

Abstract
We investigated truncated glioma-associated oncogene homolog 1 (TGLI1) that behaves as gain-of-function GLI1 and promotes tumor cell migration and invasion. Herein, we report that TGLI1 had a higher propensity than GLI1 to enhance glioblastoma angiogenesis and growth, both in vivo and in vitro. TGLI1 has gained the ability to enhance expression of pro-angiogenic heparanase. In patient glioblastomas, TGLI1 levels are correlated with heparanase expression. Together, we report that TGLI1 is a novel mediator of glioblastoma angiogenesis and that heparanase is a novel transcriptional target of TGLI1, shedding new light on the molecular pathways that support tumor angiogenesis and aggressive growth.
Keywords: GLI1, Hedgehog pathway, transcriptional regulation, angiogenesis, cancer
1. Introduction
The Hedgehog pathway is important for embryonic development [1, 2] and tumorigenesis [3–5]. It is also one of the most dysregulated pathways in human cancers and is considered to be an important target of anti-cancer therapy [6, 7]. The Hedgehog pathway is highly complex and can be activated following the binding of Sonic hedgehog (Shh) to its receptor patched, a repressor of a 7-transmembrane receptor smoothened, SMO. Shh’s binding to PTCH derepresses SMO which, in turn, activates nuclear import of GLI1. Nuclear GLI1 binds to the GLI1-binding element in GLI1-regulated genes, leading to their activation [8, 9].
The GLI family of zinc finger transcription factors constitutes the terminal effectors of the Shh pathway [10, 11]. Although the human GLI1 gene was initially identified an amplified gene in glioblastoma (GBM) cells [12], GLI1 gene amplification was later found to be rare in GBM and some of other cancer types [13–15]. Somatic mutations in the GLI1 gene have never been reported in any cell and tumor type. Very recently, the human GLI1 gene transcript was found to be alternatively spliced to yield two shorter isoforms, namely, GLI1ΔN identified in 2008 [16], and TGLI1 identified in our laboratory in 2009 [17]. GLI1ΔN contains a large deletion of 128 codons, lacks two functional domains and behaves as a weak GLI1 transcription factor [16]. The TGLI1 variant is uniquely distinct from the other two previously reported GLI1 variants, in that it contains a small in-frame deletion of 123 bases (41 codons) spanning the entire exon 3 and part of exon 4 of GLI1, and preserves functional domains of GLI1 and the ability to undergo nuclear import and to activate GLI1-targeted genes [17].
Additional evidence indicates that TGLI1 is highly expressed in GBM and breast cancer, but not detected in normal brain and mammary tissues [17, 18]. TGLI1 has gained the ability to regulate genes that are not targeted by GLI1, including CD24 [17]. More recently, our group further showed that TGLI1 transcriptionally activates expression of vascular endothelial growth factor-A (VEGF-A) and VEGFR2 genes in breast cancer and medulloblastoma cells [18, 19]. Using GBM and breast cancer models, we have shown that TGLI1, but not GLI1, enhances the propensity of cancer cells to migrate and invade [17, 18]. However, TGLI1 functionality relative to GLI1 remains poorly understood. To address this knowledge gap, we undertook the current study aiming to gain new insights into the relevance and role of TGLI1 in GBM, the most common brain cancer in adults and one of the deadliest and most therapeutically intractable human malignancies [20]. Of note, our results fill critical knowledge gaps in the Hedgehog-GLI1 pathway and in GBM vascularization by defining TGLI1 as a novel mediator of neoangiogenesis in cancers and potentially other diseases.
2. Materials and Methods
2.1. Patient GBM tumors, directly passaged xenografts and cell lines
Patient GBM Tumors were provided by Dr. Matthias Gromeier at Duke University, as well as, purchased from US Biomax (Rockville, MD). Patient GBM-derived xenografts and D54MG GBM cells were provided by the Preston Robert Tisch Brain Tumor Center at Duke University. Direct xenografts were derived patient GBM tumors that were excised during 2008–2010 and maintained in athymic mice as subcutaneous flank xenografts. The direct xenografts recapitulate features of patient tumors since patient GBMs lose several specific molecular features after long-term in vitro culture [21]. Human U87MG and U373MG GBM cells were obtained from American Type Culture Collection. Stable U87MG-vector, U87MG-GLI1 and U87MG-TGLI1 cells were previously established [17]. Human brain microvascular endothelial cells (HBMEC) were from Angio-Proteomie and maintained in EGM™-2 MV full medium containing 5% fetal calf serum and growth supplements. Other cell lines were cultured in DMEM with 10% fetal calf serum while the U87MG stable transfectant cell lines were additionally supplemented with 0.7 mg/ml G418. All cell lines had been tested and authenticated by their commercial sources and were used within 6 months of culturing after receipt.
2.2. Reagents and Plasmids
Expression vectors, pCMV-Tag2b, pCMV-Tag2b-GLI1, or pCMV-Tag2b-TGLI1, were generated in our laboratory [17]. All siRNAs were purchased from Thermo Scientific (Lafayette, CO) and Bioneer (Alameda, CA). Two different siRNAs were used for each target. The siRNA sequences are 5′-GCAGAAUCAUCACGAAGUG-3′ and 5′-CACCAUUGAAACCACUAGU(dTdT)-3′ (human VEGF-A siRNAs), 5′-CCUCGAAGAAGACGGCUA-3′ and 5′-CAUCAAUGGGUCGCA GUUA(dTdT)-3′ (human heparanase siRNAs), and 5′-UGGUUUACAUGUCGACUAA-3′ and 5′-CCUACGCCACCAAUU UCGU(dTdT)-3′ (non-targeting control siRNAs).
2.3. Western blotting (WB)
This was performed as described previously [18]. Antibodies used included mouse monoclonal antibodies against β-actin (Sigma) and β-tubulin (Sigma), and rabbit antibodies for heparanase (Santa Cruz, H-80), VEGF-A (Santa Cruz, sc-152), Akt/pan (4691, Cell Signaling), p-Akt/S473 (4060, Cell Signaling), mTOR (2983, Cell Signaling) and CD24 (Santa Cruz; FL-80). To detect both GLI1 and TGLI1, goat polyclonal antibody (Santa Cruz, C-18) and mouse monoclonal GLI1 antibody (Cell Signaling, #2643) were used following prolonged 5.5% SDS-PAGE.
2.4. Animal studies
Female nude mice (NCr-nu/nu Athymic; NCI-Frederick) of 6 weeks old were used. GBM xenografts were generated by subcutaneous implantation of U87MG-vector, U87MG-GLI1 and U87MG-TGLI1 cells into the right flanks of female nude mice. A total of 1 × 106 cells were used per inoculation. Six mice were included in each group. Tumor volume was determined twice weekly using calipers and the equation, volume = (length x width2)/2. All mice were euthanized at days 32–35 because most TGLI1-carrying tumors had reached the volume of 2,000 mm3, the humane endpoint. The animal study was conducted according to protocols approved by the Duke University IACUC committee.
2.5. Immunohistochemistry (IHC)
This was conducted as we described previously [17]. The slides were incubated with the following antibodies, rabbit polyclonal Ki-67 antibody (Thermo scientific, ready-to-use), rabbit polyclonal CD31 antibody (Thermo Scientific, Clone JC/70A, ready-to-use), rabbit polyclonal VEGF-A antibody (Santa Cruz, A-20, 1:50), rabbit polyclonal GLI1 antibody (Santa Cruz, H-300, 1:75), and rabbit polyclonal heparanase antibody (Santa Cruz, H-80, 1:25). CD31 IHC was used to mark tumor vasculature in which blood vessels in each of 8–10 microscopic fields were counted under a 40X objective using a microscope to derive microvessel density, via a standard method and the formula, # of vessels/mm2 [22]. Rabbit polyclonal TGLI1 splice junction-specific antibodies were produced by YenZym Antibodies, LLC (South San Francisco, CA). Scoring was performed by a pathologist. Histologic scores (H-Scores) were computed from both % positivity (A%, A=1–100) and intensity (B=0–3) using the equation, H-Score=A × B. For TGLI1 IHC, tumors were classified into four categories: negative (H-score=0), low (H-score=1–50), median (H-score=51–150) and high (H-score=151–300).
