

Abstract
As the older adult segment of the population increases, Alzheimer’s disease (AD) has emerged as a significant public health epidemic. Over the past three decades, advances in the understanding of the biology of AD have led to a somewhat unified hypothesis of disease pathogenesis that emphasizes the precipitating role of beta amyloid protein. However, several lines of evidence suggest that multiple pathologies are necessary for clinical manifestation of the disease. Our focus over the past several years has been on the contribution of small vessel cerebrovascular disease, visualized as white matter hyperintensities (WMH) on magnetic resonance imaging, to AD. White matter hyperintensity volume, particularly in parietal regions, is elevated among individuals with and at risk for AD, predicts future diagnosis of AD, predicts the rate of progression of cognitive symptoms among individuals with AD, and increases over time among individuals destined to develop AD. White matter hyperintensities may represent an independent source of impairment and/or may interact more fundamentally with “primary” AD pathology. Future work should focus on more inclusive models of that better define “normal” versus “pathological” aging.
Keywords: Alzheimer’s disease, white matter hyperintensities, cerebrovascular disease
Introduction
Alzheimer’s disease (AD) has emerged as one of the most devastating international public health epidemics. As the older adult segment of our population continues to grow disproportionately and as the biomedical sciences are producing more and more effective ways of prolonging life, diseases that occur primarily in late life, such as AD, represent a significant societal burden. There are currently no effective disease-modifying treatments or preventative strategies. Over the past decade, however, major technological advances - - from refined animal models, novel molecular approaches, in vivo biological marker development, and more granular behavioral characterization of the disease - - have resulted in a somewhat unified model of disease pathogenesis that holds promise for development of intervention strategies. This model was codified by Jack and colleagues in 2010 (1) and revised in 2013 (2). The model emphasizes the precipitating role of beta amyloid protein, which theoretically triggers a cascade of biological events, resulting in accumulation of tau protein and associated neurodegeneration and cognitive changes. In the revised version of the model (2) individual differences in comorbidities, premorbid cognitive abilities, or hypothetically protective genetic profiles are considered important confounds that help explain variability in symptomatological onset rather than playing primary roles in disease pathogenesis.
A major recent advance is that theoretical pathogenic models, such as those proposed by Jack and colleagues, can be tested operationally in humans via the direct or indirect measurement of the putative biological markers. For example, amyloid can now be measured in vivo via positron emission tomography (PET) ligand studies and cerebrospinal fluid (CSF) assays; tau can be measured in the cerebrospinal fluid (CSF) and its supposed neurodegenerative effects can be appreciated with glucose metabolic PET and measurement of regional brain atrophy with magnetic resonance imaging (MRI); and the clinical manifestation of the disease can be measured with refined neuropsychological and behavioral instruments. Indeed, the operational definitions of these pathogenic events have been translated directly into recently-proposed and implemented research diagnostic criteria for AD and its antecedent conditions. Pre-clinical AD is defined as evidence of an AD biomarker, such beta amyloid or hyperphosphorylated tau protein, in the absence of clinical symptomatology (3). Mild cognitive impairment due to AD is defined as evidence of cognitive impairment in the absence of functional decline and supported by presence of AD-related biomarkers (4). Frank AD has maintained its historical definition that includes evidence of cognitive or behavioral impairment in at least two domains that interferes with functional abilities (5). The diagnosis of probable AD can be supported by evidence of AD pathophysiology via the assessment of the biomarkers noted above (6). These diagnostic criteria have been embraced by the scientific community and are already informing major intervention efforts, such as the recently-funded
“A4 trial (7),” which is a secondary prevention trial that will enroll individuals meeting criteria for Preclinical AD by virtue of having evidence of amyloid pathology without symptoms of cognitive impairment.
Despite the advances in the codification of the hypothesized pathogenic events that lead to AD-associated dementia, several equivocal observations from the extant literature suggest that the translation of this model into diagnostic criteria is perhaps premature, incomplete, and requires further study. For example, about 30% of older adults have evidence of significant amyloid pathology but do not have significant cognitive impairment suggestive of dementia (8–11). Conversely, it is interesting to note that in a recently completed unsuccessful phase 3 trial of bapineuzumab, an anti-amyloid immunotherapy agent, in AD, over 35% of non APOE-ε4 carriers meeting inclusion criteria for the study (i.e., clinical diagnosis of mild-to-moderate AD) did not have evidence of significant amyloid pathology at study entrance (12). Positive amyloid biomarker evidence for amyloid is weakly - - at best - - correlated with meaningful clinical outcomes (13–19). It is also well-established that tau-related neurodegenerative changes are non-specific and frequently occur prior to amyloid deposition and in individuals who do not have AD (20, 21); this observation has been incorporated into Jack and colleagues’ revised pathogenic model (2) but questions the precipitating role of beta amyloid. Whether or how beta amyloid accumulation promotes additional tau deposition is unknown and there is some compelling evidence that elevation in the Aβ peptide and tau hyperphosphorylation may be linked “epiphenomenologically” through their shared association with an upstream driver (22). Although it is established that biomarker evidence of AD pathology is associated with increased risk for developing future dementia, at this time we are unable to determine what the risk of the development of dementia is within a specific period for an individual person given a specific biomarker profile. Thus, the so-called “Amyloid Hypothesis” is far from fully elucidated.
From a public health perspective, it could be argued that AD should be defined phenotypically or syndromically with greater emphasis on refining characterization of the behavioral attributes that cause the tremendous amount of individual, familial, and societal burden associated with disease. It is likely that even in the earliest stages, more sensitive and specific neuropsychological classifiers, or endophenotypes, can be developed to reliably characterize individuals with and without the disease, particularly as molecular (e.g., (23) and neuropsychological (e.g., (24)) profiles that distinguish aging from dementia with great anatomical specificity continue to be understood. There is obvious danger, which can manifest in several ways, in embracing fully a singular hypothesized pathogenic model without a healthy amount of scientific skepticism. For example, at the extreme, if the identified biological markers, such as fibrillar forms of beta amyloid, are in fact not causative but rather pathological bi-products, removal of beta amyloid may have no effect on symptoms or disease course. Indeed, anti-amyloid therapies to date have been ineffective or harmful (25) among individuals with mild-to-moderate stage AD. On the other hand, viable treatments may not be pursued if other pathological factors that predict cognitive outcomes in AD are not considered or if we assume that all relevant treatment targets have been identified. Full adoption of the current pathogenic model leads to a “diagnostic prophecy” in which the etiology of the syndrome is defined by a proposed hypothetical set of factors, as opposed to the alternative scenario in which known or undiscovered etiological factors are incorporated into a more comprehensive disease conceptualization that accounts for the symptoms that comprise the clinical syndrome. We have already seen this potential problem in the new diagnostic criteria for AD, which states that the “diagnosis of probable AD dementia should not be applied when there is evidence of…substantial concomitant cerebrovascular disease,” ((6) quotation from pg. 265) despite the well-established fact that vascular disease is specifically increased in AD and substantially related to its clinical syndrome (26). Thus, in the attempt to fully understand the causative factors involved with AD, their mediators and moderators, and subsequent identification of treatment or preventative targets, a clear phenotypic or syndromic definition of the disease should be implemented with emphasis on discovery of the factors that lead to the manifestation of that syndrome (Figure 1). There may also be benefit in greater consideration of individual and group differences not only in clinical symptoms but also in their putative biology. For example, a consistent clinical syndrome may be the result of different combinations of various pathologies across individuals and/or pathological features that contribute to a clinical syndrome may vary systematically across age, socioeconomic, or other demographic groups.
