

Abstract
The task of evaluating correlations is central to computational structural biology. The rigid-body correlation problem seeks the rigid-body transformation (R, t), R ∈ SO(3), t ∈ ℝ3 that maximizes the correlation between a pair of input scalar-valued functions representing molecular structures. Exhaustive solutions to the rigid-body correlation problem take advantage of the fast Fourier transform to achieve a speedup either with respect to the sought translation or rotation. We present PFcorr, a new exhaustive solution, based on the non-equispaced SO(3) Fourier transform, to the rigid-body correlation problem; unlike previous solutions, ours achieves a combination of translational and rotational speedups without requiring equispaced grids. PFcorr can be straightforwardly applied to a variety of problems in protein structure prediction and refinement that involve correlations under rigid-body motions of the protein. Additionally, we show how it applies, along with an appropriate flexibility model, to analogs of the above problems in which the flexibility of the protein is relevant.
Keywords: Fast Fourier methods, Rigid-body motions, Match and Fit, Exhaustive search, Fast correlations
1. Introduction
Structural interactions between proteins are responsible for their functions as building blocks in our cells. In order to understand protein interactions, it is essential to determine the three-dimensional structure of the participating protein or smaller protein subunits. Based on the analysis of data obtained via X-ray crystallography, NMR spectroscopy or Electron Microscopy (EM), the structure of proteins can be evaluated. Typical tasks in protein structure evaluation include molecular replacement to identify protein crystal structures [39], protein-protein docking to calculate the structure of newly-formed protein complexes [19] or protein match and fit to refine low resolution maps of proteins by replacement of known atomic structures [53].
One common aspect of these tasks is an optimization problem. The solution space is the set of motions and transformations the molecules or groups of atoms can undergo, while the objective function evaluates the quality of the complex with respect to the task. The vast variety of approaches to this problem differ in the choice of the solution space, description of the molecules or molecular subunits, the way the solution space is searched, and the metric used to evaluate the quality of the complexes.
In this paper we address the optimization problem by a fast Fourier-based evaluation of correlations over three dimensional rigid-body motion, i.e., given a pair of functions, we find the rigid-body transformation such that the overlap of the two functions is maximized. The primary contribution of this work is PFcorr, a Fourier-based approach to solve multi-dimensional rotational correlation problems. PFcorr addresses two major deficiencies/drawbacks in prior Fourier-based approaches; we discuss these drawbacks in the following section, which also provides a brief overview of present work in this area. Section 3 provides necessary mathematical preliminaries to compute rigid-body correlations, which are described in detail in Section 4. We continue by giving a practical description of a matching/correlation procedure which can be used as a step-by-step guideline using PFcorr. This includes sampling considerations and numerical results. In Section 5, we apply PFcorr to the complementary problem of flexible multi-dimensional correlations, which takes into account the flexibility of proteins in solvent (see Figure 1.1). We solve the flexible correlation problem with a suitable parametrization of the space of flexible motions of the protein, after which each element of that space is just a rigid entity, conducive to rigid-body correlations.
Fig. 1.1.
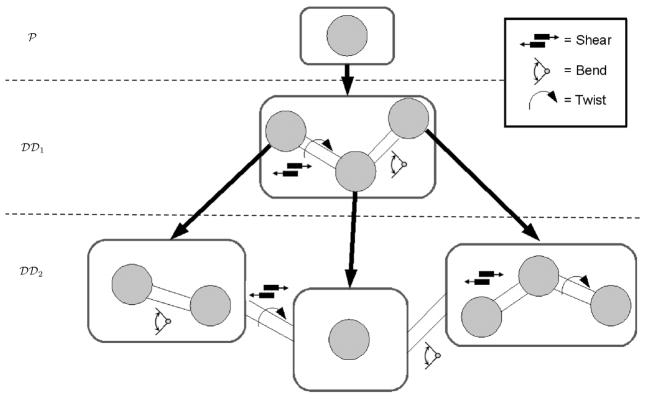
Domain-based flexible fitting as a sample application of the flexible correlation algorithm using PFcorr. The Figure chose an example of a domain decomposition with 3 hierarchical levels. The protein crystal structure
 is decomposed into domains at level
is decomposed into domains at level
 according to a chosen criterion. The domains of
are consecutively decomposed into domains at level
according to a chosen criterion. The domains of
are consecutively decomposed into domains at level
 . The motions between pairs of domains of
. The motions between pairs of domains of
 can be specified in a motion graph. They imply a set of rigid-body transformations with respect to the domains.
can be specified in a motion graph. They imply a set of rigid-body transformations with respect to the domains.
2. Related and prior work
Solving optimization problems for protein structure interpretation reduces to a correlation based scoring and search over a space of relative transformations. The objective function in general is highly non-convex, possessing several local maxima and minima. The vast number of existing solutions to the rigid-body correlation problem can be distinguished by a few basic approaches. Feature-based methods compute and correlate reduced representations of proteins. An early example of a feature-based approach is the method of vector quantization [54], in which sets of vectors are used to represent molecules. A similar approach is geometric hashing [23], whereby critical features are hashed into a table of values, and a score—related to the correlation score—measures the match between the participating proteins for a particular relative orientation. Feature-based approaches, used in docking [40] and fitting [50], result in improved performance due to the reduced search space, at the possible expense of poor resolution scaling.
Iterative approaches vary in sophistication, ranging from a simple version of steepest ascent [30] to more powerful techniques such as Powell optimization [51]. Most such approaches result in locally optimal solutions that, depending on the initial guess, may or may not be close to the globally optimal correlation. They are thus usually used in conjunction with an exhaustive approach that provides the requisite initial guess.
Exhaustive or Fourier-based approaches exploit the fact that it is beneficial if the computation of the objective function can be done relatively fast or if the search space is restricted. In these approaches the proteins are treated as rigid bodies. The automated search of all possible motions, i.e., translations and rotations to maximize the overlap between both structures is the main task of these programs. This is done by evaluating a correlation integral with respect to the motions using Fast Fourier transforms. Fourier based methods combine an accurate and exhaustive search with reduced computational cost, and have thus proven quite popular as a search scheme, see, e.g. [3, 7, 16, 26, 31, 37, 52, 57]. Many, if not most, existing Fourier-based docking algorithms use a regular discrete three-dimensional cartesian grid onto which the molecules are projected. The correlation score of these discretised and suitably weighted structures serves as the objective function for the optimization problem. The correlation score between pairs of grid cells is computed via fast Fourier transforms, thus implicitly searching over the three dimensional space of translations. The remaining rotational degrees of freedom however need to be incorporated into a global search. Such an approach has been first published by [19] in 1992. Since then, this approach has been adapted and improved many times. An overview of these translational grid-based FFT search schemes can be found in [9]. In recent approaches, the equispaced grid has been replaced with a non-equispaced Cartesian one, as in [3], or a polar one, as in [10, 13, 38, 44]. Fast translational matching exploits the fact that for each rotation, the objective function is a correlation integral, and can be computed by fast Fourier transforms. On the other hand, the space of rotations is still subject to exhaustive search.