2.6. Total RNA Extraction, Reverse-transcription (RT) and Polymerase Chain Reaction (PCR)
Total RNA isolation and RT were conducted using SV Total RNA Isolation System (Promega) and Superscript II First-Strand cDNA synthesis system (Invitrogen), retrospectively. The forward and reverse primers used for the PCR were: 5′-TGTTCAACTCGATGACCC-3′ and 5′-GTCATGGGGACCACAAGG-3′ (exons 1–4 of GLI1 and TGLI1), 5′-GGCGGCACCACCATGTACCC-3′ and 5′-AGGGGCCGGACTCGTCATACT-3′ (β-actin), 5′-GGTCAGCCTCGAAGAAAGAC-3′ and 5′-TAGCAGTCCGTCCATTCAAA-3′ (human heparanase), 5′-ATGGGCAGAGCAATGGTGGCCA-3′ and 5′-AGAGTGAGACCACGAAGAGACT-3′ (human CD24), and 5′-CACCATCGACAGAACAGTCC-3′ and 5′-GAATCCAATTCCAAGAGGGA-3′ (human VEGF-A).
2.7. Tubule Formation Assay
Tubule formation assay was performed using the In Vitro Angiogenesis Kit (Trevigen), as we previously described [18]. A total of 5,000–5,500 human brain microvascular endothelial cells were seeded into each coated well. Conditioned medium was collected from tumor cells that have been starved in EBM-2 Basal Medium for 24 hrs. After incubation at 37°C for 4–6 hrs, endothelial cells were examined for capillary-like network formation and photographed under a light microscope. Tubule formation was quantified by measuring total tubule length and total number of branch points in triplicate wells, using the NIH Image J software.
2.8. ELISA that measures VEGF-A concentrations in culture medium
This was conducted using the VEGF-A ELISA Kit from R&D, according to manufacturer’s instructions and our previous report [18]. GBM cells seeded in 24-well culture plates were incubated in the EBM-2 Basal Medium at 37°C. After 24 hrs, conditioned medium was collected and centrifuged at 1,200 xg for 10 min to remove cell debris. The supernatants were then subjected to ELISA in triplicates. Absorbance at 540 nm (and 450 nm as normalization background) was determined using the Synergy HT Multi-mode Microplate Reader (BioTek). VEGF-A concentrations were computed with reference to standard curves derived from purified VEGF-A supplied in the ELISA Kit.
2.9. Chorioallantoic Membrane (CAM) assay [18]
U373MG GBM cells were transfected with pCMV-Tag2b, pCMV-Tag2b-GLI1, or pCMV-Tag2b-TGLI1 plasmids for 48 hours. Harvested cells were then mixed with an equal volume growth factor reduced Matrigel (BD Biosciences, Bedford, MA). Aliquots (40 μL) of the cell suspension (2×106 cells/embryo) were implanted on 9-day old chorioallontoic membranes of foghorn chicken embryos (Charles River). Eight to twelve embryos were used for each group. After four days, matrigel plugs and surrounding chorioallontoic membrane were collected and photographed with a Zeiss camera. Images were used to count vessel branch points. Branch points were normalized to area of tissue.
2.10. Invasion Assay
This was conducted as we previously described using the InnoCyte™ Quantitative Cell Invasion Assay kit (EMD) [17]. Briefly, 1.75×105 cells were placed in the upper chamber of re-hydrated inserts with an 8-μm pore size polycarbonate membrane coated with a uniform layer of basement membrane matrix on the upper surface. Following incubation for 48 hrs, The inserts containing the breast cancer cells invaded through the basement membrane matrix were stained in 0.5% crystal violet. Cell proliferation rates were simultaneously determined [17, 23, 24] to derive “invasiveness:proliferation” ratios that indicate net invasiveness.
2.11. Chromatin Immunoprecipitation (ChIP) assay
A ChIP Assay kit (Upstate) was used. A rabbit polyclonal GLI1 antibody (Santa Cruz, H-300) that recognizes the COOH-terminal region (amino acids 781–1080 present in both GLI1 and TGLI1 proteins) of the human TGLI1/GLI1 proteins was used in these experiments. For both promoters, the targeted regions were located in the promoter regions that have been shown to drive their transcriptional activities. The quantitative VEGF-A ChIP assay was conducted as we previously described [18]. Primer sequences for amplifying the human heparanase gene promoter are 5′-CTCGAGGGTCAGAGGGATAC-3′ (forward) and 5′-TCCTGACGCACGTGTTCT-3′ (reverse). Rabbit normal IgG served as negative immunoprecipitation controls. Input chromatins were used as loading controls for PCR.
2.12. Luciferase reporter constructs and luciferase assay
Construction of the GLI1- and TGLI1-expressing plasmids was described in our previous study [17]. The pGL3-HPA1-Luc reporter construct was engineered to contain −693 to +374 bp promoter region of the human heparanase gene. The pGL3-VEGF-A-Luc reporter contains the promoter region of the human VEGF-A gene (−900 to +100 bp) was cloned into the pGL3-basic Firefly luciferase vector (Promega), as described in our previous study [18]. A Renilla luciferase expression vector, pRL-CMV, was used to control for transfection efficiency. Forty-eight hrs after transfection, the cells were lysed and luciferase activity measured using the Firefly and Renilla Luciferase Assay Kit (Biotium), as we previously described [17, 18]. Relative promoter activity was computed by normalizing the Firefly luciferase activity against that of the Renilla luciferase.
2.13. Cell proliferation assay
Tumor cells in exponential growth were seeded in 96-well culture plates and transfected with indicated siRNAs. After 48 hrs, the cells were subjected to cell survival analyses using the CellTiter Blue Cell Viability Assay (Promega). Briefly, 25 μl of the CellTiter Blue reagent was added to each well containing 100 ul media, incubated for 4 hrs at 37°C, and then the absorbance measured at 560 nm/590 nm using a plate reader (Synergy-HT, BIO-TEK, Winooski, VT). Triplicate wells were used for each treatment and three independent experiments were performed to derive means and standard deviations.
2.14. Statistical analyses
The student t-test and multiple regression analysis were performed using STATISTICA (StatSoft) and Microsoft Excel.
3. Results
3.1. TGLI1 protein is highly expressed in patient GBM samples, patient GBM-derived direct xenografts and has a higher propensity than GLI1 to enhance in vivo growth and vascularity of GBM xenografts
In our 2009 study [17], we reported TGLI1 transcript to be expressed in the majority of GBM cell lines, but not in normal brain or other normal tissues. Here, we further showed that TGLI1 protein is readily detected in patient GBMs and patient GBM-derived xenografts (Fig. 1A). Previous reports have shown that direct xenografts recapitulate features of the patient’s original tumor [21]. As shown in Fig. 1A, TGLI1 (146 kD) and GLI1 (150 kD) proteins were separated by prolonged SDS-PAGE followed by WB with an antibody that recognizes both GLI1 and TGLI1. Stable transfectant cells expressing TGLI1 and GLI1 were used to indicate differential mobility of the two GLI1 isoforms. Notably, GBM tumors differ in their TGLI1 level relative to GLI1 and in some cases, TGLI1 expression is comparable to or higher than GLI1.
Figure 1. TGLI1 is highly expressed in GBM and enhances in vivo growth and vascularity of GBM xenografts.

A, TGLI1 protein was detected in patient GBMs sand direct xenografts. Resolution of TGLI1 and GLI1 proteins was enabled via prolonged 5.5% SDS-PAGE and WB with a GLI1 antibody. Stable GBM cells with TGLI1 and GLI1 were used to indicate the two GLI1 isoforms.
