimulated association of p85 with IRS1 or IRS2, resulting in enhancement of Akt1 phosphorylation and activation. Deleting its binding sites for IRS1 impaired the ability of SH2-B to promote tyrosine phosphorylation of IRS1 and IRS1 association with p85.
SH2-B is expressed at high levels in leptin target tissues including the hypothalamus, liver, skeletal muscles, adipose tissues, and immune cells (data not shown). The mouse SH2-B gene is located on the distal arm of chromosome 7, which contains a locus involved in a multifactorial model of obesity (44-46). Moreover, SH2-B knockout mice exhibit leptin resistance and obesity, indicating that SH2-B mediates leptin signaling and action in animals.2 Therefore, the JAK2/SH2-B/IRS proteins/PI 3-kinase pathway identified in this work may be involved in mediating leptin regulation of feeding and energy homeostasis.
We propose a model of SH2-B-mediated activation of the PI 3-kinase/Akt pathway in response to leptin (Fig. 7). Leptin binds and activates its long form receptor LRb, resulting in activation and autophosphorylation of JAK2. SH2-B binds via its SH2 domain to JAK2 and further increases JAK2 activity, thus globally increasing tyrosine phosphorylation of JAK2 substrates including IRS1 and IRS2 (Mechanism 1). SH2-B also binds simultaneously to IRS1 or IRS2 via its PH and/or SH2 domain, thus promoting a JAK2/SH2-B/IRS1 or IRS2 complex. Appropriate proximity between JAK2 and IRS1 or IRS2 induced by SH2-B facilitates tyrosine phosphorylation of IRS1 and IRS2 by JAK2 (Mechanism 2). Tyrosine-phosphorylated IRS1 and IRS2 may be a rate-limiting step of activation of the PI 3-kinase pathway in response to leptin. APS also bound to IRS1 and IRS2 (data not shown) and may facilitate tyrosine phosphorylation of IRS1 and IRS2 by JAK2 in a similar fashion. Moreover, SH2-B and APS homo- and hetero-multimerize in cells (47). Multimerization may not only increase the affinity of multimeric SH2-B or APS for JAK2 and IRS proteins but also recruit multiple copies of JAK2 and IRS proteins to the SH2-B/APS complexes, further increasing the efficiency of JAK2 activation and its phosphorylation of IRS1 and IRS2.
Fig. 7. A Model of SH2-B action.
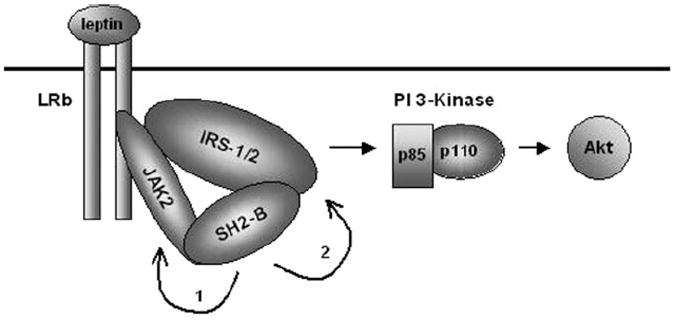
SH2-B binds to and potentiates activation of JAK2, globally enhancing the activation of pathways downstream of JAK2 (mechanism 1). SH2-B binds simultaneously to both JAK2 and IRS1 or IRS2, mediating a JAK2/SH2-B/IRS1 or IRS2 complexes. Consequently, SH2-B mediates specifically tyrosine phosphorylation of IRS1 and IRS2, resulting in activation of the PI 3-kinase pathway (mechanism 2).
In summary, we demonstrated that SH2-B bound simultaneously to both JAK2 and IRS proteins, resulting in activation of the PI 3-kinase pathway in response to leptin and GH. The PI 3-kinase pathway is required not only for leptin regulation of energy homeostasis in the hypothalamus (14, 32) but also for its action in peripheral tissues (30, 31, 48). SH2-B may serve as a potential drug target for therapeutic intervention of leptin resistance and obesity.
Acknowledgments
We thank Dr. Cai Li (Touchstone Center for Diabetes Research, The University of Texas Southwestern Medical Center, Dallas, TX) for providing the cDNA of mouse LRb. We thank David Morris and Drs. Decheng Ren, John Williams, and Michael Wang for helpful discussion.