While the optimal matching solution exists in a highly localized region of relative translations, the range of relative rotations varies widely for each translation. The disparity between size and sampling density of translational and rotational search spaces motivates this work. We present a fast rotational correlation matching that extends the methods of [38] and [44], which use spherical harmonic functions and classical orthogonal polynomials to model molecular shapes. We employ algorithms to compute the fast Fourier transform on the rotation group to solve the matching problem. In this work, instead of correlating functions defined on the unit cube, we use functions defined on ℝ3 but split into ℝ+ ×
 . We exploit the fact that correlations of functions defined on
can be computed by means of Fourier transforms on the rotation group. This enables efficient computation of the objective function over rotational degrees of freedom instead of translational degrees of freedom.
. We exploit the fact that correlations of functions defined on
can be computed by means of Fourier transforms on the rotation group. This enables efficient computation of the objective function over rotational degrees of freedom instead of translational degrees of freedom.
A three-dimensional translation can be expressed as a translation along the z-axis followed by two rotations, one about the y-axis and one about the z-axis. Hence, it has two rotational degrees of freedom and one translational. Combining this in a motion, we have five rotation angles that describe a motion and one absolute value of a translation along one axis. If we are able to speed up the computation for the rotations by correlating functions on the sphere, we get an improved complexity for five of the six degrees of freedom instead of the previous three. This approach has been suggested in [21] for protein fitting and can also be found in [10].
The essential mathematical tool used in this work for protein fitting is the fast calculation of the discrete Fourier transform on the rotation group SO(3). An implementation of such an algorithm can be found in [34]. For completeness, we also mention the related work of [15] and [6]. The paper of [15] uses representation theory of the rotation group for approaching optimization problems in the cryoEM setting, as we do. However the nature of our optimization problem is fundamentally different than theirs. The paper of [6] develops discrete Fourier transforms on the motion group SE(3), and applies it to topics ranging from workspace density of robotic manipulators to conformational statistics of macromolecules. This is quite different to our use of the fast discrete Fourier transform on the rotation group SO(3), to provide an efficient solution to our optimization problem.
Current exhaustive techniques suffer from two main drawbacks. The first drawback relates to local refinement. Depending as they do on the equispaced FFT, exhaustive techniques cannot be gracefully used to refine existing solutions. Say we wish to improve a matching pose, obtained using a translational FFT speedup with a certain grid size. If we redo the experiment with half the grid length of the previous computation, the three dimensional FFT becomes eight times as expensive, but more importantly, it spends much of its time at points on the new grid already excluded by the initial experiment. A similar argument applies to rotational speedups; in both these approaches, the concept of a local refinement is largely absent.
A second drawback relates to the question of uniform sampling in rotational space. While sampling in translational space is straightforward, involving Cartesian grids with uniform, possibly differing grid-sizes in each independent direction, the notions of uniformity and direction do not translate easily to the rotational space SO(3). In particular, equispaced Euler angular grids do not result in equispaced SO(3) samples as is illustrated in Figure 2.1. Due to this, rotational FFT-based techniques are destined to oversample certain regions of SO(3) while leaving others wholly unexamined.
Fig. 2.1.

(A) A uniform grid with respect to three Euler Angles grid with angular resolution 20° in each angle leads to (B) a non-uniform sampling of SO(3), with very high angular resolution in certain regions and holes in certain others. By contrast, (C) a highly non-uniform Euler angular grid leads to (D) a more uniform sampling of SO(3). (C) and (D) were obtained by the techniques in [28]; they contain fewer samples, exhibit a separation very close to the required angular resolution of 20°, and are highly uniform with respect to suitable metrics on SO(3). We discuss sampling on SO(3) in Section 4.1.
2.1. Proteins and flexibility
One main point of criticism of rigid-body fitting methods is that proteins undergo conformational changes during the induced fit, i.e., they not only move with respect to each other but also deform, shear or bend. Flexibility often involves movements between large rigid parts of the protein, called domains, flexible loops on the molecular surface and large side chain at active sites. A commonly used method to deal with flexibility is using multi-copying approaches or multi-term potentials. Due to the vastness of the space of flexible motions, protein flexibility can be practically dealt with by (A) conducting all-atomistic local searches, as in the case of molecular dynamical algorithms [17, 18, 24, 42, 43], (B) Building a coarse-grained representation of the protein, also known as a domain decomposition [1, 11, 33, 41], or (C) A combination of the strategies in (A) and (B) [47, 48, 59].
Domain-based approaches have so far lacked a search scheme that takes advantage of the translational or rotational speedups that FFT-based approaches can afford. This has to do with the issue of focusing: in uniform FFT-based techniques, there is no way to restrict the search space to a small area of interest that can be occupied by a single domain rather than the entire protein. By contrast, searching over the entire space for each domain is both time-consuming and results in spurious and geometrically implausible false positives, and sifting through these grows rapidly inefficient as the number of domains increases. This is also why domain-based flexibility algorithms such as those in [46, 47, 48] prefer Monte-Carlo-based or steepest-ascent-based search schemes.
2.2. Our contributions
We address the drawbacks mentioned in Section 2 with a pair of rotationally exhaustive, non-equispaced techniques to compute rigid-body correlations. The resulting family of techniques, which we call PFcorr, has the following properties:
Sampling robust. The technique is capable of efficiently computing correlations over arbitrary samples of rigid body motions ℝ3 × SO(3).
Compatible. It can be used along with existing equispaced FFT-based techniques.
General. It unifies the rotationally-exhaustive paradigms in [12, 22, 36, 38].
PFcorr thus provides an alternative to existing rigid-body correlation techniques.
The second half of this work presents an algorithm that uses PFcorr to explore correlations in multi-domain search spaces. The non-uniformity inherent to PFcorr implies that these correlations can be focused in a specific subset of ℝ3 ×SO(3), while its exhaustive nature guarantees that it is not sensitive to local optima. We believe that the above properties, along with its speed, make PFcorr a realistic and in many ways preferable alternative to existing correlation search schemes.
One of the two halves of PFcorr depends on looking up certain matrices describing the influence of a translation on the obtained series expansions. The high complexity of computing these matrix entries means that they often have to be precomputed and stored. We outline an efficient algorithm, based on polynomial update rules, that, while not obviating the need for precomputation and storage, has nevertheless a lower complexity than existing algorithms.
Finally, this work also aims to be a self-contained overview of correlation techniques that depend on expressing the input scalar valued functions in terms of rotationally invariant bases. In particular, we prove all relevant properties inherent to our mathematical framework.
3. Background
In this Section we give some necessary definitions and background information needed for the PFcorr algorithms. Note that we defer most multi-line proofs to the Appendix.
Let A, B: ℝ3 ↦ ℂ be a pair of scalar-valued functions. We define the rigid-body correlation problem as follows.
Definition 3.1
For two functions A: ℝ3 ↦ ℂ and B : ℝ3 ↦ ℂ we define
| (3.1) |
as the rigid-body correlation between A and B for a given set S = {(Ri, tj)}, Ri ∈ SO(3), tj ∈ ℝ3 of rigid-body motions. The rigid-body correlation problem is to maximize C(Ri, tj) over the set S.
The rigid-body correlation problem is a non-convex geometric optimization problem. The several problem domains in computational biology to which it applies can be distinguished by their choice of A and B. In protein-protein docking, for instance, A and B are affinity functions that represent a relevant property, such as shape or electrostatics, of the underlying protein; in protein-density map fitting, A is a blurred representation of the atoms of the protein, while B is the density map itself.
