

Abstract
According to Stephen Jay Gould, “we have a strong preference for seeing trends as entities moving somewhere.” However, trends may instead be the product of relative expansions and contractions of different subpopulations constituting the system. Variation in attributes of coronary heart disease cases during the decline in coronary heart disease mortality suggests a change in the primary source-subpopulation of cases over time. It is proposed that an early 20th-century expansion of a coronary heart disease–prone subpopulation, characterized by high serum-cholesterol phenotype and high case-fatality—which contributed to most of the coronary heart disease cases and deaths during the 1960s—may have been a late result of the 1918 influenza pandemic. The same unusual immune response to infection that in 1918 killed preferentially men, whites, and those born from 1880 to 1900 (20–40 years old) may have “primed” survivors of those birth cohorts to late coronary heart disease mortality. Ecologic evidence in favor of a birth cohort and geographic association between both epidemics is presented. Cross-reactive auto-immune response upon reinfection could explain the excess coronary heart disease deaths reported during influenza epidemics from the late 1920s onward. Mimicry between the viral hemagglutinin and the apolipoprotein B or the low-density lipoprotein receptor could be the link between infection and hypercholesterolemia. The extinction of those birth cohorts would result in a relative increase in cases coming from a 2nd subpopulation, which was characterized by insulin resistance and chronic expression of low-grade inflammation markers and was comparatively less vulnerable to die acutely from coronary heart disease.
Key words: Cardiovascular diseases/history; coronary disease/epidemiology/mortality; demography; epidemic; history of medicine, 20th cent.; inflammation/immunology; inflammation mediators/immunology; influenza/epidemiology/immunology/pathology; risk factors; United States/epidemiology; population surveillance
How absurd … was the classical view of the heart as the “spring of all heat.” Knowing so little, the ancients should have attempted no explanation of the matter; whereas we, today, with all our facts, are now able to show that bodily heat is simply the mechanic result of the velocity of blood in its circulation.
—Hermann Boerhaave Institutiones Medicae, 1708
This paper attempts to reinterpret trends associated with coronary heart disease (CHD) over the last few decades—trends in death rates, clinical course, pathogenesis, and main risk factors—in the light of the Darwinian idea of variation 1,2 and then, in close agreement with this reinterpretation, to propose an association between the 1918 influenza pandemic and the 20th-century CHD epidemic.
The 20th century's rise and fall in CHD mortality was a worldwide phenomenon described by the World Health Organization in 1969 as “the greatest epidemic mankind has faced.” 3 In the United States, its emergence was retrospectively traced to 1925. 4 At its height, during the 1960s, CHD accounted for 35% of overall deaths in the United States. 5 Between 1970 and 2000, the CHD death rate declined 58%. If it had remained at its 1960s level, CHD would have accounted for 1,329,000 deaths in 2000, as opposed to the 514,000 actually reported. 6 Reasons for the decline are not yet clear.
During the 30-year decline, the attributes of CHD, the pathogenic concepts in regard to the disease, and the chief risk factors associated with CHD all experienced interesting modifications:
There was a continuing improvement in survivorship. Declines in case-fatality for myocardial infarction and reinfarction are well documented. 7,8 The age of patients presenting with myocardial infarction and unstable angina increased, which suggests that CHD is now delayed and possibly less severe in its initial clinical presentation. 9
-
The main pathogenic idea regarding causation changed from degeneration to inflammation. During the 1960s, hypercholesterolemia had been considered the hallmark of atherosclerosis, a condition understood as a bland lipid-storage disease. 10 The acute complications of atherosclerosis were attributed to high-grade coronary stenosis. This concept was reassessed during the 1990s. Inflammation, rather than plaque size, was established as the fundamental determinant of plaque instability and thrombosis. 10 In addition, low-grade chronic inflammation has been found to predict cardiovascular events, independent of the severity of the atherosclerotic burden. 10,11 According to Ridker and colleagues, 11 C-reactive protein may now be a stronger predictor of cardiovascular events than low-density lipoprotein (LDL) serum cholesterol levels. This shift from the degenerative to the inflammatory paradigm has favored a rediscovery of an 1858 description of “atheromatous affections of arteries” written by Rudolf Virchow: 12
One who knows that the fatty degeneration is here only a termination, and that the process is really a formative one, inasmuch as it begins with a proliferation—he can readily imagine the possibility of another termination, namely ossification …. As soon as the real ossification exists, we cannot help regarding the process as one which has arisen out of irritation of the parts stimulating them to new, formative actins; so far therefore it comes under our ideas of inflammation, or at least of those processes which are extremely nearly allied to inflammation. Average levels of distinct metabolic markers associated with the occurrence of CHD changed in opposite directions during the decline 13–16 (Fig. 1). National surveys conducted between 1960 and 2000 indicate that mean serum cholesterol levels in adults in the United States have consistently declined in all age and sex groups over the last 4 decades regardless of dietary habits. 15,17 In addition, during the last 30 years, 18,19 insulin resistance and its associated features—hypertriglyceridemia, low levels of high-density lipoproteins (HDL), moderate increases in “small-dense” LDL, hypertension, and obesity—have emerged as a “second atherogenic phenotype,” 20 to become the main determinant of CHD during the last decade. 21 An increase in the prevalence of obesity is well documented during this period. 16 Interestingly, obesity, diabetes, and hypertension (in addition to smoking and acute infections) were also the main attributes associated with atherosclerosis according to Osler, 22 in 1908 (before the CHD epidemic).