Figure 1.
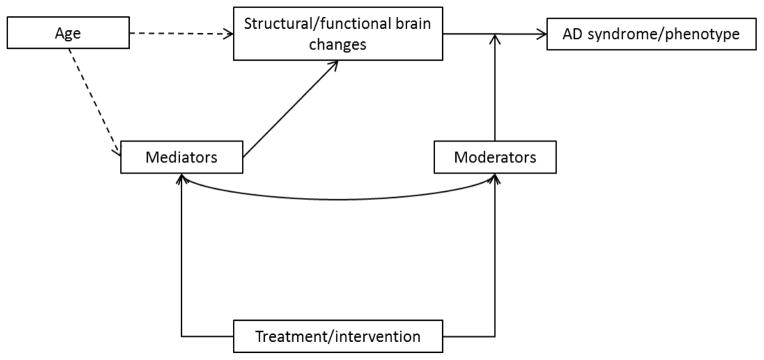
Framework for understanding contributors to the Alzheimer’s disease phenotype. By defining AD as a syndrome, we are able to identify structural and functional brain changes that predict onset of symptoms, severity of symptoms, and progression of symptoms. Scrutiny of structural and functional alterations associated with the AD syndrome can occur at multiple levels using a variety of disciplines (e.g., cell biology, neuroimaging) and across species. The extent to which age is a necessary or causative factor in AD has not been established entirely but it is noteworthy that symptom onset occurs after age 65 for the vast majority of “sporadic” forms of the disease, which comprise over 90% of AD cases. Mediating factors—the mechanisms that underlie the relevant structural and functional brain changes or through which aging impacts those changes—and moderating factors—those factors that mitigate the effects of relevant structural and functional brain changes on their clinical outcomes—can be identified as reasonable targets for treatment or prevention strategies. The model stipulates that treatment or prevention strategies target factors that ultimately impact the AD syndrome either directly (i.e., through the mediators) or indirectly (i.e., through the moderators). The line connecting the moderators and mediators is to indicate that these factors may interact with each other and are not necessarily independent.
From its earliest description (27) AD has been characterized as a mixed pathology disease comprising amyloid plaques and neurofibrillary tangles, without well-established causal relationships between the two. With this historical backdrop in mind, we have embraced the idea that additional pathologies may be relevant for risk and/or clinical expression of the disease. Indeed, accumulating evidence from multiple sources indicates that factors associated with poor cognitive aging, in the absence of frank dementia, may play a primary role in the pathogenesis and progression of AD. The epidemiological literature suggests that potentially modifiable vascular risk factors, such as hypertension, diabetes, insulin resistance, obesity/overweight, and hyperlipidemia, are at the top of this list (28–40). Although epidemiological studies that associate risk factor data to clinical outcomes provide important information, they speak little to the proximal brain factors that are more directly involved with pathogenesis. We have turned to neuroimaging to better understand those factors.
White matter hyperintensities
Over the past few decades, there has been an explosion of neuroimaging research applied to questions about cognitive aging and dementia. These efforts have been bolstered not only by improvements in instrumentation that allow for direct study of morphological and functional properties of the aging brain in vivo, but also by more sophisticated pre- and post-processing analytic streams and application of modern statistical approaches. Structural magnetic resonance imaging (MRI) in particular can be useful for the assessment of macrostructure (e.g., volumetry, cortical thickness, frank pathology) and microstructure (e.g., fiber tract integrity or subtle abnormalities to pathology that affects myelin). Though much work has highlighted the importance of gross structural or volumetric loss for cognitive aging and dementia, more recent work has highlighted the importance of subtle markers of small vessel disease. White matter hyperintensities (WMH) (Figure 2) are areas of increased signal seen on T2-weighted MRI, including fluid attenuated inversion recovery (FLAIR) images, thought to reflect the degree and distribution of small vessel occlusive disease (40). They are typically distributed in periventricular regions with confluent extension into deeper cortical regions but may have a somewhat punctate distribution as well.
Figure 2.
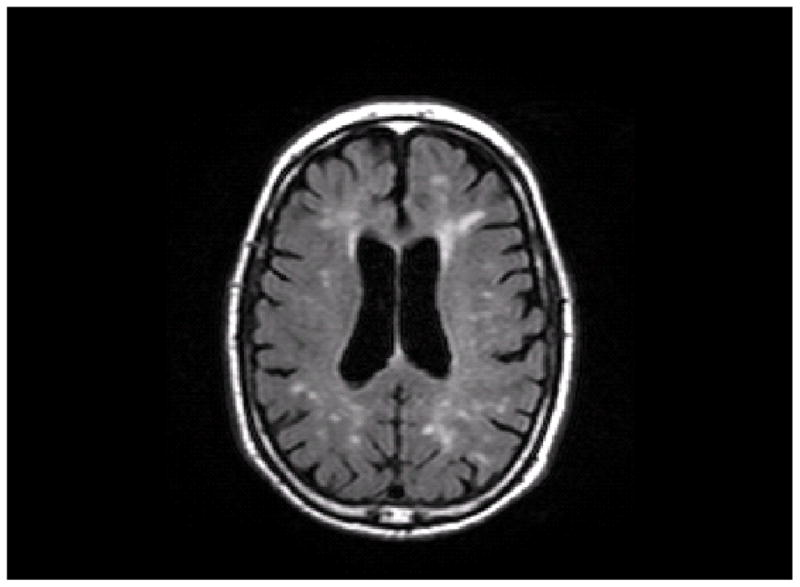
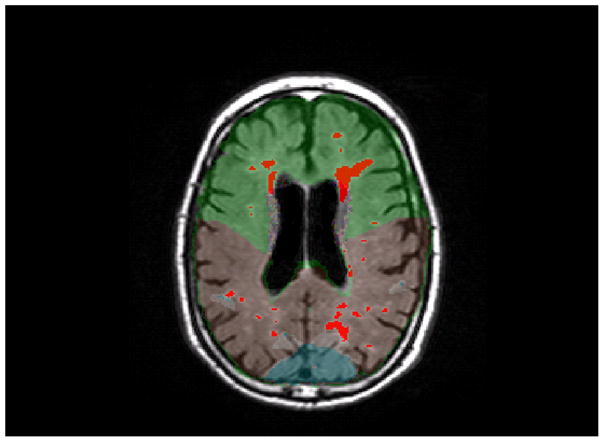
An axial slice from a T2-weighted FLAIR image. A, This image shows the unlabeled MRI scan. B, This image shows WMH labeled in red and a lobar atlas superimposed on the image. Frontal lobe is labeled in green, parietal lobe is labeled in brown, and occipital lobe is labeled in blue (temporal lobe is not visible at this level).