The objective function (3.1) can be efficiently calculated by expanding it in a series of orthogonal basis functions. The starting point of this approach is a coordinate transform of vectors x ∈ ℝ3 from Cartesian to spherical coordinates. The inner product of two square-integrable functions f, g : ℝ3 ↦ ℂ parameterized in spherical coordinates is given by
| (3.2) |
We now consider the orthogonal bases for the two components of the product space separately. Let ξ ∈
and let (ϕ, θ) ∈ [0, 2π) × [0, π] be its coordinates. For any l ∈ ℕ0 and m = −l, … l the spherical harmonics of degree l are defined as
where are associated Legendre polynomials, cf. [45], that arise as the derivatives of ordinary Legendre polynomials Pl(x).
The spherical harmonics satisfy the orthogonality relation
| (3.3) |
Secondly, we employ a weighted version of the Laguerre polynomials denoted by . These functions have been used to describe the radial part of the orbitals of hydrogenic atoms and are also known as radial wavefunctions, see [2, pp. 368 3] for general informations. In [36] these functions have been employed in the context of six-dimensional rigid-body docking.
Definition 3.2
For , l, k ∈ ℕ0, k > l, the weighted Laguerre polynomials are given by
using the Laguerre polynomials , see [45]. For , l, k ∈ ℕ0, k > l, the functions satisfy
| (3.4) |
Based on the previous orthogonality relations (3.3) and (3.4), we see that the functions for k, l ∈ ℕ, k > l ≥ |m| are orthonormal with respect to the inner product from (3.2). This follows immediately by
| (3.5) |
Moreover, these products of functions constitute an orthogonal basis of the space of square-integrable functions on ℝ3. Therefore, we find a unique series expansion of the two given functions A(x) and B(x) in terms of these functions as
| (3.6) |
with coefficients
| (3.7) |
and analogously for B(x).
Typically the initial data for A(x) and B(x) will be obtained by an EM or read in from a database as an atomic structure in terms of a collection of atoms and charges. Either way the methods only provide a finite number of samples of the unknown functions A and B. Hence the integral (3.7) will be approximated by a suitable quadrature rule. In PFcorr we use a combination of the Clenshaw-Curtis formula for the spherical part cf. [8, pp. 86] and a Gauss-Legendre formula for the radial part cf. [8, pp. 222]. Alternatives to such deterministically sampled quadrature schemes are quasi Monte-Carlo methods or Monte-Carlo methods. Since this is not the focus of this work we omit further details on quadrature and merely comment on the error induced by step.
Lemma 3.3
Let A : ℝ3 ↦ ℂ be a complex scalar-valued, 2-Lipschitz continuous function with finite support on the domain Ω ⊂ ℝ3. For a given spherical grid with maximum grid-diameter h1, for small h the coefficients âklm can be computed with an error E = Ch|Ω| for a constant C ∈ ℝ.
3.1. Multi-basis framework
As a first step in solving the rigid-body correlation problem in Equation 3.1, A and B are represented in terms of orthogonal basis functions. PFcorr offers two distinct choices of how to proceed.
-
Mixed bases. The standard approach uses the expansion (3.6) and approximates it by
(3.8) and analogously for BL(x). For convenience, we shall omit the subscript L from now on.
-
Pure spherical basis. This slightly modified approach following [22] and [12], divides the three dimensional space into discrete spherical slices and uses only a spherical basis expansion in terms of spherical harmonics on each slice with fixed radius r. The pure spherical representation of a scalar valued function A : ℝ3 ↦ ℂ for a given radial coordinate r is given by
(3.9) with coefficients(3.10) where Ar(u) = A(r, u).
The next step in solving the rigid-body correlation problem from Definition 3.1 involves applying a motion to the functions A and B. We assume that A and B are rigid bodies, and restrict the motion to rotations and translations in three-dimensional space.
3.2. Rotating basis expansions of scalar-valued functions
We shall now examine how a function expanded as in (3.8) behaves under the application of a rotation. We conveniently employ the representation property of spherical harmonics stating for arbitrary rotations R ∈ SO(3) that
| (3.11) |
where is a Wigner-D function [49].
The Wigner-D functions with degree l and orders m, n with max{|m|, |n|} ≤ l are given by the explicit expression
where α, γ ∈ [0, 2π) and β ∈ [0, π] are the Euler angle decomposition of a rotation R ∈ SO(3) and are the Wigner-d functions
| (3.12) |
are the Jacobi polynomials and
Note that is a polynomial of degree l if m + n is even. Otherwise, it is a polynomial of degree l − 1 times a factor of (1 − x2)1/2.
By virtue of (3.11), applying an arbitrary rotation R ∈ SO(3) to the given function A(x) will yield
Note that the rotation does not affect the radial parts of the function as a rotation preserves distance. Hence, a similar result holds for the radial-basis independent coefficients âlm.
Lemma 3.4
Given two functions A : ℝ3 ↦ ℂ and B : ℝ3 ↦ ℂ expanded in terms of a mixed basis as given in (3.8) the pure rotational correlation can be obtained by evaluating
| (3.13) |
for arbitrary choices of R ∈ SO(3). This is a direct result from using the orthogonality property (3.5) with the basis expansions of A(x) and B(RTx) in
Lemma 3.5
Given two functions A : ℝ3 ↦ ℂ and B : ℝ3 ↦ ℂ expanded in terms of a pure spherical basis as given in (3.9) the pure rotational correlation can be obtained by evaluating
| (3.14) |
for arbitrary choices of R ∈ SO(3).
3.3. Fourier Transforms on the rotation group SO(3)
To efficiently calculated the correlations (3.13), (3.13), we will use the Fast SO(3) Fourier Transform. For details on the algorithm we refer the reader to [34]. Here we simply outline the basic idea and show how it can be applied to compute our scoring function.
The space of square integrable functions in SO(3) is denoted L2 (SO(3)) and defined via the standard inner product
A convenient orthogonal basis for L2(SO(3)) are the Wigner-D functions which satisfy the orthogonality condition
Definition 3.6 (NDSOFT)
The nonequispaced discrete SO(3) Fourier transform (NDSOFT) is defined as the evaluation of the sums
| (3.15) |
for given Fourier coefficients and nodes (αq, βq, γq).
We outline our strategy for the fast approximate algorithm, a detailed description of this algorithm, called the nonequispaced fast Fourier transform (NFSOFT) can be found in [34]. We can rearrange (3.15) to
We can then calculate new coefficients
from the coefficients
in
 (L3 log2
L) arithmetic operations to rewrite the inner most sum for m, n = −L, …, L using the Chebyshev polynomials of first kind Tl(x),
(L3 log2
L) arithmetic operations to rewrite the inner most sum for m, n = −L, …, L using the Chebyshev polynomials of first kind Tl(x),
| (3.16) |
where χ = [m + n odd]. We are now able to replace the Chebyshev polynomials of first kind with complex exponentials,
We can compute the coefficients
from the coefficients
with
(L3) arithmetic operations. The obtained form is now ready to be inserted into (3.15) to become
| (3.17) |
This is a plain three-dimensional Fourier sum and we can use the NFFT algorithm to evaluate it with
(L3 log L + Q) operations, where Q is the number of nodes at which we evaluate the function; see [35]. Hence, the application of a NFSOFT results in
(L3 log2
L + Q) operations.