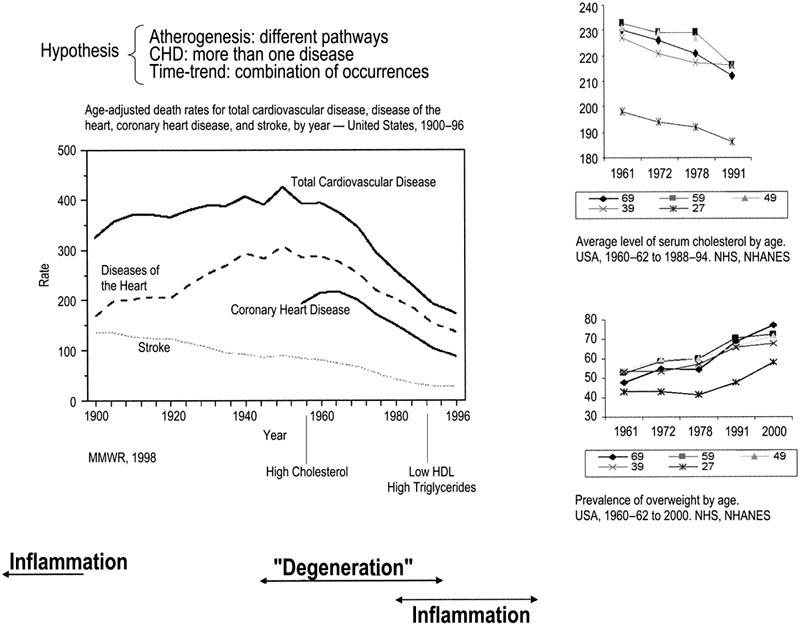
Fig. 1 Time trend in cardiovascular disease deaths and concurrent trends in average serum cholesterol levels and overweight during the decline in CHD deaths in the United States.
CHD = coronary heart disease; MMWR = Mortality and Morbidity Weekly Report; NHS = National Health Survey; NHANES = National Health and Nutrition Examination Survey
Trends in average serum cholesterol level and mean population weight during the last 40 years have been interpreted as representing a movement of the whole distribution of individual values in the same direction. According to Stephen Jay Gould, “we have a strong preference for seeing trends as entities moving somewhere.” 1 However, trends may instead be the product of relative expansions and contractions of different subpopulations constituting the system. 2 “The ensemble changes as a whole, by a change in the proportional representation of different subpopulations, which are themselves unchanging in their properties.” 1 In the case of the serum cholesterol trend, seeing the decline in average values as indicative of a movement of the whole distribution toward lower serum cholesterol levels implies looking for causal explanations, such as diet, that affect the entire population. Alternatively, interpreting the decline as a retraction of a subpopulation with intrinsically high average levels of serum cholesterol (and a correspondent relative expansion of its complement, the subpopulation with low average levels of serum cholesterol), suggests other questions, such as why (and how) this subpopulation decreased after the 1960s, and more importantly, why (and how) it grew so fast during the 1st half of the 20th century. Has this happened before? And last but not least, may CHD cases that originate from different subpopulations be considered the same disease?