White matter hyperintensities were initially described as hypodensities on computed tomography and with the advent of widespread clinical use of MRI scanning for clinical purposes, it became evident that they are quite common among older adults, though with a tremendous amount of variability in volume and distribution. Earlier clinical observations classified these lesions as either “leukoaraiosis,” suggesting that they represent pathological damage to the white matter, or as “unidentified bright objects” (UBOs), suggesting that they were radiological artifacts with little clinical significance (41, 42). It wasn’t until the 1990s that systematic study of WMH demonstrated that they are associated with poor cognitive and functional outcomes (43). Since that time, there have been myriad studies linking the severity of WMH to a reduction or decline cognitive functioning in older adults.
It is unclear exactly what causes WMH. The increased signal observed on T2-weighted MRI scans is due to reduced relaxation rate from increases in extracellular spaces and/or gliosis that restrict fluid locally (44). In theory, any process that causes local damage to white matter can manifest as WMH on MRI scans. Neuropathological correlates studies, which have tried to relate common histological features to burden of WMH in life, have shown that the severity of WMH are related to various pathological markers including arteriolosclerosis, decreased myelin pallor, and complete and incomplete infarction (45–47). Together with epidemiological studies linking mid-life and accumulating vascular risk factors to the severity of WMH in later life(48), these studies help establish that WMH represent brain pathology that is somehow age-dependent and reflective accumulating “subclinical” vascular disease. There is little evidence that WMH burden decreases once the pathology has begun. Thus, for all intents and purposes, we treat WMH severity as a marker of pathology that tends to emerge later in life.
Interest in the involvement of WMH in AD emerged from consistent reports of a reliable association between age and WMH severity and between WMH severity and cognitive functioning among non-demented individuals (49). Earlier reports tended to implicate WMH in executive functioning and working memory (50). Given the epidemiological literature linking the same vascular factors that increase risk for development of WMH to the risk for development of AD, we were interested in understanding whether WMH burden plays a specific role in AD in addition to its role in cognitive aging. Alzheimer’s disease occurs primarily in the context of aging, but whether pathological changes increase risk for AD, interact with primary AD pathology, or are important for disease course remain open questions. Over the past several years we have examined the role of WMH in cognitive aging and AD, the mediators of WMH, and factors that modify their impact on clinical expression (51).
Measuring WMH
By far, the most common approach to staging WMH has been to use well-validated rating scales, such as those developed by Scheltens (52) and Fazekas (53). Rating scales require a fair degree of operator training and establishment of intra- and inter-rater reliabilities, but experienced operators are able to produce evaluations that have tremendous utility in large-scale neuroimaging studies. However, severity rating scales do not consider the rich volumetric/parametric information contained within neuroimaging and have limited utility for fully quantitative analyses. Several laboratories have established methods to measure WMH quantitatively. In our own approach (51, 54), we remove the skulls from FLAIR images, apply Gaussian curves to each cerebral hemisphere and determine the mean and standard deviation of voxel intensities. Next we apply a WMH seed that labels intensity values greater than a pre-determined threshold value, usually about 2.5SD above the mean image intensity value. Using a 10-point connectivity scheme, the algorithm searches for and labels voxels that fall within 5% of the seed mean and labels voxels that fall within that range, continuing iteratively. The summation of labeled voxels multiplied by voxel dimensions yields a total WMH volume. By spatially fitting an anatomical atlas (55) to each image in native space, we are able to determine WMH volume in each cerebral lobe, including frontal, temporal, parietal, and occipital lobes. Further, in cases where questions about “periventricular” versus “deep” WMH arise, we are able to determine the three-dimensional distance from the walls of the lateral ventricles of each labeled WMH voxel by segmenting the lateral ventricles. Finally, all segmented images are visually inspected by an experienced operator and manually edited in cases where non-WMH voxels are improperly labeled or where WMH are under labeled. Manual approaches to deriving volumetric WMH data have also been developed (54) and a multimodal fuzzy logic classification scheme for voxel labeling has been developed for when FLAIR data are not available (56).
White matter hyperintensities in AD
Much of our work examining the role of WMH in cognitive aging and AD has taken place in the context of the Washington Heights Inwood Columbia Aging Project (WHICAP) (57). WHICAP is an ongoing community-based study of aging and dementia comprising older adults from Northern Manhattan that began in 1992. Two cohorts have been recruited, beginning in 1992 and 1999, and a third cohort is currently being recruited. A unique aspect of the study is that it comprises a racially and ethnically diverse group of older adults, including about an equal proportion of Whites, African Americans, and Latinos, that are characteristic of the population in northern Manhattan but also representative of the increasingly diverse population of older adults in general. Beginning in 2005, we began systematically collecting high-resolution MRI scans on individuals who did not have dementia at the previous study visit and who were otherwise not contraindicated (58). We collected 769 MRI scans. Fifty-two participants met clinical diagnostic criteria for AD at the visit most proximal to the MRI scan; thus, our final imaging sample comprised 717 non-demented participants and 52 participants with dementia. Beginning in 2010, we began to collect repeat MRI scans on available participants for whom baseline MRI scans were acquired. Analyses are ongoing.