4. Rigid-body correlations
Although not immediately apparent, the idea of exploiting the rotational invariance of the spherical harmonics that serve as basis functions in the Fourier expansion of a functions in L2(
) has some advantages over translation-invariant Fourier expansion in [3, 7].
The key idea is to first express the three-dimensional translation in terms of two rotations and a translation in one dimension. Hence, this translation will have two rotational degrees of freedom and one translational. A three dimensional translation t ∈ ℝ3 of a object can be uniquely expressed as t = r RZ(ϕ)RY (θ)ez for ϕ ∈ [0, 2π), θ ∈ [0, π] and r ∈ ℝ+ where ez = (0, 0, 1)T. Combining, this with the three independent rotation parameters of the object, we have five rotation angles that describe a motion and one absolute value of a translation along one axis. Consequently, are able to speed up the computation for the rotations by spherical Fourier transforms and obtain an improved complexity for five of the six degrees of freedom of rigid-body correlations instead of the previous three. For U = RZ(ϕ)RY (θ), we have
| (4.1) |
with R̃ = UTR. A similar approach has been previously suggested in [21] for protein matching. Here we will focus on its efficient computation. A schematic depiction can be found in Figure 4.1.
Fig. 4.1.
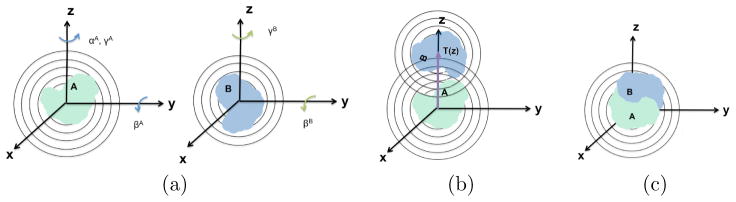
Schematic of the rigid-body correlation search scheme introduced in this work. Here A, B are two complex or real scalar-valued functions. In (a) the initial positions of A and B are given in different coordinate frames. Both functions are rotated. A is manipulated by a three-dimensional rotation, B only by a two dimensional rotation. In (b) A and B are translated to share the same origin. A set of a translation along the z-axis is searched until the best arrangement (c) is found.
After having considered the effects of rotations, it remains for us to examine the effect of the single one dimensional translation, say along the z-axis. In spherical coordinates a translation of the vector x about zez is given by x – zez = rzuz with
and
. We point out that the longitudal angle ϕ does not change during a translation along the z-axis. The effect of a translation along the z-axis on the ℝ+ ×
basis functions can be expressed in terms of translation matrix (T-matrix) elements
as described in [36] as
| (4.2) |
Note that the T-Matrices apply only to mixed basis expansions (3.8); for pure spherical basis expansions, the coefficients âlm for each radial slice with radius r have to be recomputed after each translation t ∈ ℝ3.
These translation coefficients are expressed as
| (4.3) |
where
Moreover denotes the Wigner 3-j symbol and (·)m is the Pochhammer symbol.
Directly computing T-Matrix entries in Equation 4.3 for fixed k, l, k′, l′, m takes
(L3Nt) steps, where Nt is the number of translations in one dimension. The overall complexity is thus L5·
(L3Nt) =
(L8Nt). An important contribution of the PFcorr algorithm is the fast and efficient computation of the T-Matrix entries in
(L7+L6Nt) steps. Details of this speedup can be found in the Appendix.
Having collected all the ingredients we state the following important Theorem.
Theorem 4.1
For a fixed cut-off degree L ∈ ℕ0 and two given functions
the objective function (3.1) can be evaluated by computing
for arbitrary choices of R ∈ SO(3) and t ∈ ℝ3. Its proof can be found in the Appendix.
Following an observation in [12], it is not as efficient to use the pure spherical basis expansions to express a general rigid-body correlation. Instead, Equation 3.14 is used along with a scan of the translational degrees of freedom, in which the basis coefficients are recomputed for each distinct t ∈ ℝ3. Hence we omit mentioning the case of pure spherical expansions here.
We conclude this section with some notes on the complexity of the evaluation of the introduced expansions. If we use the NFSOFT [34] to compute Equations (3.13) then the pure rotational correlation in Lemma 3.4 can be computed in steps using the following recipe, where NR is the number of distinct rotation angles per rotational degree of freedom, i.e., per Euler angle.
Recipe 1
Evaluate
| (4.4) |
for different choices of Euler angles.
Rearrange the multiple summations such that the sum over k becomes the innermost sum.
-
Compute
in
(L4) steps. -
Use the SO(3) Fourier transform to compute the remaining sums
in steps, where NR is the number of unique Euler angles per rotation axis.
In a similar fashion, the pure rotational correlation in Lemma 3.5 can be computed in
(L3 log2
L+NR + L3I) steps where I is the complexity of computing the integral ∫R+â(r)b̂(r)r2dr for a given pair of scalar-valued functions â, b̂: ℝ+ ↦ ℂ. Since there are
(L3) integrals ∫ℝ+â(r) b̂(r)r2dr we get the aforementioned complexity.
Let us now consider general rigid-body motion. The general rigid-body correlation in Theorem 4.1 can be computed in steps using the outlined a way to speed up computations of the translation matrix entries (4.3) and the NFSOFT, where and are the number of rotations of A and B respectively, and Nt is the number of one-dimensional translations. The computation is performed according to the following recipe.
Recipe 2
Evaluate
for NR different choices of R and Nt different choices of one-dimensional translations z ∈ ℝ
-
Compute
in
(L6) steps. -
Compute
using a modification of the NFSOFT in steps.
- Compute
-
Compute
using the standard NFSOFT [34] in steps.
Hence, the overall cost is , i.e., .
With these recipes established, we now outline algorithms to perform fast rigid-body correlations given a pair of scalar-valued functions as input. Algorithm 1 uses the mixed basis, while Algorithm 2 uses the pure spherical harmonic basis.
Algorithm 1.
Fast Rotational Correlation with mixed radial/spherical basis functions

|
Algorithm 2.
Fast Rotational Correlations with pure spherical harmonic basis functions

|
4.1. Rigid-body correlations: numerical results and discussion
There are three sources of error in PFcorr. The first is the expansion error, i.e., the error induced by truncating the basis expansion at a finite value of L. The second is the representation error, i.e., the error induced in numerically integrating the coefficients in Equation (3.7). The third is the NFFT error, i.e., the error induced by approximating the exponential sums by the NFFT.
Following [12, 36, 38], the first two sources of error can be respectively mitigated by choosing an expansion degree between 20 ≤ L ≤ 25, and using a single-point quadrature rule. We provide further evidence in Section 4.2 of the former assertion.
The NFFT approximates exponential sums with a kernel basis expansion, providing a choice of several kernels, and several parameters govern the actual error of the expansion. In our implementation, we choose the Gaussian kernel with an oversampling factor of 3, see [35], resulting in the errors in Table 4.1. On more information about the error of the NFFT and the NFSOFT we refer to [35] and [34], respectively. Note that, in solutions to the correlation problem, the absolute value of the correlation is less important than the value relative to other rigid-body rotations, i.e., the ability of the search scheme to discriminate between two different rigid-body motions. A measure of this ability is presented in Section 4.2 in the context of sampling arbitrary subsets of the motion group SE(3).