B, TGLI1-expressing GBM xenografts were significantly more aggressive in growth than the control and those with GLI1 (*,**, p<0.05). N=6 per group. Student t-test was conducted to determine p-values.
C, TGLI1-carrying GBM xenografts were highly vascularized. Three representative GBM xenografts are shown.
D, TGLI1-expressing xenografts contained significantly more blood vessels that those with GLI1. IHC was conducted to detect CD31 and derive microvessel density using a standard method [22, 25]. Student t-test was conducted to determine p-values. Right lower panels show high-resolution images.
E, TGLI1-expressing tumors were more proliferative than control and those with GLI1. IHC was conducted to detect Ki-67. Student t-test was used to determine p-values. Representative Ki-67-stained xenografts are shown in the right panels. Arrows point to brown Ki-67-positive tumor nuclei.
F, No significant difference in Akt activation status was observed among the three U87MG lines, as indicated by western blotting.
We inoculated three stably transfectent U87MG cell lines with vector control, GLI1 or TGLI1 into the flanks of nude mice (six per group) and determined tumor volume twice weekly until the humane endpoint. The growth curves (Fig. 1B) indicate that TGLI1-expressing GBM xenografts were significantly more aggressive in growth than the control and those with GLI1. Notably, all three groups of xenografts had a similar growth rate up to day 21 post inoculation and began to differ substantially in their volume after day 24. As shown by representative images in Fig. 1C, TGLI1-carrying GBM xenografts were larger and more vascularized than those with vector control and GLI1. To confirm this interesting observation, we conducted IHC to detect the vascular endothelial marker CD31 in order to mark tumor vasculature. Based on CD31 IHC results, we then computed microvessel density in the tumors using a previously described method [22, 25]. The results (Fig. 1D; left panel) clearly indicate that the TGLI1-expressing xenografts contained significantly more blood vessels than those expressing GLI1 or the control vector. Representative CD31-stained sections are shown in the right panels of Fig. 1D in which lower panels show high-resolution images.
Next, we determined whether the TGLI1-expressing tumors were more proliferative than those with GLI1 or the control vector. For this, we conducted IHC to detect Ki-67, a proliferative marker expressed only in actively proliferating cells but not in quiescent cells. As shown in Fig. 1E, TGLI1-expressing GBM xenografts contained significantly more Ki-67-positive cells than the control and those with GLI1 (left panel). Representative Ki-67-stained xenografts are shown in the right panels of Fig. 1E in which arrows point to brown Ki-67-positive tumor nuclei. As shown in Fig. 1F, we further determined Akt activation status and observed no significant differences, suggesting that Akt may not be an important factor for TGLI1-associated tumor growth or angiogenesis. Collectively, results in Fig. 1 indicate that TGLI1-expressing GBM tumors are more aggressively growing and more vascularized in comparison to those with GLI1 or with very low levels of GLI1 and TGLI1.
3.2. TGLI1-expressing tumor cells contain a higher propensity than control and those with GLI1 to promote tubule formation of human brain microvascular endothelial cells (HBMEC) and in vivo angiogenesis of chick embryos
Conditioned medium from U87MG-TGLI1 cells strongly promoted in vitro angiogenesis of HBMEC cells (Fig. 2A) and human umbilical vein endothelial cells, HUVEC (Supplemental Fig. 1). In the tubule formation assay that mimics in vivo angiogenesis, the extent of in vitro angiogenesis was quantified by two measurements, namely, total tubule length and branch points. These interesting findings were confirmed using another GBM cell line, U373MG. As shown in Fig. 2B, we transiently transfected the TGLI1, GLI1 and control vectors into U373MG cells and confirmed transfection effectiveness by western blotting. Using tubule formation assay (Fig. 2C), we observed that TGLI1 expression rendered U373MG cells more effective in promoting tubule formation of HBMEC cells.
Figure 2. TGLI1-expressing tumor cells strongly promote tubule formation of vascular endothelial cells and in vivo angiogenesis of chick embryos.

Three independent experiments were conducted and the student t-test was conducted to determine p-values.
A, Conditioned medium from U87MG-TGLI1 cells strongly promoted in vitro angiogenesis of HBMEC cells. The tubule formation assay was performed. Total tubule length (relative to vector control) and branch points were determined.
B, Western blots indicate that U373MG cells were effectively transfected to express TGLI1 or GLI1.
C, U373MG cells expressing TGLI1 strongly promote in vitro angiogenesis of HBMEC cells compared to those with the control vector and GLI1.
D,E, CAM assay showed that TGLI1-expressing U373MG GBM cells strongly promoted angiogenesis of chick embryos. In each transplantation, 8–12 CAMs were analyzed.
Further, we conducted the CAM assay using U373MG cells to determine the extent to which TGLI1 and GLI1 regulate angiogenesis in vivo. U373MG GBM cells transiently transfected with pCMV-Tag2b, pCMV-Tag2b-GLI1, or pCMV-Tag2b-TGLI1 plasmids were harvested cells and mixed with an equal volume growth factor reduced Matrigel. Aliquots (40 μL) of the cell suspension (2×106 cells/embryo) were implanted on 9-day old chorioallontoic membranes of foghorn chicken embryos. Eight to twelve embryos were used for each group. After four days, matrigel plugs and surrounding chorioallontoic membrane were collected and photographed with a Zeiss camera. Images were used to count vessel branch points. Branch points were normalized to area of tissue. The results of the CAM study indicated that TGLI1-expressing U373MG cells strongly promoted angiogenesis of chick embryos to a greater degree than those with GLI1 and control vector (Fig. 2D). GLI1 also rendered chick embryos more angiogenic compared to the vector control. Fig. 2E shows representative images indicating that TGLI1 expression in U373MG cells had resulted in extensive vascular sprouting and immature vasculature in chick embryos. Taken together, results in Fig. 2 indicate that TGLI1-expressing GBM cells contain a higher propensity to promote tubule-forming ability of HBMEC cells and in vivo angiogenesis of chick embryos than control cells and those with GLI1.
3.3. Heparanase gene expression is enhanced by TGLI1 in GBM cell lines and xenografts
To identify angiogenesis-promoting factors that are upregulated by TGLI1, we concentrated our efforts on heparanase because of its known role in glioma vascularity [26]. Heparanase is a multi-functional protein that: i) cleaves heparan sulfate chains of extracellular matrix proteins, leading to disruption of basement membrane structure and subsequent migration of endothelial cells to tumors, ii) releases heparan sulfate-bound angiogenic factors bFGF and VEGF, iii) generates heparan sulfate that enhances bFGF:FGFR binding [27, 28], and iv) increases VEGF-A expression via its non-enzymatic action [26]. Endogenous heparanase has been shown to exert non-enzymatic activity to increase VEGF-A expression in rat glioma cells [26].
As shown in Fig. 3A, heparanase gene transcript and protein are highly expressed in U87MG-TGLI1 xenografts but remain undetectable in U87MG-GLI1 tumors. Results of six representative xenografts are shown. These observations were confirmed by heparanase IHC in Fig. 3B. Furthermore, we examined the effects of transient expression of TGLI1 versus GLI1 on heparanase levels in U373MG and D54MG cells, and the results (Fig. 3C) showed that transient elevation of TGLI1, but not GLI1, led to increased heparanase gene expression in both cell lines. Consistent with our prior observations [17, 18], TGLI1 expression rendered U373MG and D54MG cells highly invasive (Fig. 3D). It is worth noting that D54MG cells were extracted from direct xenografts serially maintained in nude mice which showed higher levels of endogenous TGLI1 (Fig. 3C) and intrinsic invasiveness (Fig. 3D), relative to U87MG and U373MG cells.
Figure 3. Heparanase gene expression is enhanced by TGLI1.