Previous work has suggested that WMH are more severe among individuals with diagnosed AD relative to demographically-similar but neurologically-healthy controls in hospital or clinic-based settings (59–61). In WHICAP, we first asked the question whether WMH as a marker of small vessel cerebrovascular disease was associated with subtypes of mild cognitive impairment (MCI), including amnestic and non-amnestic MCI (62). Amnestic MCI refers to cognitive impairment in domains that include memory but the absence of functional impairment severe enough to qualify for diagnosis of dementia. Individuals with amnestic MCI are considered to be in the earliest stages of AD and at particularly elevated risk for future development of AD. Individuals with non-amnestic MCI, or cognitive impairment in domains that do not include memory functioning, are at relatively lower risk for future development of AD but still have cognitive impairment. We hypothesized that WMH burden would be greatest among individuals with non-amnestic MCI because previous work generally linked WMH to non-memory domains and because the cognitive impairment seen in amnestic MCI is most often attributed to AD pathology rather than vascular pathology. Though frank brain infarction was related to non-amnestic MCI, contrary to our hypothesis, individuals with amnestic MCI had the greatest WMH burden and individuals with non-amnestic MCI were intermediate. The study provided the first indication that, in this community-based cohort of older adults, WMH seemed to have a specific association with AD by virtue of being more severe among those at greatest risk for development of AD in the future. We followed this study by examining the regional distribution of WMH as a function of diagnostic group (i.e., unimpaired, amnestic MCI, non-amnestic MCI, and AD) (63). We found an interesting pattern of results. First, consistent with previous reports (64), WMH are mostly distributed in frontal and parietal lobes. Second, all three impaired groups, including amnestic MCI, non-amnestic MCI, and AD patients, had increased WMH burden in the frontal lobes relative to controls, but the amnestic MCI and AD patients had selectively increased WMH in parietal regions. Parietal lobe WMH volume severity also discriminated individuals with amnestic MCI from controls better than a marker of hippocampal atrophy, ostensibly an AD-related neurodegenerative biomarker that is present prior to cognitive impairment (1, 2). We obtained similar findings when examining the association between regional WMH volume and diagnosis in a cohort of older African Americans (65). These observations join an emerging literature showing a greater posterior involvement of WMH in AD (66, 67).
Whether neuroimaging data acquired at one point in time contains useful prognostic information to predict future diagnosis or clinical course in AD remains an extremely important clinical question. Indeed, non-demented older adults with higher WMH burden are at greater risk for the development of AD and MCI (68–71). We examined whether the regional distribution of WMH predicted incident AD among non-demented individuals in the WHICAP study (72) and found that WMH volume in the parietal lobes specifically predicted time to incident AD. Given the prevailing models of AD pathogenesis, we hypothesized that measures of hippocampal atrophy would independently predict incident AD but that regionally distributed WMH volume would provide additional prognostic information. Parietal lobe WMH volume specifically predicted time to incident AD, but a relative measure of hippocampal volume did not. In the context of our previous work (73), which showed that WMH burden interacted with total brain atrophy to predict rate of cognitive decline among individuals with prevalent AD, the findings suggest that in community-dwelling older adults, regionally distributed WMH may be early harbingers of AD pathogenesis. In contrast, the severity of WMH appears to be more specifically tied to normal aging; frontal lobe WMH, for example, are specifically associated with mortality (74). We are currently completing longitudinal WMH analyses that show a normal aging-related increase in WMH in anterior regions but an AD-specific increase in WMH in posterior regions among individuals who later develop AD (75). Taken together, our work shows that WMH volume, particularly when distributed in parietal regions, is elevated among individuals with AD, is elevated among individuals at risk for the development of AD, predicts future diagnosis of AD, predicts the rate of progression of cognitive symptoms among individuals with AD, and increases over time among individuals destined to develop AD.
Why might WMH and AD be related?
Given the consistently-observed association between WMH and clinical AD, the determination of how the two are related remains critical. There are at least three non-mutually exclusive possibilities. First, WMH represent a second, independent pathological “hit” that lowers diagnostic threshold for AD or contributes additively to disease presentation. In this scenario, WMH are simply due to perfusion or general vascular abnormalities and reflect ischemic change in the brain and do not promote AD-related pathology (or vice versa). Second, the pathology underlying WMH may be heterogeneous as a function of regional distribution and may interact with or reflect primary AD pathology. In this scenario, measured pathology in areas identified radiologically as WMH may vary across location and diagnosis and WMH (or their etiological factors) may interact mechanistically with AD pathology. Third, WMH and AD may be related to each other epiphenomenologically through their shared association with some third set of factors. We have been pursuing the first two possibilities by systematically examining the mediators of WMH and how WMH and markers of AD pathology may interact.
Consistent with epidemiological studies we confirmed that an increase in the number of vascular risk factors is associated with severity of WMH in our own community-based study (58). We also showed main and interaction effects with race/ethnicity, such that both African Americans and Latinos had increased WMH volume relative to Whites and the association between vascular disease history and WMH burden was greatest among African Americans. We have hypothesized that, given its association with AD, increased WMH burden may be one key variable that helps explain racial and ethnic disparities in AD incidence and prevalence. Incomplete brain perfusion or autoregulatory breakdown is another key factor that may promote deposition of WMH. In our community sample, we demonstrated that both high blood pressure level and fluctuations in blood pressure over time are associated with increased WMH burden (76). High blood pressure and measures of systemic hypoperfusion are related to severity of WMH (77–79) and individuals with metabolic syndrome, defined as a syndrome comprising dyslipidemia, hypertension, and hyperglycemia, is associated with increased WMH in temporo-parietal regions (80). By combining FLAIR MRI data with measures of cerebral blood flow, we showed that areas appearing as WMH had diminished blood flow relative to grey matter and normal appearing white matter (81), which is consistent with an earlier report showing that regions with normatively lower perfusion values are most vulnerable to development of WMH (82). Thus, there is fairly strong support that diminished perfusion abnormalities and perhaps compromised cerebral autoregulation are in the causal pathway to the development of WMH.
How or whether WMH and AD interact on a more mechanistic level is a more difficult question to answer. It is interesting to note that WMH tend to localize in areas with the greatest amount of AD pathology and metabolic dysfunction (83). Pathogenic models of AD implicate parenchymal deposition of Aβ protein and declining plasma levels of Aβ42 that are associated with increased risk for AD development, presumably due to oligomerization and deposition of Aβ peptides in senile plaques (84). However, it is also quite possible that vascular deposition of Aβ may also be a primary driver of the disease. In vitro studies show that the number of perivascular spaces in the white matter, not grey matter, correlate with amount of Aβ in overlying cortex and associated arteries (85). Vascular Aβ may interfere with the ability of blood vessels to shunt deposited Aβ peptides through the periarterial spaces and the white matter of AD (86–89). It is conceivable that increased WMH burden among patients with AD reflects accumulation of vascular Aβ to some degree. Indeed, cerebral amyloid angiopathy (CAA), which reflects the deposition of Aβ in cerebral arterioles, is present in the vast majority of individuals with AD whose brain tissue is examined at autopsy. With use of T2*-weighted gradient echo MRI, CAA manifests radiologically as cerebral microbleeds distributed in lobar regions. White matter hyperintensities are more severe in the presence of clinical CAA or microbleeds and individuals with microbleeds are more likely to have progressive WMH (82, 90, 91). In patients with AD, microbleeds are distributed mostly in posterior regions, similar to the distribution of WMH (92). In our own work, we showed that individuals with two or more lobar microbleeds, highly suggestive of CAA, had more severe WMH in parietal lobes specifically (93).