Table 4.1.
(A) Relative errors between the directly computed correlation and the correlation as computed by Recipes 1 and 2 for two real-valued functions A, B: ℝ3 → ℝ at varying expansion degrees over 500 randomly-generated rigid-body rotations. For L > 12, the direct correlation is exceedingly slow to compute therefore the relative error is omitted here. (B) The maximum complex value of the correlation score as a different error measure. Note that we used real valued input functions, and hence this table demonstrates the numerical error due to cut-offs. The above experiments were conducted with mixed bases; similar results hold for pure spherical bases, as the speedup scheme for these bases is the same as that used for Recipe 1.
| (A) | ||
|---|---|---|
| L | Recipe 1 | Recipe 2 |
| 4 | 1.93e-5 | 1.26e-4 |
| 6 | 1.37e-5 | 1.44e-4 |
| 8 | 1.61e-5 | 1.22e-4 |
| 10 | 1.61e-5 | 1.34e-4 |
| 12 | 1.72e-5 | 1.32e-4 |
| (B) | ||
|---|---|---|
| L | Recipe 1 | Recipe 2 |
| 4 | 1.71e-10 | 1.84e-10 |
| 6 | 1.53e-10 | 1.76e-10 |
| 8 | 1.72e-10 | 1.71e-10 |
| 10 | 1.77e-10 | 1.85e-10 |
| 12 | 1.41e-10 | 1.81e-10 |
We provide timing information in Figure 4.2. All timing information is from a single-threaded, dual core Macbook Pro at 2.3 GhZ with 8GB of RAM. For Recipe 1, we see that linear scaling with respect to the number of rotations and the quartic scaling with respect to expansion degree as predicted are reproduced by the implementation. For Recipe 2, the scaling with respect to expansion degree is not very important, as typically L6, the leading expansion term, is much less than L4NRB, which, for practical correlation problems, is in turn less than the product of the number of rotations NRA NRB. We hence examine how Recipe 2 scales with respect to the product NRA NRB; Figure 4.2(C), shows that the scaling is linear, as expected.
Fig. 4.2.

Time taken by each of the algorithms in Section 4. (A) Time requirements of the algorithms from Recipe 1 as a function of the number of rotations at fixed degree L = 25. Note that in Recipe 1 we search over three rotational degrees of freedom. (B) Time requirements of the algorithms from Recipe 2 as a function of the number of rotations at fixed degree L = 25. Note that in Recipe 2 we search over five rotational degrees of freedom. (B) Time requirements of the algorithms from Recipe 1 as a function of the maximum degree of the series expansion L at a fixed number of 30.000 rotations. (D) Time requirements of the T-Matrix computation as a function of the maximum degree of the series expansion L for the direct computation and the speedup.
For the T-Matrix computation (Figure 4.2(D)), a dramatic speedup with respect to the direct algorithm is observed in L ≥ 10 regime, where the L7 v/s L8 scaling is apparent. However, for typical values of L (see following paragraph), the computation times are still too slow to be usable in the inner loop of any Fourier-based correlation approach, including our own. Like prior work that uses the T-Matrix (See the introduction for an overview), we thus prefer to precompute and store T-Matrix entries for given values of z and λ (See Equation 4.3).
From a practical standpoint, our rigid-body correlation search is seen to be a viable, if somewhat slower, alternative to existing rigid-body correlation search techniques. Most of the degradation in performance is due to the NFFT, which uses, in its implementation, an oversampled FFT to enable the non-uniformity inherent to it. Following [38], we choose L to typically lie between 20 and 25, in which case typical run times for an exhaustive correlation involving about 1.5 · 107 distinct rigid-body samples lie between 2 and 3 minutes. We also note that, other than the argument in Section 4.2, there is no reason to prefer the non-uniformity inherent to PFcorr and, if performance is a concern, each of the steps involving the NFFT can be replaced by the equispaced FFT.
4.2. Sampling arbitrary subsets of the motion group SE(3); addressing the drawbacks of existing techniques
The main advantage of PFcorr is in sampling arbitrary (finite) subsets of the space of rigid body motion in three dimensions SE(3) = ℝ3 × SO(3). In our implementation (See Figure 4.3 for an example of its use in rigid-body fitting), this is as simple as specifying a set of rigid-body motions on which correlations are to be performed. By contrast, all prior techniques require an equispaced angular grid for rotational search, a property that results in a highly non-uniform search of the space of rotations (See Drawback 2 in the introduction). For exhaustive correlations between a pair of scalar-valued functions, one typically employs uniform sampling of the space of rotations SO(3). As we mention in the introduction, most of the uncertainty in the rigid-body correlation problem lies in the space of rigid-body rotations, and it is thus more important to sample this space exhaustively. There are several existing techniques that, given an angular sampling criterion, provide a set of samples that are uniform with respect to accepted metrics of uniformity [14, 28, 55]. We use the approach from [28], in which the metrics of local separation and global coverage compete to provide a set of highly uniform samples in SO(3). See also Figure 2.1.
Fig. 4.3.
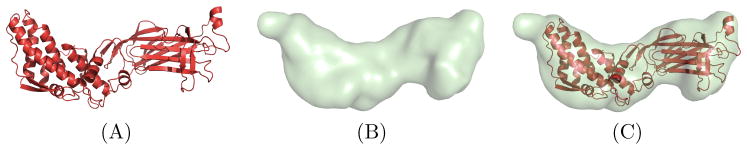
An example of a rigid-body fitting exercise. Here an atomic structure of the Rotavirus subunit is fit into a segmented 3D EM map. (A) Atomic structure of the rotavirus (PDB ID 1QHDa [27]). (B) Bilaterally smoothed rotavirus subunit segmented from EMD 1461 [58] at 3.8 Å using the techniques in [56]. (C) The atomic structure is placed into the 3D EM map subunit using the algorithms in PFcorr.
The ability to sample and correlate over arbitrary subsets of SE(3) is only useful if, at any expansion degree, the fineness of the rotational sampling does not exceed the accuracy with which âklm and b̂klm represent A and B respectively (See Equation (3.7)). Such a scenario would give rise to correlations that are so close to each other as to be essentially indistinguishable, and would result in a set of correlations clustered around the average correlation. To measure this tendency, we compute the Z-score , a measure of the distance of each individual correlation from the average. The results, in Figure 4.4, indicate that the top-ranking Z-score increases with increase in degree, as expected, leveling off at L ≥ 20, where the error due to floating-point calculations begins to rival the error due to representation, and that even at very low expansion degrees, the top-ranking Z-score is 3 standard deviations from the mean, indicating a very high confidence. Figure 4.4 also presents another argument as to why the regime 20 ≤ L ≤ 25 is best, as the latter provides a balance between the errors of representation and floating-point computation. For additional information on the Z-score measure see e.g. [32].
Fig. 4.4.
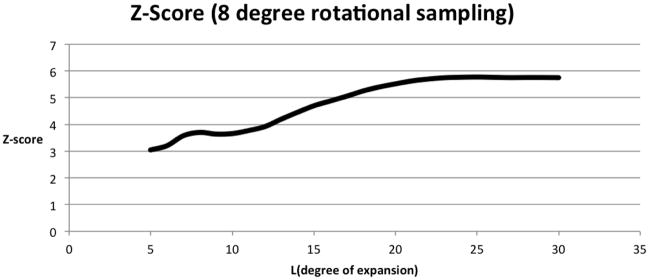
Average top-ranking Z-Scores for Recipe 1 at varying degrees. The rotational sampling fineness is fixed at 8° per Euler angle.