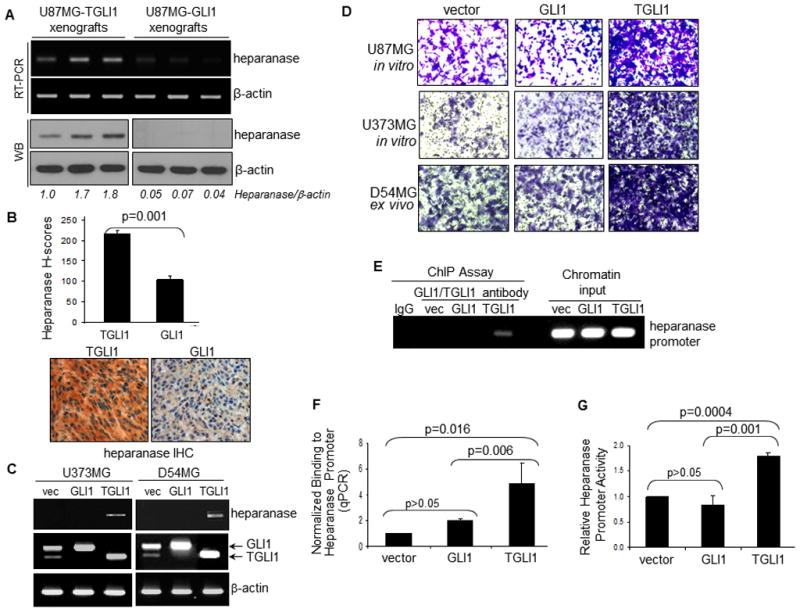
Six tumors were analyzed in each group. Student’s t-test was conducted to determine p-values
A, Heparanase gene was highly expressed in U87MG-TGLI1 xenografts but undetectable in U87MG-GLI1 tumors. Results of six representative tumors are shown.
B, Heparanase is expressed at a higher level in U87MG-TGLI1 xenografts than in U87MG-GLI1 tumors, as shown by IHC.
C, Transient elevation of TGLI1, but not GLI1, expression leads to increased levels of heparanase gene expression in U373MG and D54MG cells. RT-PCR was conducted. D54MG cells were extracted from xenografts serially maintained in nude mice which showed endogenous expression of both TGLI1 and GLI1.
D, TGLI1 expression rendered U373MG and D54MG highly invasive. Invasion transwell assay was performed in which tumor cells that have invaded through coated basement membranes were stained by crystal violet blue. D54MG cells were extracted from xenografts serially maintained in nude mice which showed higher levels of endogenous TGLI1 and intrinsic invasiveness, relative to U87MG and U373 MG cells.
E,F, TGLI1, but not GLI1, strongly binds to the human heparanase gene promoter, as shown by the ChIP assay (panel E and quantitative ChIP assay (panel F). The stable U87MG cell lines were used. TGLI1-GLI1 PCR signals were normalized against those of the vector control and the loading controls (chromatin input). Three independent experiments were conducted.
G, Heparanase gene promoter activity is higher is GBM cells with TGLI1 than those with GLI1. Means and standard deviations were derived from results of three independent experiments.
To determine whether the human heparanase gene is a transcriptional target of TGLI1, we conducted the ChIP assay to measure the extent to which intracellular TGLI1 and GLI1 differ in their ability to bind to the human heparanase gene promoter. The stable U87MG cell lines were used. ChIP assay in Fig. 3E showed that TGLI1, but not GLI1, strongly binds to the heparanase gene promoter, in which TGLI1-GLI1 PCR signals were normalized against those of the vector control and chromatin input (loading controls). Quantitative ChIP assay in Fig. 3F clearly demonstrated that TGLI1 contained a significantly higher affinity to the heparanase gene promoter than GLI1 (p=0.006). In the ChIP assay, we targeted the promoter region that has been shown to drive its transcriptional activity. Interestingly, VEGF-A and heparanase promoters do not contain consensus GLI1-response elements, consistent with the lack of GLI1 binding to the promoters. Next, we examined whether TGLI1 binding to the human heparanase gene promoter leads to promoter activation. For this, we engineered a heparanase promoter-controlled luciferase reporter (pGL3-HPA1-Luc), transfected it into the three U87MG stable transfectant cell lines and measured luciferase activity. The results (Fig. 3G) show that TGLI1 had a higher ability to transactivate the heparanase gene promoter than GLI1. Collectively, the results in Fig. 3 indicate that the human heparanase gene is a novel transcriptional target of TGLI1, but not GLI1, and that heparanase gene expression is positively associated with TGLI1 in cell lines and xenografts of GBM.
3.4. TGLI1-carrying GBM xenografts and cell lines express high levels of VEGF-A
We next investigated the role of VEGF-A in TGLI1-mediated GBM angiogenesis because VEGF-A is the ligand of VEGFR1/2 and the binding to the receptors activates vascular proliferation [29, 30]. This investigation was promoted by our recent report showing that TGLI1 enhances VEGF-A expression in breast cancer cells [18]. However, to date, the TGLI1-VEGF link has not been investigated in vivo. Here, we observed that VEGF-A transcript (Fig. 4A) and protein (Fig. 4B) levels were significantly higher in TGLI1-expressing GBM xenografts than those with GLI1. Results of six representative xenografts are shown. In agreement with our previous report showing CD24 as a transcriptional target of TGLI1 [17], CD24 is highly expressed in TGLI1-carrying tumors but not in those with GLI1. To confirm these observations, we conducted IHC to detect VEGF-A in GBM xenografts (N=6 per group), and the results (Fig. 4C) indicate that TGLI1-carrying tumors expressed more VEGF-A than those with GLI1. Representative tumors immunostained for VEGF-A are shown in Fig. 4D. We also immunostained the tumor sections for TGLI1/GLI1 using an Ab that recognizes both GLI1 and TGLI1, and the results show that both groups of xenografts expressed similar levels of transgenes, as indicated by TGLI1/GLI1-positive brown nuclei.
Figure 4. TGLI1-carrying xenografts of GBM express high levels of VEGF-A.
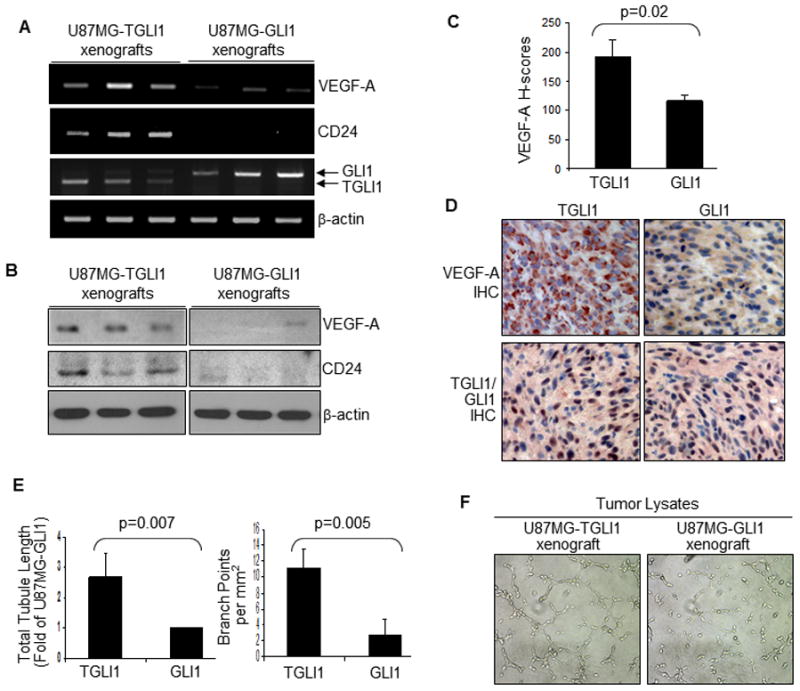
A,B, TGLI1-carrying GBM xenografts contained a higher level of VEGF-A than those with GLI1. Panel A, RT-PCR. Panel B, WB. Results of six representative tumors are shown. CD24 is highly expressed in TGLI1-carrying tumors but not in those with GLI1, which is in agreement with our previous report [17].