When one looks at Fig. 1 with this new interpretation of trends in mind, it is possible to attribute the observed changes in risk factors profile, pathophysiology, and clinical course of CHD to a change in the source-subpopulation of cases: a continuous fall in the size of the CHD-prone subpopulation, characterized by a high serum-cholesterol phenotype and high case-fatality—which contributed to most CHD cases and deaths up to the 1960s—and a resulting relative increase in cases from a 2nd subpopulation, which was characterized by insulin resistance and chronic expression of low-grade inflammation markers and was comparatively less vulnerable to die acutely from CHD.
Still to be answered is the question, What would have caused a temporary expansion of a high average-serum-cholesterol subpopulation, the source of the majority of cases identified during the 20th-century CHD epidemic?
It has been shown that some infections may increase vulnerability to environmental challenges, 23–27 such as further infections 24,25 and high-fat diets. 26,27 A major infectious event worldwide that immediately preceded the rise in CHD mortality was the 1918 influenza pandemic. Twenty-five percent of the United States population (at least 25 million people) had overt influenza during that pandemic, which resulted in at least 500,000 excess deaths from influenza and pneumonia. 28 The unique characteristics of the Spanish flu of 1918 (H1N1 subtype) in comparison with the 1958 (H2N2 subtype) and 1968 (H3N2 subtype) influenza pandemics 28,29 were 1) unusually high morbidity and mortality rates; 2) high male-to-female and white-to-black morbidity and mortality ratios; and 3) the highest morbidity and mortality burden among young adults. According to expert opinion, death was not due to direct viral damage, but rather to the strength of the immune-inflammatory response to infection, which was greatest in robust, young, white, male adults. 28 We propose that whatever immune-inflammatory mechanism caused a sex and age pattern of mortality in 1918–19 also “primed” survivors in a similar fashion, predisposing them to future development of CHD. 30 If that is the case, then the relative distribution of influenza-related deaths from ages 15 to 49 in 1918–19 (a proxy for the distribution of some particular kind of immune-inflammatory response to infection across the range of exposed birth cohorts) should predict the occurrence of CHD mortality in survivors from the correspondent birth cohorts (from about 1870–1915) in the years to come. It is further hypothesized that the reported geographic variation in time of onset of the decline in CHD death rates depended on the varying persistence of H1N1 viruses across the United States, and through their effect, on a lower-level CHD “initiation” taking place in later-born cohorts.
Methods
This paper presents ecologic associations between influenza and CHD occurrences across birth cohorts and across geographic areas of the United States, in support of the hypotheses presented above. A detailed description of our methods has been published elsewhere. 30
Influenza Data. The 1918–19 influenza and pneumonia death rates were adjusted to reflect the ages that the deceased would have been in 1920, so that the age-specific rates could be plotted for birth cohorts according to the usual center of 10-year birth-cohort intervals (July 1 of years ending with 0 or 5).
Coronary Heart Disease Mortality Data. From 1960 to 1985, CHD mortality rates were calculated on the basis of gender- and age-specific data referent to the total United States population (all races), which were obtained from the Division of Vital Statistics of the National Center for Health Statistics. 31 To document birth-cohort trends over the course of the ascendant limb of the CHD epidemic curve, we used published tables on the numbers of deaths ascribed to coronary heart disease (and population), by age and sex, referent only to Seattle–King County (State of Washington). 32 In this subset of data, the assigned cause of death resulted from a review and tabulation of all death certificates registered in that area from 1920 to 1955, at every 5th year, in accordance with the 1955 international standards (International Classification of Diseases-7). Coronary heart disease mortality was then tabulated according to 10-year age and birth-cohort strata. To make the total CHD mortality burden comparable across different distributions of age in successive birth cohorts, we defined, separately for men and women, a referent birth cohort having a mid-period number of survivors at each successive 10-year age interval (from ages 40–49 to 80+) equal to the number of individuals at the respective 10-year age strata of the 1940 United States total population. 33 We then estimated the age-strata-specific standardized number of CHD deaths for each successive birth cohort. Finally, we graphically summed the age-strata-specific standardized numbers of deaths within each birth cohort to obtain a standardized estimate of the total CHD mortality burden for each 10-year birth cohort.