We examined WMH in the Alzheimer’s Disease Neuroimaging Initiative, which has acquired multi-model imaging on older individuals with MCI, AD, and without neurological illness (56). We sought to determine how WMH and PET derived amyloid positivity impacts the clinical expression of AD. Both amyloid and WMH were independently associated with clinical AD diagnosis and WMH volume discriminated between those with and without clinical AD with good specificity and excellent sensitivity. White matter hyperintensity volume and amyloid levels were inversely related to each other while among MCI subjects both amyloid status and WMH volume predicted who would later convert to clinical AD. We also showed that regional WMH volume was more strongly associated with entorhinal cortex neurodegenerative changes than measures of amyloid from the cerebrospinal fluid (94). Taken together, our observations suggest that WMH are at least an independent source of impairment, and possibly interact with AD pathology. Our studies were carried out in epidemiological contexts, in which medical comorbidities are relatively unconstrained, but also in clinical contexts in which the “purest” AD patients were included.
Conclusion
We, and other groups, have established an important role of regionally distributed WMH in the clinical expression of AD and possibly in its pathogenesis. To the extent that WMH represent solely small vessel cerebrovascular disease secondary to perfusion abnormalities, this recent work is consistent with the idea that presence of small vessel disease in AD is the normative rule not the exception (95). White matter hyperintensities, particularly when distributed in posterior regions, may also have an amyloidogenic origin, suggesting a mechanistic interaction with “primary” AD pathology. Alzheimer’s disease is likely more complex than single-factor pathogenic models would suggest and much work is needed to understand the heterogeneous factors that lead to syndromic presentation across individuals.
Future work needs to focus on disentangling the relative contributions of various pathologies to disease presentation and understanding interactions among the pathologies that comprise the brains of older adults. Regarding the former, it is essential that we examine factors that promote cerebrovascular damage, such as autoregulatory dysfunction, together with multi-modal MRI data, data reflecting AD biomarkers, and refined neuropsychological data. In addition to these types of studies, we have begun a neuroimaging-guided histopathological examination of WMH and normal appearing white matter in postmortem tissue (96). By obtaining postmortem MRI scans on autopsied brains, we are able to define radiological abnormalities in the white matter and use those to guide our pathological examination of underlying tissue. This approach allows us to examine the pathology of WMH and normal white matter tissue as a function of region (frontal or parietal lobes) and clinical or pathological diagnosis. Longitudinal studies are required to answer questions about whether progression or accumulation of WMH leads to AD and newer, higher-resolution neuroimaging techniques are necessary to characterize in vivo changes in the aging brain with greater precision.
Acknowledgments
Work presented here was supported in part by grants from the National Institutes of Health (AG034189, AG037212, AG028786, AG029949, AG024708), Alzheimer’s Association, Mary E. Groff Surgical Medical Research and Education Charitable Trust, and Columbia University.
Footnotes
Compliance with Ethics Guidelines
Conflict of Interest
Adam M. Brickman has received travel/accommodations expenses covered or reimbursed from the International Neuropsychological Society (as a board member) and the Alzheimer’s Association.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1•.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet neurology. 2010 Jan;9(1):119–28. doi: 10.1016/S1474-4422(09)70299-6. This paper presents a comprehensive hypothesis regarding the cascade of biological events that contribute to the pathogensis of AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2•.Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet neurology. 2013 Feb;12(2):207–16. doi: 10.1016/S1474-4422(12)70291-0. This paper is an updated version of a pathogenic model for Alzheimer’s disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2010 May;7(3):280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2010 May;7(3):270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984 Jul;34(7):939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 6.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2010 May;7(3):263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sperling R, Donohue M, Aisen P. The A4 trial: Anti-amyloid treatment of asymptomatic Alzheimer’s disease. Alzheimer’s & Dementia. 2012;8(4, Supplement):425–6. [Google Scholar]
- 8.Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, et al. Frequent Amyloid Deposition Without Significant Cognitive Impairment Among the Elderly. Archives of neurology. 2008 Nov 1;65(11):1509–17. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006 Aug 8;67(3):446–52. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 10.Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009 Apr 21;106(16):6820–5. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lockhart A, Lamb JR, Osredkar T, Sue LI, Joyce JN, Ye L, et al. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain. 2007 Oct;130(Pt 10):2607–15. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- 12.Vellas B, Carrillo MC, Sampaio C, Brashear HR, Siemers E, Hampel H, et al. Designing drug trials for Alzheimer’s disease: What we have learned from the release of the phase III antibody trials: A report from the EU/US/CTAD Task Force. Alzheimer’s & Dementia. 2013;9(4):438–44. doi: 10.1016/j.jalz.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Rodrigue KM, Kennedy KM, Devous MD, Sr, Rieck JR, Hebrank AC, Diaz-Arrastia R, et al. beta-Amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology. 2012 Feb 7;78(6):387–95. doi: 10.1212/WNL.0b013e318245d295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Archives of neurology. 2008 Nov;65(11):1509–17. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourgeat P, Chetelat G, Villemagne VL, Fripp J, Raniga P, Pike K, et al. Beta-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology. 2010 Jan 12;74(2):121–7. doi: 10.1212/WNL.0b013e3181c918b5. [DOI] [PubMed] [Google Scholar]