C,D, TGLI1-carrying xenografts expressed more VEGF-A than those with GLI1, as shown by IHC. Six tumors per group were analyzed. Student t-test was conducted to determine the p-value. Panel D shows two representative tumors. TGLI1- and GLI1-positive nuclei are marked by brown signals.
E,F, TGLI1-expressing GBM xenografts contained a higher propensity to promote in vitro angiogenesis of HBMEC cells as shown by the tubule formation assay. Total proteins extracted from the xenografts were added to endothelial cells. Three independent experiments were conducted and the student t-test was conducted to determine p-values.
We further determined whether TGLI1-expressing GBM xenografts contain a higher propensity to promote tubule formation of HBMEC cells, thus mimicking in vivo angiogenesis. For this, we extracted total proteins from the xenografts, added the extracts to HBMEC cells being cultured on basement matrix extracts, and monitored the ability of the endothelial cells to form capillary-like tubular structures. As shown in Fig. 4E, HBMEC cells exposed to extracts from U87MG-TGLI1 xenografts formed more capillary structures compared to those from U87MG-GLI1 tumors. As shown by representative images in Fig. 4F, the tumor lysates from the U87MG-TGLI1 xenografts displayed a significantly higher propensity than those from the U87MG-GLI1 tumors to promote in vitro angiogenesis of HBMEC cells in both total tubule length and branch points. This observation combined with those with the conditioned medium in Fig. 2, together, suggesting that TGLI1-expressing GBM cells/tumors express higher levels of VEGF-A intracellularly and consequently, secreted higher amount of VEGF-A to stimulate angiogenesis.
We further show in Fig. 5A that VEGF-A transcripts levels were enhanced by TGLI1 expression in U87MG, U373MG and D54MG GBM cell lines. Using ChIP (Fig. 5B) and luciferase reporter (Fig. 5C) assays respectively, we further showed that TGLI1 binds to and transactivates the VEGF-A gene promoter more strongly than GLI1. In ChIP assays, we targeted the promoter region that has been shown to drive its transcriptional activity. As shown by ELISA in Fig. 5D, TGLI1 expression rendered GBM cells to secrete more VEGF-A. Collectively, the results in Figs. 4 and 5 indicate for the first time that the human VEGF-A gene is expressed at high levels in TGLI1-expressing GBM xenografts and cell lines.
Figure 5. VEGF-A expression is elevated in TGLI1-expressing in human GBM cell lines.
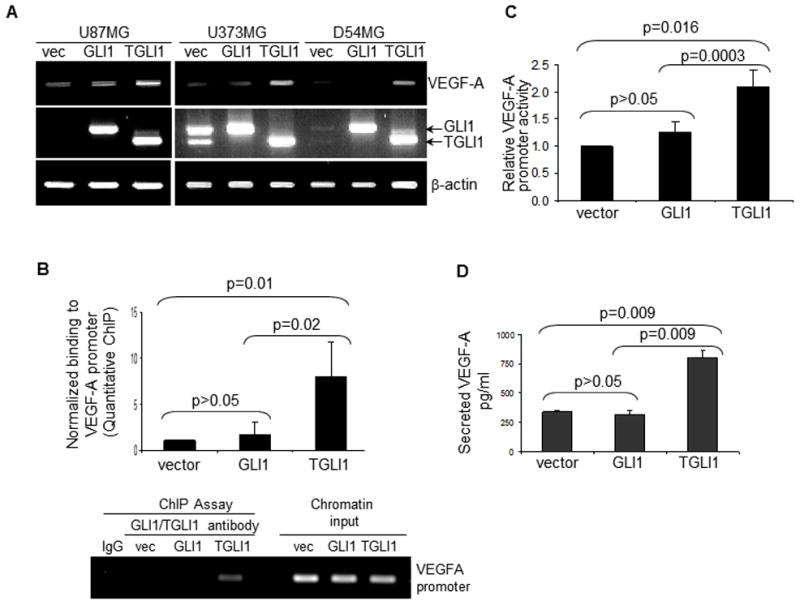
Three independent experiments were conducted to derive means and standard deviations. Student’s t-test was used to compute p-values.
A, VEGF-A expression is enhanced by TGLI1 in three human GBM cell lines, as indicated by RT-PCR.
B, TGLI1 strongly binds to the VEGF-A promoter, as indicated by the ChIP assay. The quantitative VEGF-A ChIP assay was conducted.
C, Human VEGF-A gene promoter activity is enhanced by TGLI1 expression.
D, TGLI1 expression rendered GBM cells secrete more VEGF-A, as shown by ELISA.
3.5. Double expression knockdown of both heparanase and VEGF-A significantly suppresses TGLI1-mediated in vitro angiogenesis of HBMECs
Since both heparanase and VEGF-A can promote angiogenesis and both genes can be upregulated by TGLI1, we determined the effects of dual downregulation of both genes on TGLI1-mediated angiogenesis. In these studies, we transfected U87MG-TGLI1 cells with heparanase and VEGF-A siRNAs, alone and in combination, and with control non-targeting siRNA. We also transfected U87MG-GLI1 cells with the control siRNA which served as the baseline control for the in vitro angiogenesis assay. Effectiveness and specificity of the siRNAs were indicated by the WB results in Fig. 6A (Supplemental Fig. 2). Notably, U87MG-TGLI1 cells with dual knockdown had reduced levels of heparanase and VEGF-A proteins to the levels that are comparable to those in U87MG-GLI1 cells. Next, we determined the ability of the conditioned medium from the transfected cells to regulate in vitro angiogenesis of HBMEC cells. As illustrated in Fig. 6B–D, double knockdown led to a significant reduction (~65%) of tubule formation while single knockdowns significantly reduced (~50%), but did not completely prevent in vitro angiogenesis. It is also noticeable that the doubly knock downed U87MG-TGLI1 cells behaved similarly to U87MG-GLI1 cells in modulating tubule formation. To determine whether the knockdowns affected U87MG cell proliferation, we conducted cell proliferation assay and did not find significant differences in proliferation rates. These results were further confirmed using additional siRNAs targeting different regions of heparanase and VEGF-A (Supplementary Fig. 2). Together, the results in Fig. 6 indicate that expression upregulation of both heparanase and VEGF-A is essential for TGLI1-mediated in vitro angiogenesis of HBMEC.
Figure 6. Double expression knockdown of heparanase and VEGF-A significantly reduces TGLI1-mediated in vitro angiogenesis of HBMEC cells.

A, U87MG-TGLI1 cells with dual knockdown contained low levels of heparanase and VEGF-A and the levels are equivalent to those in U87MG-GLI1 cells. Transfected cells were subjected to WB to determine the knockdown effectiveness. U87MG-GLI1 cells transfected with the control siRNA served as baseline control for the in vitro angiogenesis assay.
B, U87MG-TGLI1 cells with double knockdown abrogated their ability to promote tubule formation.
C,D, Quantitation results showing total tubule length (panel C) and branch points (panel D), relative to control siRNA-transfected U87MG-TGLI1 cells. Means and standard deviations were derived from data of three independent experiments. Student t-test was used to determine p-values.
3.6. TGLI1 splice junction-specific antibodies were developed and used to determine that TGLI1 was highly expressed in nearly half of the patient GBM samples we analyzed
We have found that commercially available antibodies we analyzed to date recognized both GLI1 and TGLI1 proteins. To our best knowledge, the existence of a TGLI1 Ab has not been reported. Thus, there is a need to develop TGLI1 junction-specific Abs. To meet this need, we recently developed rabbit polyclonal TGLI1-specific Abs via custom Ab service by YenZym Antibodies LLC. Briefly, a 15-residues peptide corresponding to the splice junction of exons 2–4 within TGLI1, but absent in GLI1, was used to immunize rabbits. Serum was subjected to several rounds of affinity column purifications followed by validations using ELISA and IHC. As shown by IHC in Fig. 7A, the TGLI1 splice junction-specific Ab yielded specific signals in a TGLI1-overexpressing xenograft (left panel). Middle panel shows that the TGLI1 signals were specifically blocked by the TGLI1 peptide used to raise the Abs. Right panel indicates that the TGLI1 Abs did not yield signals in a GLI1-overexpressing xenograft with very low TGLI1, indicating selectivity.