Geographic Correlation. We used the Spearman correlation coefficient to quantify the association across geographic divisions of the United States between the longer persistence of H1N1 influenza viruses (estimated by total excess death rate from influenza and pneumonia measured during the whole of each epidemic that occurred from 1931–1940 34) and the delayed beginning of decline in CHD death rates (estimated by the proportion of Metropolitan State Economic Areas in which the decline in CHD mortality had already begun in 1968 35).
Results
Birth-Cohort Trends. Figure 2 graphically compares the relative mortality associated with the 1918–19 pandemic with the CHD epidemic from 1920 to 1985 across successive birth cohorts, separately for men and women. The solid line connects points representing the distribution of mortality from influenza and pneumonia in 1918–19 for birth cohorts roughly corresponding to those with 10 to 50 years of age in 1918–1919 (see Fig. 1). The vertical bars display the standardized number of CHD deaths, shaded to represent deaths in different age strata, among the same birth cohorts. Pandemic-related death increases at the same rate as observed CHD death for cohorts born in the last third of the 19th century, regardless of sex. Both distributions attain their heights in cohorts born just before 1900 and then both start to fall, towards the later-born cohorts. In cohorts born after 1900, a greater CHD mortality than expected from the pandemic mortality curve can be seen, and this excess is somewhat larger for men.
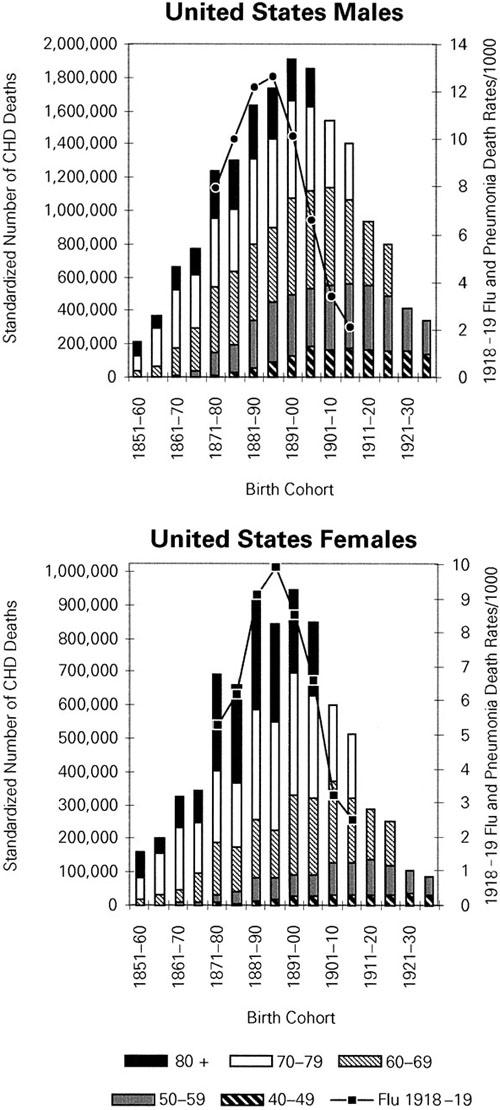
Fig. 2 Relative distributions of 1918–19 influenza and pneumonia death rates (line) and of 1920 to 1985 CHD standardized* number of deaths by age (bars) in successively born cohorts, according to age and sex—United States.
*Standard population: 1940 United States population, by sex and age strata.
CHD = coronary heart disease
Geographic Variability in Onset of CHD Decline. As seen in Figure 3, after 1930, H1N1 influenza-related excess deaths varied considerably across the geographic divisions of the United States, being less in the Northeast, Mid-Atlantic, East North Central, South Atlantic, and Pacific divisions compared with the West North Central, East South Central, West South Central, and Mountain divisions. The onset of decline in CHD mortality also varied considerably, having begun in 100% of the Northeast and 98.5% of the Pacific Metropolitan State economic areas in 1968, but in only approximately 60% of metropolitan areas of the West North Central and East South Central divisions.