- 16.Hedden T, Van Dijk KR, Becker JA, Mehta A, Sperling RA, Johnson KA, et al. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J Neurosci. 2009 Oct 7;29(40):12686–94. doi: 10.1523/JNEUROSCI.3189-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009 May;132(Pt 5):1310–23. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007 Nov;130(Pt 11):2837–44. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 19.Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiology of aging. 2010 Aug;31(8):1275–83. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 20.Braak H, Del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011 Feb;121(2):171–81. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- 21.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiology of aging. 1997 Jul-Aug;18(4):351–7. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 22.Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron. 2008 Nov 26;60(4):534–42. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pavlopoulos E, Jones S, Kosmidis S, Close M, Kim C, Kovalerchik O, et al. Molecular Mechanism for Age-Related Memory Loss: The Histone-Binding Protein RbAp48. Sci Transl Med. 2013 Aug 28;5(200):200ra115. doi: 10.1126/scitranslmed.3006373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brickman AM, Stern Y, Small SA. Hippocampal subregions differentially associate with standardized memory tests. Hippocampus. 2011 Sep;21(9):923–8. doi: 10.1002/hipo.20840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castellani RJ, Perry G. Pathogenesis and disease-modifying therapy in Alzheimer’s disease: the flat line of progress. Arch Med Res. 2012 Nov;43(8):694–8. doi: 10.1016/j.arcmed.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain. 2013 Sep;136(Pt 9):2697–706. doi: 10.1093/brain/awt188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castellani RJ, Perry G. Pathogenesis and Disease-modifying Therapy in Alzheimer’s Disease: The Flat Line of Progress. Archives of Medical Research. 2012;43(8):694–8. doi: 10.1016/j.arcmed.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 28.Elkins JS, O’Meara ES, Longstreth WT, Jr, Carlson MC, Manolio TA, Johnston SC. Stroke risk factors and loss of high cognitive function. Neurology. 2004 Sep 14;63(5):793–9. doi: 10.1212/01.wnl.0000137014.36689.7f. [DOI] [PubMed] [Google Scholar]
- 29.Kilander L, Nyman H, Boberg M, Hansson L, Lithell H. Hypertension is related to cognitive impairment: a 20-year follow-up of 999 men. Hypertension. 1998 Mar;31(3):780–6. doi: 10.1161/01.hyp.31.3.780. [DOI] [PubMed] [Google Scholar]
- 30.Kivipelto M, Helkala EL, Hanninen T, Laakso MP, Hallikainen M, Alhainen K, et al. Midlife vascular risk factors and late-life mild cognitive impairment: A population-based study. Neurology. 2001 Jun 26;56(12):1683–9. doi: 10.1212/wnl.56.12.1683. [DOI] [PubMed] [Google Scholar]
- 31.Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, et al. Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. BMJ (Clinical research ed. 2001 Jun 16;322(7300):1447–51. doi: 10.1136/bmj.322.7300.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knopman D, Boland LL, Mosley T, Howard G, Liao D, Szklo M, et al. Cardiovascular risk factors and cognitive decline in middle-aged adults. Neurology. 2001 Jan 9;56(1):42–8. doi: 10.1212/wnl.56.1.42. [DOI] [PubMed] [Google Scholar]
- 33.Luchsinger JA, Patel B, Tang MX, Schupf N, Mayeux R. Measures of adiposity and dementia risk in elderly persons. Archives of neurology. 2007 Mar;64(3):392–8. doi: 10.1001/archneur.64.3.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005 Aug 23;65(4):545–51. doi: 10.1212/01.wnl.0000172914.08967.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology. 2004 Oct 12;63(7):1187–92. doi: 10.1212/01.wnl.0000140292.04932.87. [DOI] [PubMed] [Google Scholar]
- 36.Luchsinger JA, Tang MX, Stern Y, Shea S, Mayeux R. Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort. American journal of epidemiology. 2001 Oct 1;154(7):635–41. doi: 10.1093/aje/154.7.635. [DOI] [PubMed] [Google Scholar]
- 37.Swan GE, DeCarli C, Miller BL, Reed T, Wolf PA, Jack LM, et al. Association of midlife blood pressure to late-life cognitive decline and brain morphology. Neurology. 1998 Oct;51(4):986–93. doi: 10.1212/wnl.51.4.986. [DOI] [PubMed] [Google Scholar]
- 38.Helzner EP, Luchsinger JA, Scarmeas N, Cosentino S, Brickman AM, Glymour MM, et al. Contribution of vascular risk factors to the progression in Alzheimer disease. Archives of neurology. 2009 Mar;66(3):343–8. doi: 10.1001/archneur.66.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet neurology. 2011 Sep;10(9):819–28. doi: 10.1016/S1474-4422(11)70072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorelick PB, Scuteri A, Black SE, DeCarli C, Greenberg SM, Iadecola C, et al. Vascular Contributions to Cognitive Impairment and Dementia. Stroke; a journal of cerebral circulation. 2011 Sep 1;42(9):2672–713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kertesz A, Black SE, Tokar G, Benke T, Carr T, Nicholson L. Periventricular and subcortical hyperintensities on magnetic resonance imaging: 'rims, caps, and unidentified bright objects'. Archives of neurology. 1988;45(4):404–8. doi: 10.1001/archneur.1988.00520280050015. [DOI] [PubMed] [Google Scholar]
- 42.Román GC. Senile dementia of the binswanger type: A vascular form of dementia in the elderly. Jama. 1987;258(13):1782–8. doi: 10.1001/jama.1987.03400130096040. [DOI] [PubMed] [Google Scholar]
- 43.DeCarli C, Murphy DG, Tranh M, Grady CL, Haxby JV, Gillette JA, et al. The effect of white matter hyperintensity volume on brain structure, cognitive performance, and cerebral metabolism of glucose in 51 healthy adults. Neurology. 1995 Nov;45(11):2077–84. doi: 10.1212/wnl.45.11.2077. [DOI] [PubMed] [Google Scholar]
- 44.Bronge L, Wahlund LO. White matter changes in dementia: does radiology matter? The British journal of radiology. 2007 Dec;80(Spec No 2):S115–20. doi: 10.1259/bjr/35265137. [DOI] [PubMed] [Google Scholar]
- 45.Erten-Lyons D, Woltjer R, Kaye J, Mattek N, Dodge HH, Green S, et al. Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology. 2013 Sep 10;81(11):977–83. doi: 10.1212/WNL.0b013e3182a43e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jagust WJ, Zheng L, Harvey DJ, Mack WJ, Vinters HV, Weiner MW, et al. Neuropathological basis of magnetic resonance images in aging and dementia. Annals of neurology. 2008 Jan;63(1):72–80. doi: 10.1002/ana.21296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fazekas F, Kleinert R, Offenbacher H, Schmidt R, Kleinert G, Payer F, et al. Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology. 1993 Sep;43(9):1683–9. doi: 10.1212/wnl.43.9.1683. [DOI] [PubMed] [Google Scholar]
- 48.Launer LJ. Epidemiology of white matter lesions. Top Magn Reson Imaging. 2004 Dec;15(6):365–7. doi: 10.1097/01.rmr.0000168216.98338.8d. [DOI] [PubMed] [Google Scholar]