Figure 7. TGLI1 splice junction-specific antibodies were developed and used to determine that TGLI1 was highly expressed in nearly half of the patient GBM samples we analyzed.
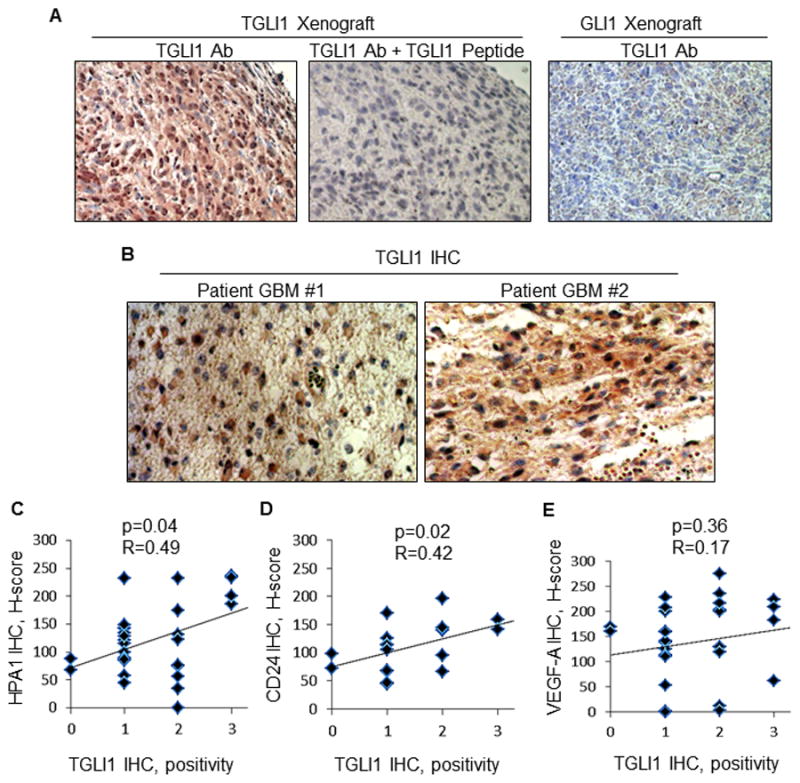
A, The TGLI1 splice junction-specific Abs yielded specific signals in a TGLI1-overexpressing xenograft (left panel). The TGLI1 signals were specifically blocked by the TGLI1 peptide used to raise the Abs (mid-panel). TGLI1 Abs did not yield signals in a GLI1-overexpressing xenograft with very low TGLI1, indicating selectivity (right panel).
B, Analysis of 30 patient GBM samples for TGLI1 expression frequency using IHC showed that 47% of the tumors expressed median (++) and high (+++) levels of TGLI1. Two representative GBM cases with high TGLI1 are shown.
C–E, TGLI1 expression was positively correlated with both CD24 (panel C and heparanase (panel D) levels, but not that of VEGF-A (panel E). Multiple regression analysis was conducted to determine R and p values.
Using the TGLI1 Abs, we analyzed 30 patient GBM samples for TGLI1 expression frequency using IHC. The results show that 47% of the tumors expressed median (++) and high (+++) levels of TGLI1. Two representative GBM cases are shown in Fig. 7B that express high levels of TGLI1. We further analyzed the tumor cohort for expression of three known TGLI1 target genes and correlated their levels with TGLI1 expression. As shown in Fig. 7C–E, multiple regression analysis indicated that TGLI1 expression is positively correlated with CD24 (panel C; p=0.04; R=0.49) and heparanase (panel D; p=0.02; R=0.42) levels, but not that of VEGF-A (panel E; p=0.56; R=0.17). We speculate that TGLI1 may better correlate with secreted VEGF-A rather than intracellular VEGF-A. Taken together, these results show that TGLI1 is highly expressed in nearly half of patient GBM samples we had analyzed and that TGLI1 expression is positively associated with levels of CD24 and heparanase.
4. Discussion
Kinzler and Vogelstein first reported the identification of the human GLI1 gene in 1987 [12] and characterized it as a member of the Kruppel family of zinc finger proteins in 1988 [10]. In the 1988 milestone study, the authors detected a shorter transcript in a human embryonal carcinoma cell line using northern blotting with a GLI1 probe [10]. Following an examination of the GLI1 genomic sequence in the cells, the authors concluded that no somatic abnormality was present in the GLI1 gene and did not further pursue the origin of the shorter transcript. In 2009, our laboratory unexpectedly isolated the TGLI1 isoform from a GBM cell line and showed it to be a product of an alternatively spliced GLI1 variant that contains an in-frame deletion of 123 bases corresponding to the entire exon 3 and part of exon 4 of the GLI1 gene [17]. In light of these reports, we speculate that the previously observed shorter GLI1 transcript [10] is that of TGLI1.
The 41 amino acid deletion in TGLI1 results in a loss of 4.4 kD from the full-length GLI1. Because of this small difference in molecular weight (146 kD for TGLI1; 150 kD for GLI1) and the fact that commercially available GLI1 antibodies detect both isoforms, we speculate that some of the previously reported GLI1 functionality may be shared by TGLI1 and GLI1 or attributed to TGLI1, but not GLI1. This speculation is supported by the results of our current study and two previous studies [17, 18] showing that TGLI1 gains the ability to upregulate VEGF-A and heparanase gene expression and thereby, enhances GBM angiogenesis, as well as, CD24 expression leading to enhanced migration and invasion. Also supporting this hypothesis is the notion that TGLI1 retains some functionality of GLI1 including the ability to undergo nuclear import and activate GLI1-regulated promoters and genes [17]. For the above mentioned reasons, we believe our findings present a provocative paradigm in which some of the known Hedgehog-GLI1 functionality is attributed to TGLI1, but not GLI1.
Notably, VEGF-A and heparanase promoters do not contain consensus GLI1-response elements. This is consistent with the lack of GLI1 binding to the promoters. This also suggests that TGLI1 has gained the ability to binds to the promoters, directly or indirectly. If TGLI1 directly binds to the promoters, TGLI1 may recognize a TGLI1-response element. If TGLI1 indirectly binds to the promoters, it may have gained the ability to interact with other transcription factors to co-regulate the transcription. These possibilities warrant future investigations.
Extremely high level of neoangiogenesis is one of the distinct histopathological features that separate GBM from other gliomas. GBM is among the most angiogenic solid malignancies. This feature enables GBMs to overcome the limitations in nutrients and oxygen supply when the tumor reaches a size of about 1–2 mm in diameter, leading to uncontrolled growth [31]. Excessive neovascularization in GBM tumors results in excessive leakiness of the neovasculature and this facilitates the high level of infiltration of tumor endothelium and GBM cells into parenchyma [31, 32]. Consequently, our results are of importance because they provide evidence suggesting that TGLI1 can be an important mediator of GBM angiogenesis, a feature central to its growth and infiltration. This link between TGLI1 and angiogenesis is also significant given the fact that sonic hedgehog has been shown to be involved in vascularity, although the exact downstream players are still not well defined [33–36]. Of note, Shh’s effects on vascularity could potentially be attributed to GLI1, GLI2, GLI3 and/or TGLI1 given our results demonstrating that Shh can activate TGLI1 transcriptional activity [17].