5. Flexible correlations: main results
We present an algorithm (Algorithm 3) for domain-based protein matching. This algorithm, given as input
A protein
,A hierarchical domain decomposition, defined in Section 5.1, of
,A scalar-valued function B: ℝ3 ↦ ℝ representing a stationary target, and,
A scalar-valued representation A of
,
produces as output the optimal correlation between A and B under rigid-body motions of the domains of
. Algorithm 3 makes use of the ability of PFcorr to uniformly sample arbitrary subsets of ℝ3
× SO(3).
5.1. Domain-based protein flexibility framework
We assume a generic framework for domain-based protein flexibility. This framework consists of ideas from domain-decomposition of proteins that have existed in various forms over the past decade (see especially [25]), as well as a set of techniques, described, for instance, in [4], to assign motions to each of these domains.
Let a protein crystal structure
comprise a set of atoms. Designate a subset of
as a domain D. A domain decomposition of
is a set
 = {Di}, 1 ≤ i ≤ n
= {Di}, 1 ≤ i ≤ n , where Di is a domain. A hierarchical domain decomposition
, where Di is a domain. A hierarchical domain decomposition
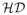 = {
}, 1 ≤ i ≤ n
= {
}, 1 ≤ i ≤ n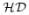 is a set of domain decompositions
such that each domain in
is a subdomain of some domain in
is a set of domain decompositions
such that each domain in
is a subdomain of some domain in
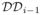 (See, for example, [5]). For each
of the hierarchical domain decomposition
, a motion graph MG specifying relative motions between domains of
can be specified. The motion graph consists of a set of edges Fij, called flexors, between pairs of domains i, j that undergo relative motion. The geometric properties of each flexor imply a set of rigid-body transformations (
), k ∈ {1 … NT} applied to Dj relative to Di [4].
(See, for example, [5]). For each
of the hierarchical domain decomposition
, a motion graph MG specifying relative motions between domains of
can be specified. The motion graph consists of a set of edges Fij, called flexors, between pairs of domains i, j that undergo relative motion. The geometric properties of each flexor imply a set of rigid-body transformations (
), k ∈ {1 … NT} applied to Dj relative to Di [4].
Algorithm 3.
Greedy multi-domain matching

|
5.2. Algorithm for flexible matching
Algorithm 3 applies to a particular domain decomposition of
, i.e, it applies to a particular index in the hierarchical domain decomposition of
. It uses the ability of PFcorr to sample arbitrary subsets of SE(3) to match representations of domains Ai ∈ A to a target scalar-valued function B: ℝ3 ↦ ℝ. Note by contrast that a classic equispaced Fourier-based correlation scheme would not be able to perform the tasks in Algorithm 3 without also producing several results that do not belong to the chosen subset of SE(3). This focusing property enables PFcorr to combine the merits of both local and global optimization schemes in the following sense. The algorithm is local in that it is restricted to the chosen subset of SE(3), but global in that it samples that subset exhaustively. It thus combines the speed of a local search without being sensitive, as local search algorithms are, to local optima.
6. Conclusion
We have presented PFcorr, a non-uniform correlation search scheme. PFcorr displays the following properties: (A) It is sampling robust, making searches over arbitrary subsets of SE(3) efficient while retaining the capabilities of classical exhaustive Fourier-based search schemes, (B) It is compatible with existing equispaced FFT-based techniques, in the sense that its non-equispaced nature is desirable but not necessary, and (C) Its algorithms extend to the rotationally exhaustive paradigms in [12, 22, 36, 38]. We have also presented an algorithm to compute translation matrix entries for SO(3) that achieves a better scaling than existing direct algorithms. Finally, we have presented an algorithm for multi-dimensional flexible correlations that leverages the sampling robustness of PFcorr. PFcorr applies to several fields within computational biology, including, most notably, molecular fitting and docking, where the above properties make it a natural and efficient tool for correlation-amenable search. A link to download PFcorr can be found at http://www.ices.utexas.edu/CVC/software/.
Acknowledgments
This work was supported in part by grants from the NIH R01 GM074258, R01-GM073087, R01-EB004873. Much of the work on this paper was accomplished when Benedikt Bauer and Antje Vollrath were visiting the University of Texas at Austin. Our in-house molecular modeling, image processing and visualization software tool, called VolumeRover, was used for producing the figures in this paper. The PFcorr library and the VolumeRover programs with associated documentation and tutorials can be freely downloaded from our CVC center’s software website (http://www.ices.utexas.edu/CVC/software/). We owe sincere thanks to Muhibur Rasheed and Deukhyun Cha for their immense help with software testing, debugging, documentation and distribution. To compute non-equispaced Fourier Transforms PFcorr depends on the NFFT software library [20].
Appendix
Here we give additional details on the mathematical background of the used algorithms.
T-Matrices Computation
The translation coefficients are polynomials of degree
Let d = 2k − l + 2k′ − l′ − 4, n = min(p, l + l′) − s and Then Equation (4.3) can be arranged to obtain
where
and s is even if and only if d is even.
The coefficients αp can be computed for all p in
(L3) steps. For fixed k, l, k′, l′, m, the T-Matrix polynomial can be computed in
(LNt). The complexity for fixed k, l, k′, l′, m is hence
(L3 +LNt), resulting in an overall complexity of
(L8 + L6Nt).
A polynomial can be evaluated at a set of equispaced arguments with
(L) multiplications. Applying Nuttall’s update rule for polynomials [29] reduces these multiplications to additions without altering the number of operations required. This affords a small speedup.
T-Matrices Computation Speedup
If is precomputed for all m, the other terms in Equation (4.3) have to be calculated only once for fixed k, l, k′, l′. In the first step, we compute
for all m and fixed n, l, n′, l′. The summation over j and the computation of
each takes
(L) steps, implying a complexity of
(L2) for each bn, and a complexity for all m of
(L3).
In the second step we compute the T-Matrix entries .
Since the above calculation has to be done for all k, l, k′, l′ and for Nt translations, the overall complexity for
is now
(L7Nt), instead of
(L8).
Computation of the coefficients can also be sped up. Only these coefficients and the boundary of the innermost sum depend on k and k′. If k and k′ are switched, the boundary of the sum does not change, so for switched k and k′ only the value changes. In the first step
is computed for all j, n, l, l′. In the second step
and tk′l,kl′ respectively are computed. In the third step
and respectively are computed.
Moreover, the symmetry property [36] implies . Hence, the dynamic programming approach above allows us to calculate and by calculating .
The complexity of the approach of representing the T -coefficients as a polynomial can be reduced by using the speed-up by dynamic programming as explained above. To achieve the reduction in the complexity we consider the calculation of αp. Instead of computing αp directly, first
is precomputed. Due to the summation and the parameters s and p, this computation has the complexity
(L3) Afterwards the αp
are computed This has the complexity
(L2), implying a complexity of
(L3) for the precomputation of αp for all m. The total computation of the αp for all m is hence
(L3 + L3) =
(L3).