Footnotes
This work was supported by Career Development Award 7-03-CD-11 from the American Diabetes Association, Grant RO1 DK 065122 from NIDDK, National Institutes of Health, and a Pilot and Feasibility Grant from the Michigan Diabetes Research and Training Center funded by Grant NIH5P60 DK20542 from NIDDK, National Institutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: LRb, long isoform receptor; IRS, insulin receptor substrate; GH, growth hormone; MEF, mouse embryonic fibroblast; PI, phosphatidylinositol; Stat, signal transducers and activators of transcription; HEK, human embryonic kidney; GST, glutathione S-transferase.
D. Ren, C. Duan, M. Li, and L. Rui, manuscript in preparation.
References
- 1.Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. J Clin Invest. 2001;108:1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI, Morgenstern JP. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 4.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 5.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Woolf EA, Monroe CA, Tepper RI. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 6.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 7.Banks AS, Davis SM, Bates SH, Myers MG., Jr J Biol Chem. 2000;275:14563–14572. doi: 10.1074/jbc.275.19.14563. [DOI] [PubMed] [Google Scholar]
- 8.Eyckerman S, Broekaert D, Verhee A, Vandekerckhove J, Tavernier J. FEBS Lett. 2000;486:33–37. doi: 10.1016/s0014-5793(00)02205-5. [DOI] [PubMed] [Google Scholar]
- 9.Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers MG., Jr J Biol Chem. 2000;275:40649–40657. doi: 10.1074/jbc.M007577200. [DOI] [PubMed] [Google Scholar]
- 10.Cui Y, Huang L, Elefteriou F, Yang G, Shelton JM, Giles JE, Oz OK, Pourbahrami T, Lu CY, Richardson JA, Karsenty G, Li C. Mol Cell Biol. 2004;24:258–269. doi: 10.1128/MCB.24.1.258-269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, Fu XY. Proc Natl Acad Sci U S A. 2004;101:4661–4666. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG., Jr Nature. 2003;421:856–859. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 13.Bjorbaek C, Uotani S, da Silva B, Flier JS. J Biol Chem. 1997;272:32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- 14.Zhao AZ, Huan JN, Gupta S, Pal R, Sahu A. Nat Neurosci. 2002;5:727–728. doi: 10.1038/nn885. [DOI] [PubMed] [Google Scholar]
- 15.Anderwald C, Muller G, Koca G, Furnsinn C, Waldhausl W, Roden M. Mol Endocrinol. 2002;16:1612–1628. doi: 10.1210/mend.16.7.0867. [DOI] [PubMed] [Google Scholar]
- 16.Carvalheira JB, Ribeiro EB, Folli F, Velloso LA, Saad MJ. Biol Chem. 2003;384:151–159. doi: 10.1515/BC.2003.016. [DOI] [PubMed] [Google Scholar]
- 17.Kellerer M, Koch M, Metzinger E, Mushack J, Capp E, Haring HU. Diabetologia. 1997;40:1358–1362. doi: 10.1007/s001250050832. [DOI] [PubMed] [Google Scholar]
- 18.Kim Y-B, Uotani S, Pierroz DD, Flier JS, Kahn BB. Endocrinology. 2000;141:2328–2339. doi: 10.1210/endo.141.7.7536. [DOI] [PubMed] [Google Scholar]
- 19.White MF. Am J Physiol. 2002;283:E413–E422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- 20.White MF. Mol Cell Biochem. 1998;182:3–11. [PubMed] [Google Scholar]
- 21.White MF. Diabetologia. 1997;40(Suppl. 2):S2–S17. doi: 10.1007/s001250051387. [DOI] [PubMed] [Google Scholar]
- 22.Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 23.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 24.Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M. Curr Biol. 1997;7:776–789. doi: 10.1016/s0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- 25.Wick MJ, Dong LQ, Riojas RA, Ramos FJ, Liu F. J Biol Chem. 2000;275:40400–40406. doi: 10.1074/jbc.M003937200. [DOI] [PubMed] [Google Scholar]