Our findings indicate that TGLI1 expression is higher in patient GBM tumors than in cultured GBM cell lines and that GBM cells serially passaged in nude mice maintained TGLI1 expression. This interesting observation is consistent with the notion that GBM specimens tend to lose their malignant features and molecular hallmarks after in vitro culturing. For example, cultured GBM cells are less invasive and losing gene amplification, such as, that of EGFR [37]. An important future task will be to further validate this interesting observation and elucidate the molecular mechanisms that promote TGLI1 expression in vivo.
In light of the positive role that TGLI1 plays in tumor neovascularization, TGLI1 has the potential to be an attractive target for novel anti-angiogenic therapy, not only for GBM, but also other cancers and diseases with excessive vascularity as the main pathobiological feature. This implication is worth investigating because anti-angiogenic treatment has been used to treat several angiogenesis-related ocular diseases, breast cancer and GBM [38, 39]; however, the therapy has yielded mixed results. In GBM patients, Avastin resistance is common and current efforts are being directed at finding Avastin-based combination therapy and other anti-angiogenesis agents for these patients [40, 41]. In light of these notions, our findings provide a strong rationale for inhibiting TGLI1 signaling as a new targeted therapy.
Several important future tasks are prompted by the findings in this study. The major ones are the following: 1) exploration of whether TGLI1 is associated with glioma stemness given that anchorage-independent growth is one of the unique features of cancer stem cells [42] and that GLI1 has been shown to be important for the proliferation of progenitor cells [43, 44], 2) investigation of the role of TGLI1 in tumorigenesis; 3) further characterization of the molecular mechanisms by which TGLI1 regulates gene expression, including, identification of consensus TGLI1-binding element, which will help with the identification of additional TGLI1-regulated genes and TGLI1 functionality; 4) elucidation of how TGLI1 expression is regulated which is currently unknown [45] and addressing this knowledge gap will help derive a means of blocking the alternative splicing event that produces TGLI1; 5) the regulation of TGLI1 by the non-canonical pathways that are known to regulate GLI1, including, Ras-MEK, Akt [46, 47] and transforming growth factor-β [48] that activate GLI1, as well as, PKA [49], PKC-β, Notch-HES1 [51] and p53 [52] that suppress GLI1; 6) investigating role of TGLI1 using orthotopic xenograft animal models and transgenic mouse models; and 7) deciphering the importance of heparanase as a mediator of TGLI1-associated tumor angiogenesis.
Supplementary Material
Acknowledgments
This study was supported by the NIH grant K01-CA118423, and W81XWH-11-1-0600 from the U.S. Department of Defense, the Pediatric Brain Tumor Foundation, the Beez Foundation and the Intramural Division of Surgical Sciences Dani P. Bolognesi, Ph.D. Award and Clarence Gardner, Ph.D. Award (to H.-W. L). We also thank Dr. Matthias Gromeier at Duke University for providing protein lysates of primary GBM specimens.
Footnotes
6. Conflict of Interest Statement
The authors declared no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 2.Roelink H, Augsburger A, Heemskerk J, Korzh V, Norlin S, Ruiz i Altaba A, Tanabe Y, Placzek M, Edlund T, Jessell TM, et al. Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell. 1994;76:761–775. doi: 10.1016/0092-8674(94)90514-2. [DOI] [PubMed] [Google Scholar]
- 3.Dahmane N, Sanchez P, Gitton Y, Palma V, Sun T, Beyna M, Weiner H, Ruiz i Altaba A. The Sonic Hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development (Cambridge, England) 2001;128:5201–5212. doi: 10.1242/dev.128.24.5201. [DOI] [PubMed] [Google Scholar]
- 4.Rao G, Pedone CA, Del Valle L, Reiss K, Holland EC, Fults DW. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene. 2004;23:6156–6162. doi: 10.1038/sj.onc.1207818. [DOI] [PubMed] [Google Scholar]
- 5.Chen BY, Liu JY, Chang HH, Chang CP, Lo WY, Kuo WH, Yang CR, Lin DP. Hedgehog is involved in prostate basal cell hyperplasia formation and its progressing towards tumorigenesis. Biochemical and biophysical research communications. 2007;357:1084–1089. doi: 10.1016/j.bbrc.2007.04.091. [DOI] [PubMed] [Google Scholar]
- 6.Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H, Koorstra JB, Habbe N, Karikari C, Mullendore M, Gabrielson KL, Sharma R, Matsui W, Maitra A. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Molecular cancer therapeutics. 2008;7:2725–2735. doi: 10.1158/1535-7163.MCT-08-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carpenter R, Lo HW. Hedgehog Pathway and GLI1 Isoforms in Human Cancer. Discovery Medicine. 2012;13 [PMC free article] [PubMed] [Google Scholar]
- 8.Kinzler KW, Vogelstein B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Molecular and cellular biology. 1990;10:634–642. doi: 10.1128/mcb.10.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon JW, Gilbertson R, Iannaccone S, Iannaccone P, Walterhouse D. Defining a role for Sonic hedgehog pathway activation in desmoplastic medulloblastoma by identifying GLI1 target genes. International journal of cancer. 2009;124:109–119. doi: 10.1002/ijc.23929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinzler KW, Ruppert JM, Bigner SH, Vogelstein B. The GLI gene is a member of the Kruppel family of zinc finger proteins. Nature. 1988;332:371–374. doi: 10.1038/332371a0. [DOI] [PubMed] [Google Scholar]
- 11.Zhu H, Lo HW. The Human Glioma-associated Oncogene Homolog 1 (GLI1) Family of Transcription Factors in Gene Regulation and Diseases. Current Genomics. 2010;11:238–245. doi: 10.2174/138920210791233108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kinzler KW, Bigner SH, Bigner DD, Trent JM, Law ML, O’Brien SJ, Wong AJ, Vogelstein B. Identification of an amplified, highly expressed gene in a human glioma. Science (New York, NY. 1987;236:70–73. doi: 10.1126/science.3563490. [DOI] [PubMed] [Google Scholar]
- 13.Bigner SH, Wong AJ, Mark J, Muhlbaier LH, Kinzler KW, Vogelstein B, Bigner DD. Relationship between gene amplification and chromosomal deviations in malignant human gliomas. Cancer genetics and cytogenetics. 1987;29:165–170. doi: 10.1016/0165-4608(87)90045-8. [DOI] [PubMed] [Google Scholar]
- 14.Forus A, Florenes VA, Maelandsmo GM, Meltzer PS, Fodstad O, Myklebost O. Mapping of amplification units in the q13–14 region of chromosome 12 in human sarcomas: some amplica do not include MDM2. Cell Growth Differ. 1993;4:1065–1070. [PubMed] [Google Scholar]
- 15.Wong AJ, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:6899–6903. doi: 10.1073/pnas.84.19.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimokawa T, Tostar U, Lauth M, Palaniswamy R, Kasper M, Toftgard R, Zaphiropoulos PG. Novel human glioma-associated oncogene 1 (GLI1) splice variants reveal distinct mechanisms in the terminal transduction of the hedgehog signal. The Journal of biological chemistry. 2008;283:14345–14354. doi: 10.1074/jbc.M800299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lo HW, Zhu H, Cao X, Aldrich A, Ali-Osman F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009;69:6790–6798. doi: 10.1158/0008-5472.CAN-09-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao X, Geradts J, Dewhirst MW, Lo HW. Upregulation of VEGF-A and CD24 gene expression by the tGLI1 transcription factor contributes to the aggressive behavior of breast cancer cells. Oncogene. 2012;31:104–115. doi: 10.1038/onc.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han W, Carpenter RL, Lo HW. TGLI1 upregulates expression of VEGFR2 and VEGF-A, leading to a robust VEGF-VEGFR2 autocrine loop and cancer cell growth. Cancer Hallmarks. 2013;1:28–37. [Google Scholar]