The subsequent computation of
is for fixed k, l, k′, l′, m and all m is
(L2Nt). Therefore the overall complexity for fixed k, l, k′, l′ and all m is
(L3 + L2Nt). Thus, for all k, l, k′, l′ the complexity is
(L4)
(L3 + L2Nt) =
(L7 + L6Nt).
Proof of Lemma 3.3
Let Ω be subdivided in N grid-cells Ωi with centers xi, volume Vi and diameter di. The approximation error in the ith grid-cell is given by
Expanding A(x) in a Taylor series about xi, we get
for some constant c, due to A(x) being 2-Lipschitz continous on Ω. Thus the error across all grid cells is the sum . Since the maximum diameter of the grid-cells is proportional to the grid fineness h, we have E ≤ Ch|Ω| for a fixed constant C.
Proof of Theorem 4.1
Consider a rotation R ∈ SO(3) that is applied to the molecule A. By the representation property of spherical harmonics 3.11 the affinity function becomes
The molecule B will be rotated by U = RZ (ϕ)RY (θ) and translated by the vector (0, 0, z)T. Using (4.2), this yields the series expansion
After inserting both of the above expansions of the affinity functions into the correlation integral (4.1), we are now able to use the orthonormality property
to simplify the correlation integral to
If we now approximate the infinite sums by sums with a certain maximum degree L we obtain the formula from Theorem 4.1.
Footnotes
The grid-diameter is the diameter of the smallest ball that the grid-cell can be enclosed in.
Contributor Information
CHANDRAJIT BAJAJ, Email: bajaj@cs.utexas.edu.
BENEDIKT BAUER, Email: bauer@evolbio.mpg.de.
RADHAKRISHNA BETTADAPURA, Email: brrk@utexas.edu.
ANTJE VOLLRATH, Email: a.vollrath@tu-bs.de.
References
- 1.Abyzov A, Bjornson R, Felipe M, Gerstein M. RigidFinder: A fast and sensitive method to detect rigid blocks in large macromolecular complexes. PROTEINS: Structure, Function, and Bioinformatics. 2010;78:309–324. doi: 10.1002/prot.22544. [DOI] [PubMed] [Google Scholar]
- 2.Atkins P, de Paula J. Physical Chemistry for the Life Sciences. Oxford University Press; 2006. [Google Scholar]
- 3.Bajaj C, Chowdhury R, Siddahanavalli V. F2Dock: Fast fourier protein-protein docking. IEEE/ACM Trans Comput Biol Bioinf. 2011;8(1):45–58. doi: 10.1109/TCBB.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bajaj C, Chowdhury RA, Siddavanahalli V. Technical report. The University of Texas; 2007. F3dock: A fast, flexible and fourier-based approach to protein-protein docking. [Google Scholar]
- 5.Bettadapura R, Vollrath A, Bajaj C. Technical Report 12–29. University of Texas; Austin: Jul, 2012. Pfflexfit: Hierarchical flexible fitting in 3d em. [Google Scholar]
- 6.Chirikjian G, Ebert-Uphoff I. Numerical convolution on the euclidean group with applications to workspace generation. IEEE Trans on Robotics and Automation. 1998;14(1):123–136. [Google Scholar]
- 7.Chowdhury R, Rasheed M, Keidel D, Moussalem M, Olson A, Bajaj C. Protein-protein docking with f2dock 2.0 and gb-rerank. PLoS ONE. 2013;8(3):e51307. doi: 10.1371/journal.pone.0051307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis PJ, Rabinowitz P. Methods of Numerical Integration. 2. Academic Press Inc; 1984. [Google Scholar]
- 9.Eisenstein M, Katchalski-Katzir E. On proteins, grids, correlations, and docking. C R Biol. 2004;327:409–420. doi: 10.1016/j.crvi.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Esquivel-Rodriguez J, Kihara D. Fitting multimeric protein complexes into electron microscopy maps using 3d zernike descriptors. J Phys Chem B. 2012;116:6854–6861. doi: 10.1021/jp212612t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flores S, Lu L, Yang J, Carriero N, Gerstein M. Hinge atlas: relating protein sequence to sites of structural flexibility. BMC Bioinformatics. 2007;8:167–186. doi: 10.1186/1471-2105-8-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garçon JI, Kovacs JA, Abagyan R. Adp em: Fast exhaustive multi-resolution docking with high-throughput coverage. Bioinformatics. 2007;23(4):427–433. doi: 10.1093/bioinformatics/btl625. [DOI] [PubMed] [Google Scholar]
- 13.Garzon J, Lopéz-Blanco J, Pons C, Kovacs JA, Abagyan R, Fernandez-Recio J, Chacon P. FRODOCK: a new approach for fast rotational protein-protein docking. Bioinformatics. 2009;25:2544–2551. doi: 10.1093/bioinformatics/btp447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gräf M, Potts D. Sampling sets and quadrature formulae on the rotation group. Numer Funct Anal Optim. 2009;30:665–688. [Google Scholar]
- 15.Hadani R, Singer A. Representation theoretic patterns in three dimensional cryo-electron microscopy I - the intrinsic reconstitution algorithm. Annals of Mathematics. 2011 doi: 10.4007/annals.2011.174.2.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halperin I, Ma B, Wolfson H, Nussinov R. Principles of docking: An overview of search algorithms and a guide to scoring functions. PROTEINS: Struct Funct Genet. 2002;47:409–443. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 17.Holm L, Sander C. Parser for protein folding units. Proteins. 1994 Jul;19(3):256–268. doi: 10.1002/prot.340190309. [DOI] [PubMed] [Google Scholar]
- 18.Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. Namd2: Greater scalability for parallel molecular dynamics. J Comput Phys. 1999;151(1):283–312. [Google Scholar]
- 19.Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem A, Aflalo C, Vakser I. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc Nat Acad Sci USA. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keiner J, Kunis S, Potts D. Using NFFT3 - a software library for various nonequispaced fast Fourier transforms. ACM Trans Math Software. 2009;36:Article 19, 1–30. [Google Scholar]
- 21.Kovacs JA, Chacón P, Cong Y, Metwally E, Wriggers W. Fast rotational matching of rigid bodies by fast Fourier transform acceleration of five degrees of freedom. Acta Crystallogr Sect D. 2003;59:1371–1376. doi: 10.1107/s0907444903011247. [DOI] [PubMed] [Google Scholar]
- 22.Kovacs JA, Wriggers W. Fast rotational matching. Acta Crystallographica Section D. 2002 Aug;58(8):1282–1286. doi: 10.1107/s0907444902009794. [DOI] [PubMed] [Google Scholar]
- 23.Lamdan Y, Wolfson H. Geometric hashing: a general and efficient model-based recognition scheme. Proceedings of the IEEE International Conference on Computer Vision; 1988. pp. 238–249. [Google Scholar]
- 24.Leech J, Prins J, Hermans J. Smd: visual steering of molecular dynamics for protein design. IEEE Computational Science and Engineering. 1996;3(4):38–45. [Google Scholar]
- 25.Maiorov VN, Abagyan RA. A new method for modeling large-scale rearrangements of protein domains. Proteins. 1997;27:410–424. doi: 10.1002/(sici)1097-0134(199703)27:3<410::aid-prot9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 26.Mandell JG, Roberts VA, Pique ME, Kotlovyi V, Mitchell JC, Nelson E, Tsigelny I, Eyck LFT. Protein docking using continuum electrostatics and geometric fit. Protein Engineering Design and Selection. 2000;14(2):105–113. doi: 10.1093/protein/14.2.105. [DOI] [PubMed] [Google Scholar]