- 49.Gunning-Dixon FM, Brickman AM, Cheng JC, Alexopoulos GS. Aging of cerebral white matter: a review of MRI findings. International journal of geriatric psychiatry. 2009 Feb;24(2):109–17. doi: 10.1002/gps.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gunning-Dixon FM, Raz N. The cognitive correlates of white matter abnormalities in normal aging: a quantitative review. Neuropsychology. 2000 Apr;14(2):224–32. doi: 10.1037//0894-4105.14.2.224. [DOI] [PubMed] [Google Scholar]
- 51.Brickman AM, Muraskin J, Zimmerman ME. Structural neuroimaging in Alzheimer’s disease: do white matter hyperintensities matter? Dialogues in clinical neuroscience. 2009;11(2):181–90. doi: 10.31887/DCNS.2009.11.2/ambrickman. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheltens P, Barkhof F, Leys D, Pruvo JP, Nauta JJ, Vermersch P, et al. A semiquantative rating scale for the assessment of signal hyperintensities on magnetic resonance imaging. Journal of the neurological sciences. 1993 Jan;114(1):7–12. doi: 10.1016/0022-510x(93)90041-v. [DOI] [PubMed] [Google Scholar]
- 53.Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. Ajr. 1987 Aug;149(2):351–6. doi: 10.2214/ajr.149.2.351. [DOI] [PubMed] [Google Scholar]
- 54.Brickman AM, Sneed JR, Provenzano FA, Garcon E, Johnert L, Muraskin J, et al. Quantitative approaches for assessment of white matter hyperintensities in elderly populations. Psychiatry research. 2011 Aug 30;193(2):101–6. doi: 10.1016/j.pscychresns.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Admiraal-Behloul F, Olofesen H, Van den Heuvel DM, Schmitz N, Reiber JH, Van Buchem MA. Fully automated lobe delineation for regional white matter lesion load quantification in a large scale study. Proceedings International Society for Magnetic Resonance in medicine. 2004:138. [Google Scholar]
- 56•.Provenzano FA, Muraskin J, Tosto G, Narkhede A, Wasserman BT, Griffith EY, et al. White Matter Hyperintensities and Cerebral Amyloidosis: Necessary and Sufficient for Clinical Expression of Alzheimer Disease? JAMA Neurol. 2013 Feb 18;:1–7. doi: 10.1001/jamaneurol.2013.1321. In this paper we showed that both WMH and amyloid are associated with Alzheimer’s disease and that among individuals with evidence of cerebral amyloidosis, those with higher WMH burden were more likely to manifest clinical symptoms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang MX, Cross P, Andrews H, Jacobs DM, Small S, Bell K, et al. Incidence of Alzheimer’s disease in African-Americans, Caribbean Hispanics and Caucasians in northern Manhattan. Neurology. 2001;56:49–56. doi: 10.1212/wnl.56.1.49. [DOI] [PubMed] [Google Scholar]
- 58.Brickman AM, Schupf N, Manly JJ, Luchsinger JA, Andrews H, Tang MX, et al. Brain morphology in older African Americans, Caribbean Hispanics, and whites from northern Manhattan. Archives of neurology. 2008 Aug;65(8):1053–61. doi: 10.1001/archneur.65.8.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheltens P, Barkhof F, Valk J, Algra PR, van der Hoop RG, Nauta J, et al. White matter lesions on magnetic resonance imaging in clinically diagnosed Alzheimer’s disease. Evidence for heterogeneity. Brain. 1992 Jun;115 (Pt 3):735–48. doi: 10.1093/brain/115.3.735. [DOI] [PubMed] [Google Scholar]
- 60.Kalaria RN. The role of cerebral ischemia in Alzheimer’s disease. Neurobiology of aging. 2000 Mar-Apr;21(2):321–30. doi: 10.1016/s0197-4580(00)00125-1. [DOI] [PubMed] [Google Scholar]
- 61.Rezek DL, Morris JC, Fulling KH, Gado MH. Periventricular white matter lucencies in senile dementia of the Alzheimer type and in normal aging. Neurology. 1987 Aug;37(8):1365–8. doi: 10.1212/wnl.37.8.1365. [DOI] [PubMed] [Google Scholar]
- 62.Luchsinger JA, Brickman AM, Reitz C, Cho SJ, Schupf N, Manly JJ, et al. Subclinical cerebrovascular disease in mild cognitive impairment. Neurology. 2009 Aug 11;73(6):450–6. doi: 10.1212/WNL.0b013e3181b1636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brickman AM, Provenzano FA, Muraskin J, Guzman VA, Manly JJ, Schupf N, et al. Distribution of MRI-defined white matter hyperintensities in mild cognitive impairment [abstract] Journal of the International Neuropsychological Society. 2011 Feb;17(S1) [Google Scholar]
- 64.Gootjes L, Teipel SJ, Zebuhr Y, Schwarz R, Leinsinger G, Scheltens P, et al. Regional distribution of white matter hyperintensities in vascular dementia, Alzheimer’s disease and healthy aging. Dementia and Geriatric Cognitive Disorder. 2004;18(2):180–8. doi: 10.1159/000079199. [DOI] [PubMed] [Google Scholar]
- 65.Meier IB, Manly JJ, Provenzano FA, Hector J, Wasserman BT, Louie K, et al. White matter predictors of cogntiive functioning in older adults. Annual meeting of the International Neuropsychological Society; February, 2011; Boston, MA. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66•.Yoshita M, Fletcher E, Harvey D, Ortega M, Martinez O, Mungas DM, et al. Extent and distribution of white matter hyperintensities in normal aging, MCI, and AD. Neurology. 2006 Dec 26;67(12):2192–8. doi: 10.1212/01.wnl.0000249119.95747.1f. This is one of the earlier papers to show the extent and distribution of white matter hyperintensities among individuals with and at risk for AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leys D, Pruvo JP, Parent M, Vermersch P, Soetaert G, Steinling M, et al. Could Wallerian degeneration contribute to “leukoaraiosis” in subjects free of any vascular disorder? Journal of Neurology, Neurosurgery & Psychiatry. 1991 Jan;54(1):46–50. doi: 10.1136/jnnp.54.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prins ND, van Dijk EJ, den Heijer T, Vermeer SE, Koudstaal PJ, Oudkerk M, et al. Cerebral white matter lesions and the risk of dementia. Archives of neurology. 2004 Oct;61(10):1531–4. doi: 10.1001/archneur.61.10.1531. [DOI] [PubMed] [Google Scholar]
- 69.Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM. Silent brain infarcts and the risk of dementia and cognitive decline. The New England journal of medicine. 2003 Mar 27;348(13):1215–22. doi: 10.1056/NEJMoa022066. [DOI] [PubMed] [Google Scholar]
- 70.Wolf H, Ecke GM, Bettin S, Dietrich J, Gertz HJ. Do white matter changes contribute to the subsequent development of dementia in patients with mild cognitive impairment? A longitudinal study. International journal of geriatric psychiatry. 2000 Sep;15(9):803–12. doi: 10.1002/1099-1166(200009)15:9<803::aid-gps190>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 71.Smith EE, Egorova S, Blacker D, Killiany RJ, Muzikansky A, Dickerson BC, et al. Magnetic resonance imaging white matter hyperintensities and brain volume in the prediction of mild cognitive impairment and dementia. Archives of neurology. 2008 Jan;65(1):94–100. doi: 10.1001/archneurol.2007.23. [DOI] [PubMed] [Google Scholar]