- 20.Chen TC, Wang W, Golden EB, Thomas S, Sivakumar W, Hofman FM, Louie SG, Schonthal AH. Green tea epigallocatechin gallate enhances therapeutic efficacy of temozolomide in orthotopic mouse glioblastoma models. Cancer Lett. 2011;302:100–108. doi: 10.1016/j.canlet.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 21.Hodgson JG, Yeh RF, Ray A, Wang NJ, Smirnov I, Yu M, Hariono S, Silber J, Feiler HS, Gray JW, Spellman PT, Vandenberg SR, Berger MS, James CD. Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro-oncology. 2009;11:477–487. doi: 10.1215/15228517-2008-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo HW, Cao X, Zhu H, Ali-Osman F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin Cancer Res. 2008;14:6042–6054. doi: 10.1158/1078-0432.CCR-07-4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu H, Cao X, Ali-Osman F, Keir S, Lo HW. EGFR and EGFRvIII interact with PUMA to inhibit mitochondrial translocalization of PUMA and PUMA-mediated apoptosis independent of EGFR kinase activity. Cancer Letters. 2010;294:101–110. doi: 10.1016/j.canlet.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J, Lund-Smith C, Borboa A, Gonzalez AM, Baird A, Eliceiri BP. Glioma-induced remodeling of the neurovascular unit. Brain research. 2009;1288:125–134. doi: 10.1016/j.brainres.2009.06.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zetser A, Bashenko Y, Edovitsky E, Levy-Adam F, Vlodavsky I, Ilan N. Heparanase induces vascular endothelial growth factor expression: correlation with p38 phosphorylation levels and Src activation. Cancer research. 2006;66:1455–1463. doi: 10.1158/0008-5472.CAN-05-1811. [DOI] [PubMed] [Google Scholar]
- 27.Vlodavsky I, Goldshmidt O, Zcharia E, Atzmon R, Rangini-Guatta Z, Elkin M, Peretz T, Friedmann Y. Mammalian heparanase: involvement in cancer metastasis, angiogenesis and normal development. Seminars in Cancer Biology. 2002;12:121–129. doi: 10.1006/scbi.2001.0420. [DOI] [PubMed] [Google Scholar]
- 28.Vreys V, David G. Mammalian heparanase: what is the message? Journal of cellular and molecular medicine. 2007;11:427–452. doi: 10.1111/j.1582-4934.2007.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Zhang N, Dai B, Liu M, Sawaya R, Xie K, Huang S. FoxM1B transcriptionally regulates vascular endothelial growth factor expression and promotes the angiogenesis and growth of glioma cells. Cancer Res. 2008;68:8733–8742. doi: 10.1158/0008-5472.CAN-08-1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson JC, McFarland BC, Gladson CL. New molecular targets in angiogenic vessels of glioblastoma tumours. Expert reviews in molecular medicine. 2008;10:e23. doi: 10.1017/S1462399408000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bello L, Giussani C, Carrabba G, Pluderi M, Costa F, Bikfalvi A. Angiogenesis and invasion in gliomas. Cancer treatment and research. 2004;117:263–284. doi: 10.1007/978-1-4419-8871-3_16. [DOI] [PubMed] [Google Scholar]
- 33.Kanda S, Mochizuki Y, Suematsu T, Miyata Y, Nomata K, Kanetake H. Sonic hedgehog induces capillary morphogenesis by endothelial cells through phosphoinositide 3-kinase. The Journal of biological chemistry. 2003;278:8244–8249. doi: 10.1074/jbc.M210635200. [DOI] [PubMed] [Google Scholar]
- 34.Nagase T, Nagase M, Machida M, Fujita T. Hedgehog signalling in vascular development. Angiogenesis. 2008;11:71–77. doi: 10.1007/s10456-008-9105-5. [DOI] [PubMed] [Google Scholar]
- 35.Chinchilla P, Xiao L, Kazanietz MG, Riobo NA. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle. 2010;9:570–579. doi: 10.4161/cc.9.3.10591. [DOI] [PubMed] [Google Scholar]
- 36.Harris LG, Pannell LK, Singh S, Samant RS, Shevde LA. Increased vascularity and spontaneous metastasis of breast cancer by hedgehog signaling mediated upregulation of cyr61. Oncogene. 2011 doi: 10.1038/onc.2011.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bigner SH, Humphrey PA, Wong AJ, Vogelstein B, Mark J, Friedman HS, Bigner DD. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990;50:8017–8022. [PubMed] [Google Scholar]
- 38.Lakka SS, Rao JS. Antiangiogenic therapy in brain tumors. Expert review of neurotherapeutics. 2008;8:1457–1473. doi: 10.1586/14737175.8.10.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, Vredenburgh J, Huang J, Zheng M, Cloughesy T. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 40.Sathornsumetee S, Desjardins A, Vredenburgh JJ, McLendon RE, Marcello J, Herndon JE, Mathe A, Hamilton M, Rich JN, Norfleet JA, Gururangan S, Friedman HS, Reardon DA. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro Oncol. 2010;12:1300–1310. doi: 10.1093/neuonc/noq099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE, 2nd, Marcello J, Norfleet JA, McLendon RE, Sampson JH, Friedman HS. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol. 2010;96:219–230. doi: 10.1007/s11060-009-9950-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Charafe-Jauffret E, Ginestier C, Birnbaum D. Breast cancer stem cells: tools and models to rely on. BMC cancer. 2009;9:202. doi: 10.1186/1471-2407-9-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merchant A, Joseph G, Wang Q, Brennan S, Matsui W. Gli1 regulates the proliferation and differentiation of HSCs and myeloid progenitors. Blood. 2010;115:2391–2396. doi: 10.1182/blood-2009-09-241703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang HH, Chen BY, Wu CY, Tsao ZJ, Chen YY, Chang CP, Yang CR, Lin DP. Hedgehog overexpression leads to the formation of prostate cancer stem cells with metastatic property irrespective of androgen receptor expression in the mouse model. J Biomed Sci. 2011;18:6. doi: 10.1186/1423-0127-18-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaphiropoulos P. Genetic variations and alternative splicing. The Glioma associated oncogene 1, GLI1, Frontiers in Genetics. 2012;3 doi: 10.3389/fgene.2012.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, Beermann F, Ruiz IAA. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5895–5900. doi: 10.1073/pnas.0700776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. The Journal of biological chemistry. 2007;282:14048–14055. doi: 10.1074/jbc.M611089200. [DOI] [PubMed] [Google Scholar]
- 48.Fan Q, He M, Sheng T, Zhang X, Sinha M, Luxon B, Zhao X, Xie J. Requirement of TGFbeta signaling for SMO-mediated carcinogenesis. The Journal of biological chemistry. 2010;285:36570–36576. doi: 10.1074/jbc.C110.164442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheng T, Chi S, Zhang X, Xie J. Regulation of Gli1 localization by the cAMP/protein kinase A signaling axis through a site near the nuclear localization signal. The Journal of biological chemistry. 2006;281:9–12. doi: 10.1074/jbc.C500300200. [DOI] [PubMed] [Google Scholar]
- 50.Cai Q, Li J, Gao T, Xie J, Evers BM. Protein kinase Cdelta negatively regulates hedgehog signaling by inhibition of Gli1 activity. The Journal of biological chemistry. 2009;284:2150–2158. doi: 10.1074/jbc.M803235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klopp AH, Lacerda L, Gupta A, Debeb BG, Solley T, Li L, Spaeth E, Xu W, Zhang X, Lewis MT, Reuben JM, Krishnamurthy S, Ferrari M, Gaspar R, Buchholz TA, Cristofanilli M, Marini F, Andreeff M, Woodward WA. Mesenchymal Stem Cells Promote Mammosphere Formation and Decrease E-Cadherin in Normal and Malignant Breast Cells. PLoS One. 2010;5:e12180. doi: 10.1371/journal.pone.0012180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stecca B, Ruiz i Altaba A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009;28:663–676. doi: 10.1038/emboj.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.