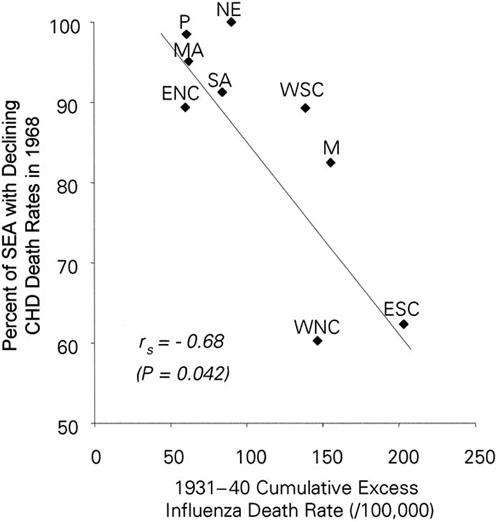
Fig. 3 Spearman (rs) correlation between persistence of H1 influenza activity after 1930 (measured by 1931–40 influenza excess death rates) and ordering of the beginning of the decline in coronary heart disease (CHD) deaths (measured by proportion of metropolitan SEA with declining CHD death rates in 1968 and earlier), across the geographic divisions of the United States.
ENC = East North Central; ESC = East South Central; M = Mountain; MA = Mid-Atlantic; NE = Northeast; P = Pacific; SA = South Atlantic; SEA = State Economic Areas; WNC = West North Central; WSC = West South Central
A notable negative correlation exists between excess influenza and pneumonia deaths after 1930 and the early decline in CHD death across the United States (rs = −0.68; P = 0.042).
Discussion
Age-related influenza and pneumonia mortality during the 1918–1919 pandemic predicts the relative distribution of CHD deaths across the correspondent birth cohorts, separately in men and women. The burden of H1N1 influenza activity after 1930, measured by influenza and pneumonia death rates, shows a strong association with delayed declines in CHD mortality rates across geographic divisions. Additionally, there are important socio-demographic similarities between those most affected by the 1918–19 influenza and those who died of CHD. Coronary heart disease mortality was always higher in men than in women. 4 Male: female death ratios during the pandemic also varied from 1.2 at ages 10 to 19 years to 1.7 at ages 40 to 49 years. 28 Coronary heart disease mortality was higher in whites than in blacks from the mid-1920s until about 1963, when a crossover in death rates occurred. 36 As mentioned, one of the unique characteristics of the 1918 pandemic was its unexpectedly high white-to-black mortality ratio. Not only was death from influenza and pneumonia lower in blacks but, during the pandemic, “death-rates for all causes of blacks between 25 and 45 years of age were below those of their white counterparts, probably for the only time in the history of the nation.” 28
Thus, these epidemiologic findings support our hypothesis that sex, race, age, and geographic differentials in the burden of pathogenic effects due to H1N1 influenza infection in the United States population could help explain the main epidemiologic characteristics—sex, race, birth cohort, and geographic pattern—of the 20th-century CHD mortality epidemic.
Speculations on Pathogenic Mechanisms. The proportion of the excess deaths during influenza epidemics that were attributed to organic heart diseases grew considerably during the ascending phase of the CHD epidemic, from 1.6% in 1918–19 to 18.4% in the minor epidemics that occurred from 1920 to 1929, 37 then to 51% (cardiovascular-renal causes) of the 86,000 excess deaths registered during the 1957 to 1960 Asian influenza epidemics. 38
Gordon and Thom 39 first suggested in 1975 that the reduction in death rates from CHD could be partially attributable to the continuing decline in influenza activity and the absence of extensive influenza epidemics after 1968. If the proposed hypothesis is correct, reduction in re-exposure of H1N1-primed individuals to repeated influenza infections could have been the determinant in the change in the disease-host relationship regarding CHD progression and death.
Infection by 1 strain of influenza A virus establishes an antibody (and possibly T-cell 40) response to infections by subsequent different strains of influenza viruses that is focused on epitopes shared by the original hemagglutinin antigens, a process known as “original antigenic sin.” 29,41 In a process similar to reactivation of rheumatic heart disease upon reinfection by group A β-hemolytic streptococcus, 42 immune responses elicited at each new encounter with an influenza virus may reactivate inflammatory pathways to CHD that were originally established by a 1st encounter with a H1N1 influenza virus and some specific immune response to it. In this regard, atherosclerotic lesions are mostly localized in areas that receive the highest loads of both viruses and immuno-inflammatory products from the infected lungs—the left side of the heart, the coronary arteries, and the aortic arch with its main branches.
Aside from a reinfection-driven autoimmune reactivation of endothelial inflammation that leads to CHD acute events or chronic progression of vascular disease (or both), another possibility must be considered: that flu infection or the immune response to it interferes with lipid metabolism, thereby increasing vulnerability to high serum cholesterol levels.