- 27.Mathieu M, Petitpas I, Navaza J, Lepault J, Kohli E, Pothier P, Prasad B, Cohen J, Rey F. Atomic structure of the major capsid protein of rotavirus: implications for the architecture of the virion. EMBO J. 2001;20:1485–1497. doi: 10.1093/emboj/20.7.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitchell JC. Discrete uniform sampling of rotation groups using orthogonal images. SIAM Journal of Scientific Computing. 2007;30(1):525–547. [Google Scholar]
- 29.Nuttall AH. Efficient evaluation of polynomials and exponentials of polynomials for equispaced arguments. IEEE Transactions On Acoustics, Speech And Signal Processing. 1987;35(10):1486–1487. [Google Scholar]
- 30.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. Ucsf chimera–a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25(13):1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 31.Pierce BG, Hourai Y, Weng Z. Accelerating protein docking in zdock using an advanced 3d convolution library. PLoS One. 2011;6(9) doi: 10.1371/journal.pone.0024657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pintilie GD, Zhang J, Goddard TD, Chiu W, Gossard DC. Quantitative analysis of cryo-em density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to quantitative analysis of cryo-em density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J Struct Biol. 2010;170:427–438. doi: 10.1016/j.jsb.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poornam G, Matsumoto A, Ishida H, Hayward S. A method for the analysis of domain movements in large biomolecular complexes. PROTEINS: Structure, Function, and Bioinformatics. 2009;76:201–212. doi: 10.1002/prot.22339. [DOI] [PubMed] [Google Scholar]
- 34.Potts D, Prestin J, Vollrath A. A fast algorithm for nonequispaced fourier transforms on the rotation group. Numerical Algorithms. 2009;52(3):355–384. [Google Scholar]
- 35.Potts D, Steidl G, Tasche M. Fast Fourier transforms for nonequispaced data: A tutorial. In: Benedetto JJ, Ferreira PJSG, editors. Modern Sampling Theory: Mathematics and Applications. chapter 12. Birkhäuser; Boston: 2001. pp. 247–270. [Google Scholar]
- 36.Ritchie DW. High order analytic translation matrix elements for real six-dimensional polar Fourier correlations. J Appl Cryst. 2005;38:808–818. [Google Scholar]
- 37.Ritchie DW. Recent progress and future directions in protein-protein docking. Curr Prot Pep Sci. 2008;9:1–15. doi: 10.2174/138920308783565741. [DOI] [PubMed] [Google Scholar]
- 38.Ritchie DW, Kozakov D, Vajda S. Accelerating and focusing protein-protein docking correlations using multi-dimensional rotational 3t generating functions. Bioinformatics. 2008;24:1865–1873. doi: 10.1093/bioinformatics/btn334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rossmann M. The molecular replacement method. Acta Crystallogr Sect A. 1990;46:73–82. doi: 10.1107/s0108767389009815. [DOI] [PubMed] [Google Scholar]
- 40.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. Geometry-based flexible and symmetric protein docking. Proteins: Structure, Function, and Bioinformatics. 2005;60(2):224–231. doi: 10.1002/prot.20562. [DOI] [PubMed] [Google Scholar]
- 41.Shatsky M, Nussinov R, Wolfson H. Flexible protein alignment and hinge detection. PROTEINS: Structure, Function, and Genetics. 2002;48:242–256. doi: 10.1002/prot.10100. [DOI] [PubMed] [Google Scholar]
- 42.Skeel RD, Tezcan I, Hardy DJ. Multiple grid methods for classical molecular dynamics. Journal of Computatioanl Chemistry. 2002;23(6):673–684. doi: 10.1002/jcc.10072. [DOI] [PubMed] [Google Scholar]
- 43.Stone JE, Gullingsrud J, Schulten K. A system for interactive molecular dynamics simulation. Proceedings of the 2001 symposium on Interactive 3D graphics; ACM Press; 2001. pp. 191–194. [Google Scholar]
- 44.Sumikoshi K, Terada T, Nakamura S, Shimizu K. A fast protein-protein docking algorithm using series expansions in terms of spherical basis functions. Genome Informatics. 2005;16:161–193. [PubMed] [Google Scholar]
- 45.Szego G. Orthogonal Polynomials. 4. Amer. Math. Soc; Providence: 1975. [Google Scholar]
- 46.Topf M, Baker ML, John B, Chiu W, Sali A. Structural characterization of components of protein assemblies by comparative modeling and electron cryo-microscopy. Journal of Structural Biology. 2005;149:191–203. doi: 10.1016/j.jsb.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 47.Topf M, Lasker K, Webb B, Wolfson H, Chiu W, Sali A. Protein structure fitting and refinement guided by cryoem density. Structure. 2008;16(2):295–307. doi: 10.1016/j.str.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008 doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wigner EP, Griffin JJ. Group theory and its application to the quantum mechanics of atomic spectra. Vol. 4. Academic Press; New York: 1959. [Google Scholar]
- 50.Woetzel N, Lindert S, Stewart P, Meiler J. Bcl::em-fit: Rigid body fitting of atomic structures into density maps using geometric hashing and real space refinement. Journal of Structural Biology. 2011;3:264–76. doi: 10.1016/j.jsb.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wriggers W. Using situs for the integration of multi-resolution structures. Biophysical Reviews. 2010;2(1):21–27. doi: 10.1007/s12551-009-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wriggers W, Chacon P. Multi-resolution contour-based fitting of macro-molecular structures. Journal of Molecular Biology. 2002;317:375–384. doi: 10.1006/jmbi.2002.5438. [DOI] [PubMed] [Google Scholar]
- 53.Wriggers W, Milligan RA, McCammon JA. Situs: A package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol. 1999;125(2–3):185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- 54.Wriggers W, Milligan RA, Schulten K, McCammon JA. Self-organizing neural networks bridge the biomolecular resolution gap. Journal of Molecular Biology. 1998;287:1247–1254. doi: 10.1006/jmbi.1998.2232. [DOI] [PubMed] [Google Scholar]
- 55.Yershova A, LaValle SM. Deterministic sampling methods for spheres and SO(3). Proceedings. IEEE International Conference on Robotics and Automation; 2004. pp. 3974–3980. [Google Scholar]
- 56.Yu Z, Bajaj C. Computational approaches for automatic structural analysis of large biomolecular complexes. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2007;5(4):568–582. doi: 10.1109/TCBB.2007.70226. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Q, Bettadapura R, Bajaj C. Macromolecular structure modeling from 3dem using volrover 2.0. Biopolymers. 2012;97(9):709–731. doi: 10.1002/bip.22052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang X, Settembre E, Xu C, Dormitzer P, Bellamy R, Harrison S, Grigorieff N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. PNAS. 2008;105(6):1867–72. doi: 10.1073/pnas.0711623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng W. Accurate flexible fitting of high-resolution protein structures into cryo-electron microscopy maps using coarse-grained pseudo-energy minimization. Biophysical Journal. 2011;100:478–488. doi: 10.1016/j.bpj.2010.12.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]