- 26.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 27.Araki E, Lipes MA, Patti ME, Bruning JC, Haag B, III, Johnson RS, Kahn CR. Nature. 1994;372:186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 28.Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, Terauchi Y, Ueki K, Kaburagi Y, Satoh S, et al. Nature. 1994;372:182–186. doi: 10.1038/372182a0. [DOI] [PubMed] [Google Scholar]
- 29.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, III, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 30.Tajmir P, Ceddia RB, Li R-K, Coe IR, Sweeney G. Endocrinology. 2004;145:1550–1555. doi: 10.1210/en.2003-1128. [DOI] [PubMed] [Google Scholar]
- 31.Vecchione C, Maffei A, Colella S, Aretini A, Poulet R, Frati G, Gentile MT, Fratta L, Trimarco V, Trimarco B, Lembo G. Diabetes. 2002;51:168–173. doi: 10.2337/diabetes.51.1.168. [DOI] [PubMed] [Google Scholar]
- 32.Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Nature. 2001;413:794–795. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- 33.Burks DJ, de Mora JF, Schubert M, Withers DJ, Myers MG, Towery HH, Altamuro SL, Flint CL, White MF. Nature. 2000;407:377–382. doi: 10.1038/35030105. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki R, Tobe K, Aoyama M, Yamauchi T, Kamon J, Kubota N, Terauchi Y, Yoshimatsu H, Matsuhisa M, Nagasaka S, Ogata H, Tokuyama K, Nagai R, Kadowaki T. J Biol Chem. 2004;279:25039–25049. doi: 10.1074/jbc.M311956200. [DOI] [PubMed] [Google Scholar]
- 35.Rui L, Mathews LS, Hotta K, Gustafson TA, Carter-Su C. Mol Cell Biol. 1997;17:6633–6644. doi: 10.1128/mcb.17.11.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yousaf N, Deng Y, Kang Y, Riedel H. J Biol Chem. 2001;276:40940–40948. doi: 10.1074/jbc.M104191200. [DOI] [PubMed] [Google Scholar]
- 37.Kurzer JH, Argetsinger LS, Zhou YJ, Kouadio JL, O’Shea JJ, Carter-Su C. Mol Cell Biol. 2004;24:4557–4570. doi: 10.1128/MCB.24.10.4557-4570.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rui L, Carter-Su C. Proc Natl Acad Sci U S A. 1999;96:7172–7177. doi: 10.1073/pnas.96.13.7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rui L, Gunter DR, Herrington J, Carter-Su C. Mol Cell Biol. 2000;20:3168–3177. doi: 10.1128/mcb.20.9.3168-3177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rui L, Yuan M, Frantz D, Shoelson S, White MF. J Biol Chem. 2002;277:42394–42398. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 41.Rui L, Archer SF, Argetsinger LS, Carter-Su C. J Biol Chem. 2000;275:2885–2892. doi: 10.1074/jbc.275.4.2885. [DOI] [PubMed] [Google Scholar]
- 42.Rui L, Fisher TL, Thomas J, White MF. J Biol Chem. 2001;276:40362–40367. doi: 10.1074/jbc.M105332200. [DOI] [PubMed] [Google Scholar]
- 43.Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Proc Natl Acad Sci U S A. 1997;94:7239–7244. doi: 10.1073/pnas.94.14.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelms K, O’Neill TJ, Li S, Hubbard SR, Gustafson TA, Paul WE. Mamm Genome. 1999;10:1160–1167. doi: 10.1007/s003359901183. [DOI] [PubMed] [Google Scholar]
- 45.Warden CH, Fisler JS, Pace MJ, Svenson KL, Lusis AJ. J Clin Invest. 1993;92:773–779. doi: 10.1172/JCI116649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warden CH, Fisler JS, Shoemaker SM, Wen PZ, Svenson KL, Pace MJ, Lusis AJ. J Clin Invest. 1995;95:1545–1552. doi: 10.1172/JCI117827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qian X, Ginty DD. Mol Cell Biol. 2001;21:1613–1620. doi: 10.1128/MCB.21.5.1613-1620.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang W, Dedousis N, Bhatt BA, O’Doherty RM. J Biol Chem. 2004;279:21695–21700. doi: 10.1074/jbc.M401546200. [DOI] [PubMed] [Google Scholar]