It has been experimentally shown 27 that Marek's disease herpesvirus–inoculated chickens, whether given cholesterol supplements or not, manifested significant increases in serum cholesterol in comparison with sham-inoculated groups that were given cholesterol or not. The amino acid antigenic domains of herpes simplex virus types 1 and 2 were shown to resemble the amino acid sequence in apoE (apolipoprotein E) that is involved in the attachment of this protein to LDL receptors. 43 Pleskov and co-authors 44 described, in some strains of influenza viruses, a significant mimicry of the amino acid sequences involved in cell attachment of the viral hemagglutinin with those of apoB involved in LDL binding to high-affinity LDL receptors. Upon re-infection, co-localization of anti-apoB antibodies, at sites of viral penetration in the vascular bed, might result in intimal LDL accumulation followed by oxidation and subsequent foam cell formation. 45
Although speculative, a mechanism involving cross-reactivity between the H1N1 influenza strains and apoB-LDL or the LDL receptor could be a link between infection and hypercholesterolemia and subsequent CHD death and might shed new light on the “diet–heart” controversy. 46 Within populations, the effect of dietary fat and cholesterol intake on serum cholesterol levels may depend relatively more on the efficiency of LDL uptake, which is possibly influenced by a cross-reactive immune response to a previous H1N1 influenza infection.
Final Remarks
The influenza hypothesis discussed herein would account for an early 20th-century expansion of a coronary heart disease–prone subpopulation. Its contraction reflected the natural dying out of those primed birth cohorts from the total population. Has it happened before?
Historical records indicate a possible occurrence of a rise and fall in CHD mortality in Britain over the last third of the 18th century. 47 Heberden's original description of the anginal syndrome in 1772, a time when its cardiac origin had not yet been established, followed a period of significant influenza activity in Britain, with epidemics of flu being recorded in 1727, 1732, 1737, and 1760. 28
Might CHD cases that originate in different subpopulations be considered the same condition? If what defines a disease is its subjacent pathophysiologic process, the answer is no. Interpreting the observed trend in CHD death as a sum of trends of 2 different conditions—one related to high average-serum-cholesterol, CHD-prone late-19th-century birth cohorts, and the other related to a subpopulation characterized by hypertension, insulin resistance, and chronic low-grade inflammation—may suggest a new hypothesis regarding determinants of current cases.
Footnotes
Address for reprints: Maria Inês Reinert Azambuja, MD, PhD, Faculdade de Medicina, UFRGS, Rua Ramiro Barcelos 2600, 4° andar, Porto Alegre, RS, 90035-003 – Brazil
E-mail: miazambuja@terra.com.br
This paper has its basis in a presentation made at the First Symposium on Influenza and Cardiovascular Disease: Science, Practice, and Policy, held on 26 April 2003, at the Texas Heart Institute, Houston, Texas.
References
- 1.Gould SJ. Full house: the spread of excellence from Plato to Darwin. New York: Harmony Books; 1996.
- 2.Lewontin R. The triple helix. Gene, organism and environment. Cambridge: Harvard University Press; 2001.
- 3.World Health Organization warns heart diseases are becoming mankind's greatest epidemic. Bull Int Soc Cardiol 1969;(1):1. Cited in: Gordon T, Kannel WB. Premature mortality from coronary heart disease. The Framingham study. JAMA 1971;215:1617–25. [PubMed]
- 4.Stallones RA. The rise and fall of ischemic heart disease. Sci Am 1980;243:53–9. [DOI] [PubMed]
- 5.Havlick, RJ, Feinleib M, editors. Proceedings of the Conference on the Decline in Coronary Heart Disease Mortality, 1978. Hyattsville: National Heart, Lung and Blood Institute, National Institutes of Health. Bethesda: U.S. Department of Health, Education and Welfare, PHS, NIH Pub no 79-1610, 1979.
- 6.National Heart, Lung and Blood Institute. Morbidity & mortality: 2002 chartbook on cardiovascular, lung and blood diseases. Rockville: U.S. Department of Health and Human Services, National Institutes of Health, 2002.