- 72.Brickman AM, Provenzano FA, Muraskin J, Manly JJ, Blum S, Apa Z, et al. Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Archives of neurology. 2012 Dec;69(12):1621–7. doi: 10.1001/archneurol.2012.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brickman AM, Honig LS, Scarmeas N, Tatarina O, Sanders L, Albert MS, et al. Measuring cerebral atrophy and white matter hyperintensity burden to predict the rate of cognitive decline in Alzheimer disease. Archives of neurology. 2008 Sep;65(9):1202–8. doi: 10.1001/archneur.65.9.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wiegman AF, Meier IB, Provenzano FA, Schupf N, Manly JJ, Stern Y, et al. Regional white matter hyperintensity volume and cognition predict death in a multi-ethnic, community cohoort of older adults. Journal of the American Geriatrics Society. doi: 10.1111/jgs.12568. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brickman AM, Zarhodne LB, Guzman VA, Narkhede A, Provenzano FA, Schupf N, et al. Reconsidering harbingers of Alzheimer’s disease. Regionally distributed progression of white matter hyperintensities in the community. in preparation. [Google Scholar]
- 76.Brickman AM, Reitz C, Luchsinger JA, Manly JJ, Schupf N, Muraskin J, et al. Long-term blood pressure fluctuation and cerebrovascular disease in an elderly cohort. Archives of neurology. 2010 May;67(5):564–9. doi: 10.1001/archneurol.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alosco ML, Brickman AM, Spitznagel MB, Garcia SL, Narkhede A, Griffith EY, et al. Cerebral Perfusion is Associated With White Matter Hyperintensities in Older Adults With Heart Failure. Congest Heart Fail. 2013 Jul;19(4):E29–34. doi: 10.1111/chf.12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alosco ML, Brickman AM, Spitznagel MB, Griffith EY, Narkhede A, Raz N, et al. Independent and interactive effects of blood pressure and cardiac function on brain volume and white matter hyperintensities in heart failure. J Am Soc Hypertens. 2013 Sep-Oct;7(5):336–43. doi: 10.1016/j.jash.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jefferson AL, Tate DF, Poppas A, Brickman AM, Paul RH, Gunstad J, et al. Lower cardiac output is associated with greater white matter hyperintensities in older adults with cardiovascular disease. Journal of the American Geriatrics Society. 2007 Jul;55(7):1044–8. doi: 10.1111/j.1532-5415.2007.01226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Portet F, Brickman AM, Stern Y, Scarmeas N, Muraskin J, Provenzano FA, et al. Metabolic syndrome and localization of white matter hyperintensities in the elderly population. Alzheimers Dement. 2012 Oct;8(5 Suppl):S88–95 e1. doi: 10.1016/j.jalz.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brickman AM, Zahra A, Muraskin J, Steffener J, Holland CM, Habeck C, et al. Reduction in cerebral blood flow in areas appearing as white matter hyperintensities on magnetic resonance imaging. Psychiatry research. 2009 May 15;172(2):117–20. doi: 10.1016/j.pscychresns.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Holland CM, Smith EE, Csapo I, Gurol ME, Brylka DA, Killiany RJ, et al. Spatial Distribution of White-Matter Hyperintensities in Alzheimer Disease, Cerebral Amyloid Angiopathy, and Healthy Aging. Stroke; a journal of cerebral circulation. 2008 Feb 21; doi: 10.1161/STROKEAHA.107.497438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 84.Schupf N, Tang MX, Fukuyama H, Manly J, Andrews H, Mehta P, et al. Peripheral Abeta subspecies as risk biomarkers of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2008 Sep 16;105(37):14052–7. doi: 10.1073/pnas.0805902105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roher AE, Kuo YM, Esh C, Knebel C, Weiss N, Kalback W, et al. Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer’s disease. Molecular medicine (Cambridge, Mass. 2003 Mar-Apr;9(3–4):112–22. [PMC free article] [PubMed] [Google Scholar]
- 86.Weller RO, Cohen NR, Nicoll JA. Cerebrovascular disease and the pathophysiology of Alzheimer’s disease. Implications for therapy. Panminerva medica. 2004 Dec;46(4):239–51. [PubMed] [Google Scholar]
- 87.Niwa K, Carlson GA, Iadecola C. Exogenous A beta1–40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J Cereb Blood Flow Metab. 2000 Dec;20(12):1659–68. doi: 10.1097/00004647-200012000-00005. [DOI] [PubMed] [Google Scholar]
- 88.Preston SD, Steart PV, Wilkinson A, Nicoll JA, Weller RO. Capillary and arterial cerebral amyloid angiopathy in Alzheimer’s disease: defining the perivascular route for the elimination of amyloid beta from the human brain. Neuropathology and applied neurobiology. 2003 Apr;29(2):106–17. doi: 10.1046/j.1365-2990.2003.00424.x. [DOI] [PubMed] [Google Scholar]
- 89.Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996 Mar 14;380(6570):168–71. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- 90.Maia LF, Vasconcelos C, Seixas S, Magalhaes R, Correia M. Lobar brain hemorrhages and white matter changes: Clinical, radiological and laboratorial profiles. Cerebrovascular diseases (Basel, Switzerland) 2006;22(2–3):155–61. doi: 10.1159/000093245. [DOI] [PubMed] [Google Scholar]
- 91.Chen YW, Gurol ME, Rosand J, Viswanathan A, Rakich SM, Groover TR, et al. Progression of white matter lesions and hemorrhages in cerebral amyloid angiopathy. Neurology. 2006 Jul 11;67(1):83–7. doi: 10.1212/01.wnl.0000223613.57229.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pettersen JA, Sathiyamoorthy G, Gao FQ, Szilagyi G, Nadkarni NK, St George-Hyslop P, et al. Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Archives of neurology. 2008 Jun;65(6):790–5. doi: 10.1001/archneur.65.6.790. [DOI] [PubMed] [Google Scholar]
- 93.Meier IB, Narkhede A, Provenzano FA, Luchsinger JA, Manly JJ, Willey JZ, et al. Lobar microbleeds are associated with white matter hyperintensities and memory in older adults [abstract] Journal of the International Neuropsychological Society. 2011 [Google Scholar]
- 94.Guzman VA, Carmichael OT, Schwarz C, Tosto G, Zimmerman ME, Brickman AM, et al. White matter hyperintensities and amyloid are independently associated with entorhinal cortex volume. Alzheimer’s and Dementia. doi: 10.1016/j.jalz.2012.11.009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van der Flier WM, Barkhof F, Scheltens P. Shifting paradigms in dementia: toward stratification of diagnosis and treatment using MRI. Annals of the New York Academy of Sciences. 2007 Feb;1097:215–24. doi: 10.1196/annals.1379.013. [DOI] [PubMed] [Google Scholar]
- 96.Provenzano FA, Cortes ER, Dashnaw S, Brickman AM. Neuroimaging-guided pathological examination of white matter hyperintensities in aging. Annual Meeting of the International Neuropsychological Society; February, 2011; Boston, MA. 2011. [Google Scholar]