- 7.Elveback LR. Coronary heart disease in Rochester, Minn., 1950-75: incidence and survivorship. In: Feinleib M, Havlick RJ, Thom T, editors. Proceedings of the Conference on the Decline in Coronary Heart Disease Mortality, 1978. Hyattsville: National Heart, Lung and Blood Institute, National Institutes of Health. Bethesda: U.S. Department of Health, Education and Welfare, PHS, NIH Pub no 79-1610; 1979. p. 116-23.
- 8.Rosamund WD, Chambless LE, Folsom AR, Cooper LS, Conwill DE, Clegg L, et al. Trends in the incidence of myocardial infarction and in mortality due to coronary heart disease, 1987 to 1994. N Engl J Med 1998;339:861–7. [DOI] [PubMed]
- 9.Pepine CI. Changing myocardial infarction population characteristics: reasons and implications. Am Heart J 1997:134 (2 Pt 2):S1-4. [DOI] [PubMed]
- 10.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 2002;105:1135–43. [DOI] [PubMed]
- 11.Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med 2002;347:1557–65. [DOI] [PubMed]
- 12.Virchow R. Cellular pathology. As based upon physiological and pathological histology. Lecture XVI—Atheromatous affection of arteries. 1858. Cited in: Nutr Rev 1989; 47:23–5. [DOI] [PubMed]
- 13.Decline in deaths from heart disease and stroke—United States, 1900-1999. MMWR Morb Mortal Wkly Rep 1999; 48(30):649–56. [PubMed]
- 14.Pastor PN, Makuc DM, Reuben C, Xia H. Chartbook on trends in the health of Americans. Health, United States, 2002. Hyattsville, Maryland: National Center for Health Statistics; 2002.
- 15.Serum cholesterol levels among persons 20 years of age and over, according to sex, age, race, and Hispanic origin: United States, 1960–62, 1971–74, 1976–80, and 1988–94. In: Pastor PN, Makuc DM, Reuben C, Xia H. Chartbook on trends in the health of Americans. Health, United States, 2002. Hyattsville: National Center for Health Statistics; 2002. p. 211.
- 16.Healthy weight, overweight and obesity among persons 20 years of age and over, according to sex, age, race, and Hispanic origin: United States, 1960–62, 1971–74, 1976–80, and 1988–94. In: Pastor PN, Makuc DM, Reuben C, Xia H. Chartbook on trends in the health of Americans. Health, United States, 2002. Hyattsville: National Center for Health Statistics; 2002. p. 213.
- 17.Taubes G. Nutrition. The soft science of dietary fat. Science 2001;291:2536–45. [DOI] [PubMed]
- 18.World Health Organization. Cardiovascular disease risk factors: new areas of research. WHO Technical Report Series 841. Geneva: WHO; 1994. [PubMed]
- 19.Welborn TA, Wearne K. Coronary heart disease incidence and cardiovascular mortality in Busselton with reference to glucose and insulin concentrations. Diabetes Care 1979;2:154–60. [DOI] [PubMed]
- 20.Ziegler O, Guerci B, Drouin P. The “second atherogenic phenotype” or the role of insulin resistance in vascular risk [in French]. Arch Mal Coeur Vaiss 1998;91 Spec No 5:33–9. [PubMed]
- 21.Rapp RJ. Hypertriglyceridemia: a review beyond low-density lipoprotein. Cardiol Rev 2002;10:163–72. [DOI] [PubMed]
- 22.Osler W. Diseases of the arteries. In: Osler W, McCrae T, editors. Modern medicine: its theory and practice in original contributions by American and foreign authors. Vol 4. Philadelphia: Lea & Febiger; 1908. p. 426-47. Cited in: Nieto FJ. Infections and atherosclerosis: new clues from an old hypothesis? Am J Epidemiol 1998;148:937–48. [DOI] [PubMed]
- 23.Evans AS, Brachman PS. Emerging issues in infectious disease epidemiology. J Chron Dis 1986;39:1105–24. [DOI] [PubMed]
- 24.Morens DM. Antibody-dependent enhancement of infection and the pathogenesis of viral disease. Clin Infect Dis 1994;19:500–12. [DOI] [PubMed]
- 25.Ochiai H, Kurokawa M, Kuroki Y, Niwayama S. Infection enhancement of influenza A H1 subtype viruses in macrophage-like P388D1 cells by cross-reactive antibodies. J Med Virol 1990;30:258–65. [DOI] [PubMed]
- 26.Hajjar DP, Fabricant CG, Minick CR, Fabricant J. Virus-induced atherosclerosis. Herpesvirus infection alters aortic cholesterol metabolism and accumulation. Am J Pathol 1986:122:62–70. [PMC free article] [PubMed]
- 27.Njenga MK, Dangler CA. Intimal lipid accretion and elevated serum cholesterol in Marek's disease virus-inoculated chickens. Vet Pathol 1996;33:704–8. [DOI] [PubMed]
- 28.Crosby AW. America's forgotten pandemic: the influenza of 1918. New York: Cambridge University Press; 1990.
- 29.Dowdle WR. Influenza A virus recycling revisited. Bull World Health Organ 1999;77:820–8. [PMC free article] [PubMed]
- 30.Azambuja MI, Duncan BB. Similarities in mortality patterns from influenza in the first half of the 20th century and the rise and fall of ischemic heart disease in the United States: a new hypothesis concerning the coronary heart disease epidemic. Cad Saude Publica 2002;3:557–77. [DOI] [PubMed]
- 31.National Center for Health Statistics, Vital Statistics of the United States, Vol. II, “Mortality”. Published and unpublished data – selected years. Washington Public Health Services; 1990.
- 32.Ravenholt RT. Historical epidemiology and grid analysis of epidemiologic data. Am J Public Health 1966;52:776–90. [DOI] [PMC free article] [PubMed]
- 33.US Bureau of the Census. Estimates of the United States population and of the components of change, by age, color and sex: 1940–1950. Current Popul Rep Popul Estim Proj 1954;P25-98:6–8.
- 34.Gover, M. Influenza and pneumonia mortality in a group of 90 cities in the United States, August 1935-March 1943, with a summary for August 1920-March 1943. Public Health Rep 1943;58:1033–61.
- 35.Wing S, Hayes C, Heiss G, John E, Knowles M, Riggan W, Tyroler HA. Geographic variation in the onset of decline of ischemic heart disease mortality in the United States. Am J Public Health 1986;76:1404–8. [DOI] [PMC free article] [PubMed]
- 36.Gillum RF. Coronary heart disease in black populations. Am Heart J 1982;104(4 Pt 1):839–51. [DOI] [PubMed]
- 37.Collins SD. Excess mortality from causes other than influenza and pneumonia during influenza epidemics. Public Health Rep 1932;47:2159–79.
- 38.Eickhoff TC, Sherman IL, Serfling RE. Observations on excess mortality associated with epidemic influenza. JAMA 1961;176:776–82. [DOI] [PubMed]
- 39.Gordon T, Thom T. The recent decrease in CHD mortality. Prev Med 1975;4:115–25. [DOI] [PubMed]
- 40.Klenerman P, Zinkernagel RM. Original antigenic sin impairs cytotoxic T lymphocyte responses to viruses bearing variant epitopes. Nature 1998;394:482–5. [DOI] [PubMed]
- 41.Hennessy AV, Davenport FM. Epidemiologic implications of the distribution by age of antibody response to experimental influenza virus vaccines. J Immunol 1958:80:114–21. [PubMed]
- 42.Guilherme L, Cunha-Neto E, Tanaka AC, Dulphy N, Toubert A, Kalil J. Heart-directed autoimmunity: the case of rheumatic fever. J Autoimmun 2001;16:363–7. [DOI] [PubMed]
- 43.Becker Y. Computer prediction of antigenic and topogenic domains in HSV-1 and HSV-2 glycoprotein B (gB). Virus Genes 1992;6:131–41. [DOI] [PubMed]
- 44.Pleskov VM, Bannikov AI, Zaitsev IuV. The receptor-mediated endocytosis of influenza viruses and low-density lipoproteins by tissue cells [in Russian]. Vopr Virusol 1994;39:121–5. [PubMed]
- 45.Steinberg D, Witzum JL. Lipoproteins and atherogenesis. JAMA 1990;264:3047–52. [PubMed]
- 46.Blackburn H, Jacobs D. Sources of the diet-heart controversy: confusion over population versus individual correlations. Circulation 1984;70:775–80. [DOI] [PubMed]
- 47.Azambuja MI. Rise and fall in ischemic heart disease mortality—it may have happened before. Rev Saude Publica 1995;29:440–3. [DOI] [PubMed]